

Abstract
Adaptations to the chronic administration of opioids reduce the utility of these drugs in treating pain and support addiction. Recent genetics-based approaches have implicated the β2 adrenergic receptor (β2-AR) in controlling some of these responses. We do not know, however, whether this receptor can modulate tolerance, dependence or changes in gene expression caused by chronic opioid administration. For our studies we used C57BL/6 mice and β2-AR knockout mice in the FVB background. Morphine dose response relationships were established both prior to and after chronic morphine treatment. In some cases, the selective β2-AR antagonist butoxamine was administered along with or after morphine. Physical dependence was assessed using naloxone-precipitated withdrawal. The expression of calcitonin gene related peptide (CGRP) and substance P (SP) were measured in spinal cord and dorsal root ganglion (DRG) tissues using both real-time PCR and enzyme-linked immunoassay (ELISA). Both the co-administration of butoxamine with morphine and the administration of butoxamine after chronic morphine reversed morphine tolerance. Morphine failed to cause tolerance in β2-AR knockout mice. Physical dependence was reduced under the same circumstances. The chronic administration of butoxamine with morphine reduced or eliminated the normally observed up-regulation of CGRP and SP in spinal cord and DRG tissues. Our results suggest that the β2-AR modulates both opioid tolerance and physical dependence. Activation of β2-ARs appears to be required for some of the key neurochemical changes which characterize chronic opioid administration. Therefore β2-AR antagonists show some promise as agents to enhance chronic opioid analgesic therapy.
Keywords: Drug abuse, morphine, butoxamine, null mutant, gene expression
Introduction
Adaptations to the chronic administration of opioids both limit the clinical utility of this class of drugs, and contribute to opioid addiction. Tolerance, for example, reduces the long term analgesic effects of opioids in patients managed for chronic pain, and impairs our ability to treat pain after injury or surgical procedures [8,20,36]. Physical dependence is another adaptation to chronic opioid administration which constitutes a barrier to the treatment of opioid abuse [20,36]. More recently, opioid-induced hyperalgesia (OIH) has been recognized as a factor which accompanies chronic opioid administration and which complicates other aspects of medical care such as perioperative pain management [6,34,35]. The syndrome of OIH is characterized by the progressive onset of hyperalgesia in the context of chronic opioid management [6].
Genetic investigations have become increasingly common as an approach to identifying key genes regulating tolerance and dependence. Using one dozen or more strains of inbred mice, several reports are available demonstrating a high degree of heritability of both tolerance and dependence [21,22,30]. Recently our laboratory used a panel of inbred mice coupled with an in silico haplotypic mapping algorithm to associate the beta-2 adrenergic receptor (β2-AR) gene with OIH [33]. However, we did not determine whether β2-AR could regulate opioid tolerance and dependence. Other genetic evidence suggests that opioid tolerance, dependence and hyperalgesia may have some common mechanistic underpinnings [32].
Independent of the genetic investigations, there is strong reason to suspect that β2-AR activity regulates opioid tolerance and dependence. For example, using non-selective (β1+ β2) adrenergic receptor blockers, Kihara and Kaneto demonstrated an inhibition of the development of morphine tolerance in mice [24]. A subsequent report suggested that mice need to have an intact adrenal medulla in order for the β-adrenergic receptor mediated opioid tolerance to occur [42]. Left unclear was the receptor subtype responsible for this effect, whether acute β-adrenergic antagonism could restore morphine sensitivity in animals with established tolerance, and whether any of these observations could be confirmed using non-pharmacological means. Complimentary experiments have shown sensitization of adenylate cyclase to β-AR stimulation and alterations in GTP binding protein expression and function indicating that chronic opioid exposure may affect this signaling system at multiple levels [2–5].
Animal behavioral data concerning the ability of non-selective β-AR blockers to reduce naloxone precipitated opioid withdrawal are conflicting [9,18]. Again, the concerted use of selective β2-AR antagonists and non-pharmacological protocols to evaluate the role of β2-AR in opioid withdrawal has not been reported. In addition, non-selective β-AR antagonists have been used to reduce withdrawal symptoms in small human trials with both positive [14,15,19] and negative [38] results.
In light of the confirmed genetic finding of β2-AR regulation of OIH and the suggestive but not conclusive information from the literature also supporting the idea that β2-AR might regulate opioid tolerance and withdrawal, we devised series of experiments using a selective β2-AR antagonist and β2-AR null mutant mice to clearly define the role of these receptors in opioid tolerance and withdrawal. In an attempt to define the mechanism of this modulation we measured dorsal root ganglion and spinal cord substance P and calcitonin gene related peptide levels. Both these peptides have demonstrated roles in supporting tolerance, dependence and OIH [25,43].
Materials and Methods
2.1 Animals
All animal experiments were done after approval of protocols by our Institutional Animal Care and Use Committee and complied with the Guide for the Care and Use of Laboratory Animals available through the National Academy of Sciences.
Inbred mice
Male C57Bl/6 were obtained from Jackson Labs (Bar Harbor, ME) at 7–8 weeks of age. Mice were kept a further 7–10 days from the date of arrival in our animal care facility prior to use to allow for acclimation. Mice were housed 8 per cage under pathogen-free conditions and were provided food and water ad libitum with a 12:12h light:dark cycle.
Transgenic mice
Male Adrb2 null mutant mice congenic in the FVB background as well as matched FVB wild type mice were obtained from Dr. Andrew Patterson at 7–8 weeks of age. Husbandry conditions were otherwise identical to those used for the other inbred strains. The original derivation of this strain is well documented [10].
2.2 Drug administration
Morphine administration
Acute morphine analgesic effects were measured after the subcutaneous injection of 50μl of 0.9% NaCl containing various concentrations of morphine sulfate (Sigma Chemical, St. Louis, MO). The chronic treatment protocol consisted of administering morphine to mice subcutaneously 20mg/kg twice per day on days 1–3 and 40mg/kg twice per day on day 4 at 8:00 and 17:00 similar to our previous protocols for generating tolerance, physical dependence and opioid-induced hyperalgesia [12,32,33]. For tolerance and dependence determinations, mice were assessed 18 hours after the final dose of morphine as we have done previously [12,32,33].
Naloxone administration
For the determination of physical dependence, naloxone (Sigma Chemical) 10mg/kg was injected s.c. in 50μl NaCl 18 hours after the final dose of morphine.
Butoxamine administration
The selective β2-AR antagonist butoxamine was purchased from Sigma Chemical and dissolved in 0.9% NaCl. To assess acute effects, butoxamine 2 mg/kg was injected subcutaneously 25 min before behavioral measurements were made or along with the first dose injection in morphine dose-response experiments. We established previously that this allowed full effect of the drug to be observed [33]. For chronic administration butoxamine was injected concurrently with morphine over the standard 4 day treatment period.
2.3 Expression studies
Tissue harvest for expression studies
Mice were sacrificed at specific time points by CO2 asphyxiation. Spinal cord lumbar segments (L3-S1) were harvested by extrusion and rapid dissection on a pre-chilled surface. Dorsal root ganglia were dissected using low power binocular magnification. Tissue was then quick frozen in liquid nitrogen and stored at −80°C until use.
Substance P (SP) and CGRP expression – mRNA levels
The isolation of RNA and quantification using real time PCR were performed as described previously for spinal cord samples [27,28]. The isolation of total RNA was performed using the RNeasy Mini Kit (Qiagen, Valencia, CA) according to manufacturer’s instructions. The purity and concentration was determined spectrophotometrically. Subsequently, cDNA was synthesized from this total RNA using random hexamer priming and a First Strand cDNA Synthesis Kit (Invitrogen, Carlsbad, CA). Briefly, 1μg of total RNA was mixed with 4 μl of 10x RT buffer, 8 μl of 25 mM MgCl2, 4 μl 0.1M DTT, 1 μl RNasin, 2 μl SSII (50 u/μl), 5 μl hexomers and RNase-free water to 40 μl. Incubation was then carried out at 42°C for 60 minutes followed by heat inactivation at 70°C. Finally 1 μl RNase H was added to each reaction and incubated at 37°C for 20minutes to degrade the RNA.
For real-time quantitative PCR, reactions were conducted in a volume of 4 μl using the Sybr Green I master kit (PE applied Biosystems, Foster City, CA). Briefly, 2 μl of a mixture of 2x sybr green and primers (see Table 1) was loaded with 2 μl diluted cDNA template in each well. Following this, 8μl mineral oil was loaded in each well to prevent loss of solution. Using an ABI prism 7900HT system PCR was carried out using the parameters 52°C, 5min→ 95, 10min then [95°C, 30s→ 60°C, 60s] for 40 cycles. Samples were analyzed in triplicate. Melting curves were performed to document single product formation, and agarose electrophoresis confirmed appropriate product size. 18s RNA was used as an internal control. The 18s primers were purchased from Ambion (Austin, TX).
Table 1. Primer Sequences.
Primer sequences for PPT-A and α-CGRP. Provided in the table are the forward and reverse primers used in real time PCR experiments. Also listed is the accession number used to generate the primer sequence or the literature reference for the source of the primers.
| Gene Name | Forward(5′→3′) | Reverse(5′→3′) | Product Size (bp) | Source Accesion #, Reference |
|---|---|---|---|---|
| PPT-A | tcttttttctcgtttccactcaa | cattaatccaaagaactgctga | 180 | NM009311 |
| α-CGRP | ctccaggcagtgcctttga | caggtggcagtgttgcagg | 199 | [44] |
Quantification was accomplished according to the standard curve method as described by the PCR system manufacturer (PE Applied Biosystems). In order to achieve the same PCR efficiency for each analyte, serial dilution of cDNA was used to construct standard curve for α-CGRP, PPT-A and 18s RNA which was used as internal control. The r2 values for the standard curves of the test genes approached 1.0 suggesting the same amplification efficiency in the PCR reactions under these conditions. The expression level of specific genes were normalized to the level of 18S expression in each sample.
Substance P (SP) and CGRP expression – peptide levels
For experiments in which spinal cord SP or CGRP peptide levels were determined, spinal cord segments were minced in 0.5 ml of 3:1 ethanol/0.7 M HCl and homogenized using a rotary homogenizer. The extracts were heated to 95°C for 10 minutes and centrifuged at 20,000 X g for 30 minutes. The supernatants were then aliquoted, lyophilized and stored at −80° until use. The SP and CGRP content of the samples were assayed in duplicate using enzyme immunoassay (EIA) kits following the manufacturers directions (SP, Assay Designs, Ann Arbor, MI; CGRP, Cayman Chemical, Ann Arbor, MI). Total protein concentrations were also determined in the crude supernatants using the DC Protein Assay kit (Bio-Rad, Hercules, CA).
Immunohistochemistry
The localization of expression of β2-AR, CGRP and SP utilized fluorescence confocal microscopy as we have described in detail previously [29,31]. Mice used in these experiments were first asphyxiated using CO2 and perfused by intracardiac injection of 10ml of 0.9% NaCl. This was followed by perfusion with 20 ml of 4% paraformaldehyde in 0.1 M phosphate-buffered saline. The spinal dorsal root ganglia (DRG) were then dissected under low power magnification and fixed in 4% paraformaldehyde for 4 h at room temperature followed by overnight incubation in 30% sucrose at 4°C. The tissues were then be embedded in OCT medium, and 30 μm sections made on a cryostat with subsequent processing on slides. Blocking took place overnight at 4°C in tris buffered saline (TBS) containing 5% dry milk, followed by exposure to one or more of the following primary antibodies: anti β2-AR/fluorosein conjugate, 1:250, Vector Labs (Burlingame, CA); anti CGRP, 1:500, Sigma Chemical; anti SP, 1:500, Santa Cruz Biotechnology (Santa Cruz, CA). Sections were then be rinsed and transferred to milk-TBS containing either CY3 conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA) or Texas Red conjugated secondary antibodies (Vector Labs) and incubated for another 1 hour. After washing, coverslips were applied. Confocal laser-scanning microscopy was carried out using a Bio-Rad BHT 1024/Nikon Diaphot confocal microscope equipped with a krypton/argon light source. Control experiments included incubation of slices in primary or secondary antibody-free solutions both of which lead to low intensity non-specific staining patterns in preliminary experiments. The specificity of the β2-AR antibody was confirmed by Western blotting.
2.4 Behavioral assays
Morphine dose-response
Cumulative morphine dose-response curves were constructed using the tail flick assay and methods similar to those we described previously [12,32,33]. For these measurements mice were gently restrained within a cone shaped tube made of cotton toweling. Using a tail-flick apparatus (Columbus Instruments, Columbus, OH), tail flick latency was measured with 0.1 second precision. A 10 second cutoff time was used to prevent permanent tissue damage. Two measurements were made per mouse with the light beam focused on two different points 1cm apart on the tail. The lamp intensity was the same for all animals which resulted in baseline tail flick measurements of about 3 seconds for the mouse strains used. For the assessment of tolerance, these dose-response experiments followed 18 hours after the final dose of morphine given as part of the chronic morphine administration protocol. The cumulative doses of morphine used were 0,1,2,4,8,16 and 32 mg/kg. When used, butoxamine 2mg/kg was injected with the first cumulative morphine injection and repeated every 2 hours to maintain effect. Tail flick latency was determined 25 minutes after morphine administration as previous experiments established 25 minutes to be the time at which peak morphine effect was achieved [12]. The parameter %MPE (percent maximal possible effect) was determined according to the following formula:
Physical dependence
Physical dependence on morphine was measured using methods previously reported by our laboratory which were modified from those described by Kest et al. [22,30,32]. Mice were treated with morphine for 4 days according to the standard morphine administration protocol for use in this paradigm. Dependence was assessed 18 hours after the final dose of morphine was given. After naloxone administration, mice were placed in clear plastic cylinders. The number of jumps made during the following 15 minutes were counted.
2.6 Statistical analysis
All data are displayed as the means +/− SEM unless otherwise noted. Dose-response data was fitted using a sigmoidal function with variable slope (Prism 4, GraphPad Software, San Diego, CA). Pairs of dose response curves were compared using the F-test. Simple comparisons of 2 groups involved two-tailed t-testing. Where more than 2 groups were employed, ANOVA analysis was used with the Tukey test applied post-hoc.
Results
The effects of β2-AR blockade on acute morphine antinociception
Though the goals of these studies pertained primarily to assessing the effects of β2-ARs on adaptations to chronic morphine administration, it was necessary that we first assess the acute effects of β2-AR blockade. Figure 1 panel A displays the morphine dose response curves for mice in the tail flick test in which cumulative doses of morphine were given with or without butoxamine pretreatment. The data reveal a very slight left shift of the morphine dose response curve when butoxamine is present (ED 3.97 versus 3.02 mg/kg, p<0.05, Table 2). Preliminary experiments established that butoxamine did not change baseline tail flick latency, and we previously demonstrated that this drug does not change thermal paw flick latency or mechanical nociceptive thresholds in this strain of mice[33]. The dose of butoxamine chosen was near maximal in reversing the thermal and mechanical hyperalgesia present after chronic opioid administration [28].
Figure 1.
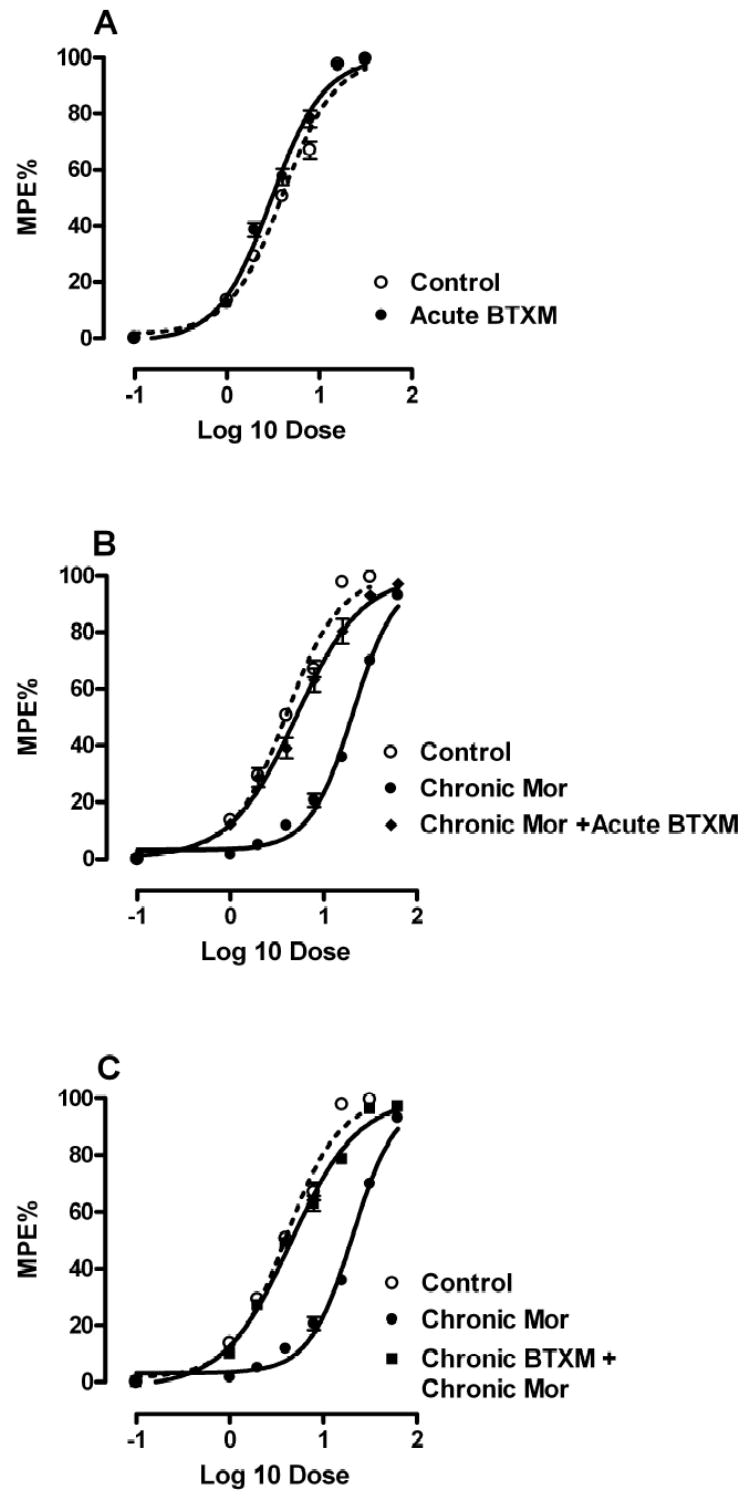
Morphine dose-response relationships in the tail flick assay for mice treated with a β2-AR antagonist. Panel A presents the dose-response relationships of opioid naïve mice given escalating doses of morphine along with saline pretreatment or pretreatment with 2mg/kg butoxamine. Panel B presents the dose-response relationship for morphine naïve mice, mice treated chronically with morphine prior to dose-response assessment, or mice treated chronically with morphine and then pretreated with butoxamine immediately before morphine dose-response assessment. Panel C presents the dose-response relationship for morphine naïve mice, mice treated chronically with morphine prior to dose-response assessment, or mice treated chronically with both morphine and butoxamine. For each group, N=8 mice.
Table 2. Morphine Dose-Response Relationships.
The calculated ED50 values and 95% confidence intervals for the data presented in Figure 1. The F-test was used to compare dose-response relationships.
| Strain | Butoxamine Treatment | Naive ED50 (mg/kg) | Chronic Mor ED50 (mg/kg) |
|---|---|---|---|
| C57 | - | 3.97 (3.51–4.492) | 20.60(19.4–21.0) |
| C57 | Acute | 3.02(.72–3.36) | 5.08(4.29–6.02) |
| C57 | Chronic | 4.47(4.00–5.00) | |
| FVB | - | 7.01(5.96–8.24) | 12.18 (9.90–14.98) |
| FVB | |||
| β2 KO | - | 9.75(8.54–11.13) | 10.61(9.10–12.39) |
The effects of β2-AR blockade on acute morphine antinociception in morphine tolerant mice
We next treated C57BL/6 mice with morphine for 4 consecutive days in a chronic morphine dosing paradigm. Figure 1 panel B shows the large rightward shift in the morphine dose-response relationship after this treatment protocol, and Table 2 provides the calculated ED50 values for these curves (p<0.05). Also provided are data for a separate group of tolerant mice in which butoxamine was administered immediately prior to dose-response testing. These data demonstrated that the acute administration of butoxamine to morphine tolerant mice strongly reverses morphine tolerance.
The effects of β2-AR blockade during chronic opioid administration
Although the acute administration of butoxamine to opioid tolerant mice reversed tolerance, we went on to determine if the co-administration of a β2-AR antagonist along with morphine would prevent tolerance from occurring. Figure 1 panel C and the data in Table 2 demonstrates that in fact tolerance is completely prevented under these conditions. The ED50 valued for morphine dose-response was not significantly shifted if butoxamine was administered with each dose of morphine (p>0.05). The morphine dose-response experiments were done 18 hours after the last combined dose of morphine and butoxamine, a time at which approximately 6 drug half lives had passed making it unlikely that residual drug was affecting the dose-response results.
Morphine tolerance in β2-AR null mutant mice
Because even selective pharmacological agents might exert non-specific effects on tolerance, we tested the ability of morphine to cause analgesic tolerance in β2-AR null mutant mice. The background strain for these null mutants was FVB, a strain showing an intermediate tendency to display opioid tolerance in our paradigm when compared with a panel of 12 other strains [30]. In Figure 2 and Table 2 data are displayed showing that no measurable tolerance to morphine occurs in FVB β2-AR null mutant mice after 4 days of opioid administration (p>0.05), while a significant shift was seen for the FVB wild type mice (p<0.05). Thus the data from these null mutant mice are consistent with the pharmacological observations using butoxamine.
Figure 2.
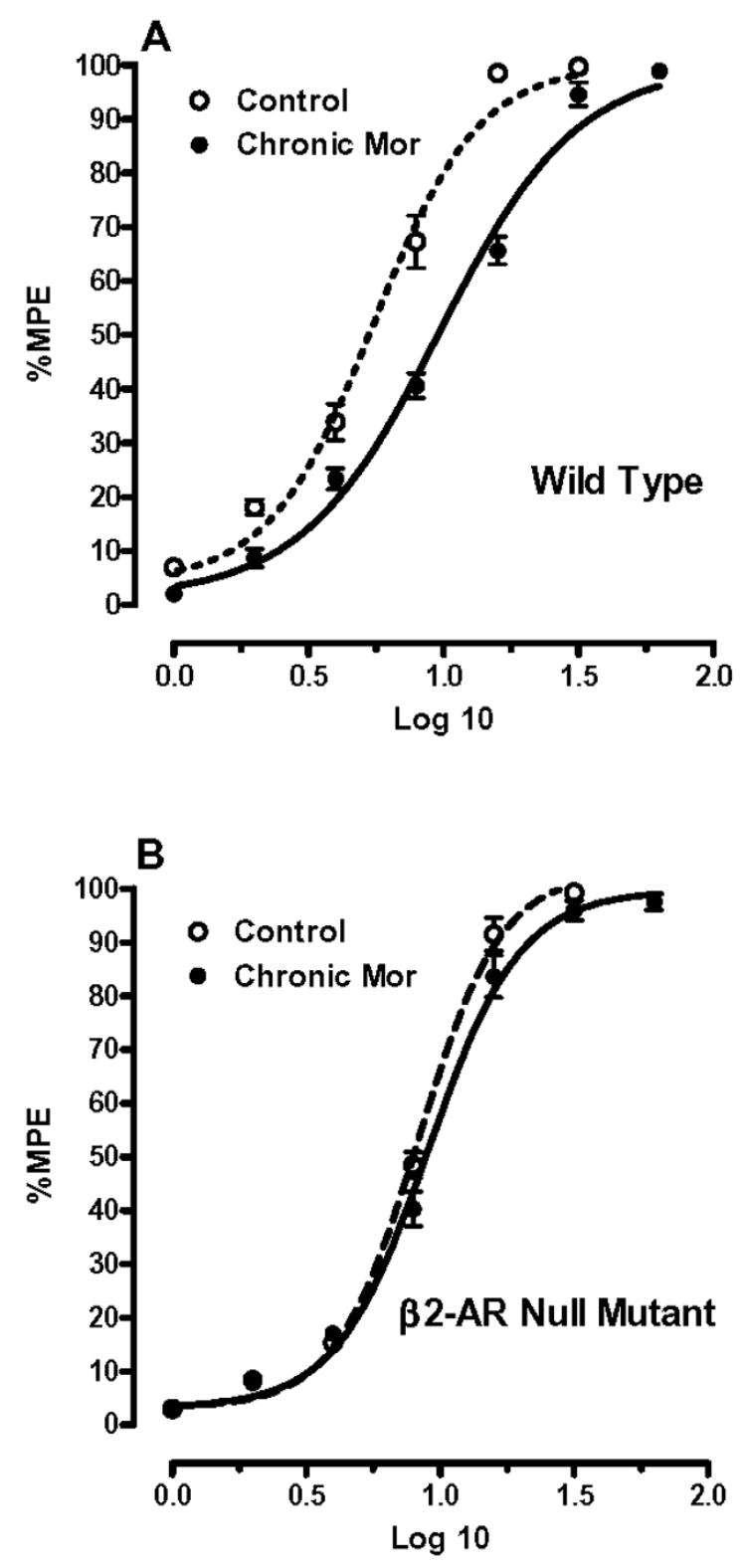
Morphine dose-response relationships for FVB wild type and FVB-β2-AR null mutant mice. Panel A shows the dose-response FVB wild type relationships in opioid naïve mice and mice treated chronically with morphine. Panel B demonstrates these dose-response relationships for FVB-β2-AR null mutant mice. For each group, N=8 mice.
The effects of β2-AR blockade and gene deletion on physical dependence
Our group and others have noted the genetic correlation between strains of mice which develop opioid tolerance with those that develop physical dependence [30]. These data have been interpreted to indicate that common mechanisms underlie the two phenomena. We therefore hypothesized that β2-AR blockade or gene deletion would reduce or eliminate physical dependence on morphine. Among withdrawal behaviors in rodents, jumping is widely considered the most sensitive and reliable index of withdrawal intensity and is the most commonly used [22,30,32]. The data displayed in Figure 3 demonstrate that the injection of the opioid receptor antagonist naloxone to morphine tolerant C57BL/6 mice leads to robust jumping behavior over the ensuing 15 minute interval. Our data also demonstrate that the acute administration of butoxamine to the already opioid dependent mice eliminates most of this jumping behavior. Likewise, the chronic co-administration of butoxamine with morphine reduces the naloxone precipitated jumping to a similar extent as acute butoxamine administration. In panel B of Figure 3 it is demonstrated that the β2-AR null mutant mice display far less physical dependence than their FVB wild type littermates consistent with the pharmacological results.
Figure 3.

Assessment of physical dependence on morphine after chronic administration. In panel A data are provided showing the number of jumps made by the groups of morphine dependent mice after the injection of 10mg/kg naloxone. The leftmost column contains data from C57BL/6 mice treated chronically with morphine prior to naloxone injection. Also presented in Panel A are data from mice treated chronically with morphine and given butoxamine one time prior to naloxone administration, and mice treated chronically with both morphine and butoxamine. In panel B, FVB wild type and FVB-β2-AR null mutant mice were treated chronically with morphine followed by naloxone administration. For each group, N=8 mice. Data are expressed as the means ± S.E.M. **P<0.01; ***P<0.001.
The distribution of expression of CGRP and SP in the DRG
Many investigators have functionally linked the primary afferent CGRP and substance P peptides to nociception, opioid tolerance and physical dependence [7,13,25,26,37]. We hypothesized that β2-AR is co-expressed on primary afferent neurons expressing these peptides. Figure 4 presents immunohistochemical data in which C57BL/6 DRG sections were double labeled with an anti-β2-AR antibody and either an anti-CGRP of anti-SP antibody. While β2-AR staining was widely observed in DRG neurons, CGRP expression was more limited. Cells staining positively for CGRP also stained positively for β2-AR in >90% of the cells examined. A larger percentage of DRG neurons stained positively for SP as compared with CGRP, and SP staining neurons nearly always co-expressed β2-AR. The DRG peptide staining patterns were similar to other recent reports [41,45].
Figure 4.
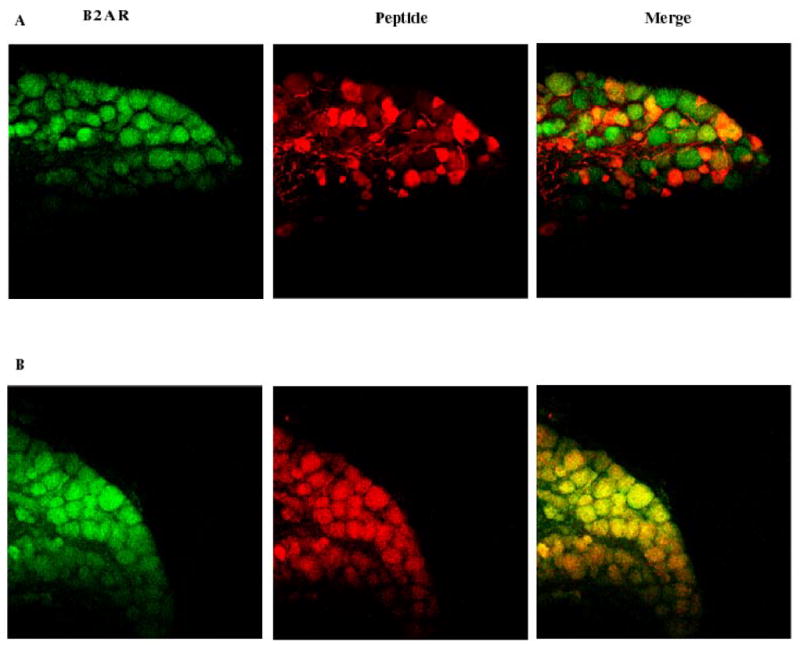
Immunohistochemical localization of CGRP and SP in DRG neurons. Panel A presents the results of DRG staining for β2-AR and CGRP. Panel B presents results from separate sections in which staining was carried out for both β2-AR and SP. In both panels the sections were viewed under 20X magnification.
The expression of neurotransmitters in DRG and spinal cord tissue after chronic opioid administration
We next hypothesized that our data demonstrating that little tolerance and physical develops with the chronic co-administration of morphine and the β2-AR antagonist butoxamine might be related to less up-regulation of expression of these peptides. Figure 5 provides data demonstrating that mRNA coding for αCGRP (the primary neuronal form of CGRP in mice [40]) and pre-protachykinin (PPT, the substance P precursor) are strongly up-regulated by chronic morphine treatment in dissected lumbar DRGs and lumbar spinal cord tissue (Figure 5 panels A and B). There was far less message for these peptide neurotransmitters in the spinal cord tissue as opposed to the DRG primary sensory neuronal tissue. In mice treated with butoxamine along with morphine, little increase in mRNA levels was found.
Figure 5.
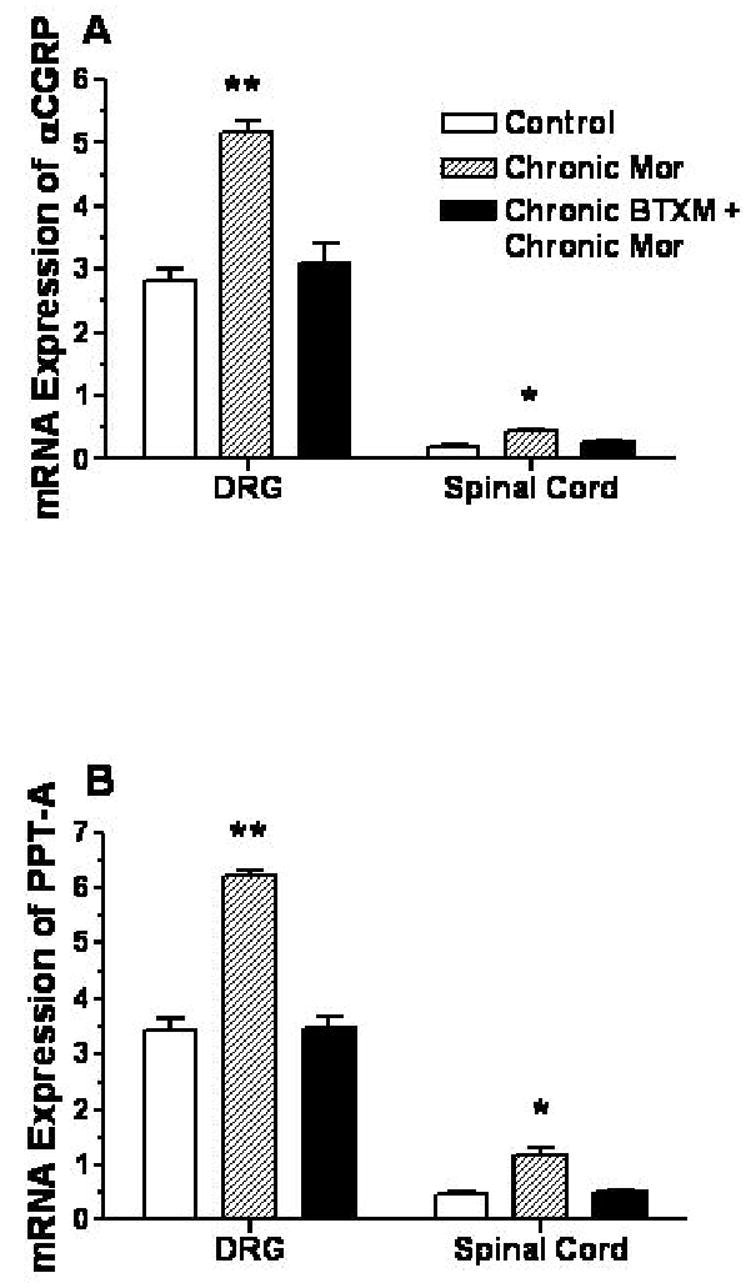
The expression of α-CGRP and SP precursor (preprotachykinin-A, PPT-A) mRNA in dorsal root ganglia (DRGs) and spinal cord tiussue. In these experiments C57BL/6 mice were opioid naïve or treated chronically with morphine or morphine and butoxamine. Panel A provides the data from assays of αCGRP. Panel B provides data of PPT-A. N=6 mice for each group. Data are expressed as the means ± S.E.M. *P<0.05; **P<0.01.
We went on to examine actual peptide levels since changes mRNA abundance do not necessarily accurately reflect changes in the ultimate peptide or protein abundance. We focused on spinal cord tissue in these assays since it is the primary afferent nerve terminals where we expected to find enhanced peptide neurotransmitter levels. Figure 6 presents data showing that while spinal cord CGRP and substance P levels were strongly up-regulated in morphine treated C57BL/6 mice, the co-administration of butoxamine reduced this morphine induced up-regulation.
Figure 6.
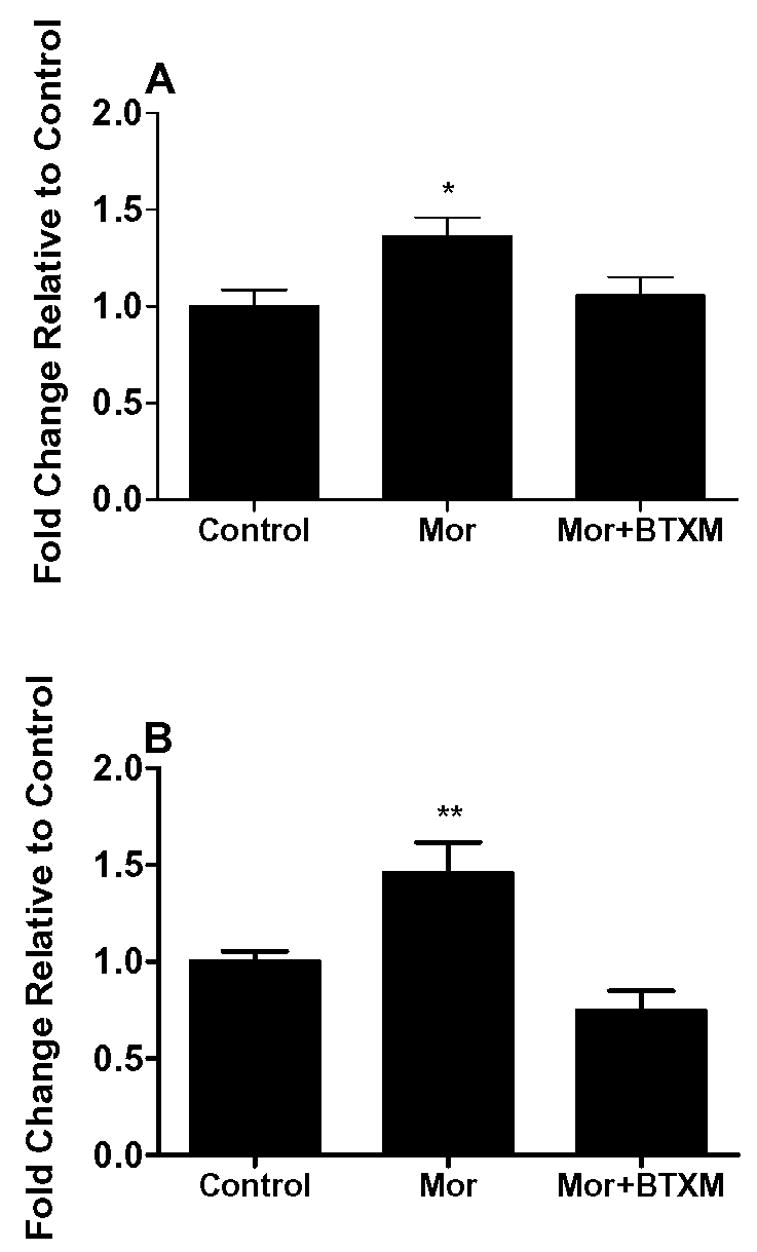
The expression of CGRP and SP peptides in spinal cord tissue. In these experiments C57BL/6 mice were opioid naïve or treated chronically with morphine or morphine and butoxamine. Spinal cord peptide levels were measured with ELISA assays. N=6 mice for each group. Data are expressed as the means ± S.E.M. *P<0.05; **P<0.01.
Discussion
In these investigations we set out to determine if β2-AR support analgesic tolerance and physical dependence, highly clinically relevant adaptations to chronic opioid administration. To test this, we employed both pharmacological reagents and null mutant mice. The results consistently showed that β2-AR activity was required for the full manifestation of both phenomena. In light of the demonstrated involvement of SP and CGRP in tolerance, dependence and OIH, we evaluated the ability of β2-AR blockade or gene deletion to reduce morphine induced up-regulation of these peptides in the DRG and spinal cord. Again, β2-AR activity was seen to be necessary for peptide up-regulation. These results are consistent with our recent genetics based report concerning the role of β2-AR in supporting OIH [33].
Analgesic tolerance is defined in terms of the need to increase drug doses to maintain the targeted effect. Many mechanisms for opioid tolerance have been presented over the past 3 decades, and dozens of molecules have been implicated. One core controversy surrounds the idea that a change in the intrinsic analgesic efficacy of the opioid underlies tolerance versus the hypothesis that enhanced nociceptive signaling explains the need for increased doses of opioid analgesics [25,37]. While these ideas are not necessarily mutually exclusive, we feel that our data fit best with the latter hypothesis. Not only have we shown that β2-AR supports OIH in addition to tolerance and dependence, but several other reports demonstrate how β2-AR activation enhances nociceptive signaling. For example, Khasar et al. demonstrated that both epinephrine and the more selective β-AR agonist isoproterenol caused a mechanical hyperalgesia when injected into the hindpaws of rats. This sensitization was blocked by β-AR antagonists [23]. In this study the authors went on to provide evidence that the mechanical nociceptive sensitization might be related to the sensitization of small diameter dorsal root ganglion neurons. In a later study, Aley et al. reproduced the earlier data and provided further evidence that β2-AR was the likely receptor subtype responsible for the sensitization [1]. In additional studies, cultured dorsal root ganglion neurons were found to respond to β2-AR stimulation with phosphorylation of extracellular signal-related kinase (ERK) [1]. This type of phosphorylation has been linked to enhanced nociception by many laboratories. Thus the up-regulation of the β2-AR signaling system during chronic opioid administration may act through enhanced nociceptive signaling to result in a shift in the morphine dose-response relationship.
The acute blockade of β2-AR both reversed morphine tolerance and strongly reduced the jumping behavior characteristic of opioid withdrawal. Regardless of the up-regulation of the β2-AR signaling system components, changes in central or peripheral gene expression or any of the other events known to take place as the result of chronic morphine exposure, acute β2-AR activation appears to be required for full expression of these traits. Thus while adaptive changes in neurotransmitter levels such as shown in this study or alterations in ion channels, receptors or second messenger system components as demonstrated by others may prime animals to demonstrate tolerance and dependence, β2-AR still retains control over the expression of those neuroadaptive changes. This observation is important as the most likely clinical uses for β2-AR antagonists to combat OIH, tolerance or dependence involve administration of the drugs after patients have been exposed to opioids.
Of these adaptations, it is only for physical dependence where any β2-AR –related clinical trials data exists. Early studies suggested that the intensity of the opioid withdrawal syndrome could be reduced by the administration of non-selective beta adrenergic receptor blockers [14,15,19]. A subsequent study by Resnick, however, failed to replicate these findings [38]. Furthermore, β-AR antagonists have not emerged as popular tools for the treatment of the opioid abstinence syndrome. While it is true that selective β2-AR antagonists are not clinically available and have not been evaluated in the treatment of opioid withdrawal, tolerance or OIH, it must be acknowledged that human adaptations to chronic opioid administration may not be as responsive to acute β2-AR inhibition as is the case with rodents.
However, the data from our studies also address the regulation of the actual adaptive processes themselves. For this part of the studies we chose to follow the nociception related neurotransmitter peptides substance P and CGRP. These peptides are primarily synthesized in afferent sensory neurons, and are released into spinal cord tissue [17,39,46]. As already discussed, enhanced nociception underlies both OIH and tolerance to some extent. Previous reports indicated that the levels of expression and release of these peptides are enhanced with chronic morphine administration, and that this enhanced expression and release of nociceptive peptides supports tolerance and OIH [16,25,37]. Another report provided data demonstrating that the spinal blockade of substance P and CGRP antagonists significantly reduced naloxone induced withdrawal symptoms in morphine dependent rats [44]. In fact we observed as hypothesized that DRG levels of the mRNA species coding for these peptides was sharply increased after 4 days of morphine exposure as were the actual peptide levels in spinal cord tissue. The co-administration of the β2-AR blocker butoxamine nearly completely prevented tolerance from occurring and reduced naloxone-induced jumping behavior to the same degree as when the butoxamine was acutely administered. Moreover, the morphine stimulated up-regulation of substance P and CGRP at both the mRNA precursor and peptide levels was sharply reduced when butoxamine was co-administered with morphine during the chronic treatment phase. Thus β2-AR blockade inhibits the actual neuroadaptive processes underlying morphine tolerance and dependence.
Our studies in the context of the many other reports cited in this discussion clearly indicate that translational studies using β-AR antagonists should be undertaken. Though no selective β2-AR antagonist is available for clinical use at present, several non-selective drugs are in use and might be valuable in these studies. The large and growing problem of opioid abuse in the US and other Western nations along with the burgeoning use of opioids for the treatment of chronic pain places increasing importance on discovering methods which might enhance the long term value of opioid analgesics in the management of pain, or reduce the physical dependence which commonly occurs when these drugs are used for licit or illicit purposes. The recent development of a clinical model for OIH and tolerance involving the 1 month administration of morphine might be of use in these studies [11].
Acknowledgments
The authors would like to thank Dr. Andrew Patterson for supplying the null mutant mice and for his input concerning beta adrenergic receptor pharmacology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Aley KO, Martin A, McMahon T, Mok J, Levine JD, Messing RO. Nociceptor sensitization by extracellular signal-regulated kinases. J Neurosci. 2001;21:6933–6939. doi: 10.1523/JNEUROSCI.21-17-06933.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ammer H, Schulz R. Adenylyl cyclase supersensitivity in opioid-withdrawn NG108-15 hybrid cells requires Gs but is not mediated by the Gsalpha subunit. J Pharmacol Exp Ther. 1998;286:855–862. [PubMed] [Google Scholar]
- 3.Ammer H, Schulz R. Alterations in the expression of G-proteins and regulation of adenylate cyclase in human neuroblastoma SH-SY5Y cells chronically exposed to low-efficacy mu-opioids. Biochem J. 1993;295 (Pt 1):263–271. doi: 10.1042/bj2950263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ammer H, Schulz R. Chronic morphine treatment increases stimulatory beta-2 adrenoceptor signaling in A431 cells stably expressing the mu opioid receptor. J Pharmacol Exp Ther. 1997;280:512–520. [PubMed] [Google Scholar]
- 5.Ammer H, Schulz R. Retinoic acid-induced differentiation of human neuroblastoma SH-SY5Y cells is associated with changes in the abundance of G proteins. J Neurochem. 1994;62:1310–1318. doi: 10.1046/j.1471-4159.1994.62041310.x. [DOI] [PubMed] [Google Scholar]
- 6.Angst MS, Clark JD. Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology. 2006;104:570–587. doi: 10.1097/00000542-200603000-00025. [DOI] [PubMed] [Google Scholar]
- 7.Belanger S, Ma W, Chabot JG, Quirion R. Expression of calcitonin gene-related peptide, substance P and protein kinase C in cultured dorsal root ganglion neurons following chronic exposure to mu, delta and kappa opiates. Neuroscience. 2002;115:441–453. doi: 10.1016/s0306-4522(02)00452-9. [DOI] [PubMed] [Google Scholar]
- 8.Carroll IR, Angst MS, Clark JD. Management of perioperative pain in patients chronically consuming opioids. Reg Anesth Pain Med. 2004;29:576–591. doi: 10.1016/j.rapm.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 9.Chipkin RE, Dewey WL, Harris LS, Lowenthal W. Effect of propranolol on antinociceptive and withdrawal characteristics of morphine. Pharmacol Biochem Behav. 1975;3:843–847. doi: 10.1016/0091-3057(75)90115-x. [DOI] [PubMed] [Google Scholar]
- 10.Chruscinski AJ, Rohrer DK, Schauble E, Desai KH, Bernstein D, Kobilka BK. Targeted disruption of the beta2 adrenergic receptor gene. J Biol Chem. 1999;274:16694–16700. doi: 10.1074/jbc.274.24.16694. [DOI] [PubMed] [Google Scholar]
- 11.Chu LF, Clark DJ, Angst MS. Opioid tolerance and hyperalgesia in chronic pain patients after one month of oral morphine therapy: a preliminary prospective study. J Pain. 2006;7:43–48. doi: 10.1016/j.jpain.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 12.Davies MF, Haimor F, Lighthall G, Clark JD. Dexmedetomidine fails to cause hyperalgesia after cessation of chronic administration. Anesth Analg. 2003;96:195–200. doi: 10.1097/00000539-200301000-00041. table of contents. [DOI] [PubMed] [Google Scholar]
- 13.Gardell LR, Wang R, Burgess SE, Ossipov MH, Vanderah TW, Malan TP, Jr, Lai J, Porreca F. Sustained morphine exposure induces a spinal dynorphin-dependent enhancement of excitatory transmitter release from primary afferent fibers. J Neurosci. 2002;22:6747–6755. doi: 10.1523/JNEUROSCI.22-15-06747.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grosz HJ. Effect of propranolol on active users of heroin. Lancet. 1973;2:612. doi: 10.1016/s0140-6736(73)92433-1. [DOI] [PubMed] [Google Scholar]
- 15.Grosz HJ. Narcotic withdrawal symptoms in heroin users treated with propranolol. Lancet. 1972;2:564–566. doi: 10.1016/s0140-6736(72)91959-9. [DOI] [PubMed] [Google Scholar]
- 16.Gu G, Kondo I, Hua XY, Yaksh TL. Resting and evoked spinal substance P release during chronic intrathecal morphine infusion: parallels with tolerance and dependence. J Pharmacol Exp Ther. 2005;314:1362–1369. doi: 10.1124/jpet.105.087718. [DOI] [PubMed] [Google Scholar]
- 17.Hammond DL, Ruda MA. Developmental alterations in nociceptive threshold, immunoreactive calcitonin gene-related peptide and substance P, and fluoride-resistant acid phosphatase in neonatally capsaicin-treated rats. J Comp Neurol. 1991;312:436–450. doi: 10.1002/cne.903120310. [DOI] [PubMed] [Google Scholar]
- 18.Harris GC, Aston-Jones G. Beta-adrenergic antagonists attenuate somatic and aversive signs of opiate withdrawal. Neuropsychopharmacology. 1993;9:303–311. doi: 10.1038/npp.1993.66. [DOI] [PubMed] [Google Scholar]
- 19.Hollister LE, Prusmack JJ. Propranolol in withdrawal from opiates. Arch Gen Psychiatry. 1974;31:695–698. doi: 10.1001/archpsyc.1974.01760170083013. [DOI] [PubMed] [Google Scholar]
- 20.Jarzyna D. Opioid tolerance: a perioperative nursing challenge. Medsurg Nurs. 2005;14:371–376. quiz 377. [PubMed] [Google Scholar]
- 21.Kest B, Hopkins E, Palmese CA, Adler M, Mogil JS. Genetic variation in morphine analgesic tolerance: a survey of 11 inbred mouse strains. Pharmacol Biochem Behav. 2002;73:821–828. doi: 10.1016/s0091-3057(02)00908-5. [DOI] [PubMed] [Google Scholar]
- 22.Kest B, Palmese CA, Hopkins E, Adler M, Juni A, Mogil JS. Naloxone-precipitated withdrawal jumping in 11 inbred mouse strains: evidence for common genetic mechanisms in acute and chronic morphine physical dependence. Neuroscience. 2002;115:463–469. doi: 10.1016/s0306-4522(02)00458-x. [DOI] [PubMed] [Google Scholar]
- 23.Khasar SG, McCarter G, Levine JD. Epinephrine produces a beta-adrenergic receptor-mediated mechanical hyperalgesia and in vitro sensitization of rat nociceptors. J Neurophysiol. 1999;81:1104–1112. doi: 10.1152/jn.1999.81.3.1104. [DOI] [PubMed] [Google Scholar]
- 24.Kihara T, Kaneto H. Important role of adrenergic function in the development of analgesic tolerance to morphine in mice. Jpn J Pharmacol. 1986;42:419–423. doi: 10.1254/jjp.42.419. [DOI] [PubMed] [Google Scholar]
- 25.King T, Ossipov MH, Vanderah TW, Porreca F, Lai J. Is paradoxical pain induced by sustained opioid exposure an underlying mechanism of opioid antinociceptive tolerance? Neurosignals. 2005;14:194–205. doi: 10.1159/000087658. [DOI] [PubMed] [Google Scholar]
- 26.Li X, Clark JD. Hyperalgesia during opioid abstinence: mediation by glutamate and substance p. Anesth Analg. 2002;95:979–984. doi: 10.1097/00000539-200210000-00035. table of contents. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Lighthall G, Liang DY, Clark JD. Alterations in spinal cord gene expression after hindpaw formalin injection. J Neurosci Res. 2004;78:533–541. doi: 10.1002/jnr.20274. [DOI] [PubMed] [Google Scholar]
- 28.Li X, Shi X, Liang DY, Clark JD. Spinal CK2 regulates nociceptive signaling in models of inflammatory pain. Pain. 2005;115:182–190. doi: 10.1016/j.pain.2005.02.025. [DOI] [PubMed] [Google Scholar]
- 29.Liang D, Li X, Clark JD. Increased expression of Ca2+/calmodulin-dependent protein kinase II alpha during chronic morphine exposure. Neuroscience. 2004;123:769–775. doi: 10.1016/j.neuroscience.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 30.Liang DY, Guo T, Liao G, Kingery WS, Peltz G, Clark JD. Chronic pain and genetic background interact and influence opioid analgesia, tolerance, and physical dependence. Pain. 2006;121:232–240. doi: 10.1016/j.pain.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 31.Liang DY, Li X, Clark JD. Formalin-induced spinal cord calcium/calmodulin-dependent protein kinase II alpha expression is modulated by heme oxygenase in mice. Neurosci Lett. 2004;360:61–64. doi: 10.1016/j.neulet.2004.02.050. [DOI] [PubMed] [Google Scholar]
- 32.Liang DY, Liao G, Lighthall GK, Peltz G, Clark DJ. Genetic variants of the P-glycoprotein gene Abcb1b modulate opioid-induced hyperalgesia, tolerance and dependence. Pharmacogenet Genomics. 2006;16:825–835. doi: 10.1097/01.fpc.0000236321.94271.f8. [DOI] [PubMed] [Google Scholar]
- 33.Liang DY, Liao G, Wang J, Usuka J, Guo Y, Peltz G, Clark JD. A genetic analysis of opioid-induced hyperalgesia in mice. Anesthesiology. 2006;104:1054–1062. doi: 10.1097/00000542-200605000-00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mao J. Opioid-induced abnormal pain sensitivity. Curr Pain Headache Rep. 2006;10:67–70. doi: 10.1007/s11916-006-0011-5. [DOI] [PubMed] [Google Scholar]
- 35.Mao J. Opioid-induced abnormal pain sensitivity: implications in clinical opioid therapy. Pain. 2002;100:213–217. doi: 10.1016/S0304-3959(02)00422-0. [DOI] [PubMed] [Google Scholar]
- 36.Mitra S, Sinatra RS. Perioperative management of acute pain in the opioid-dependent patient. Anesthesiology. 2004;101:212–227. doi: 10.1097/00000542-200407000-00032. [DOI] [PubMed] [Google Scholar]
- 37.Ossipov MH, Lai J, King T, Vanderah TW, Porreca F. Underlying mechanisms of pronociceptive consequences of prolonged morphine exposure. Biopolymers. 2005;80:319–324. doi: 10.1002/bip.20254. [DOI] [PubMed] [Google Scholar]
- 38.Resnick RB, Kestenbaum RS, Schwartz LK, Smith A. Evaluation of propranolol in opiate dependence. Arch Gen Psychiatry. 1976;33:993–997. doi: 10.1001/archpsyc.1976.01770080111011. [DOI] [PubMed] [Google Scholar]
- 39.Rossler W, Gerstberger R, Sann H, Pierau FK. Distribution and binding sites of substance P and calcitonin gene-related peptide and their capsaicin-sensitivity in the spinal cord of rats and chicken: a comparative study. Neuropeptides. 1993;25:241–253. doi: 10.1016/0143-4179(93)90109-n. [DOI] [PubMed] [Google Scholar]
- 40.Schutz B, Mauer D, Salmon AM, Changeux JP, Zimmer A. Analysis of the cellular expression pattern of beta-CGRP in alpha-CGRP-deficient mice. J Comp Neurol. 2004;476:32–43. doi: 10.1002/cne.20211. [DOI] [PubMed] [Google Scholar]
- 41.Shi TJ, Li J, Dahlstrom A, Theodorsson E, Ceccatelli S, Decosterd I, Pedrazzini T, Hokfelt T. Deletion of the neuropeptide Y Y1 receptor affects pain sensitivity, neuropeptide transport and expression, and dorsal root ganglion neuron numbers. Neuroscience. 2006;140:293–304. doi: 10.1016/j.neuroscience.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 42.Takahashi M, Okada T, Kaneto H. Differential roles of the adrenal gland in the suppression of morphine antinociceptive tolerance development by alpha- and beta-adrenergic blockers. Jpn J Pharmacol. 1991;55:555–558. doi: 10.1254/jjp.55.555. [DOI] [PubMed] [Google Scholar]
- 43.Trang T, Quirion R, Jhamandas K. The spinal basis of opioid tolerance and physical dependence: Involvement of calcitonin gene-related peptide, substance P, and arachidonic acid-derived metabolites. Peptides. 2005;26:1346–1355. doi: 10.1016/j.peptides.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 44.Trang T, Sutak M, Quirion R, Jhamandas K. The role of spinal neuropeptides and prostaglandins in opioid physical dependence. Br J Pharmacol. 2002;136:37–48. doi: 10.1038/sj.bjp.0704681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Uehara K, Yamagishi S, Otsuki S, Chin S, Yagihashi S. Effects of polyol pathway hyperactivity on protein kinase C activity, nociceptive peptide expression, and neuronal structure in dorsal root ganglia in diabetic mice. Diabetes. 2004;53:3239–3247. doi: 10.2337/diabetes.53.12.3239. [DOI] [PubMed] [Google Scholar]
- 46.Zhang X, Nicholas AP, Hokfelt T. Ultrastructural studies on peptides in the dorsal horn of the spinal cord--I. Co-existence of galanin with other peptides in primary afferents in normal rats. Neuroscience. 1993;57:365–384. doi: 10.1016/0306-4522(93)90069-r. [DOI] [PubMed] [Google Scholar]