

Abstract
CpG island hypermethylation occurs in most cases of cancer, typically resulting in the transcriptional silencing of critical cancer genes. Procainamide has been shown to inhibit DNA methyltransferase activity and reactivate silenced gene expression in cancer cells by reversing CpG island hypermethylation. We report here that procainamide specifically inhibits the hemimethylase activity of DNA methyltransferase 1 (DNMT1), the mammalian enzyme thought to be responsible for maintaining DNA methylation patterns during replication. At micromolar concentrations, procainamide was found to be a partial competitive inhibitor of DNMT1, reducing the affinity of the enzyme for its two substrates, hemimethylated DNA and S-adenosyl-l-methionine. By doing so, procainamide significantly decreased the processivity of DNMT1 on hemimethylated DNA. Procainamide was not a potent inhibitor of the de novo methyltransferases DNMT3a and DNMT3b2. As further evidence of the specificity of procainamide for DNMT1, procainamide failed to lower genomic 5-methyl-2′-deoxycytidine levels in HCT116 colorectal cancer cells when DNMT1 was genetically deleted but significantly reduced genomic 5-methyl-2′-deoxycyti-dine content in parental HCT116 cells and in HCT116 cells where DNMT3b was genetically deleted. Because many reports have strongly linked DNMT1 with epigenetic alterations in carcinogenesis, procainamide may be a useful drug in the prevention of cancer.
Epigenetic alterations are increasingly recognized as valuable targets for the development of cancer therapies. They not only occur early in carcinogenesis but also are found in virtually all cases of cancer (1-4). Importantly, epigenetic alterations do not involve changes in the DNA sequence and thus are potentially reversible. Of the epigenetic changes seen in cancer, the most extensively studied is the increase of CpG dinucleotide methylation at CpG islands in the proximal promoter regions of genes. This change in DNA methylation characteristically results in the transcriptional silencing of important cancer genes such as tumor suppressors and caretaker genes (5).
5-Azacytidine and its deoxy derivative 5-aza-2′-deoxycytidine were synthesized over 40 years ago as potential chemotherapeutic agents (6). Further investigation revealed that 5-azacytidine can induce DNA demethylation (7), eventually leading to its successful development as a treatment for myelodysplastic syndrome (8). More recently, 2-pyrimidone-1-β-d-riboside (Zebularine), an orally administrable drug that is stable in aqueous solutions (9), has been found to effectively induce the reactivation of hypermethylated genes (10, 11). However, nucleoside analogs carry considerable concerns about toxicity. To inhibit DNA methyltransferases (DNMTs),2 nucleoside analogs must be incorporated into DNA, where DNMT attack of the abnormal base results in the formation of a covalent complex that cannot be resolved (reviewed in Refs. 12, 13). Incorporation of nucleoside analogs into DNA results in a permanent alteration of the genome, often leading to mutagenesis in surviving daughter cells (14-16), thus raising the possibility of future cancer development. Moreover, the therapeutic index of nucleoside analogs is limited by life-threatening toxicities such as neutropenia and thrombocytopenia (17).
These concerns have led to consideration of non-nucleoside inhibitors of DNA methyltransferases. Of particular interest is procainamide, a non-nucleoside drug approved by the U. S. Food and Drug Administration for the treatment of cardiac arrhythmias that was originally shown by Cornacchia et al. (18) to reduce the genomic 5-methylcy-tosine content of Jurkat cells. Further investigation revealed that procainamide inhibited DNA methyltransferase activity (19) and reactivated genes silenced by promoter CpG island hypermethylation (20, 21).
Unlike nucleoside analogs, the target of procainamide and its mechanism of action are unclear. Procainamide and the related compound procaine bind to CG-rich sequences (22-24), a property that is purported to mediate their abilities to demethylate DNA. We set out to investigate the specific target and mechanism of procainamide inhibition of methyltransferase activity. Here, we report that procainamide specifically inhibits the maintenance methyltransferase activity of DNMT1, mainly by reducing the affinity of the enzyme for both DNA and S-adenosyl-l-methionine (AdoMet). Inhibition of DNMT1 maintenance methyltransferase activity was associated with a profound decrease in the processivity of the enzyme.
MATERIALS AND METHODS
Cell Culture, Genomic 5-Methyl-2′-deoxycytidine Quantification by Mass Spectrometry, and Sodium Bisulfite Genomic DNA Sequencing
HCT116 colon cancer cells were obtained from ATCC (Manassas, VA), and HCT116 DNMT1−/− and HCT116 DNMT3b−/− colon cancer cells were a kind gift from Dr. Stephen Baylin (Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University, Baltimore, MD). Cells were grown in McCoy’s 5A medium (Invitrogen) supplemented with 10% fetal bovine serum. Cells were treated with PBS, pH 7.4, or 0.5 mm procainamide (Sigma) for 96 h, and genomic DNA was isolated using the DNeasy Tissue kit (Qiagen, Valencia, CA). Quantification of genomic 5-methyl-2′-deoxycytidine content (m5dC) was performed by high performance liquid chromatography/mass spectrometry as described previously (25). Genomic DNA from cells was also subjected to bisulfite genomic sequencing analysis. 1 μg of genomic DNA was modified with sodium bisulfite using the CpGenome DNA modification kit (Serologicals Co., Norcross, GA). PCR primers (5′-TAGTTTTT-TAGGTTTTAGTTGTTTAGGAG-3′) and (5′-ACTCCTCCAACTC-CTACTCCTT-3′) were used to amplify a 299-bp CpG island surrounding the TIMP3 translation start site (PCR protocol: 1 × 95 ºC for 2 min; 30 × 95 ºC for 30 s, 54 ºC for 30 s, 72 ºC for 30 s; 1 × 72 ºC for 10 min). The PCR product was subcloned into the pCR®2.1-TOPO® vector (Invitrogen) for dideoxy sequencing.
Methylation-sensitive Southern Blotting
MCF-7 breast cancer cells obtained from ATCC were grown in minimum essential medium (Invitrogen) supplemented with 10% fetal bovine serum and treated with PBS, pH 7.4, 0.5 μm 5-azacytidine (Sigma) or 0.5 mm procainamide for 5 days. Genomic DNA was isolated using the DNeasy Tissue kit, digested with HpaII (New England Biolabs, Beverly, MA), and Southern blotted using standard techniques. Probes used for hybridization were derived from alphoid satellite sequences from chromosome 1 (26) and chromosome 18 (27).
Production of Recombinant Human DNMT1, DNMT3a, and DNMT3b2 in Sf9 Insect Cells and Purification by Ni2+ Immobilized Metal Affinity Chromatography
To produce recombinant DNMT1, full-length DNMT1 cDNA was amplified by RT-PCR from human brain poly(A)+ RNA (BD Clontech, Mountain View, CA). The product was subcloned into pFB6H, a modified pFastBac-1 baculovirus expression vector (Invitrogen) that contains a coding sequence for a His6 tag. This pFB6H-DNMT1 construct was used to transform DH10Bac™ Escherichia coli cells (Invitrogen) to generate an expression bacmid via site-specific transposition. The DNMT1 expression bacmid was transfected into Sf9 insect cells to produce recombinant DNMT1 baculovirus particles, which were subsequently used to infect additional Sf9 cells (1 multiplicity of infection, 48 h) for protein production. Recombinant His6-DNMT1 was recovered by immobilized metal affinity chromatography. After infected Sf9 cells were lysed in buffer W (50 mm Na2HPO4, pH 7.6, 500 mm NaCl, 1% Igepal CA-630, 10% sucrose, and 1× Complete Protease Inhibitor (Roche Applied Science)) with 10 mm imidazole by two freeze-thaw cycles, His6-DNMT1 was treated with 10 μg/ml RNase A (Qiagen) and bound to nickel-nitrilotriacetic acid-agarose (Qiagen) at 4 ºC. The beads were washed with buffer W with 20 mm imidazole to remove contaminating proteins. To remove excess salt, the beads were then washed with 50 mm Na2HPO4, pH 7.6, 10 mm NaCl, 10% sucrose, and 1× Complete Protease Inhibitor. His6-DNMT1 was eluted by adding 50 mm Na2HPO4, pH 7.6, 250 mm imidazole, 10 mm NaCl, 10% sucrose, and 1× Complete Protease Inhibitor. Spin dialysis was used to concentrate the protein and exchange the buffer to 50 mm Na2HPO4, pH 7.6, 10 mm NaCl, 1 mm EDTA, 1 mm dithiothreitol, 20% glycerol, and 1× Complete Protease Inhibitor. Protein concentration was determined using the BCA assay (Pierce, Rockford, IL). Recombinant His6-DNMT1 was stored at −80 ºC until further use.
His6-DNMT3a and His6-DNMT3b2 were expressed in Sf9 cells and purified as described above with the following modifications. Full-length DNMT3a cDNA was amplified from human testis poly(A)+ RNA (BD Clontech) by RT-PCR and subcloned into pFB6H to create pFB6H-DNMT3a. Full-length DNMT3b2 cDNA was amplified from human testis poly(A)+ RNA (BD Clontech) by RT-PCR and subcloned into pFB6H to create pFB6H-DNMT3b2. The nuclei of Sf9 cells expressing His6-DNMT3b2 were isolated by hypotonic lysis (10 mm Tris-HCl, pH 7.4, 10 mm NaCl, 3 mm MgCl2, 0.5% Igepal CA-630) before application to the nickel-nitrilotriacetic acid column to minimize binding of contaminating proteins. For His6-DNMT3a, the initial wash was performed with buffer W with 40 mm imidazole, and for His6-DNMT3b2 the initial wash was performed with buffer W with 50 mm imidazole. Analysis of recombinant enzymes by SDS-PAGE followed by Coomassie Blue R-250 staining showed that the preparations were >95% pure.
DNA Methyltransferase Activity Assay
DNA methyltransferase activity assays were performed by combining His6-DNMT (100 nm His6-DNMT1, 200 nm His6-DNMT3a, 200 nm His6-DNMT3b2) with 5′-biotinylated 45-bp unmethylated or hemimethylated oligonucleotide substrates containing various concentrations of CG (Top Strand 5′-biotin-GACGTCGTTCGTACGCTCGTTCGACTCGT-GCGACGGATCGGATTG-3′, Bottom Strand 5′-CAATCCGATC-CGTCGCACGAGTCGAACGAGCGTACGAACGACGTC-3′) (Integrated DNA Technologies, Skokie, IL) and S-adenosyl-l-[methyl-3H]methionine (3H-AdoMet) (Amersham Biosciences) in reaction buffer (20 mm Tris-HCl, pH 7.4, 1 mm EDTA, 5% glycerol, and 1 mm dithiothreitol) with or without procainamide. After incubation at 37 ºC, reactions were stopped at various time points by adding one volume of 10 mm cold S-adenosyl-l-methionine (Sigma). The reactions were bound to a SAM2® 96 Biotin Capture plate (Promega, Madison, WI). The plate was washed five times with PBS + 2 m NaCl and two times with dH2O to remove His6-DNMT and unreacted 3H-AdoMet. After drying the plate, 80 μl of Microscint-PS scintillation fluid (PerkinElmer) was added to each well, and tritium incorporation was quantified using the TopCount NXT liquid scintillation counter (PerkinElmer). All reactions were performed in sextuplicate. Data obtained were analyzed using the Enzyme Kinetics module of SigmaPlot (Systat Software, Point Richmond, CA).
Methylation Processivity Analysis of GSTP1 Promoter
Unmethylated GSTP1 promoter was generated by amplifying a 716-bp fragment from pGL3-GSTP-1, a GSTP1 promoter/luciferase reporter construct (28), with PCR primers (5′-GGCCGCTCTAGAACTAGTGGATC-3′) and (5′-CGAAGTACTCAGCGTAAGTGATGTC-3′). Hemimethylated GSTP1 promoter was generated using a protocol modified after Hermann et al. (29). Briefly, a 716-bp fragment was amplified with PCR primers (5′-phosphate-GGCCGCTCTAGAACTAGTGGATC-3′) and (5′-CGAAGTACTCAGCGTAAGTGATGTC-3′) using pGL3-GSTP-1 as template (PCR protocol: 1 × 95 ºC for 2 min; 30 × 95 ºC for 30 s, 58 ºC for 30 s, 72 ºC for 1 min; 1 × 72 ºC for 10 min). SssI methylase (New England Biolabs) was used to completely methylate the PCR product. 1 unit λ-exonuclease/μg PCR product was used to selectively digest the top strand for 60 min at 37 ºC. The primer (5′-GGCCGCTCTA-GAACTAGTGGATC-3′) was annealed to the methylated bottom strand and extended by incubating with PfuUltra polymerase (Strat-agene, La Jolla, CA) and dNTPs (Protocol: 1 × 95 ºC for 5 min; 1 × 58 ºC for 30 s; 1 × 72 ºC for 10 min). The hemimethylated GSTP1 promoter was purified by agarose gel electrophoresis.
100 nm DNMT1 was incubated with hemimethylated GSTP1 promoter fragment containing 1 μm CG and 1 μm S-adenosyl-l-methio-nine with or without procainamide at 37 ºC for 30 min. 1 μg of salmon sperm DNA was added as carrier, and the reaction was stopped by adding NaOH to a final concentration of 200 mm. For processivity analysis of unmethylated substrate, 200 nm DNMT1 was incubated with unmethylated GSTP1 promoter fragment containing 1 μm CG and 1 μm S-adenosyl-l-methionine with or without procainamide at 37 ºC for 1 h. The mixture was subjected to sodium bisulfite modification using the CpGenome DNA modification kit. PCR primers (5′-GTTGGG-GATTTGGGAAAGAGGGAAAGG-3′) and (5′-ATCTATAAAAA-CAATTATTCCAAAAACCAAAAC-3′) targeting a 444-bp region of the fragment were used to amplify GSTP1 CpG island sequences (PCR protocol: 1 × 95 ºC for 2 min; 303 95 ºC for 30 s, 58 ºC for 30 s, 72 ºC for 1 min; 1 × 72 ºC for 10 min). The PCR product was subcloned into the pCR®2.1-TOPO® vector for dideoxy sequencing. The processivity index of each clone was defined by dividing the difference between the total number of methylated CpGs and the number of gaps by the total number of methylated CpGs (Scheme 1). Gaps could be of any length and were defined as unmethylated CpGs that lie between methylated CpGs. Unmethylated CpGs that occurred at the ends of clones were counted as gaps. However, if unmethylated CpGs occurred at both ends of a clone, then only one gap was counted to avoid a negative processivity index. The efficiency of bisulfite conversion was calculated as the number of non-CpG cytosines that were converted to thymines divided by the total number of non-CpG cytosines. Clones with a bisulfite conversion efficiency of <95% were discarded and not scored. Completely unmethylated clones were also discarded and not scored. Processivity indices ranged from 0 to 1. A processivity index of 1 indicates that all CpGs of a clone are methylated. A processivity index of 0 indicates that none of the methylated CpGs of a clone were contiguous.
SCHEME 1.

Calculation of the Processivity Index for individual clones.
Fluorescence Anisotropy Binding Studies
Hairpin oligonucleotides with the sequence 5′-fluorescein-ATCGTCGTACGTTTTCGTAC-GACGAT-3′ with no methylated CpGs (unmethylated hairpin), 3 methylated CpGs at the 3′-end (hemimethylated hairpin), or 6 methylated CpGs (fully methylated hairpin) were incubated in reaction buffer with DNMT1 in the presence or absence of procainamide or excess NaCl for 1 h at 4 ºC. Measurements were taken in triplicate using a Beckman Coulter DTX 880 Multimode Detector in fluorescence polarization mode. Fluorescence anisotropies (r) were calculated as shown in Equation 1,
| (Eq. 1) |
where I∥ represents the parallel fluorescence intensity, I⊥ represents the perpendicular fluorescence intensity, and 0.62 is the G-factor that compensates for differences in sensitivities between the two planes. r was converted to fraction of hairpin oligonucleotides bound to DNMT1 (Fbound) as shown in Equation 2.
| (Eq. 2) |
where rmin is the anisotropy of free hairpin oligonucleotides and rmax is the anisotropy of the DNMT1-hairpin complex at saturation. Fbound was plotted against DNMT1 concentration and fitted using a one-site binding model according to the method of Michel et al. (30) to estimate a dissociation constant (Kd).
Statistical Analysis
Tests for statistical significance were performed using the Mann-Whitney U test.
RESULTS
Procainamide Treatment Decreased Genomic 5-Methyl-2′-deoxycy-tidine Content, CpG Methylation at Alphoid Satellite Sequences, and Gene-specific Methylation at a Promoter CpG Island
Procainamide has previously been shown to reduce genomic 5-methylcytosine content (18, 24), relieve repression of genes silenced by promoter CpG island hypermethylation (20, 21), and inhibit DNA methyltransferase activity (19). To ascertain the intracellular target of procainamide-mediated inhibition of DNA methyltransferase activity, HCT116 colon cancer cells and their isogenic derivatives lacking DNMT1 (31) or DNMT3b (32) were treated with PBS or 0.5 mm procainamide for 96 h. HPLC/mass spectrometry analysis of genomic DNA revealed that procainamide treatment decreased m5dC content in HCT116 cells by 15.5%. Procainamide treatment of HCT116 DNMT3b−/− cells decreased m5dC content by 11.5%, but treatment of HCT116 DNMT1−/− cells did not decrease m5dC content (Fig. 1A). These results suggested that DNMT1 was the intracellular target of procainamide. We then used bisulfite genomic sequencing to investigate the methylation status of the TIMP3 CpG island. When compared with PBS-treated HCT116 cells, we observed loss of CpG methylation in the procainamide-treated HCT116 cells (Fig. 1B). The net fraction of CpGs methylated decreased from 0.963 ± 0.007 (95% confidence interval) to 0.821 ± 0.0165 (95% confidence interval), which is similar to the amount of m5dC loss seen using HPLC/mass spectrometry. CpC, CpT, and CpA methylation levels were extremely low (95% confidence interval: 0.00581 ± 0.0025) and did not change significantly upon procainamide treatment. Methylation-sensitive Southern blotting of MCF-7 breast cancer cells showed that procainamide treatment reduced CpG methylation at alphoid satellite sequences from chromosomes 1 and 18 (Fig. 1C), but not to the same degree as 5-azacytidine treatment. Taken together, these results showed that in cells that express DNMT1, procainamide treatment can lead to global loss of CpG methylation with effects on both centromeric repeats as well as single copy genes.
FIGURE 1. Loss of global methylation, gene-specific methylation, and centromeric repeat methylation in cells treated with procainamide.
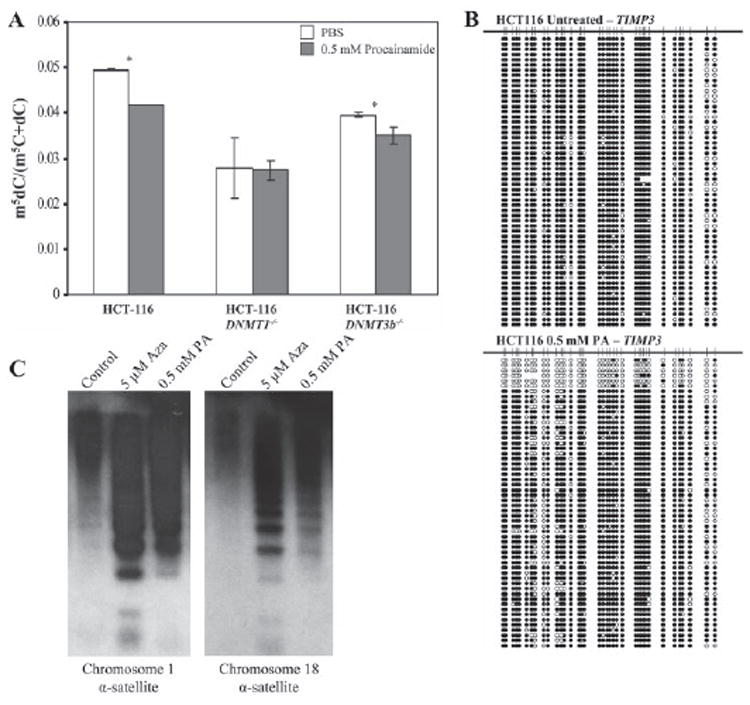
A, HCT116, HCT116 DNMT1−/−, and HCT116 DNMT3b−/− cells were treated with PBS, pH 7.4 (open bars) or 0.5 mm procainamide (shaded bars) for 96 h. Genomic DNA was isolated, and 5-methyl-2′-deoxycytidine content was quantified by high performance liquid chromatography/mass spectrometry. Error bars are ± S.D. of triplicate measurements. *, p < 0.05. B, genomic DNA isolated from HCT116 cells treated with PBS, pH 7.4, or 0.5 mm procainamide for 96 h were subjected to bisulfite genomic sequencing at the TIMP3 CpG island. C, MCF-7 cells were treated with PBS, pH 7.4, 0.5 μm 5-azacytidine, or 0.5 mm procainamide for 5 days. Methylation-sensitive Southern blotting was performed by digesting genomic DNA with HpaII followed by hybridization with probes derived from alphoid satellite sequences from chromosomes 1 and 18.
Procainamide Preferentially Inhibits DNMT1 on a Hemimethylated Oligonucleotide Substrate
DNMT1 has been reported to prefer hemimethylated over unmethylated DNA up to 40-fold in vitro (33-36). This observation, along with the discovery of a proliferating cell nuclear antigen binding domain (37) and a replication foci targeting domain (38) in DNMT1, lends credence to its proposed role in vivo as the maintenance methyltransferase responsible for copying methylation patterns during DNA replication. We investigated the ability of procainamide to inhibit DNMT1 on a hemimethylated oligonucleotide substrate and an unmethylated oligonucleotide substrate (Fig. 2). Analysis of the data showed that procainamide inhibits DNMT1 on a hemimethylated substrate with a Ki = 7.2 ± 0.6 μm. Modeling of Lineweaver-Burk plots revealed that the mode of inhibition is consistent with partial competitive inhibition (39) with an α factor of 10.6 and a β factor of 0.97. The α factor represents the change in Km when inhibitor is bound, whereas the β factor represents the change in turnover rate when the enzyme-substrate-inhibitor complex (ESI) catalyzes product formation. Therefore, procainamide increased the Km for DNA by ~10-fold without significantly decreasing the turnover rate of ESI when compared with the turnover rate of the enzyme-substrate complex (ES). Because both ES and ESI turn over at approximately the same rate, even an infinite amount of inhibitor cannot drive the velocity to zero. We found that procainamide also inhibited DNMT1 with respect to AdoMet in a partial competitive manner with an α factor of 4.5 and a β factor of 1.0.
FIGURE 2. Lineweaver-Burk plots for procainamide inhibition of DNMT1.
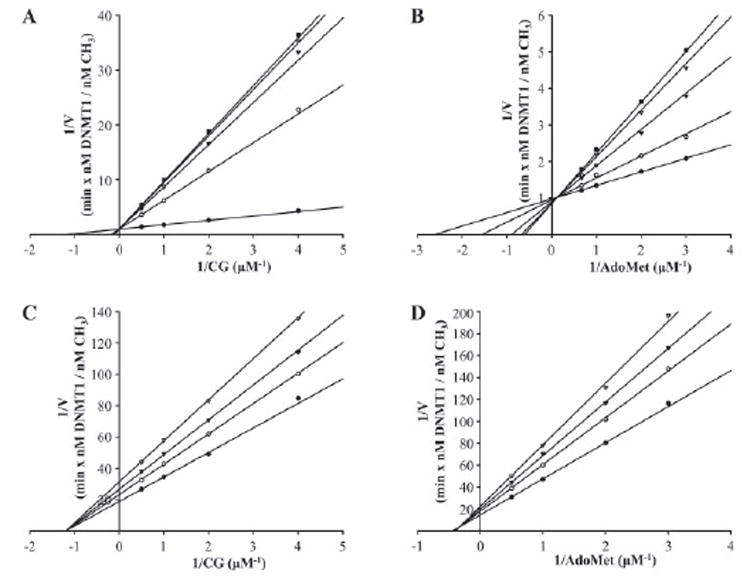
DNMT1 concentration was fixed at 100 nm for hemimethylated DNA and 200 nm for unmethylated DNA. A, procainamide inhibition of DNMT1 on hemimethylated DNA with AdoMet concentration fixed at 1 μm. Procainamide concentration was fixed at 0 μm (filled circles), 100 μm (open circles), 500 μm (filled triangles), 2 mm(open triangles), and 8 mm (filled squares). B, procainamide inhibition of DNMT1 on hemimethylated DNA with CG concentration fixed at 1 μm. Procainamide concentration was fixed at 0 μm (filled circles), 100 μm (open circles), 500 μm (filled triangles), 2 mm (open triangles), and 8 mm (filled squares). C, procainamide inhibition of DNMT1 on unmethylated DNA with AdoMet concentration fixed at 1 μm. Procainamide concentration was fixed at 0 mm (filled circles), 2 mm (open circles), 4 mm (filled triangles), and 8 mm (open triangles). D, procainamide inhibition of DNMT1 on unmethylated DNA with CG concentration fixed at 1 μm. Procainamide concentration was fixed at 0 mm (filled circles), 2 mm (open circles), 4 mm (filled triangles), and 8 mm (open triangles).
In contrast, procainamide inhibited DNMT1 on an unmethylated oligonucleotide substrate with a Ki = 4600 ± 500 μm. These data suggest that procainamide-mediated inhibition of DNMT1 is specific for its maintenance methyltransferase activity.
Procainamide Is Ineffective at Inhibiting DNMT3a and DNMT3b2
We examined the ability of procainamide to inhibit the activity of the two de novo methyltransferases, DNMT3a and DNMT3b2, on an unmethylated oligonucleotide substrate. Compared with DNMT1 on a hemimethylated substrate, DNMT3a and DNMT3b2 were not readily inhibited by procainamide. Lineweaver-Burk analysis showed that procainamide inhibited DNMT3a with a Ki = 1400 ± 200 μm and DNMT3b2 with a Ki = 10000 ± 3000 μm. Interestingly, these Ki values were in the same range as the Ki for DNMT1 on an unmethylated substrate. The calculated Ki values for the enzymes are summarized in TABLE ONE. These data reinforced the conclusion that procainamide specifically inhibits the maintenance methyltransferase activity of DNMT1.
TABLE ONE.
Ki values for recombinant DNA methyltransferases with procainamide
| Enzyme | Substrate | Ki |
|---|---|---|
| μm | ||
| DNMT1 | Hemimethylated oligo | 7.2 ± 0.6 |
| DNMT1 | Unmethylated oligo | 4600 ± 500 |
| DNMT3a | Unmethylated oligo | 1400 ± 200 |
| DNMT3b2 | Unmethylated oligo | 10000 ± 3000 |
Procainamide Inhibits the Processivity of DNMT1 on a Hemimethyl-ated Substrate
DNMT1 has recently been found to act processively on a hemimethylated substrate but not on an unmethylated substrate (29, 40). We hypothesized that the ability of procainamide to inhibit DNMT1 on a hemimethylated substrate is related to the processivity of the enzyme. To quantitatively analyze the processivity of DNMT1, we devised a processivity index that assesses the fraction of total activity that is due to methylation of contiguous CpGs. A 716-bp hemimethylated or unmethylated fragment corresponding to a segment of the GSTP1 promoter containing 62 CpGs was used as a substrate. As expected, DNMT1 acted processively on a hemimethylated substrate with a mean processivity index of 0.95. The addition of 1 mm procainamide to the reaction lowered the mean processivity index to 0.71 (Fig. 3). Inspection of the individual clones sequenced from procainamide-treated samples revealed two distinct populations: one with high activity and one with low activity. The first appeared very similar to the untreated sample containing clones with high fractions of CpGs methylated and high processivity indices. The second contained clones with markedly lower fractions of CpGs methylated; the few methylated CpGs were infrequently contiguous, resulting in low processivity indices. To test whether the reduced affinity of the enzyme for AdoMet in the presence of procainamide can result in these two populations, we analyzed the processivity indices of clones generated when the AdoMet concentration was reduced to 40 nm. We found that a similar distribution of clones was reproduced under these conditions of limiting AdoMet (Fig. 4).
FIGURE 3. Processivity analysis of DNMT1 activity on hemimethylated GSTP1 promoter.
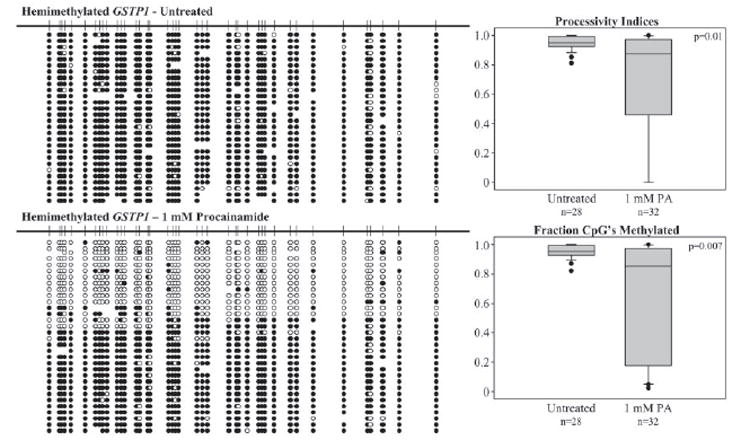
100 nm DNMT1 was incubated with 1 μm AdoMet and hemimethylated GSTP1 promoter containing 1 μm CG in the absence or presence of 1 mm procainamide. The DNA substrate was treated with sodium bisulfite, and individual clones were amplified by PCR, subcloned, and sequenced. Methylated cytosines are represented by filled circles, and unmethylated cytosines are represented by open circles. The absence of a circle at a particular position indicates that the identity of the base could not be determined. Processivity indices and fractions of CpGs methylated were calculated and plotted in box plots.
FIGURE 4. Processivity analysis of DNMT1 activity on hemimethylated GSTP1 promoter in the presence of limiting AdoMet.

100 nm DNMT1 was incubated with 40 nm AdoMet and hemimethylated GSTP1 promoter containing 1 μm CG. The DNA substrate was treated with sodium bisulfite, and individual clones were amplified by PCR, subcloned, and sequenced. Methylated cytosines are represented by filled circles, and unmethylated cytosines are represented by open circles. The absence of a circle at a particular position indicates that the identity of the base could not be determined.
Procainamide Does Not Prevent the Allosteric Activation of DNMT1 by Methylated DNA
The N terminus of DNMT1 has been proposed to stimulate the activity of DNMT1 on unmethylated DNA in the presence of methylated DNA (36, 41). To examine the ability of procainamide to inhibit this phenomenon, we analyzed the activity of DNMT1 on two different substrates, a double-stranded oligonucleotide containing 11 unmethylated CpGs and a double-stranded oligonucleotide of the same sequence containing 5 unmethylated CpGs flanked by 3 methylated CpGs on both ends. We found that the presence of 6 methylated CpGs was able to stimulate de novo methyltransferase activity 3.7-fold (Fig. 5). However, the addition of 2 mm procainamide to the reaction did not effectively inhibit allosteric activation of DNMT1 by methylated DNA. The activity was reduced by only 6%, a level unlikely to be significant because 2 mm procainamide reduced DNMT1 activity on unmethylated DNA by 10%.
FIGURE 5. Effect of procainamide on the allosteric activation of DNMT1.
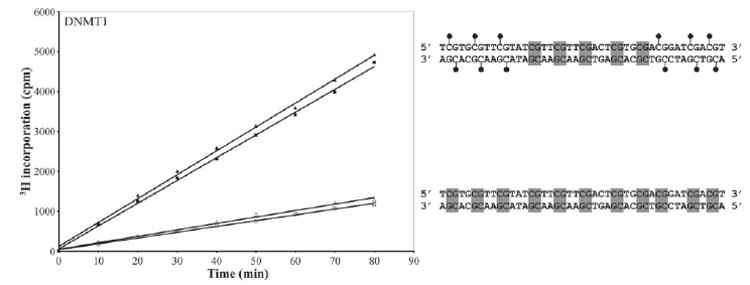
200 nm DNMT1 was incubated with 1 μm AdoMet and biotinylated oligonucleotides containing a mix of methylated CpGs and unmethylated CpGs (filled symbols) or only unmethylated CpGs (open symbols). The reactions were performed in the absence (triangles) or presence (squares) of 2 mm procainamide. Lollipops on the oligonucleotide represent methylated CpGs, and shaded boxes represent unmethylated CpGs.
Procainamide Does Not Inhibit DNMT1 Binding to DNA in the Absence of Catalytic Activity
To further investigate the mechanism by which procainamide inhibits DNMT1 on a hemimethylated template, we first asked whether DNMT1 binds differentially to hairpin oligonu-cleotides that are unmethylated, hemimethylated, or fully methylated. If DNMT1 were to bind much more tightly to hemimethylated DNA, then procainamide might specifically interfere with binding to hemimethylated DNA. Fluorescence anisotropy experiments showed that DNMT1 bound to an unmethylated hairpin at a Kd = 29.9 ± 2.7 nm, a hemimethylated hairpin at a Kd = 22.9 ± 2.1 nm, and a fully methylated hairpin at a Kd = 46.3 ± 4.8 nm (Fig. 6A). This slight difference in binding affinity of DNMT1 for unmethylated hairpin and hemimethylated hairpin is unlikely to explain the >600-fold difference between the Ki of procainamide for DNMT1 on an unmethylated oligonucleotide and a hemimethylated oligonucleotide. Next, we asked whether procainamide can inhibit DNMT1 binding to the hemimethylated hairpin in the absence of AdoMet, and thus catalytic activity. In the presence of saturating levels of DNMT1 (400 nm), even high millimolar concentrations of procainamide could not prevent the enzyme from binding to hemimethylated hairpin (Fig. 6B). In comparison, NaCl at high millimolar concentrations was effective at eliminating DNMT1 binding to the hemimethylated hairpin (IC50 ~120 mm). Thus, procainamide inhibition of DNMT1 binding to hemimethylated DNA depends on processive catalytic activity.
FIGURE 6. Effect of procainamide on the binding of DNMT1 to hairpin oligonucleotides.
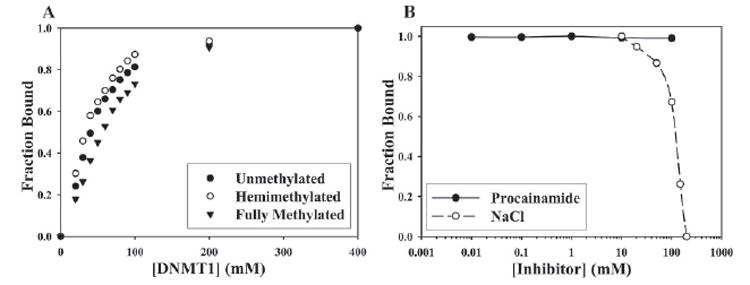
A, DNMT1 was incubated with 10 nm unmethylated hairpin (closed circles), hemimethylated hairpin (open circles), or fully methylated hairpin (closed triangles) at 4 ºC for 1 h. Fluorescence anisotropy values were obtained, converted to fraction bound, and plotted against the concentration of DNMT1 to calculate dissociation constants. B, DNMT1 at saturating levels (400 nm) was incubated with 10 nm hemimethylated hairpin in the presence of varying concentrations of either procainamide (closed circles) or NaCl (open circles). Fluorescence anisotropy values were obtained, converted to fraction bound, and plotted against inhibitor concentration to calculate an IC50 value.
DISCUSSION
The purpose of this investigation was to identify the target and mechanism of procainamide-mediated inhibition of methyltransferase activity. We discovered that the most likely target of procainamide is DNMT1, because (1) procainamide failed to reduce genomic m5dC content in HCT116 cells where DNMT1 was genetically deleted but significantly reduced genomic m5dC content in parental HCT116 cells and HCT116 cells where DNMT3b was genetically deleted, and (2) in vitro methyltransferase activity assays revealed that micromolar concentrations of procainamide effectively inhibited recombinant DNMT1 but not DNMT3a or DNMT3b2. Next, we investigated the mechanism by which procainamide inhibits DNMT1. We hypothesized that procainamide could inhibit the following features of DNMT1: high catalytic activity and processivity on hemimethylated DNA and allosteric activation by methylated DNA.
We have found that procainamide effectively inhibits DNMT1 on a hemimethylated oligonucleotide substrate but not an unmethylated oligonucleotide substrate. Furthermore, we have demonstrated that procainamide is a partial competitive inhibitor of DNMT1, increasing the Km of the enzyme for both DNA and AdoMet. The result of an apparent decrease in affinity for the substrates is the inability to act processively on the preferred substrate of the enzyme, hemimethylated DNA. For a DNA-modifying enzyme to act processively, it must be able to remain on the DNA after a catalytic cycle so that it may scan for the next site to be modified. By decreasing the affinity of DNMT1 for DNA during catalysis, procainamide may facilitate dissociation of DNMT1 from hemimethylated DNA. This might be explained by the ability of procainamide to interact directly with CG-rich DNA (22, 23). Consequently, DNMT1 is forced to rebind DNA and search for its next site to methylate, making the enzyme less processive with an apparent decrease in velocity. In vivo, the enzyme moves with the replication fork via its interaction with proliferating cell nuclear antigen, and interfering with processivity may result in the loss of methylation in significant stretches before DNMT1 can successfully reacquire its DNA substrate. The decrease in affinity for AdoMet can also affect the processivity of DNMT1. The reaction order of DNMT1 is random, but initial binding of AdoMet is preferred (42). Once DNMT1 encounters a site to methylate, if AdoMet is not already bound to the enzyme due to the presence of procainamide, then the site may be skipped and the enzyme will not act processively even if it remains bound to DNA. In support of this hypothesis, reacting DNMT1 with hemimethylated DNA in the presence of only 40 nm AdoMet generated two populations of clones that were similar to the populations in the procainamide-treated samples. The presence of a high activity population as well as a low activity population also reveals that DNMT1 truly acts in an all-or-nothing fashion.
Because procainamide is a partial competitive inhibitor of DNMT1, the velocity cannot be driven to zero even if there were an infinite amount of inhibitor present. This observation is consistent with the “plateau” that Scheinbart et al.(19) saw when they examined inhibition of DNA methyltransferase activity in cell extracts by procainamide. Additionally, Scheinbart et al. remarked that only certain DNA meth-yltransferases may be inhibited by procainamide. Our data show that the de novo methyltransferases DNMT3a and DNMT3b2 were indeed not sensitive to procainamide inhibition. However, their combined contribution to cellular DNA methyltransferase activity is low, and it is likely that the observations of Scheinbart et al. are fully explained by DNMT1.
Methylated DNA has been proposed to either participate in the allos-teric activation (36, 42) or relief of allosteric inhibition of DNMT1 (59). The allosteric effect of methylated DNA on DNMT1 was not changed by procainamide treatment, suggesting that it does not play a role in directly preventing methylation spreading, a process that was shown to occur in the promoter of GSTP1 genes transfected into prostate cancer cell lines (43). Interestingly, procainamide could not prevent DNMT1 from binding to hemimethylated DNA in the absence of AdoMet, supporting the notion that inhibition is tightly linked to processive catalytic activity. Importantly, in none of the experiments where processivity was absent did procainamide inhibit DNA methyltransferase. Because procainamide reduces the affinity of DNMT1 for both DNA and AdoMet during catalysis, it is very likely that procainamide preferentially inhibits the ternary complex containing DNMT1 and both substrates. Perhaps structural studies may fully elucidate the nature of the interaction of procainamide with DNMT1, hemimethylated DNA, and AdoMet.
We have shown that treatment of HCT116 colon cancer cells with procainamide decreased genomic m5dC content only when DNMT1 was present. Gene promoter-specific demethylation, however, may be a more complicated matter. Recently, there have been conflicting results regarding the ability of DNMT1 depletion to reverse aberrant CpG island hypermethylation in human cancer cells (31, 32, 44, 45). When Cheng et al.(10) treated T24 bladder cancer cells with Zebularine, they found a striking result. CpG-rich regions of the p16 locus retained significant levels of methylation, whereas CpG-poor regions did not. This observation was attributed to the ability of Zebularine to selectively deplete DNMT1, which is hypothesized to be singularly responsible for the maintenance of methylation in CpG-poor regions. Similarly, procainamide specifically inhibits DNMT1, and we expect procainamide-mediated demethylation of the genome to target areas whose methylation is maintained by DNMT1 alone. Interestingly, treatment of HCT116 cells with procainamide resulted in slight demethylation of the TIMP3 CpG island. The data imply first that even in HCT116 cells where DNMT1 and DNMT3b cooperate to maintain genome-wide methylation (32), methylation of the TIMP3 CpG island is sensitive to DNMT1 inhibition. Second, the presence of several alleles with almost complete demethylation suggests that inhibition of DNMT1 processivity is a likely mechanism of the effect of procainamide on DNA methylation in a cell. The functional consequences of the specificity of procainamide for DNMT1 will likely depend on the particular locus and cell line being investigated.
In the early 1980s, 5-azacytidine was used in an attempt to reactivate fetal hemoglobin expression in patients with sickle cell anemia. Therapy was successful, resulting in a significant increase in fetal hemoglobin levels and overall clinical improvement (46-49). However, because of safety concerns about the carcinogenic potential of 5-azacytidine, the trials were halted. Procainamide may therefore be a useful alternative in this situation. It has a well characterized safety profile with none of the major disadvantages of nucleoside analogs and has already been shown to reactivate genes silenced by promoter CpG island hypermethylation. It will be interesting to see whether procainamide can increase fetal hemoglobin levels.
Several studies have directly linked DNMT1 with carcinogenesis. In cell culture models, DNMT1 overexpression induced transformation of NIH 3T3 cells (50) and aberrant CpG island hypermethylation in human fibroblasts (51). Moreover, Dnmt1 expression was required in fos-mediated transformation of rodent fibroblasts (52). Animal models further support the role of DNMT1 in carcinogenesis. In a dose-dependent manner, Dnmt1 contributed to intestinal tumorigenesis in ApcMin/+ mice (53, 54). More recently, genetic disruption of a single Dnmt1 allele in a murine model of tobacco carcinogen-induced lung cancer reduced tumor formation by 50% (55). Given the importance of DNMT1 in carcinogenesis, a drug that specifically inhibits DNMT1 activity such as procainamide may be a potential agent in the prevention of cancer. In addition, further experiments with procainamide may be able to separate the enzymatic functions of DNMT1 from its other functions, such as DMAP1 binding, with respect to their roles in carcinogenesis.
A recent study by Gaudet et al.(56) showed that mice carrying both a hypomorphic Dnmt1 allele and a null allele (Dnmt1chip/−) were runted at birth and developed aggressive T cell lymphomas at 4–8 months of age. This phenotype was attributed to genomic instability promoted by genome-wide hypomethylation. These findings are of great concern when patients are treated with demethylating agents. However, the genetic dose of Dnmt1 in this setting may be crucial in determining susceptibility to genomic instability and tumor formation. For example, mice carrying two hypomorphic Dnmt1 alleles (Dnmt1chip/chip) showed no evidence of tumor formation (56). When Belinsky et al.(55) treated Dnmt1+/− mice with 5-aza-2′-deoxycytidine, they did not see the development of lymphomas. Because procainamide is a partial competitive inhibitor, there is an absolute ceiling on its ability to inhibit DNMT1 activity. Therefore, we do not expect procainamide treatment to drastically reduce DNMT1 activity to a point that will enhance genomic instability and tumor formation. Moreover, when procainamide was tested for its ability to induce genotoxic effects, it did not induce DNA fragmentation and repair, increase the frequency of 6-thioguanine resistance, or produce clastogenic effects (57, 58). It will be interesting to examine whether procainamide, a relatively safe non-nucleoside inhibitor of DNMT1 activity, can prevent the development of cancer.
Acknowledgments
We thank Dr. Stephen Baylin for the generous gift of HCT116 DNMT1−/− and HCT116 DNMT3b−/− cells.
Footnotes
This work was supported by NCI, National Institutes of Health Grant CA070196 (to W. G. N.) and NIEHS, National Institutes of Health Fellowship ES014287 (to B. H. L.).
The abbreviations used are: DNMT, DNA methyltransferase; AdoMet, S-adenosyl-l-methionine; m5dC, 5-methyl-2′-deoxycytidine; PBS, phosphate-buffered saline; HPLC, high pressure liquid chromatography.
References
- 1.Jones PA, Baylin SB. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 2.Lin X, Tascilar M, Lee WH, Vles WJ, Lee BH, Veeraswamy R, Asgari K, Freije D, van Rees B, Gage WR, Bova GS, Isaacs WB, Brooks JD, DeWeese TL, De Marzo AM, Nelson WG. Am J Pathol. 2001;159:1815–1826. doi: 10.1016/S0002-9440(10)63028-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakayama M, Bennett CJ, Hicks JL, Epstein JI, Platz EA, Nelson WG, De Marzo AM. Am J Pathol. 2003;163:923–933. doi: 10.1016/s0002-9440(10)63452-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brooks JD, Weinstein M, Lin X, Sun Y, Pin SS, Bova GS, Epstein JI, Isaacs WB, Nelson WG. Cancer Epidemiol Biomarker Prev. 1998;7:531–536. [PubMed] [Google Scholar]
- 5.Antequera F, Bird A. Proc Natl Acad Sci U S A. 1993;90:11995–11999. doi: 10.1073/pnas.90.24.11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sorm F, Piskala A, Cihak A, Vesely J. Experientia. 1964;20:202–203. doi: 10.1007/BF02135399. [DOI] [PubMed] [Google Scholar]
- 7.Jones PA, Taylor SM. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 8.Kaminskas E, Farrell A, Abraham S, Baird A, Hsieh LS, Lee SL, Leighton JK, Patel H, Rahman A, Sridhara R, Wang YC, Pazdur R. Clin Cancer Res. 2005;11:3604–3608. doi: 10.1158/1078-0432.CCR-04-2135. [DOI] [PubMed] [Google Scholar]
- 9.Kelley JA, Driscoll JS, McCormack JJ, Roth JS, Marquez VE. J Med Chem. 1986;29:2351–2358. doi: 10.1021/jm00161a034. [DOI] [PubMed] [Google Scholar]
- 10.Cheng JC, Weisenberger DJ, Gonzales FA, Liang G, Xu GL, Hu YG, Marquez VE, Jones PA. Mol Cell Biol. 2004;24:1270–1278. doi: 10.1128/MCB.24.3.1270-1278.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng JC, Matsen CB, Gonzales FA, Ye W, Greer S, Marquez VE, Jones PA, Selker EU. J Natl Cancer Inst. 2003;95:399–409. doi: 10.1093/jnci/95.5.399. [DOI] [PubMed] [Google Scholar]
- 12.Christman JK. Oncogene. 2002;21:5483–5495. doi: 10.1038/sj.onc.1205699. [DOI] [PubMed] [Google Scholar]
- 13.Gowher H, Jeltsch A. Cancer Biol Ther. 2004;3:1062–1068. doi: 10.4161/cbt.3.11.1308. [DOI] [PubMed] [Google Scholar]
- 14.Lee G, Wolff E, Miller JH. DNA Repair (Amst) 2004;3:155–161. doi: 10.1016/j.dnarep.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 15.Kelecsenyi Z, Spencer DL, Caspary WJ. Mutagenesis. 2000;15:25–31. doi: 10.1093/mutage/15.1.25. [DOI] [PubMed] [Google Scholar]
- 16.Jackson-Grusby L, Laird PW, Magge SN, Moeller BJ, Jaenisch R. Proc Natl Acad Sci U S A. 1997;94:4681–4685. doi: 10.1073/pnas.94.9.4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santini V, Kantarjian HM, Issa JP. Ann Intern Med. 2001;134:573–586. doi: 10.7326/0003-4819-134-7-200104030-00011. [DOI] [PubMed] [Google Scholar]
- 18.Cornacchia E, Golbus J, Maybaum J, Strahler J, Hanash S, Richardson B. J Immunol. 1988;140:2197–2200. [PubMed] [Google Scholar]
- 19.Scheinbart LS, Johnson MA, Gross LA, Edelstein SR, Richardson BC. J Rheumatol. 1991;18:530–534. [PubMed] [Google Scholar]
- 20.Lin X, Asgari K, Putzi MJ, Gage WR, Yu X, Cornblatt BS, Kumar A, Piantadosi S, DeWeese TL, De Marzo AM, Nelson WG. Cancer Res. 2001;61:8611–8616. [PubMed] [Google Scholar]
- 21.Segura-Pacheco B, Trejo-Becerril C, Perez-Cardenas E, Taja-Chayeb L, Mariscal I, Chavez A, Acuna C, Salazar AM, Lizano M, Duenas-Gonzalez A. Clin Cancer Res. 2003;9:1596–1603. [PubMed] [Google Scholar]
- 22.Zacharias W, Koopman WJ. Arthritis Rheum. 1990;33:366–374. doi: 10.1002/art.1780330309. [DOI] [PubMed] [Google Scholar]
- 23.Thomas TJ, Messner RP. Arthritis Rheum. 1986;29:638–645. doi: 10.1002/art.1780290508. [DOI] [PubMed] [Google Scholar]
- 24.Villar-Garea A, Fraga MF, Espada J, Esteller M. Cancer Res. 2003;63:4984–4989. [PubMed] [Google Scholar]
- 25.Agoston AT, Argani P, Yegnasubramanian S, De Marzo AM, Ansari-Lari MA, Hicks JL, Davidson NE, Nelson WG. J Biol Chem. 2005 doi: 10.1074/jbc.M501675200. [DOI] [PubMed] [Google Scholar]
- 26.Carine K, Jacquemin-Sablon A, Waltzer E, Mascarello J, Scheffler IE. Somatatic Cell Mol Genet. 1989;15:445–460. doi: 10.1007/BF01534895. [DOI] [PubMed] [Google Scholar]
- 27.Devilee P, Cremer T, Slagboom P, Bakker E, Scholl HP, Hager HD, Steven-son AF, Cornelisse CJ, Pearson PL. Cytogenet Cell Genet. 1986;41:193–201. doi: 10.1159/000132229. [DOI] [PubMed] [Google Scholar]
- 28.Lin X, Nelson WG. Cancer Res. 2003;63:498–504. [PubMed] [Google Scholar]
- 29.Hermann A, Goyal R, Jeltsch A. J Biol Chem. 2004;279:48350–48359. doi: 10.1074/jbc.M403427200. [DOI] [PubMed] [Google Scholar]
- 30.Michel SL, Guerrerio AL, Berg JM. Biochemistry. 2003;42:4626–4630. doi: 10.1021/bi034073h. [DOI] [PubMed] [Google Scholar]
- 31.Rhee I, Jair KW, Yen RW, Lengauer C, Herman JG, Kinzler KW, Vogelstein B, Baylin SB, Schuebel KE. Nature. 2000;404:1003–1007. doi: 10.1038/35010000. [DOI] [PubMed] [Google Scholar]
- 32.Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW, Baylin SB, Vogelstein B. Nature. 2002;416:552–556. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- 33.Zucker KE, Riggs AD, Smith SS. J Cell Biochem. 1985;29:337–349. doi: 10.1002/jcb.240290407. [DOI] [PubMed] [Google Scholar]
- 34.Flynn J, Glickman JF, Reich NO. Biochemistry. 1996;35:7308–7315. doi: 10.1021/bi9600512. [DOI] [PubMed] [Google Scholar]
- 35.Pradhan S, Bacolla A, Wells RD, Roberts RJ. J Biol Chem. 1999;274:33002–33010. doi: 10.1074/jbc.274.46.33002. [DOI] [PubMed] [Google Scholar]
- 36.Fatemi M, Hermann A, Pradhan S, Jeltsch A. J Mol Biol. 2001;309:1189–1199. doi: 10.1006/jmbi.2001.4709. [DOI] [PubMed] [Google Scholar]
- 37.Chuang LS, Ian HI, Koh TW, Ng HH, Xu G, Li BF. Science. 1997;277:1996–2000. doi: 10.1126/science.277.5334.1996. [DOI] [PubMed] [Google Scholar]
- 38.Leonhardt H, Page AW, Weier HU, Bestor TH. Cell. 1992;71:865–873. doi: 10.1016/0092-8674(92)90561-p. [DOI] [PubMed] [Google Scholar]
- 39.Segel IH. Enzyme Kinetics:Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems. John Wiley and Sons; New York: 1975. pp. 161–202. [Google Scholar]
- 40.Vilkaitis G, Suetake I, Klimasauskas S, Tajima S. J Biol Chem. 2005;280:64–72. doi: 10.1074/jbc.M411126200. [DOI] [PubMed] [Google Scholar]
- 41.Bacolla A, Pradhan S, Roberts RJ, Wells RD. J Biol Chem. 1999;274:33011–33019. doi: 10.1074/jbc.274.46.33011. [DOI] [PubMed] [Google Scholar]
- 42.Bacolla A, Pradhan S, Larson JE, Roberts RJ, Wells RD. J Biol Chem. 2001;276:18605–18613. doi: 10.1074/jbc.M100404200. [DOI] [PubMed] [Google Scholar]
- 43.Song JZ, Stirzaker C, Harrison J, Melki JR, Clark SJ. Oncogene. 2002;21:1048–1061. doi: 10.1038/sj.onc.1205153. [DOI] [PubMed] [Google Scholar]
- 44.Robert MF, Morin S, Beaulieu N, Gauthier F, Chute IC, Barsalou A, MacLeod AR. Nat Genet. 2003;33:61–65. doi: 10.1038/ng1068. [DOI] [PubMed] [Google Scholar]
- 45.Ting AH, Jair KW, Suzuki H, Yen RW, Baylin SB, Schuebel KE. Nat Genet. 2004;36:582–584. doi: 10.1038/ng1365. [DOI] [PubMed] [Google Scholar]
- 46.Charache S, Dover G, Smith K, Talbot CC, Jr, Moyer M, Boyer S. Proc Natl Acad Sci U S A. 1983;80:4842–4846. doi: 10.1073/pnas.80.15.4842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dover GJ, Charache SH, Boyer SH, Talbot CC, Jr, Smith KD. Prog Clin Biol Res. 1983;134:475–488. [PubMed] [Google Scholar]
- 48.Ley TJ, Anagnou NP, Noguchi CT, Schechter AN, DeSimone J, Heller P, Nienhuis AW. Prog Clin Biol Res. 1983;134:457–474. [PubMed] [Google Scholar]
- 49.Ley TJ, DeSimone J, Noguchi CT, Turner PH, Schechter AN, Heller P, Nienhuis AW. Blood. 1983;62:370–380. [PubMed] [Google Scholar]
- 50.Wu J, Issa JP, Herman J, Bassett DE, Jr, Nelkin BD, Baylin SB. Proc Natl Acad Sci U S A. 1993;90:8891–8895. doi: 10.1073/pnas.90.19.8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vertino PM, Yen RW, Gao J, Baylin SB. Mol Cell Biol. 1996;16:4555–4565. doi: 10.1128/mcb.16.8.4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bakin AV, Curran T. Science. 1999;283:387–390. doi: 10.1126/science.283.5400.387. [DOI] [PubMed] [Google Scholar]
- 53.Laird PW, Jackson-Grusby L, Fazeli A, Dickinson SL, Jung WE, Li E, Wein-berg RA, Jaenisch R. Cell. 1995;81:197–205. doi: 10.1016/0092-8674(95)90329-1. [DOI] [PubMed] [Google Scholar]
- 54.Eads CA, Nickel AE, Laird PW. Cancer Res. 2002;62:1296–1299. [PubMed] [Google Scholar]
- 55.Belinsky SA, Klinge DM, Stidley CA, Issa JP, Herman JG, March TH, Baylin SB. Cancer Res. 2003;63:7089–7093. [PubMed] [Google Scholar]
- 56.Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R. Science. 2003;300:489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 57.Mereto E, Robbiano L, Ghia M, Allavena A, Martelli A, Brambilla G. Toxicol Appl Pharmacol. 1995;131:192–197. doi: 10.1006/taap.1995.1061. [DOI] [PubMed] [Google Scholar]
- 58.Martelli A, Campart GB, Canonero R, Mattioli F, Brambilla G. Toxicol Appl Pharmacol. 1995;131:185–191. doi: 10.1006/taap.1995.1060. [DOI] [PubMed] [Google Scholar]
- 59.Svedruzic ZM, Reich NO. Biochemistry. 2005;44:9472–9485. doi: 10.1021/bi050295z. [DOI] [PubMed] [Google Scholar]