

Abstract
The core of photosystem II (PSII) of green plants contains the reaction center (RC) proteins D1D2-cytb559 and two core antennas CP43 and CP47. We have used time-resolved visible pump/midinfrared probe spectroscopy in the region between 1600 and 1800 cm−1 to study the energy transfer and charge separation events within PSII cores. The absorption difference spectra in the region of the keto and ester chlorophyll modes show spectral evolution with time constants of 3 ps, 27 ps, 200 ps, and 2 ns. Comparison of infrared (IR) difference spectra obtained for the isolated antennas CP43 and CP47 and the D1D2-RC with those measured for the PSII core allowed us to identify the features specific for each of the PSII core components. From the presence of the CP43 and CP47 specific features in the spectra up to time delays of 20–30 ps, we conclude that the main part of the energy transfer from the antennas to the RC occurs on this timescale. Direct excitation of the pigments in the RC evolution associated difference spectra to radical pair formation of 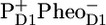 on the same timescale as multi-excitation annihilation and excited state equilibration within the antennas CP43 and CP47, which occur within ∼1–3 ps. The formation of the earlier radical pair
on the same timescale as multi-excitation annihilation and excited state equilibration within the antennas CP43 and CP47, which occur within ∼1–3 ps. The formation of the earlier radical pair 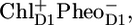 as identified in isolated D1D2 complexes with time-resolved mid-IR spectroscopy is not observed in the current data, probably because of its relatively low concentration. Relaxation of the state
as identified in isolated D1D2 complexes with time-resolved mid-IR spectroscopy is not observed in the current data, probably because of its relatively low concentration. Relaxation of the state 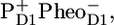 caused by a drop in free energy, occurs in 200 ps in closed cores. We conclude that the kinetic model proposed earlier for the energy and electron transfer dynamics within the D1D2-RC, plus two slowly energy-transferring antennas C43 and CP47 explain the complex excited state and charge separation dynamics in the PSII core very well. We further show that the time-resolved IR-difference spectrum of
caused by a drop in free energy, occurs in 200 ps in closed cores. We conclude that the kinetic model proposed earlier for the energy and electron transfer dynamics within the D1D2-RC, plus two slowly energy-transferring antennas C43 and CP47 explain the complex excited state and charge separation dynamics in the PSII core very well. We further show that the time-resolved IR-difference spectrum of 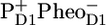 as observed in PSII cores is virtually identical to that observed in the isolated D1D2-RC complex of PSII, demonstrating that the local structure of the primary reactants has remained intact in the isolated D1D2 complex.
as observed in PSII cores is virtually identical to that observed in the isolated D1D2-RC complex of PSII, demonstrating that the local structure of the primary reactants has remained intact in the isolated D1D2 complex.
INTRODUCTION
Photosystem II (PSII) of higher plants, algae, and cyanobacteria is a transmembrane pigment-protein complex that uses light to oxidize water (produce oxygen) and reduce plastoquinone via a complex set of energy and electron transfer events. The PSII complex is comprised of a large number of subunits, but its core is formed by three pigment-binding subunits: the two antenna complexes CP43 and CP47 and the reaction center (RC) complex D1D2cytb559. During the past few years a number of crystal structures of the core of PSII from cyanobacteria have been resolved with increasingly better resolution (1–4); currently the resolution is 3.2 Å (5). From this it was established that CP43 binds 14 chlorophylls (Chls) (6) a and 3 β-carotenes (Cars), CP47 17 Chls a and 4 β-Cars, whereas the D1D2-RC contains 6 Chls a, 2 pheophytins (Pheo) a, 2 β-Cars, the primary and secondary quinones QA and QB, respectively, and the manganese (Mn)-cluster. We note that, from the crystal structure (1–5), it follows that the Chl molecules in CP43 and CP47 are relatively far (>20 Å) away from any of the RC pigments.
After excitation of the PSII core, rapid energy transfer occurs among the pigments of each of the core antennas, followed by energy transfer to the RC (7–11). When one of the pigments of the D1D2-RC is excited, an ultrafast charge separation is initiated, leading to the formation of 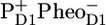 (P+H−) (6,12) On a timescale of a few hundred picoseconds the electron is transferred from the reduced pheophytin (
(P+H−) (6,12) On a timescale of a few hundred picoseconds the electron is transferred from the reduced pheophytin (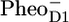 ) to the secondary acceptor, a quinone, denoted QA and subsequently in a few hundred microseconds to QB (6).
) to the secondary acceptor, a quinone, denoted QA and subsequently in a few hundred microseconds to QB (6). 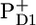 is reduced by a redox-active tyrosine, and finally the positive charge is stored in the Mn-cluster associated to the D1 protein. Two turnovers of the D1D2-RC plus the uptake of two protons generate QBH2; after each four turnovers, two water molecules are oxidized, generating four electrons, four protons, and one O2 molecule. Note, though, that in the core complexes studied in this work, the QB molecule was absent.
is reduced by a redox-active tyrosine, and finally the positive charge is stored in the Mn-cluster associated to the D1 protein. Two turnovers of the D1D2-RC plus the uptake of two protons generate QBH2; after each four turnovers, two water molecules are oxidized, generating four electrons, four protons, and one O2 molecule. Note, though, that in the core complexes studied in this work, the QB molecule was absent.
During the past two decades the precise sequence of early events in PSII has been studied in the isolated D1D2-RC using a multitude of spectroscopic techniques, including visible time-resolved pump-probe spectroscopy, fluorescence spectroscopy, low-temperature fluorescence, hole-burning, singlet-triplet spectroscopy, etc. The experimental data revealed a multitude of lifetimes associated to the charge separation and stabilization process. Energy transfer among the four Chls and two Pheos in the heart of the RC is most likely fast (hundreds of femtoseconds) (13). Recently we showed, using femtosecond visible-pump/mid-IR-probe spectroscopy, that, in isolated D1D2-RCs, charge separation occurs predominantly from the Chl (ChlD1) in the active branch to form the state 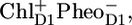 which, on a timescale of 6 ps, converts into the state
which, on a timescale of 6 ps, converts into the state 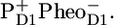 On a slower (a few hundred picoseconds) timescale, a relaxation in free energy of the radical pair causes a shift of the excited state-radical pair equilibrium toward the side of the radical pair, as monitored by a decay of fluorescence and stimulated emission (10,14,15). It was recognized that the presence of disorder severely complicates matters, making all the early events very nonexponential and allowing various paths of charge separation to compete (16–18). Later, visible pump-probe experiments essentially confirmed this picture, only the estimated rate of initial charge separation was a bit slower, possibly because of somewhat different experimental conditions (19).
On a slower (a few hundred picoseconds) timescale, a relaxation in free energy of the radical pair causes a shift of the excited state-radical pair equilibrium toward the side of the radical pair, as monitored by a decay of fluorescence and stimulated emission (10,14,15). It was recognized that the presence of disorder severely complicates matters, making all the early events very nonexponential and allowing various paths of charge separation to compete (16–18). Later, visible pump-probe experiments essentially confirmed this picture, only the estimated rate of initial charge separation was a bit slower, possibly because of somewhat different experimental conditions (19).
Early fluorescence lifetime experiments on whole thylakoids showed that the fluorescence decay of PSII was essentially biphasic with a fast phase of ∼200 ps and a slower phase of ∼500 ps to 1 ns (see review (20)). To explain this, a model was proposed in which the first stable radical pair (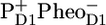 ) formed during the 200-ps phase remained in permanent equilibrium with the Chl a excited state, and consequently, the electron transfer from
) formed during the 200-ps phase remained in permanent equilibrium with the Chl a excited state, and consequently, the electron transfer from 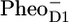 to QA in ∼0.5–1 ns gave rise to a second phase in the fluorescence decay (20). Note that this model implies ultrafast (<1 ps) energy transfer among all the components involved. Later the same model was applied by Holzwarth and colleagues to analyze the fluorescence decay of PS II cores and named the “exciton-radical pair equilibrium model” (21). The fluorescence of PSII cores was also found to decay biphasically with decay times of 40–80 ps and 200–500 ps (12,14). Recent visible pump-probe and time-resolved fluorescence measurements on PSII cores were interpreted assuming similar fast equilibration times, a dominant trapping time of ∼40 ps, and a weaker trapping component of ∼300 ps (19,22). The authors applied the exciton-radical pair model to explain their data and included ultrafast energy transfer among CP43, D1D2-RC, and CP47, the formation of the state
to QA in ∼0.5–1 ns gave rise to a second phase in the fluorescence decay (20). Note that this model implies ultrafast (<1 ps) energy transfer among all the components involved. Later the same model was applied by Holzwarth and colleagues to analyze the fluorescence decay of PS II cores and named the “exciton-radical pair equilibrium model” (21). The fluorescence of PSII cores was also found to decay biphasically with decay times of 40–80 ps and 200–500 ps (12,14). Recent visible pump-probe and time-resolved fluorescence measurements on PSII cores were interpreted assuming similar fast equilibration times, a dominant trapping time of ∼40 ps, and a weaker trapping component of ∼300 ps (19,22). The authors applied the exciton-radical pair model to explain their data and included ultrafast energy transfer among CP43, D1D2-RC, and CP47, the formation of the state 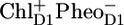 from the (delocalized) RC excited state in ∼6 ps, the subsequent formation of
from the (delocalized) RC excited state in ∼6 ps, the subsequent formation of 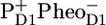 in 9 ps, and the formation of
in 9 ps, and the formation of 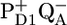 in ∼300 ps. From their fitting, they estimated a free energy drop of ∼36 mV between the RC excited state and
in ∼300 ps. From their fitting, they estimated a free energy drop of ∼36 mV between the RC excited state and 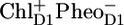 and a >70 mV drop in free energy on the transition from
and a >70 mV drop in free energy on the transition from 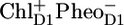 to
to 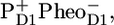 numbers that are surprisingly large compared to those estimated from the temperature-dependent triplet and fluorescent quantum yield, the RT fluorescence, and fs-IR data obtained for the isolated D1D2 complex (16,18,23). This could suggest that the contribution from the antenna pigments in the equilibrium model was not taken into account correctly. We finally note that the assumption underlying the exciton-radical pair model, namely ultrafast energy transfer among all the Chls a in the PSII core, may be seriously questioned in view of the large distance between the Chls of CP43/CP47 and the chlorins of the D1D2-RC, as observed in the PSII core structure (7,9). However, as for the D1D2-RC, the interpretation of the visible pump-probe and fluorescence results for PSII cores are seriously hindered by the spectral congestion in the 660–700 nm region and the difficulty of distinguishing excited states from charge-separated states. For that reason, we performed a visible pump/mid-IR probe study on PSII cores, with the aim of distinguishing the various excited states and charge-separated states according to their IR-signature and to resolve some of the discrepancies mentioned above. Because the spectra of Chl and Pheo in their excited, anion, cation, and neutral states have very specific signatures in the mid-IR (24–29), a more direct observation of the rates of energy transfer, PD1 oxidation, and PheoD1 reduction can be achieved when analyzing the large manifold of vibrational bands observed by using mid-IR spectroscopy.
numbers that are surprisingly large compared to those estimated from the temperature-dependent triplet and fluorescent quantum yield, the RT fluorescence, and fs-IR data obtained for the isolated D1D2 complex (16,18,23). This could suggest that the contribution from the antenna pigments in the equilibrium model was not taken into account correctly. We finally note that the assumption underlying the exciton-radical pair model, namely ultrafast energy transfer among all the Chls a in the PSII core, may be seriously questioned in view of the large distance between the Chls of CP43/CP47 and the chlorins of the D1D2-RC, as observed in the PSII core structure (7,9). However, as for the D1D2-RC, the interpretation of the visible pump-probe and fluorescence results for PSII cores are seriously hindered by the spectral congestion in the 660–700 nm region and the difficulty of distinguishing excited states from charge-separated states. For that reason, we performed a visible pump/mid-IR probe study on PSII cores, with the aim of distinguishing the various excited states and charge-separated states according to their IR-signature and to resolve some of the discrepancies mentioned above. Because the spectra of Chl and Pheo in their excited, anion, cation, and neutral states have very specific signatures in the mid-IR (24–29), a more direct observation of the rates of energy transfer, PD1 oxidation, and PheoD1 reduction can be achieved when analyzing the large manifold of vibrational bands observed by using mid-IR spectroscopy.
Earlier time-resolved mid-IR work (30,31) and time-resolved visible pump/probe experiments and the fluorescence measured using Streak camera at low temperature (8) identified a fast spectral evolution within CP43 and CP47 after a short excitation pulse as being caused by energy transfer among pigments within the same (either stromal or luminal) pigment layer (0.2–0.4ps) and trapping on a low energy state of the complex (in ∼2 ps). In both CP47 and CP43, a slower 10- to 20-ps component was present, which indicated further energy transfer to a red state. In room-temperature studies on CP47 and CP43, combining visible pump/probe and visible pump/mid-IR probe spectroscopy, this basic pattern of energy flow was confirmed (30,31). The red state in CP47 at 690 nm was found to have its keto mode at 1686 cm−1, reflecting a polar environment and/or a weak H-bond of this “red” Chl, whereas in CP43 the red state was found at 1691 cm−1. The experiments on isolated CP47 (30) and CP43 (31) showed that the initial excited state spectra were significantly different from each other and from those of the D1D2-RC (16). Moreover, the time-resolved mid-IR spectra allowed us to clearly resolve the ultrafast equilibration followed by slow “blue-to-red” energy transfer in both complexes, in agreement with earlier visible pump-probe experiments (8).
In the past, results obtained with purified D1D2-RCs have been questioned because of its biochemical state, because the purified D1D2-RC has lost the two quinones and is no longer able to oxidize water and produce oxygen. The spectroscopic properties of the triplet state as observed in D1D2-RC, unlike the state observed in PSII core, suggested strong preparation-induced shifts of the site energies of the pigments carrying the triplet state, which is localized on ChlD1 at <80 K and can be shared with PD1 at >80 K (6,10).
MATERIAL AND METHODS
The PSII core samples were isolated from spinach as previously described (32). To check quality and purity of the sample, visible absorption spectra were measured. For vis-pump/mid-IR probe experiments, the samples were concentrated to an OD of 0.5 per 20 μm−1 at 680 nm. The samples were suspended in 50 mM MES D2O buffer, pH 6.1, containing 10 mM CaCl2, 5 mM MgCl2, and 0.03% β-DM. A closed cell consisting of two CaF2 plates separated by a 20-μm spacer was filled with ∼40 μl of PSII core solution. The cell was then placed in the setup, which was contained in a flow box purged with N2.
The experimental setup consisted of an integrated Ti:sapphire oscillator-regenerative amplifier laser system (Hurricane, Spectra Physics) operating at 1 kHz and 800 nm, producing pulses of 0.6 mJ with a duration of 80 fs. A portion of the 800-nm light was used to pump a home-built noncollinear optical parametric amplifier to produce the excitation pulse at 681 nm (full width at half-maximum, 8 nm) of 125 μm diameter. A second part of the 800-nm light was used to pump an optical parametric generator and amplifier with a difference frequency generator (TOPAS, Light Conversion) to produce the mid-IR probe pulses, with the central frequency at 1680 cm−1 and with a ∼200 cm−1 bandwidth.
The visible excitation and the mid-IR probe pulses were attenuated, overlapped, and focused onto the sample with a 200 mm and 60 mm lens, respectively. The probe pulses were dispersed in a spectrograph after the sample and imaged on a 32-element MCT detector array, yielding a spectral resolution of 6 cm−1. The absorption of a polystyrene film was used for spectral calibration of the setup. To ensure the excitation of a fresh spot at every shot, the sample was continuously moved in a home-built Lissajous scanner. The polarization of the excitation pulse was set to the magic angle (54.7°) with respect to the IR probe pulses. The cross-correlation of visible and mid-IR pulses was measured in GaAs to be 150 fs.
A phase-locked chopper operating at 500 Hz was used to ensure that every second shot the sample was excited and that the change in transmission could be measured.
Experiments were repeated three times; in every experiment a fresh sample was used, and four data sets were collected. After a check for consistency, the 12 data sets containing 600 scans in total were averaged. Data analysis was performed on the raw data using global and target analysis methods (33).
RESULTS
We excited the PSII cores with 250-nJ, 100-fs, 681-nm laser pulses and measured the resulting absorption changes in the mid-IR between 1780 and 1580 cm−1. To visualize the time-dependent evolution of the measured absorption difference spectra, a sequential scheme with increasing lifetimes was fitted to the experimental data, which yielded the following lifetimes: 3 ps, 27 ps, 200 ps, and 2.5 ns. The resulting evolution associated difference spectra (EADS) are shown in Fig. 1. In the difference spectra we observe the bleaching of several keto C9=O modes of Chl or Pheo molecules between 1710 and 1640 cm−1. The exact frequency of the keto stretch of a Chl or Pheo molecule depends on the presence of a hydrogen bond and on the polarity of its environment (25,26,28). The stronger the hydrogen bond, or the larger the polarity of the environment, the lower the frequency of the C9=O mode. The 10a-ester modes of Chl and Pheo absorb in the 1710–1750 cm−1 region and are in a similar way dependent on their environment. In the excited state of Chls, the keto and ester modes downshift from 1695 cm−1 to 1660 cm−1 and from 1737 cm−1 to 1726 cm−1, respectively (16,30), whereas in the cation state of Chl, the keto band upshifts ∼25 cm−1 to 1718 cm−1 (16,24,28).
FIGURE 1.

Pump-probe measurements in the mid-IR on excitation at 681 nm, Pexc = 250 nJ. (A) Time traces detected at 1657, 1686, and 1711 cm−1. The scale is linear up to 3 ps and logarithmic thereafter. The solid line through the data points is a fit with time constants of 3 ps, 27 ps, 200 ps, and 2.5 ns; the very fast component that follows the IRF is also included. (B) EADS resulting from sequential analysis of the data using a model with increasing lifetimes.
The initial spectrum, which has a lifetime of 1–3 ps (black line, Fig. 1), represents the excited states of the PSII core chlorins as reflected by the large positive band at 1645 cm−1, typical for Chl excited states (16,30,31). A comparison of this initial excited state spectrum of PSII cores with those of the isolated D1D2-RC (16) and the core antennas CP43 (31) and CP47 (30) reveals similarities between them. The spectra show which spectral features present in the initial core spectrum are related to which subcomponent: D1D2-RC, CP43, or CP47 (see Fig. 2). The negative bands at 1705 cm−1 and 1678 cm−1 were observed in the initial spectrum of isolated D1D2-RC, and the next two negative bands at 1691 cm−1 and 1669 cm−1 and the positive band at 1698 cm−1 were observed in isolated CP47; the broad negative feature at 1745 cm−1 is probably a mixture of the bleach in excited CP47 and in CP43 at the same frequency, whereas the two positive bands at 1715 cm−1 and 1730 cm−1 were observed in both the isolated CP43 and CP47 spectra, but they were not pronounced in excited D1D2-RC. This implies that, after excitation with 681-nm light, the initial spectrum of PSII cores shows features representative for each of the three excited complexes: CP43, CP47, and RC.
FIGURE 2.

Comparison of initial spectra of core complexes and isolated PSII RC (black and red lines, respectively), excited with the same wavelength, 681 nm, and antenna CP43 and CP47 spectra (green and blue lines, respectively), excited at 585 nm.
In the next spectrum, which is formed in 1–3 ps and decays in 27 ps (red line, Fig. 1), we still observe the presence of all the excited PSII core parts, but about half the excited states have disappeared as reflected by the decay of the 1645 cm−1 band. This is most likely in large part caused by multiexcitation annihilation occurring within CP47 (30), CP43 (31), and the D1D2-RC (16), as a consequence of the relatively high excitation power we used, 250 nJ. On this timescale at the same excitation density, similar decays and spectral shifts were observed in isolated CP47 (30) and isolated CP43 (31); for instance, in CP47 after excitation, a 2-ps decay of the excited state population is observed as a result of annihilation, while the major bleaching shifts from 1686 cm−1 to lower frequency; in CP43 annihilation seems a bit faster (∼1 ps), while the spectrum does not change much, with major bleaches at 1691 cm−1 and 1677 cm−1. Furthermore, we observe a small fraction of charge separation; in particular, there is a hint of bleach at ∼1656 cm−1, the small negative band that dramatically increases in intensity as time progresses and that probably reflects a protein response to charge separation (25–29). From its amplitude we estimate that after 3 ps charge separation has occurred in ∼10% of the cores, and consequently, the 3-ps spectrum reflects mainly excited D1D2-RC, CP43, and CP47. Clear charge separation and formation of 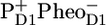 are observed after 27 ps in the green spectrum by the appearance of the negative band at 1656 cm−1. We observe a further decay of the band at 1705 cm−1, and we see the bands related to
are observed after 27 ps in the green spectrum by the appearance of the negative band at 1656 cm−1. We observe a further decay of the band at 1705 cm−1, and we see the bands related to 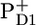 /PD1, as measured in steady-state Fourier transform infrared (FTIR) spectroscopy: positive bands at 1731 cm−1, 1712 cm−1, and 1666 cm−1, and negative bands at 1656 cm−1 and 1610 cm−1. The negative bands at 1740 cm−1 and 1722 cm−1 are related to the presence of
/PD1, as measured in steady-state Fourier transform infrared (FTIR) spectroscopy: positive bands at 1731 cm−1, 1712 cm−1, and 1666 cm−1, and negative bands at 1656 cm−1 and 1610 cm−1. The negative bands at 1740 cm−1 and 1722 cm−1 are related to the presence of 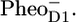 The steady-state
The steady-state 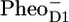 /PheoD1 difference spectrum obtained by FTIR techniques shows two strong negative bands at 1739 and 1722 cm−1 related to the 10a-carbomethoxy ester C=O group of Pheo a (27–29). Some further dynamic changes occur after 200 ps (blue line, Fig. 1), when we clearly see an increase of the band shift feature at 1666/1656 cm−1, an increase of the 1712 cm−1 band, which is probably also responsible for a decrease of the partially overlapping 1705 cm−1 band, a decay of the band at 1610 cm−1, and a disappearance of the shoulder at 1691 cm, which is related to CP43. This spectral evolution suggests that only in this final step most of the excitations disappear from the system, by trapping in the radical pair
/PheoD1 difference spectrum obtained by FTIR techniques shows two strong negative bands at 1739 and 1722 cm−1 related to the 10a-carbomethoxy ester C=O group of Pheo a (27–29). Some further dynamic changes occur after 200 ps (blue line, Fig. 1), when we clearly see an increase of the band shift feature at 1666/1656 cm−1, an increase of the 1712 cm−1 band, which is probably also responsible for a decrease of the partially overlapping 1705 cm−1 band, a decay of the band at 1610 cm−1, and a disappearance of the shoulder at 1691 cm, which is related to CP43. This spectral evolution suggests that only in this final step most of the excitations disappear from the system, by trapping in the radical pair 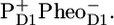 There is no indication of the state
There is no indication of the state 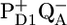 being formed, which is caused by the relatively high excitation density in our experiment, causing all RCs to be “closed” i.e., with QA in the reduced state.
being formed, which is caused by the relatively high excitation density in our experiment, causing all RCs to be “closed” i.e., with QA in the reduced state.
Although the EADS reflect the spectral evolution of the system, they do not necessarily represent “pure” states. In fact they often reflect spectroscopic mixtures of states; the spectra of the “true” states can be obtained only by applying a “realistic” kinetic model in a so-called target analysis (see below).
Target analysis
To obtain a better understanding of the energy transfer and charge separation processes in the PSII core complex, we applied a target analysis of the data and established a kinetic model for energy transfer between antennas and D1D2-RC, including the process of annihilation. The scheme of energy transfer and charge separation within the isolated PSII reaction center remained the same as the one used before and published recently (16).
The PSII core is characterized by 39 single excited states (14 Chls a in CP43 antenna, 17 Chls a in CP47, and 6 Chls a plus 2 Pheos in the D1D2-RC). In addition, at least two radical pair states must be accounted for because the 200-ps process is too slow for an energy transfer process and most likely is caused by a relaxation of the 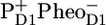 radical pair state. The experimental data only allow for a limited number of experimentally accessible parameters, and consequently we propose a simplified kinetic model, depicted in Fig. 3, which contains five compartments: three excited states and two radical pairs. The first three compartments we take to represent the CP43, CP47, and RC complexes, respectively. For each antenna there is one “equilibrated” excited state that slowly transfers energy to the next adjacent excited state RC*RP1 compartment. To describe the annihilation and fast energy transfer processes within the CP43 and CP47 antennas and the RC, each of the excited states contains an intracomplex dynamic component that takes place on a timescale of ∼2 ps.
radical pair state. The experimental data only allow for a limited number of experimentally accessible parameters, and consequently we propose a simplified kinetic model, depicted in Fig. 3, which contains five compartments: three excited states and two radical pairs. The first three compartments we take to represent the CP43, CP47, and RC complexes, respectively. For each antenna there is one “equilibrated” excited state that slowly transfers energy to the next adjacent excited state RC*RP1 compartment. To describe the annihilation and fast energy transfer processes within the CP43 and CP47 antennas and the RC, each of the excited states contains an intracomplex dynamic component that takes place on a timescale of ∼2 ps.
FIGURE 3.

Kinetic model for PSII core molecules resulting from target analysis; energy, and electron transfer rates are given in picoseconds. Note that components related to annihilation, equilibration, and intracomplex energy transfer are indicated by the circular arrow. The eigenvalues of this model are 2.5 ps, 28 ps, 40 ps, 360 ps, and 12.3 ns, in agreement with the lifetimes obtained in the measurements.
The compartment RC*RP1 represents a mixture of an RC excited state and the first radical pair RP1 identified as 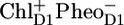 (16). Because of the complexity of the system and the low population of the
(16). Because of the complexity of the system and the low population of the 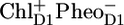 state during the experiment, including these two states separately in a model is unfeasible. The formation of the stable charge-separated state
state during the experiment, including these two states separately in a model is unfeasible. The formation of the stable charge-separated state 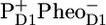 (RP2) occurs from RC*RP1. The kinetic model contains an additional radical pair RP3, which represents the relaxed form of the secondary radical pair
(RP2) occurs from RC*RP1. The kinetic model contains an additional radical pair RP3, which represents the relaxed form of the secondary radical pair 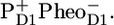
The initial 1- to 3-ps and 27-ps spectra of PSII cores resulting from the sequential analysis clearly show features that can be ascribed to specific compartments of the core complex. Nevertheless, we do not obtain meaningful estimated spectra for the excited state compartments when the target model is fitted to the data without restraint, probably because of the complexity of the IR spectra. Therefore, we use the time-resolved IR absorption difference spectra of the individual CP43 and CP47 complexes (30,31) as input for the target analysis. Compartments CP43 and CP47 are restricted to be similar within 10% to the experimental spectra of the isolated complexes (10-ps and 28-ps spectra, respectively). In addition, to add the information about the excited state dynamics, we perform a simultaneous fit of the mid-IR difference spectra with fluorescence emission data measured on PSII core complexes in the closed state (34). With this, a fit of similar quality as the one shown in Fig. 1 was obtained for the IR data and the emission data; this fit is shown in Fig. 4 C. Note that the five-compartmental model that we used yields one parameter more than when we fitted the sequential model, which is possibly a result of the inclusion of the extra number of data points from the Streak fluorescence experiment.
FIGURE 4.

(A) Species-associated difference spectra of the compartments resulting from the target analysis. The lifetimes resulting from the applied kinetic model in the absence of annihilation and fast equilibration are 2.5 ps, 28 ps, 40 ps, 360 ps, and 12 ns. (B) The concentration profiles of each of the compartments in time (linear-log scale), (C) Simultaneous fit of the fluorescence measured using Streak camera on closed PS2 cores (black line) excited with 400 nm (34) and mid-IR data (red line). The time resolution of the fluorescence experiment was ∼3.5 ps. In the emission rise, the instrument response function is included; the data were fitted with five components: 0.4 ps, 3 ps, 80 ps, 700 ps, and 6.4 ns. Note that the timescale in B and C is linear up to 30 ps and logarithmic thereafter.
The rates that are estimated from target analysis are indicated in the kinetic scheme in Fig. 3. Care was taken to fix the start values of the forward and backward rates in the energy transfer processes to a value consistent with the fact that there are twice as many Chls in each of the antennas as in the RC. The ratios of the forward and backward rates in the charge separation processes were found to be 4 and 3, respectively. The lifetimes that result from this model are shown in the first column of Table 1, which also gives the amplitude matrix of the compartments.
TABLE 1.
Amplitude matrix of the kinetic scheme shown in fig. 3
| Lifetime | CP43* | CP47* | RC*RP1 | RP2 | RP3 |
|---|---|---|---|---|---|
| 2.5 ps | −0.048 | −0.054 | 0.4 | −0.3 | 0.0028 |
| 28 ps | 0.07 | 0.08 | −0.016 | −0.15 | 0.015 |
| 40 ps | 0.016 | −0.016 | 0 | 0 | 0 |
| 360 ps | 0.12 | 0.15 | 0.066 | 0.26 | −0.6 |
| 12.3 ns | 0.08 | 0.09 | 0.05 | 0.2 | 0.6 |
Positive amplitudes denote decay of concentration of the corresponding compartment, and negative amplitudes rise, respectively. Note that the annihilation processes taking place from the three excited states in ∼2 ps, leading to a ∼50% loss of excited states, are not shown in the table.
Apart from the intracomplex equilibration and annihilation (occurring in ∼2 ps) five different species were resolved: CP43*, CP47*, RC*RP1, RP2, and RP3. The annihilation processes taking place from the three excited states in ∼2 ps, leading to a ∼50% loss of excited states, are not shown in the amplitude matrix (Table 1). The 2.5-ps component reflects the equilibration between the RC*RP1 state and its surroundings (decay of the component RC*RP1, see Table 1 and Fig. 4 B). Most of the population of RC*RP1 flows into RP2 (rise of the component RP2, see Table 1 and Fig. 4 B), 25% flows back to the antenna because RC*RP1 is connected to CP43 and CP47 via slow energy transfer processes. In the next phase of 28 ps, excited CP43 and CP47 equilibrate with RC*RP1 and RP2, and ∼25% of the excitations from the core antenna complexes have formed radical pairs. The 40-ps component in which a small fraction of energy flows from CP43 into CP47 reflects the small free energy difference that was introduced between CP43* and CP47*, with CP47* a bit lower in energy than CP43* simply because CP47 has a slightly larger number of Chls than CP43. Most of the formation of RP3 occurs during the 360-ps phase, even if the sum of the forward and backward rates is ∼200 ps, because of the equilibration of the excited state over the three excited state compartments. In 360 ps essentially RP2 decays into RP3 by transfer of excitations from the three excited state compartments. Compared to the numbers obtained for the isolated RC (16), the forward RP2→RP3 rate is a bit slower: 278 vs. 210 ps (16), whereas the backward rate is significantly slower: 868 vs. 420 ps (16). Note that in particular the value for the back rate constant is strongly determined by the fluorescence decay profile and not by the pump-probe data. A small error in the fluorescence level after 1 ns, for instance. as a result of unconnected pigments will have a strong influence on the latter value. The final component of 12 ns reflects the decay of the system to the original state, mainly via RP3, partly via the excited states that have remained on CP43, CP47, and RC*RP1.
Fig. 4 A shows the species associated difference spectra (SADS) resulting from the model applied in target analysis. Black and red spectra represent excited states of CP43 and CP47, respectively. In our model the black spectrum was linked to the 10-ps lifetime spectrum measured on the isolated CP43 complex (31), and the red spectrum to the 28-ps lifetime spectrum of CP47 (30). Because of this modeling procedure, a large part of the experimental error present in the initial spectra has been transmitted to the next spectrum, representing the RC*RP1 state (green spectrum, Fig. 4 A). Spectra measured in isolated antennas CP47 and CP43 agree very well with spectra resulting from target analysis (see Fig. 5, plots A and B), although there is one obvious discrepancy between the CP43 spectra in the region around 1700 cm−1, where we observe a shift of the peak at 1705 cm−1 in the PSII core SADS to below 1700 cm−1 in the SADS of the isolated antenna. On the other hand, comparing the CP47 spectra with those of CP43, we see that the characteristic negative band at 1696 cm−1 has a bit less intensity in the PSII core. Thus, compensation effects may have occurred that explain the difference between the CP43 SADS in PSII cores and in isolated CP43.
FIGURE 5.

Comparison of the excited state compartments measured in PSII core and in isolated antennas CP47, CP43, and D1D2-RC. Black spectra resulting from target analysis of PSII core data represent excited state of the compartments: CP47, CP43, and RC*RP1. Red spectra were measured in isolated antennas: CP47 and CP43 (A, 28-ps spectrum (30); and B, 10-ps spectrum (31), respectively). The red spectrum in plot C is the sum of PC, BC, and RP1 spectra resulting from target analysis of isolated D1D2-RC data (16).
The green spectrum (see Fig. 4 A) of the compartment we labeled RC*RP1 is clearly rich in features. We distinguish the following bands: negative bands at 1742 cm−1, 1726 cm−1, 1705 cm−1, 1680 cm−1, 1666 cm−1, 1628 cm−1 and 1614 cm−1, and positive bands at 1732 cm−1, 1715 cm−1, 1695 cm−1, 1650 cm−1, 1632 cm−1, and 1623 cm−1. On comparison of this spectrum with the sum of the earlier obtained excited state spectra of the isolated RC (denoted PC and BC in Groot et al. (16)), and of the initial radical pair state RP1, we resolved in our measurements on the isolated RC to represent 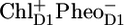 (16), we can see that there is a good agreement (see Fig. 5, plot C). This confirms that the PSII core complex can be described as being the sum of its components, and validates our approach of determining the energy transfer times to the RC by tracking the CP43, CP47 and RC-RP concentrations in time.
(16), we can see that there is a good agreement (see Fig. 5, plot C). This confirms that the PSII core complex can be described as being the sum of its components, and validates our approach of determining the energy transfer times to the RC by tracking the CP43, CP47 and RC-RP concentrations in time.
The spectra assigned to RP2 and RP3 (respectively shown in blue and cyan in Fig. 4 A) represent the secondary radical pair state 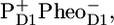 where RP3 is a relaxed form of RP2. In Fig. 6, we compare the RP3 spectrum from the PSII core complex to the radical pair spectrum RP3 from isolated D1D2-RCs (see Fig. 6 A) and with the sum of
where RP3 is a relaxed form of RP2. In Fig. 6, we compare the RP3 spectrum from the PSII core complex to the radical pair spectrum RP3 from isolated D1D2-RCs (see Fig. 6 A) and with the sum of 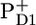 /PD1 and
/PD1 and 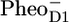 /PheoD1 spectra obtained by steady-state FTIR techniques (25,28) (see Fig. 6 B). The good agreement in both cases shows that, as expected, we can associate RP3 with
/PheoD1 spectra obtained by steady-state FTIR techniques (25,28) (see Fig. 6 B). The good agreement in both cases shows that, as expected, we can associate RP3 with 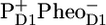 and RP2 with an unrelaxed form of
and RP2 with an unrelaxed form of 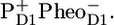 The spectra of RP2 and RP3 do not show any significant spectral differences and therefore do not bring any further information on the cause of relaxation from RP2 into RP3 on the 360-ps timescale.
The spectra of RP2 and RP3 do not show any significant spectral differences and therefore do not bring any further information on the cause of relaxation from RP2 into RP3 on the 360-ps timescale.
FIGURE 6.

Comparison of secondary radical pair RP3 obtained from the experimental PSII core data with the RP3-spectrum as measured in isolated D1D2-RC (A) and with the sum of the 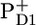 /PD1 and
/PD1 and 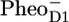 /PheoD1 spectra obtained using steady-state FTIR techniques (B).
/PheoD1 spectra obtained using steady-state FTIR techniques (B).
There is clearly a good agreement between the longest-lived EADS and the sum of 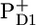 /PD1 and
/PD1 and 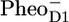 /PheoD1 spectra obtained by steady-state FTIR techniques (25,28). The
/PheoD1 spectra obtained by steady-state FTIR techniques (25,28). The 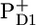 /PD1 and
/PD1 and 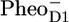 /PheoD1 spectra reported by Noguchi et al. (29) suggest an even higher degree of similarity because their
/PheoD1 spectra reported by Noguchi et al. (29) suggest an even higher degree of similarity because their 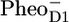 /PheoD1 spectrum shows a large amplitude of the 1664/1657 cm−1 band shift, similar to that observed in our spectrum: 1666/1656 cm−1. The magnitude of this band depends on temperature and even varies somewhat from experiment to experiment (J. Breton, unpublished results). Raman spectra of PSII reaction centers do not show any bands between 1660 and 1640 cm−1 (26), and therefore the 1664/1657 cm−1 band shift probably results from an amide C=O response to the charge separation rather than from a Chl keto. Both spectra, the
/PheoD1 spectrum shows a large amplitude of the 1664/1657 cm−1 band shift, similar to that observed in our spectrum: 1666/1656 cm−1. The magnitude of this band depends on temperature and even varies somewhat from experiment to experiment (J. Breton, unpublished results). Raman spectra of PSII reaction centers do not show any bands between 1660 and 1640 cm−1 (26), and therefore the 1664/1657 cm−1 band shift probably results from an amide C=O response to the charge separation rather than from a Chl keto. Both spectra, the 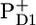 /PD1 and the
/PD1 and the 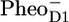 /PheoD1, have a band in this region (at 1655 and 1657 cm−1, respectively (25–29)). The origin of the band shift at 1666/1656 cm−1 is probably caused by an amide C=O response to the charge separation (26,29).
/PheoD1, have a band in this region (at 1655 and 1657 cm−1, respectively (25–29)). The origin of the band shift at 1666/1656 cm−1 is probably caused by an amide C=O response to the charge separation (26,29).
Concentration profiles of all the compartments are shown in Fig. 4 B. Note how the ratio between RC*RP1 and both CP43* and CP47* keeps on changing up to a delay time of ∼50 ps, meaning that the excited states in the three complexes are not in a quasiequilibrium during this time window. In addition, we observe that the population of the state RC*RP1 is low compared to the other excited states, which means that because of the limited signal/noise ratio the spectrum representing state RC*RP1 contains more experimental error than the other spectra.
DISCUSSION
In this work we have measured the spectroscopic mid-IR (1580–1800 cm−1) response of PSII core particles after a visible excitation pulse, with the aim of spectroscopically separating the different compartments that may contribute to the time evolution of the system. In particular, we wish to establish the time-dependent population of the excited core antenna proteins CP43 and CP47 relative to the D1D2-RC, which is very difficult to obtain from vis-vis pump-probe or visible fluorescence experiments. In contrast to visible pump-probe and fluorescence spectroscopy, in the experiments reported here we are indeed able to observe the excited state kinetics of the individual components. The measured time-dependent midinfrared difference spectra are clearly very rich in features. The multiexponential decay times of the absorption-difference spectra are in good agreement with the time constants observed earlier in femtosecond mid-IR and visible pump-probe experiments on CP47 (8,30) and CP43 (9,31) and the D1D2-RC (16,35). The aim of the target analysis, in which the experimental data are fitted to a kinetic model, is to obtain the pure spectra of the states, species associated difference spectra (SADS), which was achieved here with the exception of the spectrum representing a mixture of the reaction center excited state RC* and first radical pair 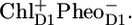 Because of the complexity of the system and the relatively low population of this state, the “final” result shown in Fig. 4 A was the best obtainable result that allowed a good fit to the data. As an input, apart from PSII core data, we used the CP47 and CP43 mid-IR spectra (measured at 28 ps and 10 ps, respectively) (30,31) and the Streak emission data obtained for closed PSII cores (34). The applied kinetic model was based on the one that we previously used to analyze the D1D2-RC data (16), and in this work it has been extended to include the energy transfer from CP43 and CP47 to the D1D2-RC and the intracomplex dynamics (annihilation, equilibration, and energy transfer) within the antenna CP43, CP47, and the D1D2-RC. The SADS resulting from the target analysis were compared to the reference spectra:
Because of the complexity of the system and the relatively low population of this state, the “final” result shown in Fig. 4 A was the best obtainable result that allowed a good fit to the data. As an input, apart from PSII core data, we used the CP47 and CP43 mid-IR spectra (measured at 28 ps and 10 ps, respectively) (30,31) and the Streak emission data obtained for closed PSII cores (34). The applied kinetic model was based on the one that we previously used to analyze the D1D2-RC data (16), and in this work it has been extended to include the energy transfer from CP43 and CP47 to the D1D2-RC and the intracomplex dynamics (annihilation, equilibration, and energy transfer) within the antenna CP43, CP47, and the D1D2-RC. The SADS resulting from the target analysis were compared to the reference spectra: 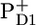 /PD1,
/PD1, 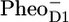 /PheoD1 (24,25,27,28) (measured with FTIR), to the CP43* (31), CP47* (30), and RC* (16) (measured on isolated antennas and RC with vis pump/mid-IR probe spectroscopy) to determine the nature of the states that were formed.
/PheoD1 (24,25,27,28) (measured with FTIR), to the CP43* (31), CP47* (30), and RC* (16) (measured on isolated antennas and RC with vis pump/mid-IR probe spectroscopy) to determine the nature of the states that were formed.
Kinetic model, amplitude matrix and target analysis
In this work we have extended our earlier kinetic scheme (16), which describes charge separation in the isolated PSII RC, to include energy transfer from the core antennas CP43 and CP47. To obtain a realistic fit to the data, the intracomplex energy transfer and annihilation events had to be explicitly included. In the structure of the PSII core only one pigment in CP43 and one pigment in CP47 are in effective contact with the pigments of the PSII core. At a center-to-center distance of ∼2 nm one may expect a local hopping rate of ∼2 ps. A good example is the bacterial LH2 complex, where in the B800 ring the “monomeric” B800s are at a distance of ∼2 nm, and the estimated B800-to-B800 hopping rate is ∼2–3 ps (36–38). Because the excitation has a (more or less) equal probability to be localized on any of the 14/17 Chls of CP43/CP47, the effective transfer rate from CP43 and CP47 to the PSII RC must be of the order of 30–50 ps. The back rates from the PSII RC to CP43/CP47 at room temperature must each be about a factor of 2 faster to account for the 2:1 pigment ratio between CP43/CP47 and the RC. We conclude that the kinetic model applied earlier describing energy transfer and charge separation within the D1D2-RC (16), together with two antenna compartments that transfer energy on a slow timescale, explains energy and electron transfer in the PSII core very well. The slow energy transfer from the core antennas to the D1D2-RC is in agreement with the structural data (7,9,39), but in contrast to the fast (< 1 ps) equilibration rate proposed from earlier photon counting emission data (22).
Identification of excited states
To identify the species representing the SADS spectra of excited state, compartments CP43*, CP47*, and RC*RP1 were compared to the spectra of the individual complexes (see Fig. 5). We of course see an excellent agreement of first two SADS with the CP43 (31) and the CP47 (30) SADS measured in the isolated antenna after the first fast relaxation (annihilation plus excited state equilibration) has taken place (see plots A and B in Fig. 5). To obtain a reasonable fit to the data, we had linked the spectra of CP43/CP47 in the core to those of the isolated complexes. There is one obvious discrepancy between the CP43 spectra in the region around 1700 cm−1, where we observe a shift of the peak at 1705 cm−1 in the PSII core SADS to below 1700 cm−1 in the SADS of the isolated antenna. On the other hand, comparing the CP47 spectra with those of CP43, we see that the characteristic negative band at 1696 cm−1 has a bit less intensity in the PSII core. Thus, compensation effects may have occurred that explain the difference between the CP43 SADS in PSII cores and in isolated CP43.
The RC*RP1 spectrum was compared to the sum of PC, BC, and RP1 spectra resulting from a target analysis of time-resolved vis pump/mid-IR probe data measured for the isolated D1D2-RC (16). These spectra predominantly represent the excited state of the D1D2-RC. However, in the experiments on PSII cores, the population of RC*RP1 is significantly lower than those of the other states (see Fig. 4 B). Moreover we “linked” the CP43 and CP47 spectra obtained in the isolated complexes to those in the PSII core, implying that all the deviations were going to be put in the RC*RP1 spectrum. For this reason the spectrum is relatively less certain than the other SADS. Nevertheless, we see many similarities between the two spectra: they share negative bands at 1726 cm−1, 1705 cm−1, 1680 cm−1, 1628 cm−1, and 1614 cm−1 and positive bands at 1715 cm−1, 1695 cm−1, 1650 cm−1, 1632 cm−1, and 1623 cm−1 (see Fig. 5, plot C). Therefore, we conclude that PC* is the major contribution to the spectrum of compartment RC*RP1. One notable difference is at 1656 cm−1, where there is a clear negative band in the PC* spectrum, whereas the RC*RP1 SADS displays a broad positive band in the same wavelength region. In the PSII core we observe a bleaching of this band only when the later species 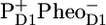 appears. One possibility is that in the D1D2-RC 681-nm excitation produces a direct charge separation because of the mixing of the first charge-separated state with the lowest excitonic state (17). In this respect it is relevant to note the difference in initial spectra measured for the D1D2-RC after “red” and “blue” excitation (see Fig. 3 a in Groot et al. (16)).
appears. One possibility is that in the D1D2-RC 681-nm excitation produces a direct charge separation because of the mixing of the first charge-separated state with the lowest excitonic state (17). In this respect it is relevant to note the difference in initial spectra measured for the D1D2-RC after “red” and “blue” excitation (see Fig. 3 a in Groot et al. (16)).
Comparison of radical pairs RP3 and steady-state PD1+/PD1 and PheoD1−/PheoD1 spectra
Fig. 6 shows a comparison of the RP3 spectrum representing the 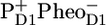 radical pair in PSII core with the SADS of the same state measured in isolated RC (A), and with the sum of
radical pair in PSII core with the SADS of the same state measured in isolated RC (A), and with the sum of 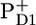 /PD1 and
/PD1 and  /PheoD1 spectra obtained by steady-state FTIR techniques (B) (25,28). The long-lived spectrum measured in intact cores of PSII shows a very good agreement with the RP3 spectrum measured in isolated RC (see Fig. 6 A), as well as with the composed steady-state
/PheoD1 spectra obtained by steady-state FTIR techniques (B) (25,28). The long-lived spectrum measured in intact cores of PSII shows a very good agreement with the RP3 spectrum measured in isolated RC (see Fig. 6 A), as well as with the composed steady-state 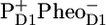 /PD1PheoD1 spectrum (see Fig. 6 B). The spectra of RP2 and RP3 do not show any significant spectral differences and therefore do not bring any further information on the cause of relaxation from RP2 into RP3 on the 360-ps timescale. This fact may suggest that the amount of protein relaxation that causes RP2 to relax into RP3 must be small, and the observed differences may arise from noise (e.g., because of the limited number of data points during the lifetime of RP3).
/PD1PheoD1 spectrum (see Fig. 6 B). The spectra of RP2 and RP3 do not show any significant spectral differences and therefore do not bring any further information on the cause of relaxation from RP2 into RP3 on the 360-ps timescale. This fact may suggest that the amount of protein relaxation that causes RP2 to relax into RP3 must be small, and the observed differences may arise from noise (e.g., because of the limited number of data points during the lifetime of RP3).
The well-pronounced bleaching of the band at 1656 cm−1 in RP2 and RP3 spectra, superimposed on a positive signal, resembles the spectrum of Chl*/Chl in CP47 (30). Possibly the band at 1656 cm−1 is also caused by a Chl molecule with strong H-bond interaction, but because no Raman bands are observed at this frequency (26,40), we speculate that it is the response of an amide C=O to the changed electron distribution of the Chl(s)+.
Other kinetic models
Recently, studies of the excited state and radical pair dynamics in PSII cores and isolated D1D2-RCs using femtosecond transient absorption spectroscopy (19) and later by picosecond fluorescence (22) have been published. The results from these studies were analyzed with the exciton-radical pair equilibrium model, which assumes the ultrafast equilibration of all the excited states before charge separation occurs, in clear opposition to the assumptions made here. One important difference between the experimental data shown here and in elsewhere (19,22) is that, according to the other authors, it was not necessary to account for multiexcitation annihilation, whereas in the data shown here annihilation is clearly present, and we took that explicitly into account. Nevertheless, the reported amplitude of the absorption changes in PSII cores (19) of the order of 10–20 mOD are on the edge of being annihilation free for just CP43 and CP47, implying that in Holzwarth et al. (19) as well, the PSII core data may have contained multiexcitation contributions. According to Holzwarth et al. (19) and Miloslavina et al. (22), the fastest <2-ps component describes an energy transfer within the antenna CP43 and CP47 and from antenna to RC. This component corresponds to our “mixed” annihilation and equilibration kinetics within the complexes, the main difference being that in our model the CP43/CP47 energy transfer to the RC is much slower and plays no role on this ultrafast timescale, in agreement with structural data and modeling. In our model there is a fast 2.5-ps equilibration between the RC*RP1 state and the RP2 (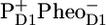 ) that is absent in their models (19,22). In the latter, all the following components must be related to the primary and secondary charge separation steps (19,22). The next 9-ps component has a significant intensity in the fluorescence decay experiments (22), whereas in Holzwarth et al. (19) there is no stimulated emission component of this lifetime. Holzwarth et al. have resolved a 30- to 40-ps component, which they ascribe to the formation of RP2 out of RP1. Because they set RP2 much lower in free energy, during this decay phase the majority of excited states disappear from the excited antenna, and in both the fluorescence and the pump-probe data, this component represents the major decay of excited states. In our work we resolved a 28-ps component, which is similar in lifetime to the 30- to 40-ps component in Holzwarth et al. (19) and Miloslavina et al. (22). However, in our model the physical origin of this component is the excited state equilibration process amomg the antenna CP43, CP47, and RC via slow energy transfer, leading to the rise of the RP2 population, which was already in a quasiequilibrium with RC*RP1. Thus, as in Holzwarth et al. (19) and Miloslavina et al. (22), excited state decay leads to 30- to 40-ps RP2 formation, but the physical reason is different: slow formation of RP2 (19,22) versus slow energy transfer from CP43*/CP47* (present work). Finally, we note that in case the equilibrated excited states of CP43 and CP47 are not at the same free energy, then in our model we would expect some additional equilibration between these two antenna complexes, basically occurring on the timescale of energy transfer between them and the RC (see the 40-ps component in the amplitude matrix, Fig. 3 B). Of course, at room temperature such a component will be small, the excited state energies of the two complexes are virtually the same, but at some lower temperature, this process may become visible and in fact be an excellent test of this kinetic model. Finally, in Holzwarth et al. (19) and Miloslavina et al. (22), the 30- to 40-ps decay is the main excited state decay, whereas in our results the dominant excited state decay occurs in 360 ps by formation of the RP3 radical pair. Because we are working under conditions of closed RCs (meaning QA is in the reduced state), this component reflects the relaxation of the radical pair
) that is absent in their models (19,22). In the latter, all the following components must be related to the primary and secondary charge separation steps (19,22). The next 9-ps component has a significant intensity in the fluorescence decay experiments (22), whereas in Holzwarth et al. (19) there is no stimulated emission component of this lifetime. Holzwarth et al. have resolved a 30- to 40-ps component, which they ascribe to the formation of RP2 out of RP1. Because they set RP2 much lower in free energy, during this decay phase the majority of excited states disappear from the excited antenna, and in both the fluorescence and the pump-probe data, this component represents the major decay of excited states. In our work we resolved a 28-ps component, which is similar in lifetime to the 30- to 40-ps component in Holzwarth et al. (19) and Miloslavina et al. (22). However, in our model the physical origin of this component is the excited state equilibration process amomg the antenna CP43, CP47, and RC via slow energy transfer, leading to the rise of the RP2 population, which was already in a quasiequilibrium with RC*RP1. Thus, as in Holzwarth et al. (19) and Miloslavina et al. (22), excited state decay leads to 30- to 40-ps RP2 formation, but the physical reason is different: slow formation of RP2 (19,22) versus slow energy transfer from CP43*/CP47* (present work). Finally, we note that in case the equilibrated excited states of CP43 and CP47 are not at the same free energy, then in our model we would expect some additional equilibration between these two antenna complexes, basically occurring on the timescale of energy transfer between them and the RC (see the 40-ps component in the amplitude matrix, Fig. 3 B). Of course, at room temperature such a component will be small, the excited state energies of the two complexes are virtually the same, but at some lower temperature, this process may become visible and in fact be an excellent test of this kinetic model. Finally, in Holzwarth et al. (19) and Miloslavina et al. (22), the 30- to 40-ps decay is the main excited state decay, whereas in our results the dominant excited state decay occurs in 360 ps by formation of the RP3 radical pair. Because we are working under conditions of closed RCs (meaning QA is in the reduced state), this component reflects the relaxation of the radical pair 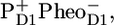 whereas in Holzwarth et al. (19) and Miloslavina et al. (22), RP3 is the radical pair
whereas in Holzwarth et al. (19) and Miloslavina et al. (22), RP3 is the radical pair 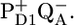 Nevertheless, it is remarkable that in the fluorescence data in Miloslavina et al. (22), there are many components slower than 30–40 ps that are totally absent in the stimulated emission decay of the pump-probe results (19). We finally note that the excited state decay as observed in the femtosecond mid-IR data is adequately described in our model and also fits the fluorescence streak data recorded for PSII cores under similar conditions.
Nevertheless, it is remarkable that in the fluorescence data in Miloslavina et al. (22), there are many components slower than 30–40 ps that are totally absent in the stimulated emission decay of the pump-probe results (19). We finally note that the excited state decay as observed in the femtosecond mid-IR data is adequately described in our model and also fits the fluorescence streak data recorded for PSII cores under similar conditions.
Consistency of PD1+PheoD1− spectra in intact core molecules and isolated RC
The identity of the rates and the mechanism of the early electron transfer processes in isolated PSII RC and intact core complex has been a matter of debate since the 1970s. The results obtained for isolated RCs have been questioned recently (7,9,41,42) because of the biochemical state of the purified D1D2-RC that, in the absence of the quinones QA and QB, can not produce a stable charge separation and consequently can not oxidize water and produce oxygen any more. The spectroscopic properties of the triplet state as observed in D1D2-RC versus the state observed in PSII core suggested strong preparation-induced shifts of the site energies of the pigments carrying the triplet state, which are localized on ChlD1 at <80 K and can be shared with PD1 at >80 K (6). Furthermore, high-resolution spectroscopy on very concentrated core PSII samples suggested that the nature of the PD1 in PSII cores might be very different from what has been reported for D1D2-RC (41). In contrast, here we find that the mid-IR spectra for the state 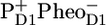 measured in PSII cores and in the D1D2-RC are virtually identical, suggesting that the D1D2-RC in its purified form represents the in vivo situation: all the important interactions between the Chl cofactors and the protein environment are still intact. Also, the fact that we could use the reaction model established for D1D2-RC (16) to fit the PSII core data point to a high degree of similarity of the PSII-RC in both preparations. The deviating observations at low temperature may be caused by the interference of Chl triplet states in the antenna, which have been shown for CP47 to be quenched for only 50% by carotenoids at T < 40K (43).
measured in PSII cores and in the D1D2-RC are virtually identical, suggesting that the D1D2-RC in its purified form represents the in vivo situation: all the important interactions between the Chl cofactors and the protein environment are still intact. Also, the fact that we could use the reaction model established for D1D2-RC (16) to fit the PSII core data point to a high degree of similarity of the PSII-RC in both preparations. The deviating observations at low temperature may be caused by the interference of Chl triplet states in the antenna, which have been shown for CP47 to be quenched for only 50% by carotenoids at T < 40K (43).
CONCLUSIONS
From the experiments presented in this manuscript we conclude that the kinetic model proposed for the reactions occurring in the isolated D1D2-RC on the basis of earlier visible pump/mid-IR probe data (16), when combined with two slow energy-transferring antenna compartments, CP43 (31) and CP47 (30), explains the observed spectral evolution in PSII cores very well. This proposed scheme is in agreement with the structural data for the PSII core. The spectra obtained for the stable radical pair 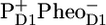 in PSII cores strongly resemble those measured earlier for the PSII RC indicating that the pigments involved in these radical pairs experience a very similar local (protein) environment.
in PSII cores strongly resemble those measured earlier for the PSII RC indicating that the pigments involved in these radical pairs experience a very similar local (protein) environment.
Acknowledgments
We thank Henny van Roon for preparing samples.
This research was supported by the Netherlands Organization for Scientific Research, NWO-ALW, and by Human Frontier Science Program award R GP0038/2006 C.
Editor: Robert Callender.
References
- 1.Zouni, A., H. T. Witt, J. Kern, P. Fromme, N. Krauss, W. Saenger, and P. Orth. 2001. Crystal structure of photosystem II from Synechococcus elongatus at 3.8 angstrom resolution. Nature. 409:739–743. [DOI] [PubMed] [Google Scholar]
- 2.Kamiya, N., and J. R. Shen. 2003. Crystal structure of oxygen-evolving photosystem II from Thermosynechococcus vulcanus at 3.7-angstrom resolution. Proc. Natl. Acad. Sci. USA. 100:98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferreira, K. N., T. M. Iverson, K. Maghlaoui, J. Barber, and S. Iwata. 2004. Architecture of the photosynthetic oxygen-evolving center. Science. 303:1831–1838. [DOI] [PubMed] [Google Scholar]
- 4.Biesiadka, J., B. Loll, J. Kern, K. D. Irrgang, and A. Zouni. 2004. Crystal structure of cyanobacterial photosystem II at 3.2 angstrom resolution: a closer look at the Mn-cluster. Phys. Chem. Chem. Phys. 6:4733–4736. [Google Scholar]
- 5.Loll, B., J. Kern, W. Saenger, A. Zouni, and J. Biesiadka. 2005. Towards complete cofactor arrangement in the 3.0 angstrom resolution structure of photosystem II. Nature. 438:1040–1044. [DOI] [PubMed] [Google Scholar]
- 6.Diner, B. A., E. Schlodder, P. J. Nixon, W. J. Coleman, F. Rappaport, J. Lavergne, W. F. J. Vermaas, and D. A. Chisholm. 2001. Site-directed mutations at D1-His198 and D2-His97 of photosystem II in synechocystis PCC 6803: Sites of primary charge separation and cation and triplet stabilization. Biochemistry. 40:9265–9281. [DOI] [PubMed] [Google Scholar]
- 7.Vasil'ev, S., P. Orth, A. Zouni, T. G. Owens, and D. Bruce. 2001. Excited-state dynamics in photosystem II: Insights from the x-ray crystal structure. Proc. Natl. Acad. Sci. USA. 98:8602–8607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Weerd, F. L., I. H. M. van Stokkum, H. van Amerongen, J. P. Dekker, and R. van Grondelle. 2002. Pathways for energy transfer in the core light-harvesting complexes CP43 and CP47 of photosystem II. Biophys. J. 82:1586–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vassiliev, S., C. I. Lee, G. W. Brudvig, and D. Bruce. 2002. Structure-based kinetic modeling of excited-state transfer and trapping in histidine-tagged photosystem II core complexes from Synechocystis. Biochemistry. 41:12236–12243. [DOI] [PubMed] [Google Scholar]
- 10.Barter, L. M. C., M. Bianchietti, Ch. Jeans, M. J. Schilstra, B. Hankamer, B. Diner, J. Barber, J. R. Durrant, and D. R. Klug. 2001. Relationship between excitation energy transfer, trapping, and antenna size in photosystem II. Biochemistry. 40:4026–4034. [DOI] [PubMed] [Google Scholar]
- 11.Van Grondelle, R., V. Sundstrom, J. P. Dekker, and T. Gillbro. 1994. Energy trapping and transfer in photosynthesis. Biochim. Biophys. Acta (Bioenerg.). 1187:1–65. [Google Scholar]
- 12.Nuijs, A. M., H. J. van Gorkom, J. J. Plijter, and L. N. M. Duysens. 1986. Primary-charge separation and excitation of chlorophyll-alpha in photosystem-ii particles from spinach as studied by picosecond absorbency-difference spectroscopy. Biochim. Biophys. Acta. 848:167–175. [Google Scholar]
- 13.Durrant, J. R., G. Hastings, D. M. Joseph, J. Barber, G. Porter, and D. R. Klug. 1992. Subpicosecond equilibration of excitation-energy in isolated photosystem-II reaction centers. Proc. Natl. Acad. Sci. USA. 89:11632–11636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schatz, G. H., H. Brock, and A. R. Holzwarth. 1987. Picosecond kinetics of fluorescence and absorbency changes in photosystem-II particles excited at low photon density. Proc. Natl. Acad. Sci. USA. 84:8414–8418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gatzen, G., M. G. Muller, K. Griebenow, and A. R. Holzwarth. 1996. Primary processes and structure of the photosystem II reaction center.3. Kinetic analysis of picosecond energy transfer and charge separation processes in the D1–D2-cyt-b559 complex measured by time-resolved fluorescence. J. Phys. Chem. 100:7269–7278. [Google Scholar]
- 16.Groot, M. L., N. P. Pawlowicz, L. J. G. W. van Wilderen, J. Breton, I. H. M. van Stokkum, and R. van Grondelle. 2005. Initial electron donor and acceptor in isolated Photosystem II reaction centers identified with femtosecond mid-IR spectroscopy. Proc. Natl. Acad. Sci. USA. 102:13087–13092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Novoderezhkin, V. I., E. G. Andrizhiyevskaya, J. P. Dekker, and R. van Grondelle. 2005. Biophys. J. 89:1464–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Groot, M. L., E. J. G. Peterman, P. J. M. van Kan, I. H. M. van Stokkum, J. P. Dekker, and R. van Grondelle. 1994. Temperature-dependent triplet and fluorescence quantum yields of the photosystem-II reaction-center described in a thermodynamic model. Biophys. J. 67:318–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holzwarth, A. R., M. G. Muller, M. Reus, M. Nowaczyk, J. Sander, and M. Rogner. 2006. Kinetics and mechanism of electron transfer in intact photosystem II and in the isolated reaction center: Pheophytin is the primary electron acceptor. Proc. Natl. Acad. Sci. USA. 103:6895–6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Grondelle, R. 1985. Excitation, energy trapping and annihilation in photosynthetic systems. Biochim. Biophys. Acta. 811:147–195. [Google Scholar]
- 21.Schatz, G. H., H. Brock, and A. R. Holzwarth. 1988. Kinetic and energetic model for the primary processes in photosystem-II. Biophys. J. 54:397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miloslavina, Y., M. Szczepaniak, M. G. Muller, J. Sander, M. Nowaczyk, M. Rogner, and A. R. Holzwarth. 2006. Charge separation kinetics in intact photosystem II core particles is trap-limited. A picosecond fluorescence study. Biochemistry. 45:2436–2442. [DOI] [PubMed] [Google Scholar]
- 23.Van Mourik, F., M. L. Groot, R. van Grondelle, J. P. Dekker, and I. H. M. van Stokkum. 2004. Global and target analysis of fluorescence measurements on photosystem 2 reaction centers upon red excitation. Phys. Chem. Chem. Phys. 6:4820–4824. [Google Scholar]
- 24.Nabedryk, E., M. Leonhard, W. Mantele, and J. Breton. 1990. Fourier-transform infrared difference spectroscopy shows no evidence for an enolization of chlorophyll-a upon cation formation either invitro or during P700 photooxidation. Biochemistry. 29:3242–3247. [DOI] [PubMed] [Google Scholar]
- 25.Breton, J., R. Hienerwadel, and E. Nabedryk. 1997. FTIR difference spectrum of the photooxidation of the primary electron donor of photosystem II. In Spectroscopy of Biological Molecules: Modern Trends. P. Carmona, R. Navarro, and A. Hernanz, editors. Kluwer, Dordrecht, The Netherlands, 101–108.
- 26.Noguchi, T., T. Tomo, and Y. Inoue. 1998. Fourier transform infrared study of the cation radical of P680 in the photosystem II reaction center: Evidence for charge delocalization on the chlorophyll dimer. Biochemistry. 37:13614–13625. [DOI] [PubMed] [Google Scholar]
- 27.Tavitian, B. A., E. Nabedryk, W. Mantele, and J. Breton. 1986. Light-induced Fourier-transform infrared (FTIR) spectroscopic investigations of primary reactions in Photosystem-I and Photosystem-II. FEBS Lett. 201:151–157. [Google Scholar]
- 28.Nabedryk, E., S. Andrianambinintsoa, G. Berger, M. Leonhard, W. Mantele, and J. Breton. 1990. Characterization of bonding interactions of the intermediary electron-acceptor in the reaction center of Photosystem-II by FTIR spectroscopy. Biochim. Biophys. Acta. 1016:49–54. [Google Scholar]
- 29.Noguchi, T., T. Tomo, and C. Kato. 2001. Triplet formation on a monomeric chlorophyll in the photosystem II reaction center as studied by time-resolved infrared spectroscopy. Biochemistry. 40:2176–2185. [DOI] [PubMed] [Google Scholar]
- 30.Groot, M. L., J. Breton, L. J. G. W. van Wilderen, J. P. Dekker, and R. van Grondelle. 2004. Femtosecond visible/visible and visible/mid-IR pump-probe study of the photosystem II core antenna complex CP47. J. Phys. Chem. B. 108:8001–8006. [Google Scholar]
- 31.Di Donato, M., R. van Grondelle, I. H. M. van Stokkum, and M. L. Groot. 2007. Excitation energy transfer in the photosystem II core antenna complex CP43 studied by femtosecond visible/visible and visible/mid-infrared pump probe spectroscopy. J. Phys. Chem. B. 111:7345–7352. [DOI] [PubMed] [Google Scholar]
- 32.Groot, M. L., R. N. Frese, F. L. de Weerd, K. Bromek, A. Petterson, E. J. G. Peterman, I. H. M. van Stokkum, R. van Grondlle, and J. P. Dekker. 1999. Spectroscopic properties of the CP43 core antenna protein of photosystem II. Biophys. J. 77:3328–3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Stokkum, I. H. M., D. S. Larsen, and R. van Grondelle. 2004. Global and target analysis of time-resolved spectra. Biochimica et Biophysica Acta (Bioenergetics). 1657:82–104. [DOI] [PubMed] [Google Scholar]
- 34.Andrizhiyevskaya, E. G., J. A. Butista, B. A. Diner, R. van Grondelle, and J. P. Dekker. 2005. Energy transfer and charge separation in the Photosystem II core complex studied by time-resolved fluorescence. In Energy Transfer and Trapping in Photosynthetic Complexes with Variable Antenna Size. PhD Thesis. Vrije Universiteit Amsterdam, Amsterdam, The Netherlands. 81–96.
- 35.Rech, T., J. R. Durrant, D. M. Joseph, J. Barber, G. Porter, and D. R. Klug. 1994. Does slow energy-transfer limit the observed time constant for radical pair formation in photosystem-II reaction centers. Biochemistry. 33:14768–14774. [DOI] [PubMed] [Google Scholar]
- 36.Monshouwer, R., I. O. de Zarate, F. van Mourik, and R. van Grondelle. 1995. Low-intensity pump-probe spectroscopy on the b800 to b850 transfer in the light-harvesting 2 complex of Rhodobacter sphaeroides. Chem. Phys. Lett. 246:341–346. [Google Scholar]
- 37.Novoderezhkin, V., R. Monshouwer, and R. van Grondelle. 1999. Exciton (de)localization in the LH2 antenna of Rhodobacter sphaeroides as revealed by relative difference absorption measurements of the LH2 antenna and the B820 subunit. J. Phys. Chem. B. 103:10540–10548. [Google Scholar]
- 38.Salverda, J. M., M. Vangris, B. P. Krueger, G. D. Scholes, A. R. Czarnoleski, V. Novoderezhkin, H. van Amerongen, and R. van Grondelle. 2003. Energy transfer in light-harvesting complexes LHCII and CP29 of spinach studied with three pulse echo peak shift and transient grating. Biophys. J. 84:450–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saito, K., T. Kikuchi, M. Nakayama, K. Mukai, and H. Sumi. 2006. A single chlorophyll in each of the core antennas CP43 and CP47 transferring excitation energies to the reaction center in Photosystem II of photosynthesis. J. Photochem. Photobiol. a. Chem. 178:271–280. [Google Scholar]
- 40.Germano, M., A. Pascal, A. Y. Shkuropatov, B. Robert, A. J. Hoff, and H. J. van Gorkom. 2002. Pheophytin-protein interactions in photosystem II studied by resonance Raman spectroscopy of modified reaction centers. Biochemistry. 41:11449–11455. [DOI] [PubMed] [Google Scholar]
- 41.Hughes, J. L., B. J. Prince, E. Krausz, P. J. Smith, R. J. Pace, and H. Riesen. 2004. Highly efficient spectral hole-burning in oxygen-evolving photosystem II preparations. J. Phys. Chem. B. 108:10428–10439. [Google Scholar]
- 42.Arskold, S. P., B. J. Prince, E. Krausz, P. J. Smith, R. J. Pace, R. Picorel, and M. Seibert. 2004. Low-temperature spectroscopy of fully active PSII cores. Comparisons with CP43, CP47, D1/D2/cyt b(559) fragments. Journal of Luminescence. 108:97–100. [Google Scholar]
- 43.Groot, M. L., E. J. G. Peterman, I. H. M. van Stokkum, J. P. Dekker, and R. van Grondelle. 1995. Triplet and fluorescing states of the CP47 antenna complex of photosystem-II studied as a function of temperature. Biophys. J. 68:281–290. [DOI] [PMC free article] [PubMed] [Google Scholar]