

Abstract
A new membrane probe, based on the perylene imide chromophore, with excellent photophysical properties (high absorption coefficient, quantum yield (QY) ≈ 1, high photostability) and excited in the visible domain is proposed for the study of membrane rafts. Visualization of separation between the liquid-ordered (Lo) and the liquid-disordered (Ld) phases can be achieved in artificial membranes by fluorescence lifetime imaging due to the different decay times of the membrane probe in the two phases. Rafts on micrometer-scale in cell membranes due to cellular activation can also be observed by this method. The decay time of the dye in the Lo phase is higher than in organic solvents where its QY is 1. This allows proposing a (possible general) mechanism for the decay time increase in the Lo phase, based on the local field effects of the surrounding molecules. For other fluorophores with QY < 1, the suggested mechanism could also contribute, in addition to effects reducing the nonradiative decay pathways, to an increase of the fluorescence decay time in the Lo phase.
INTRODUCTION
Since the postulation of the formation of lipid rafts (1), many efforts were directed to demonstrate their existence and function in living cells. Lipid rafts are membrane microdomains enriched in the liquid-ordered (Lo) phase resulting from packing of the long saturated alkyl chains of sphingolipids and cholesterol. They are thought to be segregated from the liquid-disordered (Ld) phase composed of unsaturated phospholipids. Initially, biochemical approaches based on detergent extraction were used to study the membrane rafts, but they are prone to artifacts and they do not provide information about the dynamics of molecules composing the rafts (2). Therefore, methods capable to explore rafts in situ, with few perturbations, were developed. Among them, visualization of lipid phases by fluorescence microscopy could be achieved due to the different partition characteristics of fluorophores in the various phases. In this way, Lo/Ld phase separation was demonstrated in ternary lipid mixtures forming artificial membranes such as supported mono-/bilayers (Langmuir-Blodgett films) (3–5), free-standing planar bilayers (6), and giant unilamellar vesicles (7–14). In living cells, rafts on micrometer-scale were observed but only after cellular activation (15–18). Therefore, if rafts exist in resting cells, they are supposed to be organized on the submicrometer scale (below the resolution of the optical microscopy of ≈300 nm), having a very fast exchange (in the order of hundreds of ns) of molecules with the bulk phase (18,19). Fluorescence methods which extend the resolution down to the submicron scale (hetero- and homo-fluorescence energy transfer, fluorescence correlation spectroscopy, single molecule techniques) are used to investigate molecular proximity, lateral diffusion and clustering of lipid rafts in living cells (14,20–23).
The majority of the commonly used fluorescent dyes for lipid phase characterization (diphenyl hexatriene and its derivatives, pyrene, perylene, laurdan, and recently synthesized polyene-lipids (24)) are UV-excitable probes, generating complications in microscopy due to the special optical parts that have to be used (lasers, lenses) and due to the very high autofluorescence of living cells in this spectral region. One possibility to overcome these problems is to excite them nonlinearly, in a two-photon absorption process (10,25). Phase separation was also observed using phospholipids labeled with a fluorescent marker excitable in the visible region (e.g., tetramethyl rhodamine, NBD-7-nitrobenz-2-oxa-1,3-diazol) (5,9,23). For the identification of lipid rafts, fluorescent cholesterol derivatives (dehydroergosterol, NBD-cholesterol), filipin-cholesterol fluorescent complexes, or fluorescently-labeled cholera toxin subunit B were used (26–28).
Despite this variety of fluorophores available for membrane studies, there is an ongoing search for new chromophores with better photophysical properties (high absorption coefficient in the visible domain, high quantum yield, good photostability). As living cell processes are nowadays explored with single molecule techniques, more stable fluorophores are necessary to follow the biological pathways on longer and more relevant timescales (29). Most of the commercially available dyes are photobleached in single molecule studies after 10–20 s (30). In this article, an amphiphilic perylene monoimide (PMI) derivative able to intercalate between phospholipids is proposed for the study of lipid rafts.
Initial experiments showed a very fast incorporation of the PMI derivative into living cells. Fluorescent membrane labeling occurs already after 5 min at room temperature. The time-resolved measurements presented in this article show that the amphiphilic PMI derivative has different decay times in Ld and Lo phases, allowing the visualization of lipid domains in model membranes and living cells by fluorescence lifetime imaging (FLIM). The FLIM technique is often used for membrane studies in association with fluorescence energy transfer to detect the colocalization of two biologically active molecules by monitoring the lifetime decrease of a fluorescent donor in the presence of an acceptor (31,32). In our approach, domains on micrometer scale in artificial and natural membranes can be visualized based on the different decay times of the same fluorophore. These domains can be identified even though the partition of the fluorophore is not equal among different phases, because lifetime determination is not dependent on the concentration if strong aggregation of the dye does not occur (33,34). Previous reports about the possibility to identify the different lipid phases using time-resolved methods were done with diphenyl hexatriene (DPH) and its derivative TMA-DPH, laurdan, or parinaric acids (35–40). Very recently, other groups have demonstrated the possibility to use styryl dye derivatives to visualize membrane rafts based on their spectral shifts (41) or on their different decay times observed in Lo and Ld phases (42). The latter study also applies the FLIM technique for the visualization of membrane rafts. However, the PMI derivative we use offers several advantages. First, the chromophore is more photostable than most of the commercially available dyes. Second, its quantum yield is practically equal to 1 in organic solvents, and therefore the decay time modifications that we observe in the Lo phase cannot be attributed to further reduction of the nonradiative pathways (e.g., due to bond rotation possibly leading to cis-trans isomerization). This allows us to propose a possible mechanism explaining the increase of the decay time of dyes embedded in the Lo phase, based on our previous experience with individual chromophores studied in polymer matrices (43).
MATERIALS AND METHODS
Chemicals
The structure of the fluorescent molecule used in this study, n-(2,6-diisopropylphenyl)-9-(4-carboxyphenyl)perylene-3,4-dicarboximide (PMI-COOH) is presented in Fig. 1. The synthesis of this dye was published elsewhere (44).
FIGURE 1.

Structure of the investigated perylene monoimide amphiphilic dye (PMI-COOH).
Milli-Q water (18 MΩ resistance; Millipore, Billerica, MA) was used for all experiments. All solvents were spectroscopic grade (Sigma-Aldrich Chemie, Steinheim, Germany). Phosphate buffer saline (PBS) with pH 7.4 was purchased from Sigma and reconstituted with Milli-Q water.
Small unilamellar vesicles (SUVs) were prepared from dioleoyl phosphatidylcholine (DOPC), dipalmitoyl phosphatidylcholine (DPPC), and cholesterol (Sigma). The purity of the lipids was ≥99% and they were used as received. Appropriate amounts of lipids and PMI-COOH (molar ratio 500:1) from stock solutions in chloroform were mixed and the solvent was evaporated. PBS was added to form multilamellar vesicles (lipid concentration: 0.5 mg/ml). SUVs were obtained by 10 extrusions through a polycarbonate membrane with 100-nm pore diameter. Vesicles prepared only with DOPC were obtained at room temperature. Vesicles containing DPPC were prepared at 70°C and then cooled down to room temperature. All measurements using SUVs were done at room temperature, in 1-cm cuvettes. The solutions were optically clear.
Giant unilamellar vesicles (GUVs) were prepared by the electroformation method (45,46). A microscope chamber was constructed in a home-made holder using two ITO-covered glasses and a silicone rubber spacer. A few microliters of a stock solution of lipids and fluorescent dye in chloroform were evaporated under a flow of argon gas on the surface of the bottom cover glass. Milli-Q water was added in the chamber and an AC voltage of 3 V and 10 Hz was applied for 1–2 h at 65°C. GUVs were cooled down to room temperature before measurements. For confocal laser scanning microscopy, PMI-COOH and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiD-C18) (Molecular Probes, Eugene, OR) were used to simultaneously label the different lipid phases at a dye/lipid ratio of 1:1000 (for each dye). GUVs images were taken at room temperature.
Jurkat cells were a kind gift of Prof. Zeger Debyser (Laboratory of Molecular Virology and Gene Therapy, Rega Institute, Leuven, Belgium). They were grown in RPMI 1640 medium with 10% fetal bovine serum and 1% Penicillin plus Streptomycin (Gibco, Invitrogen, Paisley, Scotland). For confocal microscopy and fluorescence lifetime imaging, cells were attached on coverslips covered with poly-L-lysine (Sigma). Washing, labeling, and visualization procedures were performed in PBS. PMI-COOH was added from a stock solution in tetrahydrofuran to a final dilution of 4 × 10−9 M in PBS supplemented with 2% bovine serumalbumin. Cell labeling was performed for 2 min at room temperature. Cholesterol depletion from membranes was done by incubating the cells for 20 min at 37°C with 10 mM methyl-β-cyclodextrin (Sigma) in PBS. Rafts coalescence was induced using a modified procedure of Janes et al. (15). Cells were incubated at room temperature with 4 μl mouse anti-CD3 antibodies (Sigma) for 10 min, followed by 2 min incubation with 2 μl anti-mouse IgG antibodies (Sigma). For colocalization experiments of PMI-COOH in rafts (see Fig. 8 D), labeled anti-mouse IgG antibodies with Alexa 647 (Molecular Probes) were used. All cell images were taken at room temperature.
FIGURE 8.

Fluorescence intensity (gray) and fluorescence lifetime images (color) obtained for PMI-COOH in: (A) normal Jurkat cell; (B) Jurkat cell after cholesterol extraction using methyl-β-cyclodextrin. (C) Jurkat cell stimulated by cross-linking membrane CD3 receptors using specific antibodies; the yellow arrow points toward a region where the PMI-COOH lifetime is different from the rest of the membrane, which can be due to the formation of a big lipid raft. The yellow rectangle indicates the area that is magnified in the fourth panel of C. The third panel of C is the distribution of the decay time values only from the plasma membrane, obtained by applying a mask to the whole cell image. (D) Experiment with two dyes on a stimulated Jurkat cell. (First panel) Fluorescence intensity image of the distribution of anti-IgG mouse antibodies labeled with Alexa 647 used to cross-link the anti-CD3 antibodies and to induce raft formation. (Second panel) Fluorescence lifetime image of PMI-COOH. (Third panel) Superposition of both channels. (Fourth panel) Enlarged picture of the area delineated by the yellow rectangle in the third panel of D. The PMI-COOH images were recorded using pulsed 488-nm excitation and the emission was collected with a 555-nm longpass filter (for one-dye experiments) or with a 580/60-nm bandpass filter (for two-dye experiments). All FLIM images are represented using the same scale bar, from 3 to 6 ns. Alexa 647 was excited with a continuous wave 633-nm laser and the emission was collected using a 670/50-nm bandpass filter. (E) Distribution of PMI-COOH decay times for images A–C: normal cell (blue); cell after cholesterol extraction (red); and stimulated cell, (green). (F) Average distributions of PMI-COOH decay times for plasma membranes of seven cells in each category: normal cells (blue), cells after cholesterol depletion (red), and stimulated cells (green).
Steady-state measurements were performed with a Perkin-Elmer Lambda 40 spectrophotometer (Wellesley, MA) for absorption spectra and with a Spex Fluorolog 3-22 fluorimeter (Jobin Yvon-Spex Instruments, Edison, NJ) for fluorescence spectra. Emission spectra were corrected according to standard procedures (47). The fluorescence quantum yield was determined using a peryleneimide, metasubstituted polyphenylene dendrimer as reference, with quantum yield ≈1 in toluene (48). This compound was chosen because of its similar structure with PMI-COOH and because of the very good superposition of the absorption and emission spectra.
The single photon timing setup (SPT) is described in detail elsewhere (49,50). In brief, the output of a mode-locked Ti:sapphire laser (Tsunami 3950D, Spectra Physics, Mountain View, CA) pumped by a diode-pumped Nd:YVO4 laser (Millenia Xs, Spectra Physics) was directed into a frequency doubler to deliver the 488-nm wavelength used for excitation. A pulse picker was used to reduce the repetition rate of the laser from 82 MHz down to 4.1 MHz. Emission light, passing through a sheet polarizer set to the magic angle (54.7°), is focused on a monochromator (9030DS, Sciencetech, London, ON, Canada) to obtain spectral separation and detected with a microchannel plate (R3809U, Hamamatsu Photonics, Hamamatsu, Japan). Acquisition was done using a TCSPC PC card (SPC 630, Becker & Hickl, Berlin, Germany). The full width at half-maximum of the experimental instrument response function (obtained with a scattering solution) was in the order of 50 ps. Decays were collected in 4096 channels with 10,000 counts in the maximum. SPT data were analyzed using a program based on a Marquardt algorithm, by convoluting the experimental instrument response function with a linear combination of exponentially decaying functions. The fitting was evaluated by the value of the reduced χ-square parameter (maximum acceptable value is considered to be 1.2) and also by the appearance of the residuals. The contributions to the total fluorescence intensity of the decay times recovered after global analysis were estimated using the relative amplitudes:
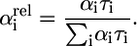 |
Confocal laser scanning microscopy images were acquired with an IX70 Olympus microscope equipped with the Fluoview FV500 scanning unit (Olympus, Tokyo, Japan). Continuous excitation at 488 nm was obtained from an Argon-ion laser (163-C1210, Spectra Physics), and at 633 nm wavelength from a Helium-Neon laser (model 117A, Spectra Physics). The excitation light was directed on the sample through an oil immersion objective lens (60×, 1.4 NA), the fluorescence being separated by a 488/543/633 nm dichroic mirror. The fluorescence was further split in two channels using a 630-nm dichroic mirror. PMI-COOH emission was collected in the first channel using a 560-nm longpass filter, while the emission from DiD-C18 was visualized through a 660-nm longpass filter in the second channel. The pinhole size was 150 μm for PMI-COOH images and 180 μm for the DiD images. Sequential scanning of the two wavelengths was applied, to avoid the fluorescence leakage of PMI-COOH into the second channel and to minimize the exposure of both dyes to unwanted excitation light.
Fluorescence lifetime imaging (FLIM) was realized using a home-made setup. The same system for excitation as for the SPT setup was used, but the light is directed on the back aperture of an Olympus IX 70 inverted microscope. An oil immersion lens (Olympus 100×; 1.3 NA) was used. Samples were mounted on a scanning stage (Physik Instrumente, Waldbronn, Germany) driven by a home-made program. The images were acquired using 256 × 256 pixels, with an integration time of 10 ms per pixel. The time necessary to acquire a FLIM image was ≈11 min. Depending on the sample, the excitation intensity was set between 850 and 6000 W/cm2 measured after the objective lens. Fluorescence was collected by the same lens, then passed through a dichroic mirror (Chroma Technology, Brattleboro, VT) and focused on a 100-μm pinhole. Two or more longpass filters (500, 515, 525, 535, and 555 nm, Chroma Technology) were used to reject the remaining excitation light and, partially, the cellular autofluorescence. The emitted light was collected by an avalanche photodiode (APD) (SPCM-AQR14, EG&G, Vaudreuil, Quebec, Canada). The signal detected by the APD was recorded with a TCSPC PC card (SPC 630, Becker & Hickl) using the FIFO mode (first-in, first-out). This allows the recording of the time interval of each photon with respect to the excitation pulse (microtime) and also of the time lag with respect to the previous detected photon (macrotime). The stored data are used in a home-made program to reconstruct the fluorescence intensity image, as well as the FLIM image. The intensity image is given by the sum of the photons accumulated in one pixel during the acquisition time. To reconstruct the FLIM image, the microtimes of photons belonging to a certain pixel are averaged, and the conversion to lifetimes was done as explained in Supplementary Material. The instrumental response function was obtained by using a solution of erythrosine in water. The lifetime distributions for separated plasma membranes in living cells were obtained by importing the FLIM images as raw data in the ImageJ program (National Institutes of Health, Bethesda, MD) and applying a mask. For the experiments with two dyes (PMI-COOH and Alexa 647), two-color excitation was used (pulsed 488 nm for PMI-COOH and continuous wave, 633 nm, for Alexa 647), the emission being split with a 630-nm dichroic mirror. The corresponding notch filter plus a 670/50-nm bandpass filter was used for the Alexa 647 detector, and two bandpass filters (580/60 nm and 580/70 nm) were used in front of the PMI-COOH detector. Leaking of PMI emission to the Alexa 647 channel was corrected by measuring a solution containing only PMI-COOH. The fluorescence intensity ratio between the two channels is multiplied with the intensity image of PMI-COOH channel, and the product is then subtracted from the intensity image obtained in the Alexa 647 channel.
Geometry optimization of molecules in the ground state (S0) has been performed using the semiempirical Hartree-Fock Austin Model 1 (AM1) technique in the Ampac package (Semichem, Shawnee, KS). For the excited state (S1), geometry optimization was done by coupling the semiempirical Hartree-Fock Austin Model 1 (AM1) method to a full configuration interaction scheme within a limited active space, as implemented in the Ampac program. ZINDO calculations applied to the optimized geometry of the S1 state (51) were used to estimate the transition dipole moments and the transition frequencies, allowing thus the calculation of radiative lifetimes for the vertical transition S1→S0. Polarizabilities were determined by a sum-over-states method over all states involved in the configuration interaction scheme in the same program. The influence of a polarizable element in the environment of a chromophore on the chromophore lifetime was determined according to Vallée et al. (43).
RESULTS AND DISCUSSIONS
Photophysical properties of PMI-COOH in organic solvents
The absorption spectrum of PMI-COOH (Fig. 2) extends in the visible region (the commonly used 488- and 514-nm lines can efficiently excite the dye), making it suitable for one-photon fluorescence microscopy experiments in living cells.
FIGURE 2.

Normalized absorption and emission spectra (the excitation wavelength was 488 nm), quantum yields, and decay times of the PMI-COOH derivative in different solvents (methyl cyclohexane MCH, diethyl ether DEE, and chloroform CHCl3).
The extinction coefficient for this PMI derivative is 40,000 M−1 cm−1 at 510 nm in diethyl ether, while the quantum yield is 1 within the experimental error (Fig. 2). A more detailed characterization of the photophysical properties of the molecule in organic solvents with different polarities and polarizabilities is presented in Supplementary Material. The molecule is essentially nonfluorescent in water. Based on the spectral and decay time changes observed in water and water mixtures with organic solvents (see Supplementary Material), we attribute the reduction of the fluorescence quantum yield to an aggregation process of PMI.
The decay time of the monomer is not very sensitive to the solvent polarity, and has a value of ∼4 ns (Fig. 2 and Supplementary Material). Therefore, the decay time changes that we observe in artificial membranes are not determined only by modifications of the bilayer hydrophobicity (i.e., polarity), but should be related to other processes (see below).
An important property of the PMI chromophore and its derivatives is the much higher photostability compared to the commercially available dyes. The high photostability was proven in single molecule studies (52).
Photophysical characteristics of PMI-COOH in artificial membranes
Because perylene is a hydrophobic molecule used for fluidity and lipid phase transition studies in artificial membranes (10,53–55), we assumed that this amphiphilic PMI derivative will have the ability to be incorporated among the phospholipids of a bilayer. This can be demonstrated if a change in the photophysical properties of the molecule (emission spectral maximum, emission polarization, decay time, etc.) is observed as a function of lipid phase.
For these experiments, SUVs with different composition were prepared. Two phospholipids were used to obtain phase coexistence at room temperature: DOPC for the Ld phase (the main transition temperature Tm = −20°C) and DPPC (Tm = 41°C) for the gel phase. Cholesterol was added to DPPC in a range of 5–30 mol % to form the Lo phase.
PMI-COOH excitation and emission spectra
The excitation spectrum in the Ld phase shows a residual vibrational structure (Fig. 3 A). In the gel phase, the intensity above 540 nm increases, while the vibrational peak at ∼525 nm is very much diminished (Fig. 3 A). Adding increasing concentrations of cholesterol to DPPC results in the gradual decreasing of the intensity above 540 nm and in the reappearance of vibrational peak at ∼525 nm (Fig. 3 B). These changes in the excitation spectra were considered to be an indication for the aggregation tendency of PMI-COOH in the gel phase SUVs at the dye/lipid ratio used (1:500). Therefore, the modifications observed in the emission spectra and the results obtained from time-resolved measurements (see below) were interpreted in terms of coexistence and/or dominance of monomers and/or aggregates in different phases.
FIGURE 3.

Excitation (A) and emission (C) spectra of PMI-COOH in SUVs composed of Ld phase (DOPC), gel phase (DPPC), and their mixtures (25, 50, and 75% DPPC). Excitation (B) and emission (D) spectra of PMI-COOH in SUVs composed of DPPC (gel phase) plus increasing amounts of cholesterol (0–30 mol %), which generate an increased proportion of the Lo phase. (E) Spectral changes in the emission of PMI-COOH between Ld phase (DOPC, black line) and Lo phase (DPPC plus 30 mol % cholesterol, red line). The excitation spectra were taken by monitoring the emission at 600 nm. The emission spectra were taken using 488 nm excitation. All spectra were recorded at room temperature.
In the case of pure phases, a strong bathochromic shift of the emission maximum is observed between the Ld (570 nm) and the gel (615 nm) phase (Fig. 3 C), supporting the strong aggregation of the dye in the gel phase, while monomers dominate in the fluid phase. This is probably due to the fact that the quite bulky PMI chromophore cannot accommodate among the rigidly organized alkyl chains in the gel phase and tends to segregate in separate domains. In the binary mixture of Ld and gel phases, the shift of the emission maximum toward longer wavelengths is observed only when the gel phase represents at least 75%, which can be explained by a preferential partition of this dye into the fluid phase (observed also in confocal microscopy using GUVs—see below) and the lower fluorescence quantum yield of the aggregates (56).
Changes are more dramatic when mixing the gel phase (DPPC) with cholesterol (Fig. 3 D). The maximum of the emission spectrum of the pure gel phase (615 nm) is blue shifted (to 556 nm) when 30 mol % cholesterol is added, suggesting that aggregates formed in the pure gel phase are more and more dissociated in the presence of higher amounts of cholesterol in the membrane. Furthermore, while in the Ld phase almost no vibrational structure can be seen, vibrational fine structure is again present in the presence of 30 mol % cholesterol.
PMI-COOH decay times
For all the investigated lipid phases, the PMI-COOH decay times could not be analyzed monoexponentially. Multiexponential or nonexponential decays are generally reported for fluorescent probes incorporated in artificial membranes (57) and Langmuir-Blodgett films (58,59). If an aggregation process is taken into consideration in our case, then the different decay times could be attributed to PMI-COOH monomers and aggregates. Monomers were considered to have a maximum contribution at 560 nm (according to their spectral characteristics), which diminishes at longer wavelengths. Aggregates show a very modest contribution at 560 nm, which increases at wavelengths longer than 610 nm. Given the fact that the fluorescence decay curves were recorded on 4096 channels, with 10,000 counts in the maximum and were analyzed by global analysis (60,61), the errors for the estimation of the decay times and of the preexponential factors are <10%, as determined from the calculation of confidence intervals.
For the Ld phase, the global analysis of the decays recorded at different emission wavelengths (Fig. 4 A) demonstrate that, for the excitation at 488 nm, the contribution of monomers (τM = 4.8 ns) to the emission is 82–95% (between 560 and 650 nm), while aggregates (τA = 7.2 ns) represent only 2.5–20%. A third component of ∼1 ns, which contribution decreases and then becomes negative at long wavelengths, can be due to a small proportion of monomers undergoing Förster type energy transfer to the aggregates present in their close proximity. This would suggest a nonhomogeneous distribution of monomers and aggregates. Indeed, at the dye/lipid ratio that we used, the average distance between the monomers and aggregates (≈160 nm) is higher than the maximum Förster distance (≈10 nm). It is also possible that the negative contribution (which was not observed in the other phases) is due to a fraction of aggregates undergoing dynamic reorganization on the timescale of their excited state lifetime.
FIGURE 4.

Contributions of the different PMI-COOH decay times measured for SUVs formed of different lipid phases as determined from global analysis (excitation at 488 nm). (A) Ld phase (DOPC) as a function of wavelength. (B) Gel phase (DPPC) as a function of wavelength. (C) Lo phase (DPPC plus 30 mol % cholesterol) as a function of wavelength. (D) DPPC and cholesterol as a function of cholesterol concentration at 560 nm.
For the gel phase (Fig. 4 B), the monomer decay time is comparable to that in the Ld phase (4.7 ns). Aggregates have a longer decay time (12 ns) and a much higher contribution (25–80% between 560 and 650 nm) compared to the Ld phase. The short decay time (1.3 ns) can again be attributed to the energy transfer process from monomers to aggregates. This should be possible if we consider the segregation of the dye molecules in separate domains, already suggested by the spectral characteristics of PMI-COOH in the gel phase (see above). The contribution of the long component decreases by reducing the dye/lipid ratio, supporting the assignment of the long decay time component to aggregates (see Supplementary Material).
We were also interested to determine the fluorescence decay times of PMI-COOH incorporated into SUVs composed of DPPC and cholesterol due to the dramatic changes in the emission spectra that were observed, and also because the association between cholesterol and saturated phospholipids triggers the formation of the particular Lo phase. When 30 mol % cholesterol is added to DPPC, there is a dominant species contributing >90% to the decay at the emission maximum (560 nm) and its contribution decreases at long wavelengths (Fig. 4 C). According to the spectral characteristics, this contribution should be attributed to a monomeric species. The intriguing aspect is the longer decay time (τM = 6.5 ns) compared to the PMI-COOH monomers (∼4 ns in organic solvents). The long and the short decay times are similar to those observed in pure DPPC, but their contribution is lower in the presence of cholesterol. To get more insight, global analysis was performed for SUVs composed of DPPC and different concentrations of cholesterol (0–30 mol %) at 560 nm (Fig. 4 D). This analysis shows a gradual increase in the contribution of the 6.5 ns component, while the contribution of the 4.7 ns component decreases when more cholesterol is added. Aggregates (12 ns component) are reduced by increasing the cholesterol concentration. These results show that cholesterol reduces aggregation and induces a particular long decay time for the PMI-COOH monomer when incorporated into the phospholipid bilayer.
Possible explanation for the increased decay time of PMI-COOH monomer in the Lo phase
As can be observed in Fig. 3 E, the emission spectrum in the Lo phase has a more defined vibrational structure, with a blue-shifted peak which can be attributed to the monomer species. However, the monomer species has a longer decay time (6.5 ns) compared to the Ld phase (4.8 ns). Other fluorophores were also reported to have longer decay times in cholesterol-containing bilayers than in pure phospholipid bilayers (37,38,62–64). Generally, this is attributed to the increased hydrophobicity of cholesterol-containing bilayers compared to the fluid bilayers, which are more permeable for the water molecules (65–67), or to an increased viscosity (68). Very recently, two groups report a spectral shift toward shorter wavelengths and an increase of the decay time of styryl derivatives in the Lo phase (41,42). Since the quantum yield of these dyes is <1 in organic solvents (69), one can assume that the increased decay time in the Lo phase could be due to the reduction of the nonradiative deactivation. Indeed, the tight molecular packing in the Lo phase can reduce nonradiative decay processes such as bond rotation in the limit leading to cis-trans isomerization, consequently increasing the fluorescence decay time. At the same time, blue spectral shifts and narrower emission bands can be observed because the surrounding molecules do not reorient around the chromophore during the excited state lifetime. In our case, the puzzling observation is that the decay time of PMI-COOH measured in organic solvents remains at ∼4 ns for a quantum yield close to 1 within the experimental error, and therefore, the value of 6.5 ns measured for the monomer in the Lo phase cannot be the consequence of further reduction of nonradiative deactivation pathways, and must be due to an increase of the radiative lifetime.
The purely radiative lifetime τ of a fluorescent molecule depends on the spontaneous emission rate Γtot by a relation of inverse proportionality:
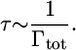 |
(1) |
In its turn, Γtot is proportional with the square of the transition dipole moment μtot of the probe molecule:
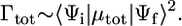 |
(2) |
This implies that a reduced transition dipole moment corresponds to the increased decay time of 6.5 ns of PMI-COOH, in comparison with the situation when the decay time is 4.8 ns. We tentatively explain the reduction of the transition dipole moment for PMI-COOH in the Lo phase by applying a microscopic model elaborated in our group for chromophores embedded in polymer matrices, seen as inhomogeneous lattices of polarizable elements (polymer chain segments) and voids (free volume) (43). The model is based on the local field effect exerted by the surrounding elements on the investigated probe. Essentially, the transition dipole moment μs of a dye (e.g., PMI-COOH) generates an electric field which induces an opposite dipole μind in the polarizable neighboring molecules (Fig. 5 A), if the neighboring molecules are favorably aligned during the excited state lifetime. This results in a reduced transition dipole moment (μtot = μs – μind) and, consequently, to an increased decay time of the dye (Eqs. 1 and 2). (For more details about this model, see (43).) Our calculations show that when an element with the polarizability of cholesterol is placed transversally at 4–5 Å close to PMI-COOH, its effect is to increase the fluorophore decay time by ≈1.3 times, which is in agreement with the ratio of the experimental values (Fig. 5, A and C). The presence of cholesterol favorably aligned at an optimal distance can be explained by the tight packing of the molecules in the Lo phase and by the possibility of hydrogen bonding between the −OH group of cholesterol and the −COOH group of the dye. The rest of the chromophore, being more hydrophobic, should sit between the fatty alkyl chains, probably parallel with the sterol ring. Furthermore, the calculated length of the dye molecule is 20 Å, very similar to the length of cholesterol (18 Å). Due to the comparable lengths and amphiphilic structures, it seems thus very probable that the PMI-COOH molecule will adopt a similar localization as cholesterol inside the bilayer (67,70). When considering an element with the polarizability of a phospholipid molecule in the vicinity of PMI-COOH in the Ld phase, its radiative lifetime should be comparable with the one in the Lo phase, which is in contradiction with the experimental results. This means that we have to take into consideration the less tight packing in the Ld phase, which generates a high proportion of free volume (∼40% as demonstrated in molecular dynamics simulation studies (67)). One has also to take into account the fact that a hydrogen bond between DOPC and PMI-COO− (dissociated at pH 7.4, which is the pH of the buffer used to prepare the SUVs) cannot take place in the Ld phase. The reason is that neither the phosphate (dissociated at pH 7.4), nor the choline forming the phospholipid headgroups can provide the necessary proton. To simulate the free volume in the Ld phase, a void was placed near the PMI-COOH molecule (Fig. 5 D), which does not influence the decay time. In the gel phase (pure DPPC), the proportion of the free volume is reduced, but the bulky PMI-COOH molecule cannot insert favorable between the highly ordered alkyl chains. This results in the aggregation of the probe, while the few monomers present either will disrupt the packing (thus creating locally an environment with a high proportion of free volume), or will prefer a region where defects already exist. This can explain why the same monomer decay time is detected in both neat DOPC and DPPC.
FIGURE 5.

Schematic representation of DPPC monolayers with PMI-COOH molecules. (A) The electrical field generated by the transition-induced dipole moment of the dye (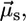 green arrow) determines an induced dipole moment in the polarizable molecule placed in its vicinity (
green arrow) determines an induced dipole moment in the polarizable molecule placed in its vicinity (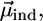 blue arrow). The resulting transition dipole moment of the dye (
blue arrow). The resulting transition dipole moment of the dye (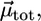 red arrow) is the sum of the two vectors and has smaller amplitude. (B) Lo phase. Due to the tight packing of molecules in this phase, a cholesterol molecule (representing the polarizable element) was placed near a PMI-COOH molecule. (C) The calculated decay time variation when cholesterol is placed near PMI-COOH, as a function of distance. The line is drawn at a distance of ∼4 Å between the centers of the molecules; smaller values are less probable due to steric effects. At a distance of 5 Å from PMI-COOH, the cholesterol molecule produces an increase in the dye lifetime of ≈1.3 times, comparable with the experimental value. (D) Ld phase. Because of the less tight packing of the molecules in this phase and of the presence of a high proportion of free volume (∼40% as demonstrated in molecular dynamics simulation studies (67)), a void is placed near the dye. This does not influence the decay time of PMI-COOH.
red arrow) is the sum of the two vectors and has smaller amplitude. (B) Lo phase. Due to the tight packing of molecules in this phase, a cholesterol molecule (representing the polarizable element) was placed near a PMI-COOH molecule. (C) The calculated decay time variation when cholesterol is placed near PMI-COOH, as a function of distance. The line is drawn at a distance of ∼4 Å between the centers of the molecules; smaller values are less probable due to steric effects. At a distance of 5 Å from PMI-COOH, the cholesterol molecule produces an increase in the dye lifetime of ≈1.3 times, comparable with the experimental value. (D) Ld phase. Because of the less tight packing of the molecules in this phase and of the presence of a high proportion of free volume (∼40% as demonstrated in molecular dynamics simulation studies (67)), a void is placed near the dye. This does not influence the decay time of PMI-COOH.
The local field effect described here can be observed for dyes with quantum yield of 1 (as PMI-COOH). For other dyes with quantum yield <1 embedded in the Lo phase, it would be difficult to discriminate between the contribution of this general effect and that of other effects reducing the nonradiative decay pathways (e.g., by tight molecular packing and increased microviscosity).
PMI-COOH distribution in different lipid phases
Because we are dealing with a new membrane probe, we investigated in a first step the distribution of PMI-COOH in different lipid phases using confocal laser scanning microscopy. GUVs were prepared from different lipid mixtures. The DiD-C18 fluorophore, which, according to literature, tend to partition preferentially (approximately three times more) in the gel phase due to the presence of the very long and saturated alkyl chains (71,12), was simultaneously used with PMI-COOH to discriminate between the lipid phases.
In the Ld phase, the two fluorophores have a uniform distribution in the GUVs membranes (data not shown). However, in a binary mixture (DOPC and DPPC) showing gel/Ld phase separation, we can visualize regions where the PMI derivative tends to accumulate (Fig. 6 A). Given the tendency of DiD-C18 to accumulate in the gel phase, it can be concluded that PMI-COOH prefers the Ld phase. This is in agreement with the spectroscopic data, which show modifications of the emission maximum only when >75% DPPC is mixed with DOPC. For those vesicles where the phase separation is not visible, the presence of domains with a size below the resolution of the optical microscope cannot be excluded.
FIGURE 6.

Confocal microscopy images representing PMI-COOH distribution (green), DiD-C18 distribution (red) and their superposition in GUVs. (A) Ld/gel phase mixture (DPPC/DOPC 0.60:0.40 molar ratio); white arrows indicate regions where PMI-COOH is preferentially accumulated (fluid phase) and yellow arrows point to regions where DiD-C18 is in higher concentration (gel phase); (B) Lo phase (DPPC/Cholesterol 0.70:0.30 molar ratio); (C) Lo/Ld phase separation (DPPC/DOPC/Cholesterol 0.56:0.24:0.20 molar ratio). The white arrow indicates a region where both PMI-COOH and DiD-C18 are concentrated; the yellow arrow points to a region where mainly PMI-COOH is distributed. (D) Three-dimensional image of two GUVs showing Lo/Ld phase separation. The same lipid composition as in panel C was used.
For the Lo phase (Fig. 6 B), differences in the partition of the two dyes are observed among the formed vesicles: some vesicles contain high concentration of both dyes, while others accumulate preferentially DiD-C18. This probably reflects the heterogeneous composition of GUVs when two or more lipids are used.
In GUVs composed of a ternary mixture showing Lo/Ld phase separation, there are regions where both DiD-C18 and PMI-COOH are concentrated (Fig. 6 C, white arrow), and other regions where mainly PMI-COOH is present (Fig. 6 C, yellow arrow). Analysis of the PMI-COOH intensity profiles along the membranes of the vesicles shows that the accumulation of the dye is 4.4 ± 1.3 times higher in one phase than in the other (estimation done on 15 vesicles). Because of the saturated alkyl chains, one would expect that DiD-C18 partitions together with DPPC in the Lo phase, leading to the conclusion that PMI-COOH also partitions more into the Lo than into the Ld phase. However, diffusion coefficients values obtained by fluorescence correlation spectroscopy (12) demonstrate that in the case of Lo/Ld phase separation, DiD-C18 tends to partition into the fluid phase (together with DOPC, not with DPPC). The situation is in contrast with the two components lipid mixture. This result is confirmed by our FLIM images showing a shorter decay time in the region where PMI-COOH accumulates preferentially, compared to the region where PMI tends to be excluded (see below).
To see if it is possible to obtain information about how the domains are distributed over the surface of GUVs in three dimensions, z scans were performed (Fig. 6 D). As the transition dipole moment of the molecule on the top of the vesicles is perpendicular to the polarization direction of the laser, the excitation of the dye should be less efficient (72). The z stacks in Fig. 6 D demonstrate that domains in which PMI preferentially accumulates could still be imaged, while the reduced intensity of the top part can be attributed to the reduced excitation efficiency.
Identification of lipid phases by fluorescence lifetime imaging (FLIM)
Even though the decays recorded in SUVs were not monoexponential, there is a dominant species in the Ld and in the Lo phase, corresponding to the monomer, whereas the other decay times have smaller weighted contributions. The very high sensitivity of the detection system used in this case (APD) allowed the labeling of GUVs with a much lower ratio dye/lipid (1 PMI-COOH for ≈125,000 lipid molecules) as compared to the ratio used for SPT measurements (1:500), thus reducing the possibility of the dye aggregation (see Supplementary Material). In these conditions, the long decay time due to aggregates and the short component due to energy transfer between monomers and aggregates can no longer be separated in the FLIM image, and only the dominant component given by the decay time of monomers will determine the contrast on the FLIM micrographs. Furthermore, the number of photons accumulated in one pixel during the acquisition time is much lower in this case than in the SPT measurements, making impossible to discriminate two or more decay times especially when they have small contributions (see Supplementary Material). Fig. 7, A and B, shows lifetime images of PMI-COOH-labeled GUVs composed of Ld and Lo phase using the same scale bar (3–6 ns). The lowest value of the decay times is observed for the Ld phase, and the highest value for the Lo phase. The distribution of the decay times obtained in each image is presented in Fig. 7 D. Each distribution was fitted using one Gaussian function, giving a maximum of 4.4 ns for the Ld phase (red curve) and of 6.4 ns for the Lo phase (blue curve). Fig. 7 C shows a FLIM image of a GUV with Lo/Ld phase separation labeled with PMI. The corresponding distribution of the decay times (green curve in Fig. 7 D), which is clearly bimodal, was fitted using two Gaussian functions, centered at ∼4.4 and 6.0 ns, comparable with the pure phases.
FIGURE 7.

Fluorescence intensity (gray) and fluorescence lifetime images (color) obtained for PMI-COOH in GUVs composed of different lipid phases: (A) Ld phase (DOPC); (B) Lo phase (DPPC plus 30 mol % cholesterol); (C) Lo/Ld phase separation (DPPC/DOPC/Cholesterol 0.56:0.24:0.20 molar ratio). The images were recorded using 488-nm excitation and the emission was collected with a 525-nm longpass filter. All FLIM images are represented using the same scale bar, from 3 to 6 ns. (D) Distribution of the decay times determined for each phase: Ld phase, red; Lo phase, blue; Lo/Ld mixture, green.
Analysis of Fig. 7, C and D, demonstrate that in the region with preferential partitioning, PMI-COOH decay times correspond to the Ld phase; but in the region that tends to exclude PMI-COOH (corresponding to the Lo phase), the decay time is increased.
At this point, we can conclude that identification of lipid domains in artificial membranes using FLIM imaging of PMI-COOH is possible. An advantage of this method is that even though the molecule does not equally partition in all lipid phases, domains can be identified because the decay time is not dependent on the concentration if no significant aggregation is present.
Identification of lipid rafts in living cells using PMI-COOH
A final validation of the PMI derivative as a probe for membrane rafts should come from experiments in living cells. Fig. 8 A shows a normal Jurkat cell labeled with 4 nM PMI-COOH for 2 min. The fluorescence is distributed both in the plasma and in the intracellular membranes. The FLIM image is represented on the same scale bar as for the GUVs, and thus the decay times of the probe located in the plasma membrane correspond to the Lo phase in artificial bilayers, while the decay times of the PMI-COOH molecules inside the intracellular membranes is similar to the Ld phase. The distribution of the decay times is represented by the blue line in Fig. 8 E, which was fitted using two Gaussian functions. The maxima are at 4.4 and 5.0 ns, with the higher value corresponding to the probe located in the plasma membrane. Considering the SPT data and the FLIM measurements on artificial bilayers, we explain the longer decay time of the probe located in the plasma membrane by its higher cholesterol content (>30 mol %) compared to the intracellular membranes (5–10 mol %) (73), and to the presence of a higher amount of sphingomyelin, which can pack favorably with cholesterol to form lipid rafts. To strengthen this hypothesis, cholesterol was depleted from cells using methyl-β-cyclodextrin. Indeed, this treatment results in a decreased decay time measured for PMI-COOH in both the plasma and the intracellular membranes (Fig. 8 B). The decay time distribution (Fig. 8 E, red line) can now be fitted with one Gaussian function centered on 3.9 ns. We notice that in this case not only the decay time distribution is shifted toward lower values compared to the normal cell, but the second peak of the distribution situated at longer values is not present anymore. In a third set of experiments, rafts on micrometer-scale were induced by cross-linking the T cell receptors using specific antibodies. The yellow arrow in Fig. 8 C is pointing toward an area with a different decay time compared to the rest of the membrane, most probably a lipid raft. To demonstrate that these regions are indeed lipid rafts, the stimulated Jurkat cells were simultaneously labeled with PMI-COOH and Alexa 647 anti-mouse IgG secondary antibodies against the primary anti-CD3 antibodies, in a two-color excitation experiment. The first panel in Fig. 8 D shows the distribution of Alexa 647 antibodies (fluorescence intensity image), and the second panel represents the FLIM image of PMI-COOH. After the superposition of the two channels, one can observe that most of the membrane regions showing a change of the PMI-COOH decay time (blue-green) corresponds to regions where the labeled antibodies accumulate (red), demonstrating that these are indeed the lipid rafts.
Because the formation of lipid rafts involves changes on the plasma membrane, average distributions of PMI-COOH decay times in several cells were constructed only for these structures (Fig. 8 F and Table 1). Although the distribution obtained for the stimulated cells is not bimodal (green), the curve is shifted compared to the normal cells (blue), showing that the formation of lipid rafts influences the decay time of the probe. A clear difference is observed between the distribution of the decay times from the normal membranes and the plasma membrane from cholesterol-depleted cells (red). These first experiments open perspectives for more in-depth studies about the possibility of visualization of lipid rafts in living cells with the PMI-COOH chromophore.
TABLE 1.
Average values of the PMI-COOH decay times in the plasma membrane of Jurkat cells (seven cells were included in each category)
| Average decay time | |
|---|---|
| Normal Jurkat cells | 5.4 ± 0.1 ns |
| Stimulated Jurkat cells | 5.1 ± 0.2 ns |
| Cholesterol-depleted Jurkat cells | 4.3 ± 0.2 ns |
Most of the methods employed to study the formation of lipid rafts are based on the preferential accumulation of a molecule (fluorescently-labeled phospholipids, cholesterol, cholera toxin, or membrane proteins) in domains that are visible on the intensity image. A second group of techniques is based on spectral changes of a dye that can partition in different domains, as shown for laurdan and di-4-ANEPPDHQ (41,74,75). More recently, FLIM was proposed as an alternative to image lipid rafts (42). Because PMI-COOH, laurdan, and di-4-ANEPPDHQ are fluorescent probes that are inserted into the membrane, providing direct information about the lipid phase, we deem it useful to compare their photophysical properties and the microscopy methods that can be applied for the study of membrane rafts (Table 2). PMI-COOH and di-4-ANEPPDHQ absorb in the visible region and have the advantage of using single photon excitation, avoiding the need of a femtosecond pulsed laser for two-photon excitation. However, a picosecond pulsed laser must be used to image the lipid rafts by the FLIM method using these two dyes. Laurdan and di-4-ANEPPDHQ display spectral changes, and the ratio image obtained by recording their fluorescence emission on two channels can partially solve the problems related to photobleaching. In-focus photobleaching is more important in the case of laurdan due to the high power used for two-photon excitation (although the out-of-focus photodestruction is reduced). A disadvantage of the method could be the important superposition of the dyes spectra in the two phases (42). From the data accumulated up to now, the three probes compared here can all provide information about the physical state of lipids in artificial bilayers, to differentiate between the plasma and the intracellular membranes, to identify changes in the stimulated cells, and to monitor the cholesterol depletion. The fact that in these first experiments we did not detect heterogeneities on the plasma membranes of resting cells as shown for laurdan and di-4-ANEPPDHQ (41,42,74,75) can be related not only to the dye, but also to the type of cells that were used. The advantages of the newly proposed PMI derivative rely on its higher extinction coefficient, its high quantum yield, and its excellent stability to photobleaching compared to other chromophores.
TABLE 2.
Comparison of the photophysical characteristics, microscopy techniques, and information obtained with three membrane probes for lipid rafts (according to (41,42,74–76))
| Laurdan | di-4-ANEPPDHQ | PMI-COOH | |
|---|---|---|---|
| Absorption wavelength: | 364 nm. | 488 nm. | 488 nm. |
| Emission wavelength: | 440 nm (gel)/490 nm (Ld). | 468 nm (Lo)/635 nm (Ld). | 560 nm. |
| Extinction coefficient: | 20,000. | 36,000. | 40,000. |
| Quantum yield: | 1 (ethanol). | ≈0.2, increasing to ≈0.7 for styryl dye derivatives in solvents with high viscosity (69). | 1 (diethylether). |
| Photostability: | Poor. | Good. | Excellent. |
| Mechanism: | Solvatochromism (solvent polarity/dielectric constant). | Solvatochromism/electrochromism (dipole potential inside the lipid bilayer). | Polarizability. |
| Microscopy technique: | Spectral changes of fluorescence intensity; two-photon excitation, femtosecond-pulsed laser. | 1. Spectral changes of fluorescence intensity; continuous-wave laser. | Fluorescence lifetime imaging; picosecond-pulsed laser. |
| 2. Second harmonic generation; two-photon excitation, femtosecond-pulsed laser. | |||
| 3. Fluorescence lifetime imaging; picosecond-pulsed laser. | |||
| Information about Ld and Lo domains in GUVs: | Yes. | 1. Yes. | Yes. |
| 2. Yes. | |||
| 3. Yes. | |||
| Information about plasma and intracellular membranes: | Yes. | 1. Yes. | Yes. |
| 3. Yes. | |||
| Information about lipid rafts in the plasma membrane: | Mouse fibroblasts. | 1. Neutrophils. | Stimulated Jurkat cells. |
| THP-1 cells. | |||
| RAW264.7 cells. | 3. HEK293 cells. | ||
| Sensitivity to cholesterol depletion: | Yes. | 1. Yes. | Yes. |
| 3. Yes. |
CONCLUSIONS
We report in this article about the possibility of visualizing the segregation between Ld and Lo phases in artificial bilayers based on the different decay times in the two phases of a new perylene imide based membrane probe. The potential of the dye for the identification of membrane rafts in living cells is demonstrated. Although in our setup following the dynamics of lipid rafts formation is limited by the relative long acquisition time, a higher temporal resolution of the process could be obtained by using PMI-COOH in a wide-field microscope with time-gated detection relative to the laser pulse (77).
The photostability of the PMI chromophore opens the possibility to study the exchange dynamics between Lo and Ld phases at the single molecule level using multiparametric detection, in which information about fluctuations in the chromophore lifetime, polarization, and diffusion properties can be combined.
Another direction opened by this study is to get a better understanding of the mechanisms responsible for the lifetime modification of the dyes embedded in the Lo phase. We proposed a possible general mechanism based on the local field effect of the nanoenvironment surrounding the dyes to explain their increased decay time in the Lo phase. This phenomenon can add to other mechanisms changing the decay time, for example suppression of the nonradiative decay pathways in the Lo phase. By understanding in depth these mechanisms, new dyes showing more important decay time change between Ld and Lo phase and less perturbing for the bilayer can be designed in future.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
Support from the Fonds voor Wetenschapplijk Onderzoek Vlaanderen (grant No. G-0229-07), the KULeuven Research Fund (grant No. GOA 2006/2), the Flemish Ministry of Education (grant No. ZWAP 04/007), and the Federal Science Policy of Belgium (grant No. IAP/VI/27) are gratefully acknowledged. This work, as part of the European Science Foundation EUROCORES Program SONS, was supported from funds by the FWO and the EC Sixth Framework Program, under contract No. ERAS-CT-2003-980409. The work was financed in part by the Impulse Initiative Cell Imaging Core of the KULeuven through a grant to J.-i.H. A.S. received a grant through a Max Planck research award received by the laboratory.
Editor: Alberto Diaspro.
References
- 1.Simons, K., and E. Ikonen. 1997. The fluid mosaic model of the structure of cell membranes. Nature. 387:569–572. [DOI] [PubMed] [Google Scholar]
- 2.Edidin, M. 2003. The state of lipid rafts: from model membranes to cells. Annu. Rev. Biophys. Biomol. Struct. 32:257–283. [DOI] [PubMed] [Google Scholar]
- 3.Dietrich, C., Z. N. Volovyk, M. Levi, N. L. Thompson, and K. Jacobson. 2001. Partitioning of Thy-1, GM1, and cross-linked phospholipids analogs into lipid rafts reconstituted in supported model membranes monolayers. Proc. Natl. Acad. Sci. USA. 98:10642–10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dietrich, C., L. A. Bagatolli, Z. N. Volovyk, N. L. Thompson, M. Levi, K. Jacobson, and E. Gratton. 2001. Lipid rafts reconstituted in model membranes. Biophys. J. 80:1417–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crane, J. M., and L. K. Tamm. 2004. Role of cholesterol in the formation and nature of lipid rafts in planar and spherical model membranes. Biophys. J. 86:2965–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samsonov, A. V., I. Mihalyov, and F. S. Cohen. 2001. Characterization of cholesterol-sphingomyelin domains and their dynamics in bilayer membranes. Biophys. J. 81:1486–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korlach, J., P. Schwille, W. W. Webb, and G. W. Feigenson. 1999. Characterization of lipid bilayer phases by confocal microscopy and fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA. 96:8461–8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feigenson, G. W., and J. T. Buboltz. 2001. Ternary phase diagram of dipalmitoyl-PC/dilauroyl-PC/cholesterol: nanoscopic domain formation driven by cholesterol. Biophys. J. 80:2775–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Veatch, S. L., and S. L. Keller. 2003. Separation of liquid phases in giant vesicles of ternary mixtures of phospholipids and cholesterol. Biophys. J. 85:3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baumgart, T., S. T. Hess, and W. W. Webb. 2003. Imaging coexisting fluid domains in biomembrane models coupling curvature and line tension. Nature. 425:821–824. [DOI] [PubMed] [Google Scholar]
- 11.Scherfeld, D., N. Kahya, and P. Schwille. 2003. Lipid dynamics and domain formation in model membranes composed of ternary mixtures of unsaturated and saturated phosphatidylcholines and cholesterol. Biophys. J. 85:3758–3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kahya, N., D. Scherfeld, K. Bacia, B. Poolman, and P. Schwille. 2003. Probing lipid mobility of raft-exhibiting model membranes by fluorescence correlation spectroscopy. J. Biol. Chem. 278:28109–28115. [DOI] [PubMed] [Google Scholar]
- 13.Kahya, N., D. Scherfeld, K. Bacia, and P. Schwille. 2004. Lipid domain formation and dynamics in giant unilamellar vesicles explored by fluorescence correlation spectroscopy. J. Struct. Biol. 147:77–89. [DOI] [PubMed] [Google Scholar]
- 14.Bacia, K., D. Scherfeld, N. Kahya, and P. Schwille. 2004. Fluorescence correlation spectroscopy relates rafts in model and native membranes. Biophys. J. 87:1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Janes, P. W., S. C. Ley, and A. I. Magee. 1999. Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J. Cell Biol. 147:447–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Janes, P. W., S. C. Ley, A. I. Magee, and P. S. Kabouridis. 2000. The role of lipid rafts in T cell antigen receptor (TCR) signaling. Sem. Immunol. 12:23–34. [DOI] [PubMed] [Google Scholar]
- 17.Subczynski, W. K., and A. Kusumi. 2003. Dynamics of raft molecules in the cell and artificial membranes: approaches by pulse EPR spin labeling and single molecule optical microscopy. Biochim. Biophys. Acta. 1610:231–243. [DOI] [PubMed] [Google Scholar]
- 18.Kusumi, A., I. Koyama-Honda, and K. Suzuki. 2004. Molecular dynamics and interactions for creation of stimulation-induced stabilized rafts from small unstable steady-state rafts. Traffic. 5:213–230. [DOI] [PubMed] [Google Scholar]
- 19.Pike, L. J. 2006. Rafts defined: a report on the Keystone Symposium on Lipid Rafts and Cell Function. J. Lipid Res. 47:1597–1598. [DOI] [PubMed] [Google Scholar]
- 20.Kenworthy, A. K., N. Petranova, and M. Edidin. 2000. High-resolution FRET microscopy of cholera toxin B-subunit and GPI-anchored proteins in cell plasma membranes. Mol. Biol. Cell. 11:1645–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Almeida, R. F. M., L. M. S. Loura, A. Fedorov, and M. Prieto. 2005. Lipid rafts have different sizes depending on membrane composition: a time-resolved fluorescence resonance energy transfer study. J. Mol. Biol. 346:1109–1120. [DOI] [PubMed] [Google Scholar]
- 22.Varma, R., and S. Mayor. 1998. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature. 394:798–801. [DOI] [PubMed] [Google Scholar]
- 23.Schütz, G. J., G. Kada, V. P. Pastushenko, and H. Schindler. 2000. Properties of lipid microdomains in a muscle cell membrane visualized by single molecule microscopy. EMBO J. 19:892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuerschner, L., C. S. Ejsing, K. Ekroos, A. Shevchenko, K. I. Anderson, and C. Thiele. 2005. Polyene-lipids: a new tool to image lipids. Nat. Methods. 2:39–45. [DOI] [PubMed] [Google Scholar]
- 25.Bagatolli, L. A., and E. Gratton. 2000. A correlation between lipid domain shape and binary phospholipids mixture composition in free standing bilayers: a two-photon fluorescence microscopy study. Biophys. J. 79:434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukherjee, S., X. Zha, I. Tabas, and F. R. Maxfield. 1998. Cholesterol distribution in living cells: fluorescence imaging using dehydroergosterol as a fluorescent cholesterol analog. Biophys. J. 75:1915–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wassif, C. A., D. Vied, M. Tsokos, W. E. Connor, R. D. Steiner, and F. D. Porter. 2002. Cholesterol storage defect in RSH/Smith-Lemli-Opitz syndrome fibroblasts. Mol. Genet. Metab. 75:325–334. [DOI] [PubMed] [Google Scholar]
- 28.Parton, R. G., and A. A. Richards. 2003. Lipid rafts and caveolae as portals for endocytosis: new insights and common mechanisms. Traffic. 4:724–738. [DOI] [PubMed] [Google Scholar]
- 29.Weiss, S. 1999. Fluorescence spectroscopy of single biomolecules. Science. 283:1676–1683. [DOI] [PubMed] [Google Scholar]
- 30.Margineanu, A., J. Hofkens, M. Cotlet, S. Habuchi, A. Stefan, J. Qu, C. Kohl, K. Muellen, J. Vercammen, Y. Engelborghs, T. Gensch, and F. C. De Schryver. 2004. Photophysics of a water-soluble rylene dye: comparison with other fluorescent molecules for biological applications. J. Phys. Chem. B. 108:12242–12251. [Google Scholar]
- 31.Verveer, P. J., F. S. Wouters, A. R. Reynolds, and P. I. H. Bastiaens. 2000. Quantitative imaging of lateral ErbB1 receptor signal propagation in the plasma membrane. Science. 290:1567–1570. [DOI] [PubMed] [Google Scholar]
- 32.Herreros, J., T. Ng, and G. Schiavo. 2001. Lipid rafts act as specialized domains for tetanus toxin binding and internalization into neurons. Mol. Biol. Cell. 12:2947–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bastiaens, P. I. H., and A. Squire. 1999. Fluorescence lifetime imaging microscopy: spatial resolution of biochemical processes in cells. Trends Cell Biol. 9:48–51. [DOI] [PubMed] [Google Scholar]
- 34.Tadrous, P. J. 2000. Methods for imaging the structure and function of living tissues and cells: 2. Fluorescence lifetime imaging. J. Pathol. 191:229–234. [DOI] [PubMed] [Google Scholar]
- 35.Barrow, D. A., and B. R. Lenz. 1985. Membrane ultrastructural domains. Resolution limits using diphenylhexatriene fluorescence decay. Biophys. J. 48:221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fiorini, R., E. Gratton, and G. Curatola. 1989. Effects of cholesterol on membrane microheterogeneity: a study using 1,6-diphenyl-1,3,5-hexatriene fluorescence lifetime distributions. Biochim. Biophys. Acta. 1006:198–202. [DOI] [PubMed] [Google Scholar]
- 37.Bernsdorff, C., R. Winter, T. L. Hazlett, and E. Gratton. 1995. Influence of cholesterol and β-sitosterol on the dynamic behavior of DPPC as detected by TMA-DPH and PyrPC fluorescence. A fluorescence lifetime distribution and time-resolved anisotropy study. Ber. Bunsenges. Phys. Chem. 99:1479–1488. [Google Scholar]
- 38.Mateo, C. R., A. U. Acuña, and J.-C. Brochon. 1995. Liquid-crystalline phases of cholesterol/lipid bilayers as revealed by the fluorescence of trans-parinaric acid. Biophys. J. 68:978–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nyholm, T., M. Nylund, A. Söderholm, and J. P. Slotte. 2003. Properties of palmitoyl phosphatidylcholine, sphingomyelin, and dihydrosphingomyelin bilayer membranes as reported by different fluorescent reporter molecules. Biophys. J. 84:987–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sinha, M., S. Mishra, and P. G. Joshi. 2003. Liquid-ordered microdomains in lipid rafts and plasma membrane of U-87 MG cells: a time-resolved fluorescence study. Eur. Biophys. J. 32:381–391. [DOI] [PubMed] [Google Scholar]
- 41.Jin, L., A. C. Millard, J. P. Wuskell, X. Dong, D. Wu, H. A. Clark, and L. M. Loew. 2006. Characterization and application of a new optical probe for membrane lipid domains. Biophys. J. 90:2563–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Owen, D. M., P. M. P. Lanigan, C. Dunsby, I. Munro, D. Grant, M. A. A. Neil, P. M. W. French, and A. I. Magee. 2006. Fluorescence lifetime imaging provides enhanced contrast when imaging the phase sensitive dye di-4-ANEPPDHQ in model membranes and live cells. Biophys. J. 90:L80–L82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vallée, R. A. L., M. Van der Auweraer, F. C. De Schryver, D. Beljonne, and M. Orrit. 2005. A microscopic model for the fluctuations of local field and spontaneous emission of single molecules in disordered media. ChemPhysChem. 6:81–91. [DOI] [PubMed] [Google Scholar]
- 44.Weil, T., M. A. Abdalla, C. Jatzke, J. Hengstler, and K. Müllen. 2005. Water-soluble rylene dyes as high-performance colorants for the staining of cells. Biomacromolecules. 6:68–79. [DOI] [PubMed] [Google Scholar]
- 45.Angelova, M. I., N. Hristova, and I. Tsoneva. 1999. DNA-induced endocytosis upon local microinjection to giant unilamellar cationic vesicles. Eur. Biophys. J. 28:142–150. [DOI] [PubMed] [Google Scholar]
- 46.Angelova, M. I., and I. Tsoneva. 1999. Interactions of DNA with giant liposomes. Chem. Phys. Lipids. 101:123–137. [DOI] [PubMed] [Google Scholar]
- 47.Lakowicz, J. R. 1986. Principles of Fluorescence Spectroscopy. Plenum Press, New York.
- 48.Lor, M., S. Jordens, G. De Belder, G. Schweitzer, E. Fron, L. Viaene, M. Cotlet, T. Weil, K. Müllen, J. W. Verhoeven, M. Van der Auweraer, and F. C. De Schryver. 2003. Direct proof of electron transfer in a rigid first generation triphenyl amine core dendrimer substituted with a peryleneimide acceptor. Photochem. Photobiol. Sci. 2:501–510. [DOI] [PubMed] [Google Scholar]
- 49.Maus, M., M. Cotlet, J. Hofkens, T. Gensch, and F. De Schryver. 2001. An experimental comparison of the maximum likelihood estimator and nonlinear least-squares fluorescence lifetime analysis of single molecules. Anal. Chem. 73:2078–2086. [DOI] [PubMed] [Google Scholar]
- 50.Cotlet, M., J. Hofkens, S. Habuchi, G. Dirix, M. Van Guyse, J. Michiels, J. Vanderleyden, and F. C. De Schryver. 2001. Identification of different emitting species in the red fluorescent protein DsRed by means of ensemble and single-molecule spectroscopy. Proc. Natl. Acad. Sci. USA. 98:14398–14403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zerner, M. C., G. H. Loew, R. F. Kirchner, and U. T. Mueller-Westerhoff. 1980. Intermediate neglect of differential-overlap technique for spectroscopy of transition-metal complexes—ferrocene. J. Am. Chem. Soc. 102:589–599. [Google Scholar]
- 52.Vosch, T., M. Coltlet, J. Hofkens, K. Van Der Biest, M. Lor, K. Weston, P. Tinnefeld, M. Sauer, L. Latterini, K. Müllen, and F. C. De Schryver. 2003. Probing Förster type energy pathways in a first generation rigid dendrimer bearing two perylene imide chromophores. J. Phys. Chem. A. 107:6920–6931. [Google Scholar]
- 53.Lakowicz, J. R., H. Cherek, and B. P. Maliwal. 1985. Time-resolved fluorescence anisotropies of diphenylhexatriene and perylene in solvents and lipid bilayers obtained from multifrequency phase-modulation fluorometry. Biochemistry. 24:376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chong, P. L., B. W. van der Meer, and T. E. Thompson. 1985. The effects of pressure and cholesterol on rotational motions of perylene in lipid bilayers. Biochim. Biophys. Acta. 813:253–265. [DOI] [PubMed] [Google Scholar]
- 55.Khan, T. K., and P. L. Chong. 2000. Studies of archaebacterial bipolar tetraether liposomes by perylene fluorescence. Biophys. J. 78:1390–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hofkens, J., M. Maus, T. Gensch, T. Vosch, M. Cotlet, F. Köhn, A. Herrmann, K. Müllen, and F. C. De Schryver. 2000. Probing photophysical processes in individual multichromophoric dendrimers by single-molecule spectroscopy. J. Am. Chem. Soc. 122:9278–9288. [Google Scholar]
- 57.Davenport, L., B. Shen, T. W. Joseph, and M. P. Straher. 2001. A novel fluorescent coronenyl-phospholipid analogue for investigations of submicrosecond lipid fluctuations. Chem. Phys. Lipids. 109:145–156. [DOI] [PubMed] [Google Scholar]
- 58.Ballet, P., M. Van der Auweraer, F. C. De Schryver, H. Lemmetyinen, and E. Vuorimaa. 1996. Global analysis of the fluorescence decays of n,n′-dioctadecyl rhodamine B in Langmuir-Blodgett films of diacyl phosphatidic acids. J. Phys. Chem. 100:13701–13715. [Google Scholar]
- 59.Laguitton-Pasquier, H., M. Van der Auweraer, and F. C. De Schryver. 1998. Bidimensional distribution of a cyanine dye in LB monolayer studied by time-resolved and spatially resolved fluorescence. Langmuir. 14:5172–5183. [Google Scholar]
- 60.Janssens, L. D., N. Boens, M. Ameloot, and F. C. De Schryver. 1990. A systematic study of the global analysis of multi-exponential fluorescence decay surfaces using reference convolution. J. Phys. Chem. 94:3564–3576. [Google Scholar]
- 61.Beechem, J. M., E. Gratton, M. Ameloot, J. R. Knutson, and L. Brand. 1991. The global analysis of fluorescence intensity and anisotropy decay data:second-generation theory and programs. In Topics in Fluorescence Spectroscopy, Vol. 2, Principles. J. R. Lakowicz, editor. Plenum Press, New York.
- 62.Konopásek, I., P. Kvasnička, E. Amler, A. Kotyk, and G. Curatola. 1995. The transmembrane gradient of the dielectric constant influences the DPH lifetime distribution. FEBS Lett. 374:338–340. [DOI] [PubMed] [Google Scholar]
- 63.Mitchell, D. C., and B. J. Litman. 1998. Effect of cholesterol on molecular order and dynamics in highly polyunsaturated phospholipid bilayers. Biophys. J. 75:896–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Arora, A., H. Raghuraman, and A. Chattopadhyay. 2004. Influence of cholesterol and ergosterol on membrane dynamics: a fluorescence approach. Biochem. Biophys. Res. Commun. 318:920–926. [DOI] [PubMed] [Google Scholar]
- 65.Finkelstein, A. 1976. Water and nonelectrolyte permeability of lipid bilayer membranes. J. Gen. Physiol. 68:127–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Carruthers, A., and D. L. Melchior. 1983. Studies of the relationship between bilayer water permeability and physical state. Biochemistry. 22:5797–5807. [Google Scholar]
- 67.Tu, K., M. L. Klein, and D. J. Tobias. 1998. Constant-pressure molecular dynamics investigation of cholesterol effects in a dipalmitoylphosphatidylcholine bilayer. Biophys. J. 75:2147–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Prendergast, F. G., R. P. Haugland, and P. J. Callahan. 1981. 1-[4-(Trimethylamino)phenyl]-6-phenylhexa-1,3,5-triene: synthesis, fluorescence properties, and use as a fluorescence probe of lipid bilayers. Biochemistry. 20:7333–7338. [DOI] [PubMed] [Google Scholar]
- 69.El-Daly, S. A., M. H. Abdel-Kader, R. M. Issa, and E.-S. A. El-Sherbini. 2003. Influence of solvent polarity and medium acidity on the UV-Vis spectral behavior of 1-methyl-4-[4-amino-styryl] pyridinium iodide. Spectrochim. Acta [A]. 59:405–411. [DOI] [PubMed] [Google Scholar]
- 70.Huang, J. 2002. Exploration of molecular interactions in cholesterol superlattices: effect of multibody interactions. Biophys. J. 83:1014–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vaz, W. L. C., and E. Melo. 2002. Fluorescence spectroscopic studies on phase heterogeneity in lipid bilayer membranes. J. Fluor. 11:255–271. [Google Scholar]
- 72.Bagatolli, L., and E. Gratton. 2000. A correlation between lipid domain shape and binary phospholipid mixture composition in free standing bilayers: a two-photon fluorescence microscopy study. Biophys. J. 79:434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cooper, G. M. 2000. The Cell: A Molecular Approach, 2nd Ed. American Society for Microbiology, Washington, DC. Sinauer Associates, Sunderland, MA.
- 74.Yu, W., P. T. C. So, T. French, and E. Gratton. 1996. Fluorescence generalized polarization of cell membranes: a two-photon scanning microscopy approach. Biophys. J. 70:626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gaus, K., E. Gratton, E. P. W. Kable, A. S. Jones, I. Gelissen, L. Kritharides, and W. Jessup. 2003. Visualizing lipid structure and raft domains in living cells with two-photon microscopy. Proc. Natl. Acad. Sci. USA. 100:15554–15559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim, H. M., H.-J. Choo, S.-Y. Jung, Y.-G. Ko, W.-H. Park, S.-J. Jeon, C. H. Kim, T. Joo, and B. R. Cho. 2007. A two-photon fluorescent probe for lipid raft imaging: C-laurdan. ChemBioChem. 8:553–559. [DOI] [PubMed] [Google Scholar]
- 77.Cole, M. J., J. Siegel, S. E. D. Webb, R. Jones, K. Dowling, M. J. Dayel, D. Parsons-Karavassilis, P. M. W. French, M. J. Lever, L. O. D. Sucharov, M. A. A. Neil, R. Juškaitis, and T. Wilson. 2001. Time-domain whole-field fluorescence lifetime imaging with optical sectioning. J. Microsc. 203:246–257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.