

Abstract
Analysis of antigen dissociation provides insight into peptide presentation modes of folded human leukocyte antigen (HLA) molecules, which consist of a heavy chain, β2-microglobulin (β2m), and an antigenic peptide. Here we have monitored peptide-HLA interactions and peptide dissociation kinetics of two HLA-B27 subtypes by fluorescence depolarization techniques. A single natural amino-acid substitution distinguishes the HLA-B*2705 subtype that is associated with the autoimmune disease ankylosing spondylitis from the non-disease-associated HLA-B*2709 subtype. Peptides with C-terminal Arg or Lys represent 27% of the natural B*2705 ligands. Our results show that dissociation of a model peptide with a C-terminal Lys (GRFAAAIAK) follows a two-step mechanism. Final peptide release occurs in the second step for both HLA-B27 subtypes. However, thermodynamics and kinetics of peptide-HLA interactions reveal different molecular mechanisms underlying the first step, as indicated by different activation energies of 95 ± 8 kJ/mol (HLA-B*2705) and 150 ± 10 kJ/mol (HLA-B*2709). In HLA-B*2709, partial peptide dissociation probably precedes fast final peptide release, while in HLA-B*2705 an allosteric mechanism based on long-range interactions between β2m and the peptide binding groove controls the first step. The resulting peptide presentation mode lasts for days at physiological temperature, and determines the peptide-HLA-B*2705 conformation, which is recognized by cellular ligands such as T-cell receptors.
INTRODUCTION
Major histocompatibility complex (MHC) class I molecules play a central role in the initiation and propagation of immune responses and the recognition of intracellular pathogens through the presentation of antigenic peptides to cytotoxic CD8+-T cells (1). The heterotrimeric MHC molecule (pMHC) consists of a highly polymorphic transmembrane heavy chain (HC), a light chain, β2-microglobulin (β2m), and a peptide (Fig. 1). Peptidic fragments of self- or non-self-proteins are loaded on HC/β2m heterodimers in the endoplasmic reticulum followed by transport of the heterotrimer to the cell surface (2). The peptides, usually 8–10 amino acids in length, present anchoring backbone atoms at or near both termini as well as MHC-binding side chains at position 2 and at the C-terminus (3). Once at the cell surface, pMHC serve as potential targets for cytotoxic T lymphocytes.
FIGURE 1.

Model of the crystal structure of the HLA-B*2705 ectodomain loaded with the m9 peptide (1JGE). The peptide is displayed in red and yellow. The yellow color marks the two main anchor residues of the peptide, pArg2 and pLys9, as well as the amino-acid positions p6 and p8, which were exchanged to cysteine and modified with the fluorescent label LY in our studies. The tryptophan residues in β2m (dark gray) and in HC (light gray) are colored in green and turquoise, respectively. Amino acid 116 at the floor of the peptide binding groove is colored in black. (Right) Closeup of the peptide binding groove.
One of the most striking features of human MHC (HLA) class I genes is their potential association to diseases (4). For example, individuals with HLA-B27 exhibit an ∼90-fold-higher risk to acquire the autoimmune disease ankylosing spondylitis (AS) than HLA-B27-negative individuals (5–8). Furthermore, even HLA-B27 subtypes may be differentially associated to AS: HLA-B*2705 is AS-associated, while HLA-B*2709 is not (9). The only difference between the HLA-B*2705 and HLA-B*2709 molecules (B*2705 and B*2709 in short) is a natural substitution of a single amino acid at the floor of the peptide-binding groove, Asp116 in B*2705 to His116 in B*2709 (10). The presence of Asp or His at HC residue 116 results in ∼80–90% of shared and 10–20% of differentially bound peptide ligands that can be eluted from the two HLA-B27 molecules (11). All B*2705 ligands not bound to B*2709 had a C-terminal basic (Arg, Lys) or Tyr residue, while a small percentage of peptides containing C-terminal Arg or Tyr are shared, i.e., bind to both subtypes (11).
The differential association of HLA-B27 subtypes to AS as well as the differential binding of subsets of natural ligands is expected to provide unique insight into the pathogenic role of HLA-B27, as the mechanism of disease association is still elusive. Consequently, elucidation of the molecular mechanisms underlying the strong association of HLA-B27 with AS requires an understanding of the structural and dynamic properties of the pMHC and interacting proteins, although other factors may also determine AS pathogenesis. The high resolution x-ray structures of B*2705 and B*2709 with the model peptide m9 (GRFAAAIAK) (which binds to both subtypes in vitro (12)) as well as thermodynamic analyses (13,14), fluorescence depolarization, and molecular dynamics simulation studies (13) provide a clue for the lack of peptides with pLys9 in B*2709. The ɛ-amino group of pLys9 forms a salt bridge with Asp116 in B*2705, whereas only a hydrogen bond exists between His116 and pLys9 in B*2709 (12). Despite the peptide's basic C-terminal Lys, the two complexes with the m9 peptide display virtually identical crystal structures (12), however, the real-time dynamic properties are clearly different (13). Among various studies of thermodynamic and kinetic properties of class I pMHCs (15–19), a few have addressed HLA-B27 molecules (13,14,20–23). These investigations included analyses of the thermal unfolding of B*2705, complexed with various peptides, using circular dichroism spectroscopy (14,20,22,23), differential scanning calorimetry (14,23), and fluorescence depolarization (13,21–23). The results of these studies show the importance of peptide residues at p1, p3, and p9 as well as of residue Cys67.
In this article, we present a systematic comparison of the thermodynamic behavior of the m9 peptide bound to B*2705 and B*2709. We compared thermodynamic parameters of peptide dissociation with those of complex unfolding by using the fluorescence anisotropy of a fluorescent label bound to the peptide and the temperature-dependent fluorescence emission of endogenous tryptophans (Fig. 1), respectively, as observables to identify the primary event in complex dissociation. Extensive investigations of peptide dissociation kinetics and their evaluation by kinetic models reveal two different mechanistic principles of m9 dissociation from B*2709 and B*2705. A specific effect of β2m on peptide presentation was observed for B*2705, suggesting long-range intermolecular peptide stabilization effects.
MATERIALS AND METHODS
Protein preparation
Complexes of the subtypes B*2705 and B*2709 with the peptide m9 (GRFAAAIAK) were prepared as described previously (12,13,24,25). The synthetic m9-peptide as well as its fluorescent derivatives labeled with Lucifer Yellow (LY, Molecular Probes, Eugene, OR) in position C6 (GRFAAC(-LY)IAK; m9-C6-LY) and C8 (GRFAAAIC(-LY)K; m9-C6-LY), respectively, were purchased from Biosynthan (Berlin, Germany). Briefly, heterotrimeric HLA-B27-m9 complexes (HC/β2m/peptide) complexes were refolded from 6 M urea in the presence of the respective peptide. This procedure also applies for the refolding of β2m alone. Refolding of pMHC with β2m-dimers, covalently linked via a disulfide bridge between two β2m monomers, was performed as described for pMHC formation with monomeric β2m, except that oxidized and reduced gluthation were employed in equimolar amounts (5 mM). After size-exclusion chromatography, the concentrations of pMHC in 10 mM phosphate buffer, pH 7.5, 150 mM NaCl were adjusted to A426 = 0.0012 OD using the absorption band of Lucifer Yellow (λmax = 426 ± 2 nm).
Thermal complex unfolding
Thermal unfolding of pMHC and isolated β2m was measured in the temperature range from 5°C to 85°C, with an average heating rate of 0.6°C per min, by observing the spectral change of the fluorescence emission of tryptophan at equilibrium conditions. Intrinsic tryptophans were excited at λex = 280 nm. Tryptophan fluorescence spectra were recorded between 300 and 450 nm using a standard fluorescence spectrometer (Spex-FluoroMax, HORIBA Jobin Yvon, Edison, NJ). The temperature-dependent unfolding was analyzed by calculating the quotient of the intensities at 360 and 320 nm (13,26). This eliminates the temperature-dependent baseline shift due to the decrease in emission intensity of tryptophan with increasing temperature. Thermodynamic parameters, such as the enthalpy and entropy changes of the unfolding process as well as melting temperatures Tm, were obtained by fitting the following equations to the emission intensity quotients I360/I320 of tryptophan using the least-square method. Either Eq. 1, representing a two-state transition for a single process, or Eq. 2, describing a three-state transition when two stepwise processes were observed, was used to fit the data (26):
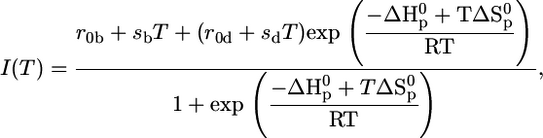 |
(1) |
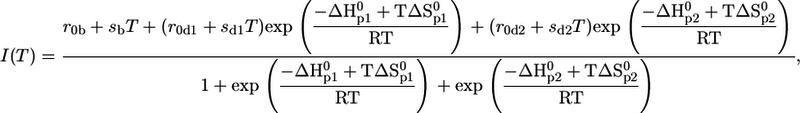 |
(2) |
with
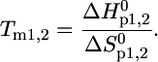 |
(3) |
These equations involve six (two-state) and 10 (three-state) fitting parameters, respectively; the intensity quotient of the folded state (native protein), r0b, and of the unfolded or dissociated state, r0d1,2; the temperature-dependence of the intensity quotient of the folded state, sb, and of the unfolded or dissociated state, sd1,2; and the enthalpy change 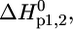 and the entropy change,
and the entropy change, 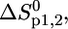 for the two- or three-state unfolding/dissociation reaction, the thermodynamic parameters being determined at the midpoint of each transition.
for the two- or three-state unfolding/dissociation reaction, the thermodynamic parameters being determined at the midpoint of each transition.
Thermodynamics of peptide dissociation
Temperature-induced peptide dissociation from the pMHC was measured as described (13) by detection of the change in the stationary anisotropy of LY covalently bound to m9 in the temperature range from 5°C to 85°C. An average heating rate of 0.6°C per min was applied. Lucifer Yellow was excited at λex = 428 nm and the steady-state anisotropy was determined within a time-correlated single photon-counting setup (27) by collecting the integral emission intensity for λ ≥ 495 nm, parallel I‖, and perpendicular I⊥, with respect to the linearly polarized excitation light. Data evaluation was performed as described above applying Eq. 1, assuming a two-state transition. The anisotropy r(T) instead of the fluorescence intensity I(T) was used as the temperature-dependent parameter in Eq. 1. The anisotropy values were calculated according to Eq. 4:
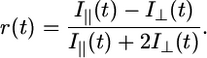 |
(4) |
Kinetics of peptide dissociation
The temporal dissociation behavior of the LY-labeled peptides, m9-C6-LY and m9-C8-LY, from the HLA-B*2709 and B*2705 complexes, was monitored by measuring the time-dependent change in the stationary fluorescence anisotropy. Lucifer Yellow was excited at λex = 428 nm, and the anisotropy as a function of time was recorded at 530 nm using a standard fluorescence spectrometer (Spex-FluoroMax, Jobin Yvon). The G-factor was G = 0.67. The anisotropy values were calculated according to Eq. 4 and automatically corrected for the G-factor during the measurement. Peptide-dissociation kinetics was recorded at five different temperatures below the transition temperature of temperature-induced peptide dissociation. The sample was equilibrated at the given temperature. To avoid rebinding of the labeled peptide, peptide dissociation from the complex was investigated with a 100-fold molar excess of unlabeled peptide m9. The measurement was started after addition of unlabeled m9.
Emission intensities at 530 nm (λexc = 426 nm) were averaged for 5 s, in a time interval of 14 or 25 s, which sets the time resolution of the experiment. The final point of the measurement was reached when the anisotropy of the sample was equal to the value of the free labeled peptide, m9-C6-LY or m9-C8-LY, at the given temperature.
Peptide-dissociation time traces were fitted with a mono- or biexponential decay law (Eq. 5) depending on the subtype, label position, and temperature:
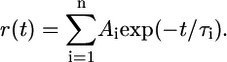 |
(5) |
A stretched exponential function was applied for peptide dissociation kinetics from B*2705 complexes at higher temperatures, with h being a heterogeneity parameter (28):
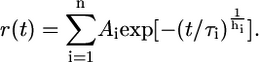 |
(6) |
RESULTS AND DISCUSSION
Spectroscopic characterization of the HLA-molecules complexed with the fluorescently labeled m9 peptide
The absorption spectrum of the HLA*B2705 and HLA*B2709 molecules complexed with the m9-C6-LY peptide is characterized by a weak absorption band centered at 428 nm, which is due to the absorption of the fluorescent label LY attached to the peptide, and a strong band at 280 nm, representing the transition of LY to higher electronic states and the absorption of tryptophan residues in the heavy chain and β2m (Fig. 2 A). The emission spectra in Fig. 2 B show the fluorescence of LY with a maximum at λmax = 532 ± 1 nm as well as the fluorescence of tryptophan with an emission maximum at λmax = 340 ± 1 nm. For comparison, the absorption and emission spectra of β2m alone, which contains two tryptophan residues (see Fig. 1), are displayed in Fig. 2, C and D.
FIGURE 2.

Sample characterization. Absorption- (A) and emission spectra (B) of the B*2705-m9-C6-LY. Absorption- (C) and emission spectra (D) of β2m. Lucifer Yellow and intrinsic tryptophans were excited at λexc = 428 nm and λexc = 280 nm, respectively. Conditions: 0.3 μM HLA-complex or 0.3 μM β2m dissolved in 10 mM phosphate buffer, 150 mM NaCl, pH 7.5.
Comparison of thermodynamic parameters describing pMHC denaturation and peptide dissociation
pMHC stability was assessed for both subtypes by investigating their thermal behavior when loaded with the m9 peptide. Temperature-induced peptide dissociation and global unfolding experiments were performed to extract thermodynamic parameters, such as the apparent enthalpy and entropy, to identify the primary event in complex unfolding. Monitoring the fluorescence polarization or anisotropy of the labeled peptide is especially suited to directly follow the mole fraction of the peptide molecules bound to the complex and the fraction of peptides free in solution (13,15,21,22).
The left column in Fig. 3 shows 1), peptide dissociation from B*2709 as observed by steady-state fluorescence anisotropy changes of the fluorescent dye LY bound to m9 at position 6 (Fig. 3 A); 2), pMHC unfolding as observed by the temperature-induced changes of the tryptophan emission (Fig. 3 B); and 3), unfolding of β2m via tryptophan emission (Fig. 3 C). A single two-state transition for peptide dissociation was observed for m9 dissociation from the B*2709-m9-C6-LY-complex with a transition temperature of Tm = 46 ± 2°C (13) (Fig. 3 A). The temperature-dependent changes in tryptophan emission, shown as the quotient of the intensities at the red and blue edges of the spectrum, 360 nm and 320 nm, reveal two transitions for complex unfolding (Fig. 3 B), whereas for β2m alone, only one transition was observed (Fig. 3 C). For a better comparison of these multiple transitions, the changes in tryptophan fluorescence and in LY anisotropy as a function of temperature are presented as their first derivatives in the middle column (Fig. 3, D–F). The bell-shaped curves represent the respective transition with the transition temperature at the maximum of the curve. The dissociation of m9-C6-LY from B*2709 (Tm = 46 ± 2°C) coincides with the first transition observed in global unfolding (Tm = 46 ± 3°C, Fig. 3, A, B, D, and E) (13), whereas the second transition (Tm = 65 ± 3°C (Fig. 3, B and E) coincides with β2m unfolding at Tm = 64 ± 3°C (Fig. 3 C). Thus, in B*2709, the release of m9 and unfolding of the heavy chain are tightly coupled, while β2m melts at higher temperatures at the intrinsic β2m melting temperature. In contrast to B*2709, unfolding of the B*2705-m9-C6-LY complex is characterized by a single transition at ∼65°C (Fig. 3 H). This transition temperature coincides with the peptide dissociation temperature (Fig. 3 G) and is also close to the temperature of β2m unfolding. For B*2705 peptide release, unfolding of heavy chain and β2m therefore seems to occur in a cooperative way (Fig. 3, G–I). Despite the differences in thermal unfolding between B*2705-m9 and B*2709-m9, our results suggest that m9 dissociation is correlated with the initial events in thermal pMHC unfolding and is tightly coupled to HC unfolding. Similar conclusions as for m9 dissociation were drawn from peptide dissociation and complex unfolding experiments of the self-peptide TIS (RRLPIFSRL) complexed with the subtypes B*2705 and B*2709 (23). In the case of the TIS-peptide (TIS-C6-LY), the transition temperatures for peptide dissociation from both subtypes (Tm = 58°C and Tm = 60°C for B*2705 and B*2709, respectively), are lower than the transition temperature of β2m unfolding (Tm = 64°C), similar to B*2709-m9.
FIGURE 3.

Comparison of peptide dissociation and complex unfolding. (A) Temperature-dependent anisotropy of the LY-labeled complex HLA-B*2709-m9-C6-LY; (B) intensity quotients I360nm/I320nm of the tryptophan fluorescence of the complexes; and (C) β2m unfolding. Data were fitted according to the two-state or three-state model. (D,G) Derivatives of the anisotropies of the LY-labeled complexes HLA-B*2709/05-m9-C6-LY and E/H derivatives of the intensity quotients I360nm/I320nm of the tryptophan fluorescence of the complexes. (F,I) Derivatives of the intensity quotients I360nm/I320nm of the tryptophan fluorescence of β2m (in both figures, the same set of data is displayed). All derivatives are fitted applying a Gaussian function. Conditions were chosen as in Fig. 2.
A closer look at the unfolding of B*2705-m9, however, reveals that peptide dissociation in B*2705-m9 could as well be initiated by the dissociation and subsequent unfolding of β2m as the transition temperature of peptide dissociation of 65°C coincides with the melting temperature of β2m. To test this model of β2m-induced peptide dissociation, we reconstituted the subtype HLA-B*2705 with a destabilized β2m molecule. In the experiment described here, destabilization was achieved by a dimeric β2m form. The β2m-dimer, consisting of two β2m-molecules covalently attached to each other via a disulfide bond, exhibits a reduced thermal stability as compared to the native β2m with a transition temperature of Tm = 58 ± 1°C (Fig. 4 A). If β2m properties determine complex unfolding and peptide dissociation for strong peptide binders, we will expect m9 dissociation from the B*2705 complex to coincide with β2m dimer melting, which occurs at a transition temperature 7°C lower than that of the monomer. Our results show that the transition temperature for m9 dissociation from B*2705 complexed with the β2m-dimer, Tm = 59 ± 2°C, is indeed ∼7°C lower than that of B*2705 complexed with the β2m-monomer, Tm = 66 ± 1°C (Fig. 4 B). Thus, the interaction of the thermally less stable β2m dimer with the HC in the HLA-B*2705-m9-C6-LY complex results in a reduction of thermal complex stability and a decrease of the transition temperature for peptide dissociation. This correlation clearly supports our hypothesis that m9 release from B*2705 depends on the properties of β2m, even though it may turn out that this correlation is not based on the reduced thermal stability of the β2m dimer, but is caused by an impaired temperature-dependent interaction of the dimer with the HC.
FIGURE 4.

Thermal stability of β2m monomer and β2m dimer. (A) Unfolding of β2m as a monomer and a covalently linked dimer in solution; melting temperatures are Tm (monomer) = 65°C and Tm (dimer) = 55°C. (B) Peptide dissociation from HLA-B*2705 with β2m as a monomer and dimer; temperatures for peptide dissociation are Tm (monomer) = 67°C and Tm (dimer) = 59°C. Conditions were chosen as in Fig. 2.
In a more general way, our results on the subtypes B*2705 and B*2709 show that both the peptide interaction with a given subtype and the properties of β2m are required for stabilization of the HC fold. For a weak peptide binder (here m9 in B*2709), peptide release causes HC unfolding and complex dissociation; but for a strong peptide binder (here m9 in B*2705), the impaired interaction of β2m with the HC leads to β2m dissociation, causing peptide dissociation and complex unfolding.
In our case, the strong binding properties of m9 to the B*2705 complex molecule are a consequence of the salt bridge between Asp116 in the F-pocket at the bottom of the peptide binding groove and the C-terminal basic Lys of the peptide (12). Systematic variation of the p9 residue in the GRAFVTIGK peptide, which shares amino-acid positions 1, 2, 7, and 9 with m9, showed that the basic amino acids Lys and Arg are by far the strongest anchors (20). Peptides with such basic C-terminal anchors account for ∼27% of the natural B*2705 ligands (11,29), a number that underscores the relevance of our results.
Kinetics of peptide dissociation
Kinetic peptide dissociation experiments were performed to further explore the differences between HLA-B*2705 and HLA-B*2709 molecules complexed with the m9 peptide to gain an understanding of the effect of the natural Asp116His exchange on peptide stability. Temperature-dependent kinetics of peptide dissociation allows for the evaluation of activation energies of peptide dissociation. We used two LY-reporter positions for peptide dissociation: p6, close to middle of the peptide and p8, close to the C-terminus. Peptide dissociation kinetics from the trimeric HLA-B27 complexes were investigated by monitoring the stationary anisotropy of LY bound to m9 as a function of time and temperature (Fig. 5). The stationary anisotropy at time zero, the starting point of the dissociation kinetics at a given temperature, displays anisotropy values between 0.06 and 0.2 in the temperature range between 22°C and 61°C, respectively. These values reflect the steric restriction of the label as well as the peptide flexibility inside the peptide binding groove at a given temperature (13). A decreasing initial anisotropy with increasing temperature in the temperature range below the transition temperature of peptide dissociation is due to faster label and peptide dynamics as well as the reduction in steric restriction caused by constituents of the peptide binding groove. At infinite times, the endpoint of the peptide dissociation time trace, the anisotropy value equals the value of the respective peptide free in solution. These anisotropy values were measured in independent experiments (data not shown) and incorporated into the anisotropy time traces as the end-value r∞ (Table 1).
FIGURE 5.

Time-dependent change in anisotropy of LY-labeled HLA-B27-complexes at different temperatures. (Top) Subtype HLA-B*2709 complexed with m9-C6-LY (A) and m9-C8-LY (B) at 22, 26, 30, 34, and 37°C. (Bottom) Subtype HLA-B*2705 complexed with m9-C6-LY (C, at 26, 30, 37, 50, and 60°C) and m9-C8-LY (D, at 26, 30, 37, 50, 55, and 61°C). Black lines represent mono- or biexponential fits (dependent on temperature, a stretched exponential had to be applied in HLA-B*2705, panel D). Colored lines represent the model fits according to Eq.12 and 17 to the respective data set. (A stretching factor 1/h was considered in Eq. 17 for data at 55 and 61°C, displayed in D.)
TABLE 1.
Parameters from the kinetic model
| Subtype | Temperature K | k1 s−1 | k−1 s−1 | k2 s−1 | r∞ | r0–r∞ | h |
|---|---|---|---|---|---|---|---|
| B*2709-m9-C6-LY | 295 | 6.67 × 10−5 | 5.00 × 10−5 | 1.22 × 10−5 | 0.030 | 0.120 | — |
| 299 | 1.18 × 10−4 | 9.09 × 10−5 | 2.78 × 10−5 | 0.028 | 0.111 | — | |
| 303 | 2.87 × 10−4 | 2.27 × 10−4 | 4.76 × 10−5 | 0.026 | 0.103 | — | |
| 307 | 5.56 × 10−4 | 4.55 × 10−4 | 1.22 × 10−4 | 0.024 | 0.099 | — | |
| 310 | 1.18 × 10−3 | 9.09 × 10−4 | 1.54 × 10−4 | 0.022 | 0.094 | — | |
| B*2705-m9-C6-LY | 299 | 1.11 × 10−3 | 9.09 × 10−4 | 1.96 × 10−7 | 0.028 | 0.110 | — |
| 303 | 1.43 × 10−3 | 2.22 × 10−3 | 1.11 × 10−6 | 0.026 | 0.112 | — | |
| 310 | 3.33 × 10−3 | 4.35 × 10−3 | 1.50 × 10−6 | 0.022 | 0.095 | — | |
| 323 | 1.11 × 10−2 | 1.33 × 10−2 | 2.38 × 10−5 | 0.017 | 0.071 | — | |
| 333 | 5.55 × 10−2 | 6.67 × 10−2 | 2.82 × 10−4 | 0.017 | 0.057 | — | |
| B*2709-m9-C8-LY | 295 | 4.54 × 10−6 | — | — | 0.028 | 0.152 | — |
| 299 | 7.69 × 10−6 | — | — | 0.026 | 0.150 | — | |
| 303 | 1.82 × 10−5 | — | — | 0.024 | 0.140 | — | |
| 307 | 4.76 × 10−5 | — | — | 0.022 | 0.131 | — | |
| 310 | 8.48 × 10−5 | — | — | 0.021 | 0.112 | — | |
| B*2705-m9-C8-LY | 299 | 2.00 × 10−7 | — | — | 0.026 | 0.152 | — |
| 303 | 1.25 × 10−7 | — | — | 0.024 | 0.137 | — | |
| 310 | 9.09 × 10−7 | — | — | 0.021 | 0.108 | — | |
| 323 | 3.23 × 10−6 | — | — | 0.017 | 0.097 | — | |
| 328 | 1.21 × 10−5 | — | — | 0.015 | 0.093 | 2 | |
| 334 | 2.38 × 10−4 | — | — | 0.015 | 0.086 | 2 |
The time traces shown in Fig. 5 reveal distinct differences both in the kinetics of peptide dissociation depending on subtype as well as reporter group position. In agreement with the thermodynamic data for peptide dissociation described in the previous section, the kinetics at a given temperature are faster by a factor of ∼100 for peptide dissociation from B*2709, the less stable subtype in complex with m9, than from the B*2705 complex (Fig. 5). At physiological temperature, the main time constants for m9-C6-LY dissociation from B*2705 and B*2709 are τ = 306 h (1.1 × 106 s) and τ = 3.9 h (1.4 × 104 s), respectively (see Table S1, Supplementary Material). Interestingly, the incorporation of LY in position p8 results in a stabilization of the complex as shown by the slower peptide-dissociation kinetics (Fig. 5). The higher initial anisotropy values for m9-C8-LY in both subtypes, however, are due to a higher steric restriction caused by constituents of the peptide-binding groove (12).
The time traces of peptide dissociation were fitted with a mono- or biexponential decay law depending on label position and temperature. In the case of m9-C6-LY, all time-traces for B*2705 and B*2709 were fitted with two exponential functions according to Eq. 5. With increasing temperature, the relative amplitude of the fast time constant increases, thereby lowering the relative amplitude of the slower time. At higher temperatures (>50°C), the discrete exponential was substituted with a stretched exponential function (Eq. 6), to yield better fit results for peptide dissociation from B*2705. The occurrence of stretched exponentials in the description of biological processes can be interpreted as a heterogeneity within the biological system that may result in a distribution of time constants (28). In contrast, the kinetics of m9-C8-LY dissociation from both subtypes are monoexponential at low temperatures, while at higher temperatures the dissociation from HLA-B*2709-m9-C8-LY could be best fitted by applying a biexponential decay function. Peptide dissociation kinetics from HLA-B*2705-m9-C8-LY could be best approximated by a stretched exponential at high temperatures (>50°C), as in the case of HLA-B*2705-m9-C6-LY.
Kinetic model of m9 dissociation
The temperature dependence of the apparent time constants and amplitudes of the data shown in Fig. 5 was analyzed using a kinetic model that was developed on the basis of our experimental observations and current ideas about peptide dissociation from MHC class I complexes (13,15,16,20). Biphasic peptide dissociation processes were already observed for MHC class I complexes (15,16), but have been controversially discussed. In HLA-A2 complexes, the observation of biexponential kinetics was interpreted as the result of two pathways for peptide dissociation, from the fully assembled heterotrimer and from a β2m-depleted HC/peptide heterodimer (15). On the other hand, biphasic peptide dissociation kinetics in HLA-A2 molecules has been interpreted to result from two conformations in the fully assembled heterotrimer (16). In extension of these previous studies, we simulated our experimental data with a kinetic model, which allows an analysis of the activation energies for peptide dissociation based on the microscopic rate constants. Since we observed at most two time constants in our data sets for peptide dissociation from the B*2705 and B*2709 complexes, the following kinetic model is sufficient to describe our data properly:
 |
(7) |
Here, m9 dissociation is interpreted as a two-step process, with the first step representing an equilibrium between the peptide-bound state (Pbound) and an intermediate state (Pintermediate), described by the rate constants k1 and k−1. The full release of the peptide (Punbound) is described by the slower irreversible process with a rate constant k2. In the experiment, rebinding of the fluorescently-labeled peptide was prevented using an excess of unlabeled peptide. The events after peptide dissociation, such as concomitant complex unfolding in the case of B*2705 or unfolding of the heavy chain and dissociation of β2m in the case of B*2709, are not included in the model, because they are not accessible via peptide dissociation kinetics.
Using Eqs. 8–17 (see Appendix), the kinetic model was applied to the data displayed in Fig. 5. The time traces were fitted at all temperatures with one set of five parameters for the dissociation of m9-C6-LY (Eq. 15: k1, k−1, k2, [Pbound]0 as the initial concentration of peptides bound to the HLA-complex (Eq. 14), and a temperature-dependent offset (r∞), which accounts for the anisotropy of the free peptide) and with one set of three parameters for the dissociation of m9-C8-LY (Eq. 17: k1, [Pbound]0 and r∞). The time constant τ1 from the biexponential least-squares fits at each temperature (Fig. 5, black traces) was taken as a first estimate for the rate constant k1 at the respective temperature. The initial value for k2 was taken from the slowest 1/e decay time of each time trace from the biexponential least-squares fit (see Table S1, Supplementary Material). The rates for k1 and k−1 were adapted to give the best fit of the data. The results of the kinetic model fits are shown in Fig. 5, as colored lines (blue, B*2705; red, B*2709). The agreement between data and model is very good, especially in view of the errors in the kinetic data points. At higher temperatures, however, a stretching factor h had to be considered for the kinetics of B*2705-m9-C8-LY to yield satisfactory fits. In Table 1, all model parameters (fit results) are summarized.
Our simple model for the temperature dependence of the peptide dissociation kinetics is successful for both subtypes B*2705 and B*2709. The model describes correctly the slower dissociation of the peptide from the subtype B*2705 as compared to B*2709. For B*2709-m9-C6-LY, we observed that the two time constants of the biexponential fit at a given temperature do not differ by more than a factor of 5, whereas, for B*2705-m9-C6-LY, the two time constants differ by more than three orders of magnitude. This difference in peptide dissociation kinetics between the two subtypes is also well reproduced by the model (Table 1). In addition, the monoexponential kinetics for m9-C8-LY is satisfactorily described by the modified model applying Eq. 17 (see Appendix).
The microscopic rate constants derived from our kinetic model were used to obtain activation energies for the two transitions in the peptide-dissociation process. The rate constants listed in Table 1 were presented in Arrhenius plots (ln k vs. 1/T), as shown in Fig. 6. Assuming a thermally activated peptide dissociation process, an activation energy was determined for each rate constant k1, k−1, and k2 from the slope of a least-squares fit for each temperature-dependent data set. The respective activation energies are summarized in Table 2. The activation energies for full peptide release, EA(krelease), are derived from k2 in the case of m9-C6-LY and k1 in the case of m9-C8-LY.
FIGURE 6.

Arrhenius plots of B*2705 and B*2709 complexed with m9-C6-LY and m9-C8-LY, respectively. Rate constants result from the kinetic model described in the text.
TABLE 2.
Activation energies from the kinetic model
| Subtype | EA(k1) kJ mol−1 | EA(k−1) kJ mol−1 | EA(krelease) kJ mol−1 |
|---|---|---|---|
| B*2709-m9-C6-LY | 147.7 ± 9.7 | 150.4 ± 8.2 | 134.3 ± 7.4 |
| B*2709-m9-C8-LY | — | — | 153.4 ± 9.2 |
| B*2705-m9-C6-LY | 93.6 ± 7.6 | 96.4 ± 8.1 | 165.3 ± 16.5 |
| B*2705-m9-C8-LY | — | — | 156.0 ± 40.2 |
By comparing EA(krelease), it can easily be seen that these values are quite similar for both subtypes and both peptide variants, since a value of ∼150 kJ/mol was obtained in all cases. This indicates that the underlying mechanism of full peptide release described by the respective microscopic rate constant (k2 in the case of m9-C6-LY and k1 in the case of m9-C8-LY) is also similar in the two subtypes and independent of the reporter group position. Based on x-ray structure analysis, arginine in position 2 of the peptide, close to the N-terminus of the peptide, provides the primary anchor of peptides bound to the B-pocket of the peptide binding groove in HLA-B27 molecules (30), while the C-terminal residue is regarded as a particularly important secondary anchor, which binds within the F pocket. The peptide dissociation process mainly reflects the unbinding of these anchors, although every other amino acid may, in principle, contribute to peptide binding stability and therefore to the peptide dissociation process. As the difference between the two HLA-B27 subtypes under investigation is a single amino-acid exchange located in the F pocket, which does not effect stable accommodation of the main anchor pArg2 of the peptide m9 in the B pocket (12), we suggest that the similar activation energies for full peptide release in both subtypes are associated with the dissociation process of the main anchor pArg2 from the binding groove.
The activation energies for the first step in temperature-induced m9-C6-LY dissociation, however, differ between the two subtypes and point to distinct underlying mechanisms. Surprisingly, EA(k1, k−1) for the less thermally stable complex B*2709-m9 is higher than for B*2705-m9. A comparison of EA(krelease) with EA(k1, k−1) for B*2709 shows that similar activation energies of ∼150 kJ/mol are found for the first and the second transition in peptide dissociation from the B*2709 complex. In addition, we also observed that the values obtained for the rate constants k1, k−1, and k2 are of the same order of magnitude (Table 1, Appendix). For B*2709, which lacks the C-terminal salt bridge between p9 and residue 116 in the F-pocket as compared to B*2705, we consider an intermediate state in m9 dissociation (Pintermediate in Eq. 7) that is characterized by a partially unbound peptide as very likely, based on the idea that peptide dissociation may start from the C-terminus (13,20). In contrast, the rate constants obtained for the first and second steps in peptide dissociation from the B*2705 complex differ by about two to three orders of magnitude. Here, a scenario of a partially unbound peptide seems very unlikely because of the strong C-terminal anchor mediated by the pLys9-Asp116 salt bridge. However, a conformational change of the peptide binding pocket, leading to an intermediate state with a different peptide binding mode (Pintermediate in Eq. 7), may account for the lower activation energy for the first step in m9 dissociation from B*2705. The idea of a conformationally altered peptide binding groove, leading to more conformational freedom of the peptide but not to a partially unbound peptide as suggested for B*2709-m9, is consistent with the smaller amplitudes of the first kinetic component (Table S1) observed for peptide release from B*2705-m9 (1.1–4% of the total amplitude) compared to B*2709-m9 (9.5–76.1% of the total amplitude). An altered peptide binding domain, probably connected to a β2m-induced conformational change (16), was already reported for HLA-A2 molecules. In addition, two different pathways for m9 dissociation from the two HLA-B27 subtypes, including the involvement of β2m-depleted HC/peptide heterodimers (15,16) cannot be ruled out. Further experiments, dissecting the role of β2m on complex stability and dynamics, were therefore performed.
Peptide dissociation kinetics in dependence of β2m concentration reveal two different pathways for B*2705 and B*2709
Since earlier studies in the field found evidence for an initial β2m release in complex unfolding and suggested a role for β2m in dampening conformational dynamics in the HLA-A2 heavy chain (15), we also monitored the kinetics of m9 dissociation in the presence of excess β2m. If there is fast initial β2m release, we would expect the addition of excess β2m to shift the β2m dissociation equilibrium toward the fully assembled trimeric HLA-complex. While there was no effect on our measured anisotropy decay curves in the case of the subtype HLA-B*2709 at 30°C (Fig. 7 B), the initial anisotropy of subtype HLA-B*2705 at 60°C as well as at physiological temperature increased after addition of excess of β2m as shown in Fig. 7 (A) (1000-fold excess) and Fig. 8 (2000-fold excess). Additionally, the mean decay constant of the concentration of bound peptide is slower with an excess of β2m. The time traces at 60°C were fitted with stretched exponentials and at 37°C a biexponential decay law was applied; the results are summarized in Table 3. Note that the value of the heterogeneity parameter h decreases with excess of β2m, indicating a reduction in heterogeneity in the trimeric complex.
FIGURE 7.

Kinetics of peptide dissociation measured via time-dependent change in anisotropy of LY-labeled HLA-B*2705-m9-C6-LY with and without β2m addition at 60°C (A). Time-dependent change in anisotropy of LY-labeled HLA-B*2709-m9-C6-LY with and without β2m addition at 30°C (B). Conditions were chosen as in Fig. 2.
FIGURE 8.

Time-dependent change in anisotropy of LY-labeled HLA-B*2705-m9-C6-LY with and without addition of β2m at physiological temperature. Conditions were chosen as in Fig. 2.
TABLE 3.
Parameters of the exponential fits of the kinetics of peptide dissociation from HLA-B*2705-m9-C6 with and without addition of β2m observed by time-dependent detection of the stationary anisotropy
| 60°C
|
37°C
|
||||||
|---|---|---|---|---|---|---|---|
| A | τ/s | h | A1/% | τ1/s | A2/% | τ2/s | |
| W/o excess β2m | 0.0875 | 4.14 × 103 | 1.50 | 2.9 | 3.60 × 102 | 97.1 | 8.32 × 105 |
| With excess β2m | 0.0881 | 7.69 × 103 | 1.21 | 4.6 | 1.39 × 104 | 95.4 | 2.30 × 106 |
For the kinetics at 60°C, stretched exponential decay and for those at 37°C, biexponential decay laws were applied.
The data presented in Figs. 7 and 8 reveal that the effect of β2m concentration on both the dissociation kinetics and the steric restriction of the fluorophore LY bound to the peptide m9 is exclusively observed for the B*2705 complexes. Therefore, β2m seems to have a stabilizing effect on the binding groove conformation of the B*2705 subtype and, through long-range interactions, is indirectly involved in the dissociation of the peptide m9 from the B*2705 complex. This result provides further implications for the nature of the initial peptide dissociation step from B*2705, as will be discussed below.
Current models of peptide release from MHC class I molecules favor either parallel (16) or branched kinetics (15). In both models, initial β2m dissociation from the heterotrimeric pMHC plays a role. Interestingly, for HLA-A2 complexes it was observed that an excess of β2m reduced the amplitude of the first phase in the biphasic peptide dissociation time trace (15), leading the authors to assume that the excess of β2m shifts the β2m dissociation equilibrium toward the fully assembled heterotrimer. Accordingly, the fast peptide dissociation phase results from initial dissociation of β2m, with subsequent rapid peptide release from the HC/peptide dimer (15). In our study, however, addition of β2m had no effect on the kinetics and the amplitudes of B*2709-m9 complexes, while for B*2705 complexes excess of β2m even slowed down peptide dissociation kinetics and increased the amplitude of the first phase, thereby increasing the steric restriction of peptide mobility by constituents of the binding groove. Assuming initial β2m dissociation before peptide release, we would expect exactly the opposite effect, i.e., a smaller amplitude for the first phase. We therefore exclude fast initial β2m release as the origin of the first peptide release component for B*2705-m9. Instead, we suggest a β2m induced conformational change of the peptide binding groove as the origin of the first phase before full dissociation of m9 from B*2705. Comparable long-range effects of β2m association with the heavy chain have been reported to alter the peptide binding domain in HLA-A2 molecules, probably by a conformational change (16). In this study, however, biphasic peptide dissociation kinetics was interpreted as a result of the fully assembled trimeric complex existing in two distinct conformations that exhibited differential stability. It was assumed that peptide dissociation from the complex with the less stable conformation involved initial β2m dissociation and subsequent fast peptide release, while dissociation from the complex with the stable conformation probably proceeded via at least one intermediate state (16). These distinct dissociation mechanisms should result in complex multiexponential kinetics, which, however, were not observed in our experiments.
Based on these considerations, we favor a model for m9 dissociation from B*2705 being a two-step process, which includes an intermediate state characterized by different interactions of β2m with the heavy chain as compared to the initial peptide bound state, leading to less restriction by the peptide binding groove (Fig. 9 A) and therefore a decrease in the anisotropy. About the nature of the different interaction between β2m and complex and possible conformational changes within β2m we can only speculate. However, our experimental investigations and theoretical work show that the peptide-binding groove is very sensitive to changes with regard to β2m surrounding and degree of interaction to the protein complex as well as β2m concentration. As, for B*2709-m9, β2m excess had no significant effect on the peptide-dissociation kinetics and the weak C-terminal interactions of m9 within the F-pocket lead to an enhanced flexibility of the peptide (13), we propose for B*2709 a two-step model with the first step representing an equilibrium between the peptide-bound state and a state with a partially unbound peptide (Fig. 9 B).
FIGURE 9.

Model for m9 peptide dissociation from HLA-B*2705 (A) and HLA-B*2709 (B).
SUMMARY
Aiming at a detailed dissection of antigen stability in two differentially autoimmune-disease-associated HLA-B27 subtypes, we have carried out various steady-state and time-resolved fluorescence depolarization experiments to study thermodynamics and kinetics of peptide dissociation using fluorescently-labeled peptides. In this report, we have focused on the peptide m9 (GRFAAAIAK). C-terminal lysine or arginine, as found in the sequence of the m9 peptide, account for ∼27% of the natural B*2705 ligands (11). Analysis of peptide-dissociation kinetics is important, as differences in peptide affinity are mainly due to differences in peptide-dissociation constants (16).
Our data reveal two different pathways for m9 dissociation from B*2705 and B*2709, underscoring the quantitative differences in the binding properties of peptides bearing a C-terminal Lys with the two differentially AS-associated subtypes. Fast peptide-release kinetics, with time constants of ∼1 h at physiological temperature as observed for B*2709, explain the low probability of native peptides with basic C-terminal anchor residues to be eluted from that B27 subtype (11,29). Simulation of the dissociation kinetics with a two-step kinetic model revealed similar activation energies of ∼130–150 kJ/mol for the two transitions observed for peptide dissociation from B*2709, dominated by the dissociation of the C-terminal and the primary R2 anchor, respectively. For the disease-associated subtype B*2705, an intermediate state in the peptide-dissociation pathway was observed, which is characterized by a β2m-coupled conformational change of the peptide binding pocket. Such allosteric mechanisms, controlling the interactions between the components of the trimeric HLA complex, not only govern its assembly and dissociation (16) but might also influence recognition by T-cell receptors. At physiological temperature, the transition to the intermediate state with altered β2m/HC interaction occurs within minutes, while final peptide release lasts for ∼10 days and thus determines the peptide-HLA-B*2705 conformation, which will be recognized by cellular ligands of the immune system, such as T-cell receptors.
The finding that β2m contributes, in a subtype-dependent fashion, to the stability of pMHC, adds not only to an understanding of the mechanism of pMHC dissociation, but may be relevant for the pathogenesis of AS as well (31). A recent study employing rats transgenic for B*2705 and human β2m (to different levels of expression) showed that β2m is intimately connected with the occurrence of a variety of diseases. Only rats with very high levels of human β2m suffered from AS-related symptoms, indicating the importance of human β2m (and B*2705 complexes with human β2m) for disease development. However, as pointed out previously (31), there are important differences between the development of AS in humans and transgenic animals, in particular with regard to the number of HLA-B27 and β2m gene copies present (each maximally two in humans, each >20 in diseased rats homozygous for the respective locus) and the time course of disease progression (decades in humans, <1 year in rats with high HLA-B27 and β2m copy numbers). Nevertheless, the successful reconstitution of the B*2705-m9 complex with covalent β2m-dimers described in our study provides, in this context, additional insight into the variety of interactions that may occur within HLA-complexes.
In summary, evidence has been presented in this study that an allosteric mechanism, intimately connected with β2m, controls m9 peptide presentation and dissociation from B*2705. The described differences in complex stability and peptide presentation modes between the differentially AS-associated B*2705 and B*2709, as well as the appearance of long-range interactions between β2m and the peptide-binding groove in the strong AS-associated subtype B*2705, are solely due to the natural polymorphism in the single amino-acid position 116 at the floor of the peptide-binding groove.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Dr. R. Misselwitz (Institut für Immungenetik) for the help with purification of HLA-B27-peptide complexes and preparation of free β2m-microglobulin and A. Eismann for excellent technical assistance. We are also grateful to Dr. A. Ziegler for his comments on the article.
This work was financially supported by the Deutsche Forschungsgemeinschaft SFB 449 (project Nos. A5 to U.A. and B6 to B.U.-Z.) and by the Volkswagen-Foundation (grant No. I/79 983 to U.A.).
APPENDIX
The kinetic model (Eq. 7) can be described with a set of differential equations for the change in concentrations of 1), peptides bound to the complex [Pbound]; 2), intermediately bound peptides [Pint]; and 3), peptides completely dissociated from the complex [Punbound]. In the case of m9-C6-LY, the following equations apply:
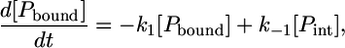 |
(8) |
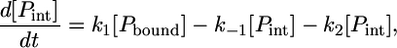 |
(9) |
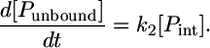 |
(10) |
Since we assume an equilibrium process of the first dissociation process (Eq. 7), i.e., [Pbound] + [Punbound] = const., the change in concentration of the intermediately bound peptide becomes zero, d[Pint]/dt = 0. An expression for the change in concentration of the bound peptide [Pbound] can then be obtained:
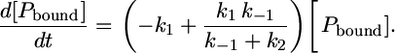 |
(11) |
Separation of variables and integration of Eq. 11 gives
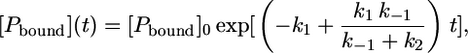 |
(12) |
with [Pbound]0 as the initial concentration within the heterotrimeric complex. As the fluorescence anisotropy directly reflects the mole fraction of peptide molecules bound to the pMHC and the fraction of peptide free in solution, the change in concentration of the bound peptide [Pbound] is monitored by the decrease in anisotropy r(t):
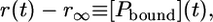 |
(13) |
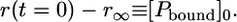 |
(14) |
Since the anisotropy of the free peptide is not zero, a corresponding temperature-dependent offset, r∞, has to be added to Eq. 12, yielding the correct concentration-time profile:
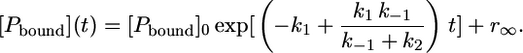 |
(15) |
For the dissociation of the peptide m9-C8-LY from B*2705 and B*2709, we observed monoexponential kinetics at moderate temperatures and biexponential kinetics at higher temperatures as well as slightly slower kinetics compared to the corresponding values observed with the peptide m9-C6-LY (Fig. 5, B and D). Under the assumption that an interaction between C-LY in position 8 of the peptide and residues of the binding groove results in a shifted equilibrium toward the bound peptide, the kinetic model described above is modified in the following way. The rate constants for the first reaction step and for the back reaction become much smaller than the rate constant for the full release of the peptide
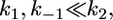 |
(16) |
so that the quotient in Eq. 14 approaches zero. Thus, only the slowest rate constant determines the speed of the reaction as described by Eq. 17:
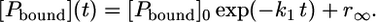 |
(17) |
Note added in proof: Under the assumption that the rates in our kinetic model follow a simple Arrhenius law and the anisotropy values of the three different states have linear temperature dependence, the system of coupled differential equations describing our kinetic model can be solved in the form of a sum of two exponential functions and the respective activation energies can be determined using a global fit to data sets at five different temperatures. The activation energies from this fit (B*2705-m9-C6-LY: EA(k1) = 118 kJ/mol, EA(k−1) = 100 kJ/mol, EA(k2) = 155 kJ/mol; B*2709-m9-C6-LY: EA(k1) = 130 kJ/mol, EA(k−1) = 145 kJ/mol, EA(k2) = 131 kJ/mol) agree with the values obtained from the simplified approach described above. The anisotropy in the intermediate state (Pmt) amounts to 93% (B*2705-m9-C6-LY) and 36% (B*2709-m9-C6-LY) of the anisotropy value obtained for the respective peptide-bound state.
Anja Winter's new address is Institute of Structural & Molecular Biology, The University of Edinburgh, Mayfield Road, Edinburgh EH9 3JR, Scotland United Kingdom.
Dr. Christine Rueckert's new address is Leibniz-Institut für Molekulare Pharmakologie, Robert-Rössle-Str. 10, D-13125 Berlin, Germany.
Editor: Brian R. Dyer.
References
- 1.Madden, D. R. 1995. The three-dimensional structure of peptide-MHC complexes. Annu. Rev. Immunol. 13:587–622. [DOI] [PubMed] [Google Scholar]
- 2.Pamer, E., and P. Cresswell. 1998. Mechanisms of MHC class I-restricted antigen processing. Annu. Rev. Immunol. 16:323–358. [DOI] [PubMed] [Google Scholar]
- 3.Saper, M. A., P. J. Bjorkman, and D. C. Wiley. 1991. Refined structure of the human histocompatibility antigen HLA-A2 at 2.6 Å resolution. J. Mol. Biol. 219:277–319. [DOI] [PubMed] [Google Scholar]
- 4.Horten, R., L. Wilming, V. Rand, R. C. Lovering, E. A. Bruford, V. K. Khodiyar, M. J. Lush, S. Povey, C. C. Talbot, Jr., M. W. Wright, H. M. Wain, J. Trowsdale, A. Ziegler, and S. Beck. 2004. Gene map of the extended human MHC. Nature Rev. Gen. 5:889–899. [DOI] [PubMed] [Google Scholar]
- 5.Khan, M. A. 2002. Update on spondyloarthropathies. Ann. Intern. Med. 136:896–907. [DOI] [PubMed] [Google Scholar]
- 6.Ramos, M., and J. A. López de Castro. 2002. HLA-B27 and the pathogenesis of spondyloarthritis. Tissue Antigens. 60:191–205. [DOI] [PubMed] [Google Scholar]
- 7.Kim, T.-H., W.-S. Uhm, and R. D. Inman. 2005. Pathogenesis of ankylosing spondylitis and reactive arthritis. Curr. Opin. Rheumatol. 17:400–405. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez-Roces, S., M. V. Alvarez, S. Gonzalez, A. Dieye, H. Makni, D. G. Woodfield, L. Housan, V. Konenkov, M. C. Abbadi, N. Grunnet, E. Coto, and C. Lopez-Larrea. 1997. HLA-B27 polymorphism and worldwide susceptibility to ankylosing spondylitis. Tissue Antigens. 49:116–123. [DOI] [PubMed] [Google Scholar]
- 9.D'Amato, M., M. T. Fiorillo, C. Carcassi, A. Mathieu, A. Zuccarelli, P. P. Bitti, R. Tosi, and R. Sorrentino. 1995. Relevance of residue 116 of HLA-B27 in determining susceptibility to ankylosing spondylitis. Eur. J. Immunol. 25:3199–3201. [DOI] [PubMed] [Google Scholar]
- 10.Fiorillo, M. T., G. Greco, and R. Sorrentino. 1995. The Asp116-His116 substitution in a novel HLA-B27 subtype influences the acceptance of the peptide C-terminal anchor. Immunogenetics. 41:38–39. [DOI] [PubMed] [Google Scholar]
- 11.Ramos, M., A. Paradela, M. Vázquez, A. Marina, J. Vázquez, and J. A. López de Castro. 2002. Differential association of HLA-B*2705 and B*2709 to ankylosing spondylitis correlates with limited peptide subsets but not with altered cell surface stability. J. Biol. Chem. 277:28749–28756. [DOI] [PubMed] [Google Scholar]
- 12.Hülsmeyer, M., R. C. Hillig, A. Volz, M. Rühl, W. Schröder, W. Saenger, A. Ziegler, and B. Uchanska-Ziegler. 2002. HLA-B27 subtypes differentially associated with disease exhibit subtle structural alterations. J. Biol. Chem. 277:47844–47853. [DOI] [PubMed] [Google Scholar]
- 13.Pöhlmann, T., R. A. Böckmann, H. Grubmüller, B. Uchanska-Ziegler, A. Ziegler, and U. Alexiev. 2004. Differential peptide dynamics is linked to major histocompatibility complex polymorphism. J. Biol. Chem. 279:28197–28201. [DOI] [PubMed] [Google Scholar]
- 14.Hillig, R., M. Hülsmeyer, W. Saenger, K. Welfle, R. Misselwitz, H. Welfle, C. Kozerski, A. Volz, B. Uchanska-Ziegler, and A. Ziegler. 2004. Thermodynamic and structural analysis of peptide- and allele-dependent properties of two HLA-B27 subtypes exhibiting differential disease association. J. Biol. Chem. 279:652–663. [DOI] [PubMed] [Google Scholar]
- 15.Binz, A.-K., R. C. Rodriguez, W. E. Biddison, and B. M. Baker. 2003. Thermodynamic and kinetic analysis of a peptide-class I MHC interaction highlights the noncovalent nature and conformational dynamics of the class I heterotrimer. Biochemistry. 42:4954–4961. [DOI] [PubMed] [Google Scholar]
- 16.Gakamsky, D. M., D. M. Davis, J. L. Strominger, and I. Pecht. 2000. Assembly and dissociation of human leukocyte antigen HLA-A2 studied by real-time fluorescence resonance energy transfer. Biochemistry. 39:11163–11169. [DOI] [PubMed] [Google Scholar]
- 17.Gakamsky, D. M., P. J. Bjorkman, and I. Pecht. 1996. Peptide interaction with a Class I major histocompatibility complex-encoded molecule: allosteric control of the ternary complex stability. Biochemistry. 35:14841–14848. [DOI] [PubMed] [Google Scholar]
- 18.Gakamsky, D. M., L. F. Boyd, D. H. Margulies, D. M. Davis, J. L. Strominger, and I. Pecht. 1999. An allosteric mechanism controls antigen presentation by the H-2K complex. Biochemistry. 38:12165–12173. [DOI] [PubMed] [Google Scholar]
- 19.Strong, R. K., M. A. Holmes, P. Li, L. Braun, N. Lee, and D. E. Geraghty. 2003. HLA-E allelic variants. J. Biol. Chem. 278:5082–5090. [DOI] [PubMed] [Google Scholar]
- 20.Dédier, S., S. Reinelt, T. Reitinger, G. Folkers, and D. Rognan. 2000. Thermodynamic stability of HLA-B*2705-peptide complexes. Effect of peptide and major histocompatibility complex protein mutations. J. Biol. Chem. 275:27055–27061. [DOI] [PubMed] [Google Scholar]
- 21.Dédier, S., S. Reinelt, S. Rion, G. Folkers, and D. Rognan. 2001. Use of fluorescence polarization to monitor MHC-peptide interactions in solution. J. Immunol. Methods. 255:57–66. [DOI] [PubMed] [Google Scholar]
- 22.Reinelt, S., S. Dédier, G. Asuni, G. Folkers, and D. Rognan. 2001. Mutation of Cys-67 alters the thermodynamic stability of the human leukocyte antigen HLA-B*2705. J. Biol. Chem. 276:18472–18477. [DOI] [PubMed] [Google Scholar]
- 23.Hülsmeyer, M., K. Welfle, T. Pöhlmann, R. Misselwitz, U. Alexiev, H. Welfle, W. Saenger, B. Uchanska-Ziegler, and A. Ziegler. 2005. Thermodynamic and structural equivalence of two HLA-B27 subtypes complexed with a self-peptide. J. Mol. Biol. 346:1367–1379. [DOI] [PubMed] [Google Scholar]
- 24.Garboczi, D. N., D. T. Hung, and D. C. Wiley. 1992. HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc. Natl. Acad. Sci. USA. 89:3429–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Menssen, R., P. Orth, A. Ziegler, and W. Saenger. 1999. Decamer-like conformation of a non-peptide bound to HLA-B*3501 due to non-standard positioning of the C-terminus. J. Mol. Biol. 285:645–653. [DOI] [PubMed] [Google Scholar]
- 26.Eftink, M. R. 1994. The use of fluorescence methods to monitor unfolding transitions in proteins. Biophys. J. 66:482–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alexiev, U., I. Rimke, and T. Pöhlmann. 2003. Elucidation of the nature of the conformational changes of the EF-interhelical loop in bacteriorhodopsin and of the helix 8 on the cytoplasmic surface of bovine rhodopsin: a time-resolved fluorescence depolarization study. J. Mol. Biol. 328:705–719. [DOI] [PubMed] [Google Scholar]
- 28.Lee, K. C., J. Siegel, S. E. D. Webb, S. Lévêque-Fort, M. J. Cole, R. Jones, K. Dowling, M. J. Lever, and P. M. W. French. 2001. Application of the stretched exponential function to fluorescence lifetime imaging. Biophys. J. 81:1265–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.López de Castro, J. A., I. Alvarez, M. Marcilla, A. Paradela, M. Ramos, L. Sesma, and M. Vázquez. 2004. HLA-B27: a registry of constitutive peptide ligands. Tissue Antigens. 63:424–445. [DOI] [PubMed] [Google Scholar]
- 30.Madden, D. R., J. C. Gorga, J. L. Strominger, and D. C. Wiley. 1992. The three-dimensional structure of HLA-B27 at 2.1 Å resolution suggests a general mechanism for tight peptide binding to MHC. Cell. 70:1035–1048. [DOI] [PubMed] [Google Scholar]
- 31.Uchanska-Ziegler, B., and A. Ziegler. 2003. Ankylosing spondylitis: a β2m-deposition disease? Trends Immunol. 24:73–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.