

Abstract
Evolutionary mechanisms of origins of new gene function have been a subject of long-standing debate. Here we report a convincing case in which positive Darwinian selection operated at the molecular level during the evolution of novel function by gene duplication. The genes for eosinophil cationic protein (ECP) and eosinophil-derived neurotoxin (EDN) in primates belong to the ribonuclease gene family, and the ECP gene, whose product has an anti-pathogen function not displayed by EDN, was generated by duplication of the EDN gene about 31 million years ago. Using inferred nucleotide sequences of ancestral organisms, we showed that the rate of nonsynonymous nucleotide substitution was significantly higher than that of synonymous substitution for the ECP gene. This strongly suggests that positive Darwinian selection operated in the early stage of evolution of the ECP gene. It was also found that the number of arginine residues increased substantially in a short period of evolutionary time after gene duplication, and these amino acid changes probably produced the novel anti-pathogen function of ECP.
Keywords: ancestral sequence, anti-pathogenic function, rapid evolution, synonymous and nonsynonymous substitution
Gene duplication and subsequent functional divergence of duplicate genes is one of the most important mechanisms for the evolution of novel gene function (1–5). However, it has been controversial whether the functional divergence occurs by positive Darwinian selection that accelerates the fixation of advantageous mutations (largely nonsynonymous or amino acid-altering mutations) (6) or by random fixation of neutral mutations, which later induce a change in gene function when the environment or the genetic background is altered (Dykhuizen–Hartl effect) (7, 8). In the latter case the rate of nonsynonymous nucleotide substitution may also be enhanced because of relaxation of functional constraints of redundant genes after gene duplication (8), but the rate will never exceed that of synonymous substitution. Therefore, it is possible to distinguish between the two hypotheses by comparing the rates of synonymous and nonsynonymous substitution. Earlier studies (9–11) have indicated that the rate of nonsynonymous substitution is often enhanced after gene duplication, but in no case was the rate significantly higher than that of synonymous substitution, leaving the role of positive selection uncertain. In this paper we present a convincing case of positive selection that occurred during the early stage of divergence of the duplicated eosinophil cationic protein (ECP) and eosinophil-derived neurotoxin (EDN) genes in primates.
EVOLUTION OF THE EDN AND ECP GENES
Novel Function of the ECP Gene.
ECP and EDN in hominoids and Old World (OW) monkeys belong to the ribonuclease (RNase) superfamily, and they are present in the large specific granules of eosinophilic leukocytes (12). EDN has high catalytic activity of RNase and nonphysiological neurotoxicity (lethal to experimental rabbits when injected into the brain) (12). The real physiological function of EDN is not well understood, but recent studies suggest that it functions as an antiviral agent through ribonucleolytic destruction of genomic RNA of retroviruses (13, 14). By contrast, ECP has very low RNase activity but is a potent toxin to various pathogenic bacteria and parasites (15, 16). Interestingly, ECP kills these pathogens by making pores in their cell membranes (17), and this activity is RNase independent (18). New World (NW) monkeys have only one gene that is homologous to the ECP and EDN genes of hominoids and OW monkeys (19). This gene is called the EDN gene, because its product has physicochemical properties similar to those of EDN and has no toxicity against bacteria and parasites, though the RNase activity is much lower than that of human EDN (16). Phylogenetic analysis of DNA sequences (19) has suggested that the ECP and EDN genes were produced by tandem gene duplication from an EDN-like ancestral gene after divergence of OW and NW monkeys but before separation of hominoids and OW monkeys (see Fig. 1). The anti-pathogen function of ECP was apparently acquired after gene duplication, because this function is not shared by EDN (12). Moreover, the evolutionary rates of the ECP and EDN genes, particularly the former, are among the highest of all primate genes so far studied (19). These observations have led to the suggestion that the ECP gene has been subject to positive Darwinian selection (19), but comparison of the ECP and EDN gene sequences does not show a rate of nonsynonymous substitution significantly higher than that of synonymous substitution. Nevertheless, we suspected that positive selection operated in a relatively short period of time after gene duplication but the subsequent purifying selection and neutral nucleotide substitutions have masked the effect of positive selection. We therefore examined the rates of synonymous and nonsynonymous substitution for each branch of the ECP/EDN gene tree in Fig. 1.
Figure 1.

Evolutionary tree of primate ECP and EDN genes. The root of this tree is located on the branch linking node a and the NW monkey tamarin if we use mouse sequences (54, 55) as outgroups. The gene duplication occurred at node a. (A) The b̂N and b̂S for each branch are presented as b̂N (×100)/b̂S (×100) above the branch. The standard errors of b̂N and b̂S are 0.0176 and 0.0151 for the branch a–b and 0.0108 and 0.0056 for the branch d–e, respectively. The statistical significance of the positiveness of b̂N − b̂S was determined by one-tailed Z test and is indicated by asterisks (∗∗, 1%). (B) The numbers of nonsynonymous (n) and synonymous (s) substitutions per sequence per branch are presented as n/s above each branch. The N/S ratio for the sequences is given above the tree. The statistical significance of the difference between n/s and N/S was tested by Fisher’s exact test (Table 1).
Test of Positive Selection by Using the Numbers of Synonymous (bS) and Nonsynonymous (bN) Nucleotide Substitutions per Site for Each Tree Branch.
A simple method to estimate the number of synonymous nucleotide substitutions per synonymous site (bS) and the number of nonsynonymous substitutions per nonsynonymous site (bN) for each branch of the phylogenetic tree is first to estimate the numbers of synonymous (dS) and nonsynonymous (dN) substitutions per site for all pairs of present-day sequences and then to estimate branch lengths (bS and bN values) by using the least-squares method for a given tree topology (20). The variances of the estimates (b̂S and b̂N) of bS and bN can be obtained from the variances and covariances of the estimates (d̂Ss and d̂Ns) of dSs and dNs by the method given in ref. 20. Once these estimates are obtained, we can test positive selection by examining the statistical significance of the difference between b̂S and b̂N for each branch.
The Nei–Gojobori (NG) method (21) is often used for estimating dS and dN, and the formulas given in ref. 22 can be used for computing the variances and covariances of d̂Ss and d̂Ns. However, the NG method is known to give biased estimates of dS and dN when the rate of transitional nucleotide change is higher than that of transversional change. For this reason, a number of authors have developed new methods to rectify this deficiency (23–26). Unfortunately, these methods are not appropriate for our purpose, because it is difficult to compute the covariances of d̂Ss and d̂Ns that are required for estimating the variances of b̂S and b̂N and testing the difference between b̂S and b̂N. Furthermore, in some of the methods (23–25), nonsense mutations are included in the category of nonsynonymous substitutions and d̂S and d̂N sometimes become negative when sequence divergence is low, whereas the other (26) is often inapplicable when closely related sequences are used. For these reasons we used a modified version of the NG method, which is presented in the Appendix.
In the present study, we used the ECP and EDN gene sequences from the human (Homo sapiens), chimpanzee (Pan troglodytes), gorilla (Gorilla gorilla), orangutan (Pongo pygmaeus), and OW monkey macaque (Macaca fascicularis), and the EDN gene sequence from the NW monkey tamarin (Saguinus oedipus) (19), for which the phylogenetic tree is known (Fig. 1). To estimate dS and dN values by the modified NG method, we need to know the ratio (R) of transitional changes to transversional changes for the data set used. For this purpose we used the methods given in refs. 27 and 28 and obtained R = 1 approximately. We therefore assumed R = 1 in the following computation. The b̂S and b̂N for each branch obtained from dS and dN values are presented in Fig. 1A. For some short branches, b̂S and b̂N became negative apparently because of sampling errors, and these estimates were assumed to be 0. A simple way to conduct a statistical test of the positiveness of b̂N − b̂S is to use the one-tail Z (or t∞) test under the assumption that the numbers of synonymous and nonsynonymous substitutions are so large that b̂S and b̂N are normally distributed (29).
Fig. 1A shows that in the EDN gene b̂S is greater than b̂N for most branches, as expected for genes that are subject to purifying selection. In the branch a–c, b̂N is greater than b̂S, but the difference is not statistically significant. In the ECP gene, b̂N is greater than b̂S for several branches, but the application of the Z test shows that the difference b̂N − b̂S is significantly greater than 0 only in the branches a–b and d–e (Fig. 1A). These results suggest that the ECP gene was subject to positive Darwinian selection in the early stage of evolution, as we suspected. However, because the b̂S values in these branches are quite small and the sequences used are relatively short (157 codons), the applicability of the Z test is questionable. When the numbers of synonymous and nonsynonymous substitutions are very small, the Z test is known to be too liberal (30). We therefore conducted an exact test proposed by Zhang et al. (30).
Test of Positive Selection by Using Synonymous (s) and Nonsynonymous (n) Substitutions per Sequence for Each Branch.
In the Zhang et al. test (30), nucleotide sequences of ancestral organisms at all ancestral nodes are inferred from the present-day sequences, and the numbers of synonymous (s) and nonsynonymous (n) substitutions per sequence rather than b̂S and b̂N are computed for each branch. The s and n values are then compared with their expected numbers under the hypothesis of neutral evolution.
To compute s and n for each branch of the tree, we first inferred the ancestral amino acid sequences by using the distance-based Bayesian method (31) under the Poisson model of amino acid substitution and then inferred the ancestral nucleotide sequences under the restriction of the inferred ancestral amino acids. We also used an empirical model of amino acid substitution (JTT model in ref. 32), but the results were essentially the same. Direct inference of ancestral nucleotide sequences by the likelihood-based Bayesian method (33) and the parsimony method (34) gave similar results, but some inferred ancestral sequences contained amino acids that do not exist in the present-day sequences. For this reason, the ancestral sequences inferred from the distance-based Bayesian method were used in further analyses. Because the extent of sequence divergence was low in the present case, the accuracy of the inference was generally very high except for a few sites (Fig. 2). The average accuracy (posterior probability) of the ancestral amino acid inference was in the range of 0.96–0.99 for the nine interior nodes. The accuracy of inference of ancestral nucleotides was 0.99–1.00 when the ancestral amino acids were given.
Figure 2.

Amino acid sequences of present-day and ancestral ECP and EDN proteins. Amino acids are presented by single-letter codes, and dots show the same amino acids as those of the sequence at node a. The arginine (R) residues of ECP and EDN are shown in bold type when they are not identical with those of the ancestral protein at node a. Ambiguous amino acid sites of ancestral sequences at nodes a, b, and c are underlined, where the posterior probability of the most likely amino acid is lower than twice the probability of the second most likely amino acid. The alternative amino acids at the underlined sites were K, R, M, T, Q, N, and T for sites 28, 72, 87, 88, 104, 111, and 113, respectively. These ambiguities occurred almost always at the sites where the orangutan ECP and EDN have the same amino acids but different ones from those of the other orthologous sequences. The overall accuracies (posterior probabilities) of the ancestral proteins were 0.96, 0.97, and 0.98, for nodes a, b, and c, respectively.
The numbers of synonymous (s) and nonsynonymous (n) substitutions for each tree branch (see Fig. 1B) were then estimated from the sequences at the two nodes of the branch by the method in refs. 21 and 35. To test the null hypothesis of equal rates of synonymous and nonsynonymous substitution (neutral evolution), we have to know the numbers of potential synonymous sites (S) and potential nonsynonymous sites (N) for the sequences compared. These numbers can easily be computed by the method described in the Appendix if the transition/transversion ratio (R) is given. In the present case R can be obtained by counting the total numbers of transitional and transversional changes that are observed in the entire phylogenetic tree. This method yielded R = 109/113 = 0.96, which is very close to the values obtained by the mathematical methods mentioned earlier. Therefore, we again used R = 1. Using this R value, we computed S and N for all present-day and ancestral sequences and obtained approximately S = 124 and n = 347 for all sequences.
Under the hypothesis of neutral evolution, the n/s ratio is expected to be equal to N/S = 2.80. There are several branches in which n/s is higher than 2.8. However, n and s are so small that the difference could be due to stochastic errors. We therefore applied Fisher’s exact test for examining the statistical significance of the difference between n/s and N/S. This test showed that the difference is significant only in the branch a–b (P = 0.0055; Table 1). In the previous large-sample Z test, branch d–e also showed a significant difference, but the present analysis shows that the large-sample test is clearly inappropriate for this branch (Fig. 1B). It is interesting, however, that both the large-sample and the small-sample tests give essentially the same result for the branch a–b.
Table 1.
Test of positive selection for the branch a–b
| Nonsynonymous | Synonymous | |
|---|---|---|
| Changes | 33 (n) | 3 (s) |
| No changes | 314 (N − n) | 121 (S − s) |
Fisher’s exact test was used to test the null hypothesis of equal rates of synonymous and nonsynonymous changes. P = 0.0055.
We also considered alternative (but less likely) amino acids for those sites where the ancestral amino acids could not be accurately inferred (Fig. 2). There are seven such sites, and the alternative amino acids are given in the legend to Fig. 2. The posterior probability of having this set of amino acids is about one sixth of that of the set of more likely amino acids presented in Fig. 2. However, if we consider this worst-case scenario (i.e., smallest n/s ratio), n and s for the branch a–b become 26 and 3, respectively, and the difference between n/s and N/S is still statistically significant (P = 0.028). These results strongly suggest that positive Darwinian selection accelerated nonsynonymous substitution in the ECP gene immediately after gene duplication.
The above test of positive selection depends on the assumption that synonymous substitutions are neutral. The rate of synonymous substitution for the EDN gene is close to the rate of nucleotide substitution for intron regions of genes and pseudogenes in primates (36). This approximate equality suggests that the synonymous substitutions in the EDN gene are more or less neutral. The rate of synonymous substitution in the ECP gene after divergence of hominoids and OW monkeys appears to be lower than that for the EDN gene, though the rates of nonsynonymous substitution of the two genes are similar to each other (Fig. 1). (The codon usage pattern is essentially the same for the two genes.) However, the synonymous substitution rate of the ECP gene seems to be normal at the early stage of evolution after gene duplication, because the s values for the branches a–b and a–c are essentially the same. Therefore, our test of positive selection for the branch a–b seems to be acceptable.
High Rate of Amino Acid Substitution in the Branch a–b.
To evaluate the absolute rate of amino acid substitution in the branch a–b, we estimated the time of occurrence of gene duplication by using data on synonymous substitutions. There are on average 22.75 synonymous substitutions in the EDN genes between the macaque and hominoid lineages (Fig. 1), which diverged about 25 million years (Myr) ago (37). The average number of synonymous substitutions for branches a–b and a–c is 2.75. Therefore, the time span of the branch a–b is estimated to be [(2 × 2.75)/22.75] × 25 = 6.0 Myr. Twenty-nine amino acid substitutions were identified for the branch a–b (Fig. 2). Therefore, a conservative estimate of the rate of amino acid substitution for the branch a–b becomes (29/157)/6 Myr = 31 ×10−9 per site per year, where 157 is the total number of amino acids of ECP. Although this estimate has a rather large sampling error, it is more than 3 times higher than the rate (9 ×10−9) of the fastest evolving protein, fibrinopeptide (35). Since the fibrinopeptide rate is considered to be close to the neutral rate (8), this finding supports positive selection involved in the evolution of ECP.
Nonrandom Amino Acid Substitution.
It is of great interest to identify the amino acid substitutions that produced the novel anti-pathogen function of ECP. ECP is known to be more basic than EDN (12, 19), and the isoelectric point (pI) for the ECPs from hominoids and OW monkeys is in the range of 9.9–10.3, whereas that for the EDNs is 8.4–8.8. These pI values were computed from the amino acid sequences by using the Lasergene software. The pI of the ancestral protein at node a was computed to be 8.4 from the amino acid sequence, and this indicates that the ancestral protein was physicochemically more similar to EDN than to ECP. The pI of the protein at node b was computed to be 10.6. This suggests that the new biochemical properties and probably the function of ECP were established before separation of hominoids and OW monkeys.
Of the 29 amino acid substitutions that occurred in the branch a–b, 12 resulted in arginines (R), the most basic amino acid (Figs. 2 and 3). If we assume that amino acid substitution occurs at random, the probability that a substitution results in arginine is p = 1/20. The probability of having 12 or more arginine substitutions out of 29 substitutions is given by the binomial formula Σi=1229(i29)pi(1 − p)29−i, which becomes 6.7 × 10−9. A similar probability (9.8 × 10−9) was obtained when the amino acid composition of the ancestral protein at node a was considered and an empirical model of amino acid substitution (JTT model) was used. These results strongly suggest that amino acid substitutions in the branch a–b were nonrandom and arginines were positively selected for. We also compared the arginine substitution pattern for the branch a–b with that for the ECP lineages that descended from node b, including all branches linking the present-day ECP genes. The total number of amino acid substitutions for these descendent lineages was 39, of which four resulted in arginines. Fisher’s exact test showed that the proportion of arginine changes for these lineages (4/39) is significantly lower than that (12/29) for the branch a–b (P = 0.003), indicating that the amino acid substitution pattern is not the same for the two evolutionary periods. Note also that the proportion of arginine changes in the EDN lineages that descended from node a is low (6/41). It is apparent that the arginine content of ECP increased substantially in a relatively short period of time and then has remained more or less constant (Fig. 3). These results suggest that the arginine changes probably generated the novel anti-pathogen toxicity. There is experimental evidence that ECP kills bacteria and parasites by making pores in their cell membranes, probably through the contact of positively charged amino acids to the membranes (17, 38).
Figure 3.

Evolution of the novel anti-pathogen toxicity and arginine changes in ECP. Each circle represents one arginine residue in ECP or EDN sequences (see Fig. 2). For the present-day proteins, only the numbers of arginines for human sequences are presented. The average number of arginines for OW monkeys and hominoids is 18.2 for ECP and 7.6 for EDN. The pI value for each protein is also presented. The numbers of amino acid changes to arginines in the branches a–b and a–c in Fig. 1 are given alongside the arrow signs. The numbers of changes from arginines to other amino acids are given in parentheses. MY, million years.
DISCUSSION
In the present paper we used two different methods for testing positive selection in ancestral sequences: estimation of bS and bN from d̂S and d̂N values (b̂N/b̂S test) and computation of s and n from ancestral nucleotide sequences (n/s test). These two methods have advantages and disadvantages. When the number of nucleotides used is large and b̂S and b̂N are relatively large, the b̂N/b̂S test is a valid statistical test. A special case of this test was recently used for the DNA sequences of the gene Acp26Aa (a gene controlling male reproduction) from four Drosophila sibling species, and positive selection was detected (39). Our reanalysis of this data set by using the n/s test gave the same conclusion. However, when the conditions mentioned above are not satisfied, the b̂N/b̂S test may become too liberal (30).
The n/s test is more complicated than the b̂N/b̂S test and requires accurate inference of ancestral sequences. When closely related sequences are used, the latter requirement is usually satisfied (31). However, when the sequences used are distantly related, two or more possible nucleotides may be inferred at a site, and this makes the n/s test less powerful. At the present time, the condition under which the n/s test becomes inefficient is unclear, and we are currently investigating this problem.
Our analysis suggested that an increase in arginine content is responsible for the evolution of anti-pathogen toxicity of ECP. However, the number of arginine residues of the orangutan ECP is somewhat smaller than that of the other ECP sequences, and it appears that some of the arginine residues at node b mutated back to the original amino acids in the orangutan lineage (Fig. 2). At first sight, this observation does not seem to conform to our conclusion, but the important factor is apparently the total charge of the ECP protein. Actually, the orangutan ECP contains two extra basic amino acids, lysines (K), and the pI value for this protein is 9.9 and is not particularly lower than that of other ECPs.
We have shown that the ECP gene rapidly evolved by positive selection after gene duplication. However, it is not very clear how the positive selection operated for this gene. One plausible scenario is that when the EDN gene was duplicated, the purifying selection for the duplicate genes was relaxed and at this stage a few neutral or nearly neutral mutations changed the function of one of the two duplicate genes in the direction of anti-pathogen toxicity. Once this change occurred, the anti-pathogen toxicity of the gene was enhanced by further mutations and natural selection due to exposure to bacteria and parasites. The enhancement of nonsynonymous substitution apparently occurred in this second stage. Another scenario is that the original EDN gene had a weak anti-pathogen activity as well as the RNase function, and gene duplication provided an opportunity for one of the two redundant genes to evolve into the ECP gene specializing in anti-pathogen toxicity. In this scenario the primordial ECP gene was functional as an anti-pathogen gene from the beginning, and no neutral mutations were necessary at the initial stage. Hughes has emphasized the importance of this type of evolution for duplicate genes (40). At this moment, it is difficult to decide between these two hypotheses.
In this study we were able to show that positive selection was involved in the evolution of the novel anti-pathogen function of ECP. To our knowledge, this is the first case in which the Dykhuizen–Hartl effect was ruled out. Previously, a number of authors reported detection of positive selection after gene duplication (6, 10, 11, 41), but none of them has seriously considered the possibility of the Dykhuizen–Hartl effect. Therefore, their conclusions are debatable. For example, Long and Langley reported an extraordinary case of origin of a new gene called jingwei (jgw) in two sibling species, Drosophila teissieri and Drosophila yakuba (10). This gene is believed to have arisen by retrotransposition of a processed mRNA of the alcohol dehydrogenase (Adh) gene and by acquisition of upstream exons, introns, and possibly a promoter region from unknown sources. These events apparently occurred in an ancestral species some time before the two species diverged. Sequence analysis of the jgw and Adh genes from D. teissieri and D. yakuba as well as of the Adh genes from related species indicated that eight nonsynonymous and no synonymous substitutions occurred in the Adh segment of jgw between the time of occurrence of the retrotransposition and the divergence of the two species. Because this pattern of nucleotide substitution is unusual for most functional genes, the authors suggested that positive selection was involved in the early stage of evolution of jgw.
It is unclear whether the 5′ region of the jgw gene was acquired immediately after the retrotransposition event or some time later, but it is possible that the Adh segment of jgw accumulated random mutations for a period of time because of relaxation of purifying selection. We therefore examined the possibility of neutral evolution in the early stage of evolution of jgw by using the n/s test. We obtained R = 0.6, N = 567, and S = 198 for the Adh segment of jgw, and the n/s test showed that an n/s ratio of 8/0 is not significantly different from the expectation of N/S = 567/198 (P = 0.090). Therefore, we cannot reject the null hypothesis of neutral evolution. The function of jgw is still unknown, but if it has gained a new function, the evolution of this gene can be explained by the Dykhuizen–Hartl effect. Furthermore, the fact that jgw has a lower degree of codon usage bias than Adh (10) suggests that the purifying selection for jgw has been less stringent than that for Adh, because the codon usage bias in Drosophila genes is mainly caused by purifying selection (42). Long and Langley also applied the McDonald–Kreitman test (43) to polymorphic sequences of this gene and showed that the ratio of nonsynonymous to synonymous substitutions between the two species is higher than that within species. They took this finding as evidence for positive selection. In the present case, however, the same results would be obtained even without positive selection if the intensity of purifying selection gradually increased after the divergence of the two species as their data suggest.
Of course, the above comments do not imply that there was no amino acid substitution aided by positive selection in the evolution of jgw. Positive selection might be common in the evolution of new gene function (44). However, it is generally difficult to rule out the possibility of the Dykhuizen–Hartl effect or relaxation of purifying selection, unless we identify the target site of selection (45) or concentrate our attention to short branches of evolutionary trees (30, 46, 47). As shown by many authors (48–50), the number of amino acid substitutions that are involved in a change of gene function is usually small, so that statistical tests of positive selection are not always powerful. In recent years, however, it has become possible to reproduce ancestral proteins by site-directed mutagenesis and examine the functional change of proteins in the evolutionary process (51–53). This approach in combination with the statistical analysis used in this paper will probably make it easier to identify positive selection.
Acknowledgments
We thank Esther Siegfried for providing computer facilities for computing the pI values and Andy Clark, Xun Gu, Sudhir Kumar, Tanya Sitnikova, and Tom Whittam for their comments on an earlier version of this paper. This work was supported by National Institutes of Health and National Science Foundation research grants to M.N.
ABBREVIATIONS
- ECP
eosinophil cationic protein
- EDN
eosinophil-derived neurotoxin
- OW
Old World
- NW
New World
- RNase
ribonuclease
- NG
Nei–Gojobori
- Myr
million years
Appendix
Modified NG Method.
In the original NG method (21) the numbers of synonymous and nonsynonymous nucleotide differences between two homologous sequences are computed for each codon site by taking into account the properties of the codons compared. The total numbers of synonymous (Sd) and nonsynonymous (Nd) differences per sequence are then obtained by the sums of the numbers of synonymous and nonsynonymous differences over all codons, respectively. We then estimate the proportions of synonymous and nonsynonymous differences by
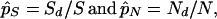 |
1 |
respectively, where S and N are the expected numbers of potential synonymous and nonsynonymous sites of a sequence and are obtained under the assumption of random nucleotide substitutions. Once p̂S and p̂N are obtained, d̂S and d̂N are computed by
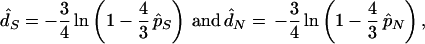 |
2 |
respectively. As mentioned earlier, when the ratio (R) of the numbers of transitional to transversional changes is higher than 0.5, the above NG method tends to give biased estimates of p̂S, p̂N, d̂S, and d̂N, because in this case S is underestimated and N is overestimated. Therefore, if we compute S and N correctly by taking into account the actual value of R, unbiased estimates of pS and pN can be obtained by Eq. 1. Similarly, bias-corrected but approximate values of d̂S and d̂N are obtained by Eq. 2 if we use newly defined p̂S and p̂N.
For estimating S and N, we use Kimura’s model of nucleotide substitution (27). In this model the rates of transitional and transversional changes are denoted by α and β, respectively. Because any nucleotide can have one transitional and two transversional changes, the proportion of transitions among the total changes is given by α/(α + 2β) = R/(1 + R), where R = α/(2β). Ina showed that the expected number of synonymous sites per codon can be expressed in terms of R for all codons (26). For example, in the case of codon TTT the number of potential synonymous sites is si = 0 + 0 + α/(α + 2β) = R/(1 + R), because only the third codon position produces synonymous changes and only one (transition) of the three possible changes is synonymous. Therefore, if we know R, we can compute si for every codon and obtain S by the sum of si over all codons and N by N = 3r − S, where r is the total number of codons. R can be estimated by various methods (e.g., see refs. 27 and 28).
Study of positive selection is usually conducted when p̂S and p̂N are relatively small, as in the present case. In such a case we may use p̂S and p̂N for estimating bS and bN. The b̂S and b̂N obtained from p̂S and p̂N may be smaller than those obtained from d̂S and d̂N, but the b̂N/b̂S ratio should be nearly the same. Furthermore, the former estimates have smaller variances than the latter. If sequence divergence is high and R is large, d̂S and d̂N obtained by Eq. 2 could both be underestimates. However, this is not a serious problem for testing positive selection, because the d̂N/d̂S ratio is hardly affected. In the data set used in this paper, the same conclusion is reached, whether p̂S and p̂N or d̂N and d̂S are used. In the case of nuclear genes, R is usually between 0.5 and 2. Therefore, the modified NG method is appropriate in most cases.
The modified NG method has advantages over other methods (23–26) in that it never gives negative estimates of dS and dN when sequence divergence is low, and the number of inapplicable cases (35) is much lower than that in other methods when sequence divergence is high. In fact, if we use p̂S and p̂N, we can avoid inapplicable cases completely. Furthermore, the sampling variances of d̂S and d̂N obtained by the modified NG method are smaller than those obtained by other methods.
Computer programs for computing all the statistical quantities required for the b̂N/b̂S and the n/s tests are available on request.
References
- 1.Bridges C B. Teaching Biol. November; 1936. 17–23. [Google Scholar]
- 2.Stephens S G. Adv Genet. 1951;4:247–265. doi: 10.1016/s0065-2660(08)60237-0. [DOI] [PubMed] [Google Scholar]
- 3.Ohno S. Sex Chromosomes and Sex-Linked Genes. New York: Springer; 1967. [Google Scholar]
- 4.Nei M. Nature (London) 1969;221:5175–5177. doi: 10.1038/221040a0. [DOI] [PubMed] [Google Scholar]
- 5.Ohno S. Evolution by Gene Duplication. New York: Springer; 1970. [Google Scholar]
- 6.Goodman M, Moore G W, Matsuda G. Nature (London) 1975;253:603–608. doi: 10.1038/253603a0. [DOI] [PubMed] [Google Scholar]
- 7.Dykhuizen D, Hartl D L. Genetics. 1980;96:801–817. doi: 10.1093/genetics/96.4.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kimura M. The Neutral Theory of Molecular Evolution. Cambridge, U.K.: Cambridge Univ. Press; 1983. [Google Scholar]
- 9.Li W-H, Gojobori T. Mol Biol Evol. 1983;1:94–108. doi: 10.1093/oxfordjournals.molbev.a040306. [DOI] [PubMed] [Google Scholar]
- 10.Long M, Langley C H. Science. 1993;260:91–95. doi: 10.1126/science.7682012. [DOI] [PubMed] [Google Scholar]
- 11.Ohta T. Genetics. 1994;138:1331–1337. doi: 10.1093/genetics/138.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snyder M R, Gleich G J. In: Ribonucleases: Structures and Functions. D’Alessio G, Riordan J F, editors. New York: Academic; 1997. pp. 425–444. [Google Scholar]
- 13.Domachowske J B, Rosenberg H F. J Leukocyte Biol. 1997;62:363–368. doi: 10.1002/jlb.62.3.363. [DOI] [PubMed] [Google Scholar]
- 14.Domachowske, J. B. & Rosenberg, H. F. (1998) J. Infect. Dis., in press.
- 15.Slifman N R, Loegering D A, McKean D J, Gleich G J. J Immunol. 1986;137:2913–2917. [PubMed] [Google Scholar]
- 16.Rosenberg H F, Dyer K D. J Biol Chem. 1995;270:21539–21544. doi: 10.1074/jbc.270.37.21539. [DOI] [PubMed] [Google Scholar]
- 17.Young J D E, Peterson C G B, Venge P, Cohn G J. Nature (London) 1986;321:613–616. doi: 10.1038/321613a0. [DOI] [PubMed] [Google Scholar]
- 18.Rosenberg H F. J Biol Chem. 1995;270:7876–7881. doi: 10.1074/jbc.270.14.7876. [DOI] [PubMed] [Google Scholar]
- 19.Rosenberg H F, Dyer K D, Tiffany H L, Gonzalez M. Nat Genet. 1995;10:219–223. doi: 10.1038/ng0695-219. [DOI] [PubMed] [Google Scholar]
- 20.Rzhetsky A, Nei M. Mol Biol Evol. 1993;10:1073–1095. doi: 10.1093/oxfordjournals.molbev.a040056. [DOI] [PubMed] [Google Scholar]
- 21.Nei M, Gojobori T. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 22.Ota T, Nei M. Mol Biol Evol. 1994;11:613–619. doi: 10.1093/oxfordjournals.molbev.a040140. [DOI] [PubMed] [Google Scholar]
- 23.Li W-H. J Mol Evol. 1993;36:96–99. doi: 10.1007/BF02407308. [DOI] [PubMed] [Google Scholar]
- 24.Pamilo P, Bianchi N O. Mol Biol Evol. 1993;10:271–281. doi: 10.1093/oxfordjournals.molbev.a040003. [DOI] [PubMed] [Google Scholar]
- 25.Comeron J M. J Mol Evol. 1995;41:1152–1159. doi: 10.1007/BF00173196. [DOI] [PubMed] [Google Scholar]
- 26.Ina Y. J Mol Evol. 1995;40:190–226. doi: 10.1007/BF00167113. [DOI] [PubMed] [Google Scholar]
- 27.Kimura M. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 28.Yang Z. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 29.Kumar S, Tamura K, Nei M. mega, Molecular Evolutionary Genetics Analysis, Version 1.02. University Park: Pennsylvania State Univ.; 1993. [Google Scholar]
- 30.Zhang J, Kumar S, Nei M. Mol Biol Evol. 1997;14:1335–1338. doi: 10.1093/oxfordjournals.molbev.a025743. [DOI] [PubMed] [Google Scholar]
- 31.Zhang, J. & Nei, M. (1997) J. Mol. Evol. 44 (Suppl. 1), S139–S146. [DOI] [PubMed]
- 32.Jones D T, Taylor W R, Thornton J M. Comput Appl Biosci. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- 33.Yang Z, Kumar S, Nei M. Genetics. 1995;141:1641–1651. doi: 10.1093/genetics/141.4.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fitch W M. Syst Zool. 1971;20:406–416. [Google Scholar]
- 35.Nei M. Molecular Evolutionary Genetics. New York: Columbia Univ. Press; 1987. [Google Scholar]
- 36.Li W-H, Ellsworth D L, Krushkal J, Chang B H-J, Hewett-Emmett D. Mol Phylogenet Evol. 1996;5:182–187. doi: 10.1006/mpev.1996.0012. [DOI] [PubMed] [Google Scholar]
- 37.Pilbeam D. Sci Am. 1984;252(3):84–96. doi: 10.1038/scientificamerican0384-84. [DOI] [PubMed] [Google Scholar]
- 38.Rosenberg H F, Ackerman S J, Tenen D G. J Exp Med. 1989;170:163–176. doi: 10.1084/jem.170.1.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsaur S-C, Wu C-I. Mol Biol Evol. 1997;14:544–549. doi: 10.1093/oxfordjournals.molbev.a025791. [DOI] [PubMed] [Google Scholar]
- 40.Hughes A L. Proc R Soc London Ser B. 1994;256:119–124. [Google Scholar]
- 41.Begun D J. Genetics. 1997;145:375–382. doi: 10.1093/genetics/145.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Powell J R, Moriyama E N. Proc Natl Acad Sci USA. 1997;94:7784–7790. doi: 10.1073/pnas.94.15.7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McDonald J H, Kreitman M. Nature (London) 1991;351:652–654. doi: 10.1038/351652a0. [DOI] [PubMed] [Google Scholar]
- 44.Walsh J B. Genetics. 1995;139:421–428. doi: 10.1093/genetics/139.1.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hughes A L, Nei M. Nature (London) 1988;355:167–170. doi: 10.1038/335167a0. [DOI] [PubMed] [Google Scholar]
- 46.Yu M, Irwin D M. Mol Phylogenet Evol. 1996;5:298–308. doi: 10.1006/mpev.1996.0025. [DOI] [PubMed] [Google Scholar]
- 47.Messier W, Stewart C-B. Nature (London) 1997;385:151–154. doi: 10.1038/385151a0. [DOI] [PubMed] [Google Scholar]
- 48.Perutz M F. Mol Biol Evol. 1983;1:1–28. doi: 10.1093/oxfordjournals.molbev.a040299. [DOI] [PubMed] [Google Scholar]
- 49.Asenjo A B, Rim J, Oprian D D. Neuron. 1994;12:1131–1138. doi: 10.1016/0896-6273(94)90320-4. [DOI] [PubMed] [Google Scholar]
- 50.Newcomb R D, Campbell P M, Ollis D L, Cheah E, Russell R J, Oakeshott J G. Proc Natl Acad Sci USA. 1997;94:7464–7468. doi: 10.1073/pnas.94.14.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jermann T M, Opitz J G, Stackhouse J, Benner S A. Nature (London) 1995;374:57–59. doi: 10.1038/374057a0. [DOI] [PubMed] [Google Scholar]
- 52.Chandrasekharan U M, Sanker S, Glynias M J, Karnik S S, Husain A. Science. 1996;271:502–505. doi: 10.1126/science.271.5248.502. [DOI] [PubMed] [Google Scholar]
- 53.Dean A M, Golding G B. Proc Natl Acad Sci USA. 1997;94:3104–3109. doi: 10.1073/pnas.94.7.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Larson K A, Olson E V, Madden B J, Gleich G J, Lee N A, Lee J J. Proc Natl Acad Sci USA. 1996;93:12370–12375. doi: 10.1073/pnas.93.22.12370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Batten D, Dyer K D, Domachowske J B, Rosenberg H F. Nucleic Acids Res. 1997;25:4235–4239. doi: 10.1093/nar/25.21.4235. [DOI] [PMC free article] [PubMed] [Google Scholar]