

Abstract
Antibody binding and neutralization are major host defenses against viruses, yet the mechanisms are often not well understood. Eight monoclonal antibodies and their Fab fragments were tested for neutralization of canine parvovirus and feline panleukopenia virus. All IgGs neutralized >90 percent of virus infectivity. Two Fabs neutralized when present at 5 nM, while the others gave little or no neutralization even at 20–100 nM. The antibodies bind two antigenic sites on the capsids which overlap the binding site of the host transferrin receptor (TfR). There was no specific correlation between Fab binding affinity and neutralization. All Fabs reduced capsid binding of virus to purified feline TfR in vitro, but the highly neutralizing Fabs were more efficient competitors. All partially prevented binding and uptake of capsids by feline TfR on cells. The virus appears adapted to allow some infectivity in the presence of at least low levels of antibodies.
Keywords: antibody, immunity, neutralization, canine parvovirus, feline panleukopenia virus
INTRODUCTION
Protection and recovery from viral infection in animals are complex processes that involve many components of the innate and adaptive immune systems, and antibodies are a critical adaptive immune response against most viruses of vertebrates (Burton, 2002). Antibody neutralization is defined as the “abrogation of virus infectivity in vitro by the binding of antibodies to the virion” (Klasse and Sattentau, 2002), and that binding may affect the virus at one or more stages in the viral infection cycle. In different models antibodies have been shown to crosslink and aggregate virions (Che et al., 1998), prevent viral attachment to cells (Knossow et al., 2002), block receptor binding by steric hindrance or by preventing viral conformational changes required for infection (Che et al., 1998; Ludert et al., 2002), inactivate the virus before cell binding occurs (reviewed by (Edwards and Dimmock, 2001b))(Edwards and Dimmock, 2001a; Gomez-Puertas et al., 1996; Osiowy and Anderson, 1995), or prevent infection after uptake into cells (Nybakken et al., 2005; Varghese et al., 2004)(reviewed in (Klasse and Sattentau, 2002)). Additional antibody-mediated mechanisms occur in vivo, where they can cause Fc-mediated phagocytosis, complement binding and activation, opsonization, and antibody-dependant cellular cytotoxicity of infected cells (Burton, 2002).
Viruses and their hosts have been co-evolving for long periods, and the antigenic sites of viral structural proteins will be those that allow highest viral fitness in the face of antibody selection. Different viruses show various levels of antibody binding and differ in their ability to accommodate antigenic variation. Some viruses (such as rhinovirus, influenza, and HIV) either come in many serotypes or are able to tolerate amino acid variation to escape from or avoid the antibody responses of their hosts (Gao et al., 2003; Holguin et al., 1997; Ledford et al., 2004; Smith et al., 2004). Other viruses (such as measles virus or polioviruses) are found as only 1 or a small number of antigenic types even after long periods of selection and are apparently unable to vary antigenically to escape preexisting antibodies (Rota et al., 1992; Rota et al., 1994; Sheshberadaran and Norrby, 1986; Sheshberadaran et al., 1985). These differences in antigenic structure and variation likely result from differences in the replication of the viruses, their modes of transmission, and the abilities of their structural proteins to accommodate antigenic change while still maintaining other viral properties (Bailey et al., 2004; Vossen et al., 2002).
Although often considered a factor in antibody neutralization, the relationships between the antibody and receptor binding sites have only been examined directly in a few cases. Some receptor binding sites are conserved and buried in clefts or other protected structures that are at least partially inaccessible to antibodies (Saphire et al., 2001), while other receptor binding sites are on prominent structures and show significant overlap with the antibody binding sites (Domingo et al., 1999; Liebermann et al., 1998).
Parvoviruses are small and have simple capsids made of essentially a single protein structure, and they do not appear to encode genes that can specifically manipulate the immune responses of their hosts. Despite this simplicity, they cause a variety of different types of disease, with acute infections in many cases. However, several of the parvoviruses form persistent infections, indicating that they are able to persist in the face of host immunity. Canine parvovirus (CPV) and feline panleukopenia virus (FPV) have 25nm non-enveloped T=1 icosahedral capsids that are assembled from 60 copies of a mixture of viral proteins. The infectious capsids contain ~55 copies of VP2, and ~5 copies of the VP1 protein which in CPV and FPV contains both the VP2 sequence and an 143 additional N-terminal residues (Tsao et al., 1991; Xie and Chapman, 1996). Full (DNA-containing) capsids have about 20 residues of some the VP2 N termini exposed on the outside of the capsid where they may be cleaved by host proteases to form VP3, and those N-termini are likely exposed through a pore at the five-fold axis of symmetry (Weichert et al., 1998; Xie and Chapman, 1996). Elaborate loops forming most of the capsid surface make up most of the functional sites of the capsid, including those involved in receptor and antibody binding (Agbandje et al., 1993; Govindasamy et al., 2003; Hueffer et al., 2003a; Strassheim et al., 1994; Tsao et al., 1991; Wikoff et al., 1994).
The cell receptor for CPV and FPV is the transferrin receptor (TfR), and appropriate TfR binding leads to cell infection (Hueffer et al., 2003b; Parker et al., 2001). The TfR assembles as a stable homodimer, and each monomer is made up of 3 domains: a protease-like domain, an apical domain, and a helical domain. Transferrin and hereditary hemachromatosis protein bind to the membrane proximal portions of the protease-like and helical domains (Cheng et al., 2004; Giannetti et al., 2003; Lawrence et al., 1999; West et al., 2001) whereas CPV and FPV bind to the apical domain (Palermo et al., 2003). Canine TfR binding to the CPV capsid is affected by residues in three positions on the threefold spike that are ~20–30Å apart, suggesting that the receptor binds to a broad surface of the capsid (Govindasamy et al., 2003; Hueffer et al., 2003a).
There is variation among the CPV- and FPV-like viruses that is associated with differences in their host ranges. The original CPV strain (termed CPV type-2 (CPV-2)) emerged in 1978 due to a small number of mutations of an FPV-like virus which altered both receptor and antibody binding sites on the capsids. A further 4 or 5 mutations in the capsid protein gene of CPV-2 resulted in the emergence of a variant virus (CPV type-2a (CPV-2a)) in 1979 which replaced CPV-2 worldwide within two years (Parrish et al., 1991; Parrish et al., 1988; Parrish et al., 1985). CPV-2a differs from CPV-2 in affinity for the feline TfR and also in antigenic structure as defined by monoclonal antibody (MAb) binding (Palermo, Hueffer, and Parrish, 2003; Parrish et al., 1991; Parrish et al., 1985). Those strains of virus also differ in canine and feline host ranges, as FPV infects cats but not dogs, CPV-2 strains infect dogs but not cats, while CPV-2a and later viruses infect both cats and dogs (Mochizuki et al., 1993; Truyen et al., 1995; Truyen and Parrish, 1992). Since 1980, variations of single residues in the capsid have become widely selected, including VP2 residue Asn426 to Asp (after 1984, designated the virus being designated CPV-2b (Parrish et al., 1991)), VP2 residue Ser297 to Ala around 1990, and residue Asp426 to Glu after 2000 (Buonavoglia et al., 2001).
The antigenic structure of CPV and FPV has been examined by MAb binding, analysis of peptide binding by polyclonal sera, cryoelectron microscopic (cryoEM) analysis of capsid:Fab complexes, and analysis of natural and selected antigenic variants (Casal et al., 1995; Hafenstein et al., 2006; Langeveld et al., 1994; Langeveld et al., 1993; Parrish and Carmichael, 1983; Rimmelzwaan et al., 1990; Strassheim et al., 1994; Truyen et al., 1995; Wikoff et al., 1994). Cross-competition studies of MAb binding and analysis of sequences that determine antigenic variations show that the epitopes can be divided among two major antigenic sites designated A and B (Parrish and Carmichael, 1983; Strassheim et al., 1994). Although site A is near the top of the threefold spike and site B is on the side (shoulder) of that structure, both epitopes are composed of multiple loops so that the binding sites are conformational in nature (Strassheim et al., 1994; Tsao et al., 1991; Wikoff et al., 1994; Xie and Chapman, 1996). Although FPV or mink enteritis virus (MEV) isolates collected over many years show little antigenic variation (Parrish and Carmichael, 1983; Parrish et al., 1984), both A and B site changes have been detected in CPV isolates from dogs and cats (Martella et al., 2004; Nakamura et al., 2001; Nakamura et al., 2003; Parrish et al., 1991; Parrish et al., 1985).
Panels of MAbs have been generated against FPV, CPV-2, and CPV-2b capsids, and all of those antibodies neutralized virus infectivity when tested as IgGs (Strassheim et al., 1994). When the antibody binding (variable) domains of two MAbs were expressed as single chain variable domains (scFvs), they neutralized infectivity, although at levels lower than the corresponding IgGs, and that neutralization was in part due to cross-linking and aggregation of the capsids by dimeric forms of the scFv (Yuan and Parrish, 2000).
Here we examine the processes of antibody neutralization of canine and feline parvoviruses. We tested 8 different MAbs recognizing either site A or B for their abilities to neutralize the viruses as IgGs and Fabs. While all IgGs neutralized the viruses, only two of the Fabs recognizing site B efficiently neutralized viral infectivity, one was weakly neutralizing, and the other Fabs showed little or no neutralization. All Fabs at least partially inhibited the binding of capsids to the purified feline TfR, but the highly neutralizing antibodies showed the greatest inhibitory effect. There were only modest differences in the affinity of binding of the Fabs, but the highly neutralizing antibodies were not uniformly of higher affinity. Efficient neutralization by Fab binding likely results from either Fab-induced differences in the capsid-TfR interaction, or to effects on later stages of infection.
RESULTS
Antibody and Fab preparation and binding
Fab fragments of MAbs prepared by papain digestion were further purified by gel chromatography to ensure that only monomeric Fab proteins were used in these studies. All the Fabs bound to CPV-2 and FPV capsids in ELISA (Fig. 1), except for the CPV-specific Fab14 which does not bind FPV (Parrish and Carmichael, 1983)(Fig. 1B). Direct calibration ELISA was used to determine the affinity of binding of the 8 Fabs to CPV-2 capsids (Fig. 2) (Fuchs et al., 1995). Seven of the Fabs showed clearl binding kinetics in the assays, and their affinities were readily determined (Table 2). Fab B showed low binding in the assays, and in the transfer assay showed increasing binding during the repeated transfers (Fig. 2C), so that its affinity could not be calculated directly but was estimated to be around 8 x 10−9 nM. The binding affinities of 7 of the Fabs tested did not differ by more than 3-fold. The intact IgGs all showed higher avidities compared to the Fabs, as would be expected from their bivalent interactions (results not show).
Figure 1.
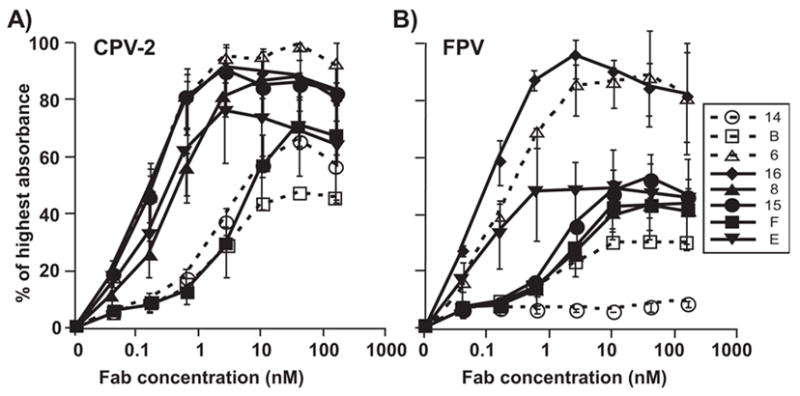
Binding of Fabs for (A) CPV or (B) FPV capsids. The Fab fragments bound were detected with either an anti-mouse or anti-rat horseradish peroxidase conjugated secondary antibody as appropriate. Error bars show the standard error of the mean, comparing the results of 3 separate experiments.
Figure 2.
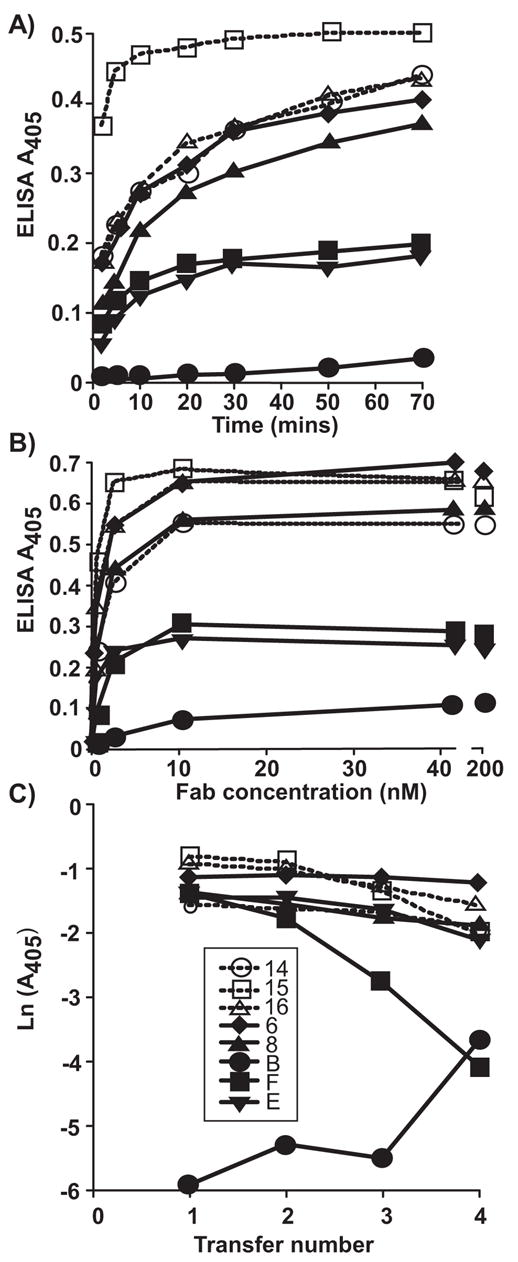
Determining the binding affinities of the 8 Fabs to the CPV capsids by measuring the kinetics of binding, the saturated binding level, and the degree of binding when the Fabs were incubated with the viral antigen and then transferred to a new well after various times; the 30 min transfer data is shown as an example. (A) The rate of Fab binding to the antigen, tested by allowing the Fabs to incubate for various times with the capsids before washing, then detecting with anti-mouse or anti-rat IgG HRPO conjugates. (B) The saturation of binding for the 8 different Fabs, showing the binding when the antigen was incubated with increasing amounts of the Fabs, allowing the saturated binding to be estimated. (C) The binding of each Fab in a transfer assay where the Fabs were incubated with the antigen for 30 min before transferring to a new well. This was repeated 4 times for various lengths of time; the 30 min data is shown here.
Table 2.
Determination of the affinities of the Fabs examined here using the direct calibration ELISA method. The three components of the analysis are given, as well as the affinity calculated as the dissociation constant. We were unable to accurately determine the affinity of Fab B using this method. ND = not determined.
| Antibody ID | Rate of complex formation (Kc, s−1) | Calibration Factor (c, M) | Transfer Factor (F) | Dissociation constant (Kd, M) |
|---|---|---|---|---|
| 14 | 1.2 × 10−3 | 7.0 × 10−10 | 0.9 | 3.4 × 10−9 |
| 15 | 3.4 × 10−3 | 1.3 × 10−9 | 0.7 | 2.1 × 10−9 |
| 16 | 1.0 × 10−3 | 8.6 × 10−10 | 0.83 | 2.6 × 10−9 |
| 6 | 1.3 × 10−3 | 4.8 × 10−11 | 0.99 | 3.2 × 10−9 |
| 8 | 1.7 × 10−3 | 1 × 10−9 | 0.82 | 3.4 × 10−9 |
| B | ND | ND | ND | ~6.1 × 10−9 |
| F | 1.7 × 10−3 | 8 × 10−9 | 0.39 | 1.5 × 10−9 |
| E | 8.5 × 10−4 | 2 × 10−9 | 0.8 | 1.7 × 10−9 |
Neutralization of FPV and CPV by IgG and Fabs
When tested as IgGs 7 of the antibodies neutralized >90% of CPV infectivity at 5nM and >99% CPV infectivity at 25nM, while MAb 14 neutralized only 88% of the infectivity at 100 nM (Fig. 3A). FPV showed similar neutralization sensitivity, and >99% of the infectivity was also neutralized by 5nM of the 7 IgGs that reacted with the virus, but not by MAb 14 (Fig. 3B). When the Fabs were tested, E and F neutralized >99% of the viral infectivity of both CPV and FPV at 5nM or lower concentrations, while Fab 16 neutralized at 25nM, and Fab 6 neutralized FPV only at 100nM (Figs. 4). By capture ELISA we determined that the capsids were present in the infectivity stocks used for the TCID50 titrations at concentrations of ~6 μg/ml, so that the Fab:capsid ratios were ~100:1 at 5 nM (data not shown).
Figure 3.
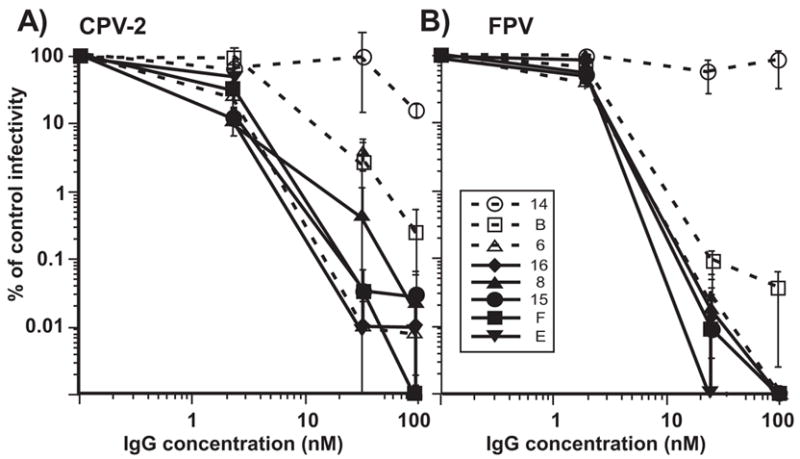
The neutralization of (A) CPV-2 and (B) FPV infectivity by various amounts of the IgGs of the 8 different MAb tested here. The IgGs were added to 10,000 TCID50 of each virus at concentrations between 0 and 100 nM, incubated for 1 h at 37°C, then the surviving virus was diluted and tested for surviving TCID50 in CRFK cells. The results are shown as the surviving infectivity in each culture compared to the titer of the control virus. Bars show the standard error of the mean, comparing the results of 3 separate experiments done on separate days.
Figure 4.
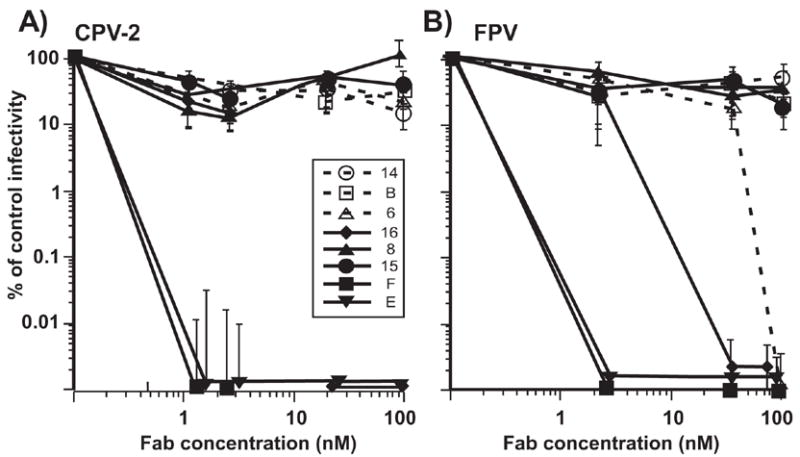
The neutralization of (A) CPV-2 and (B) FPV infectivity by the Fab fragments of the 8 different MAbs as shown for the IgGs in Figure 3. The results are shown as the surviving infectivity in each culture compared to the titer of the control virus. Bars show the standard error of the mean, comparing the results of 3 separate experiments done on separate days.
Fabs compete for receptor binding of the capsid
The CPV-2 capsids were labeled with Cy2 and bound to the purified ectodomain of feline TfR in a solid phase assay. All Fabs reduced capsid binding to the TfR at concentrations of 29 nM or greater (20 Fabs per capsid) (Fig. 5). Fabs E and F showed the greatest competition, while Fab 6 did not completely block binding even at 100nM (Fig. 5). Fab B also allowed some binding at 100 Fabs per capsid, perhaps due to its lower affinity relative to the other Fabs (Fig. 2 and Table 2).
Figure 5.
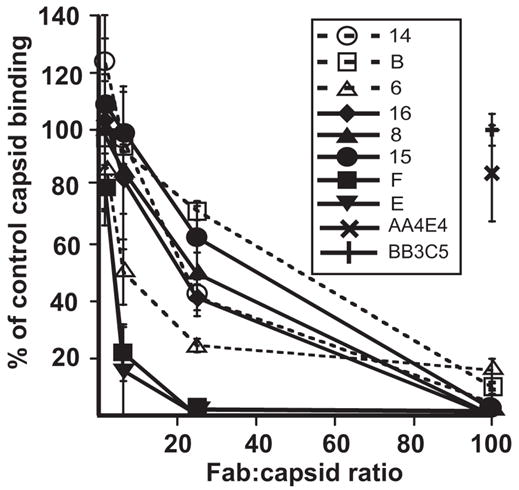
Inhibition of binding of Cy2-labelled CPV-2 capsids to the feline TfR by the 8 Fabs tested here. The capsids were incubated with varying amounts of Fab per capsid, and then added to plates coated with the purified feline TfR ectodomain, and the bound capsids detected using a fluorescence plate reader. The amount of binding is shown as the percentage of the binding of the capsids without Fab added. Control Fabs against the capsids of adeno-associated viruses types 1 and 5 were tested at only 100 Fabs per capsid. The results are shown as the mean and standard error of three separate experiments.
Fabs and viral attachment to cells
All Fabs were able to block the binding and uptake of a proportion of the capsids into TRVb cells expressing the feline TfR when tested at Fab:capsid ratios of up to 100:1 (Fig. 6). However, all Fabs reduced binding and uptake to similar levels, and no specific distinction was seen between the highly neutralizing Fabs and the less or non-neutralizing Fabs.
Figure 6.
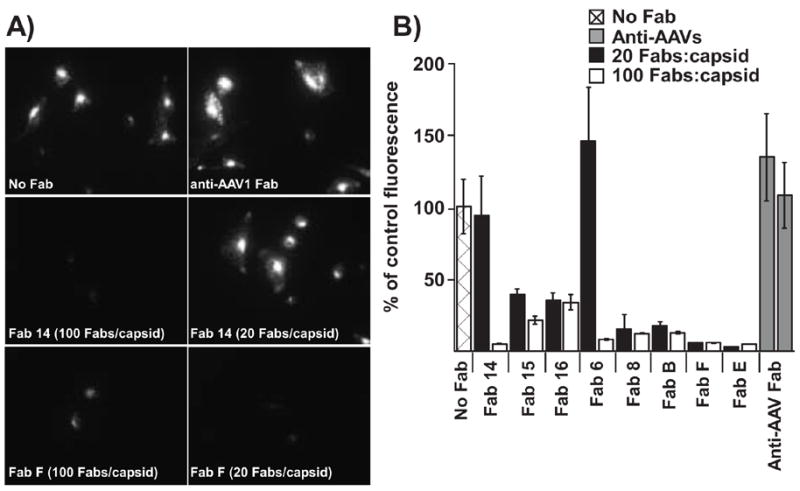
Competition by Fabs for cell binding and uptake. CPV-2 capsids were incubated with varying amounts of the Fabs for 1 h at 37°C, then bound to TRVb cells expressing the feline TfR. (A) Representative micrographs are shown for the no Fab control, and the Fabs against the A site (Fab 14) or the B site (Fab F). (B) Total average cell-associated fluorescence (per cell) was determined for at least 20 cells at each concentration. Results are plotted as a percentage of the CPV bound to cells in the absence of any added Fabs. The control anti-AAV Fabs were tested at 100 Fabs/capsid.
DISCUSSION
Here we show that the parvovirus capsid interactions with antibodies are surprisingly complex and showed clear differences in viral neutralization despite the fact that they bind with similar affinities to most or all of the 60 sites on the capsid. Studies of escape mutations and competition assays have suggested that most antibodies generated against intact capsids recognize two dominant antigenic sites, described as site A near the top of the three fold axis and site B on the shoulder of that structure (Strassheim et al., 1994). The general features of those sites are shown in Figure 7, where the residues that affect the A and B sites in previous studies are displayed, along with projected Fab density from radial sections of Fab-virus cryoEM reconstructions Fab 14 bound to the A site while Fab 15 bound to the B site. In other studies, footprints of these Fabs have been defined by cryoEM reconstruction techniques which confirm the disposition of the two binding sites (Hafenstein et al., 2006).
Figure 7.

A roadmap showing the location of surface residues of CPV-2 that affect the binding of the different antibodies to the capsid structures. A single asymmetric unit of the capsid is shown. Two antigenic sites have been previously defined, and designated as (A) and (B) through the analysis of mutations that affected antibody binding or by cross-competition. Residues that affect antibody binding to some A-site antibodies are shown in red, and those affecting B-site antibodies are shown in blue. CryoEM density corresponding to Fab14 as bound to the A site and Fab15 as it interacts with the B site are projected to the surface of the virion and indicated as red (Fab14) or blue (Fab15). The projection of Fab density obtained from virus-Fab complex shows the A and B sites as previously described, but with some possible overlap between the antibodies binding to the two sites (Hafenstein et al., 2006).
Although all 8 IgGs neutralized the viruses only two of the Fabs neutralized efficiently, and another was intermediate in neutralizing ability. Both neutralizing and non-neutralizing Fabs bound to the B site of the capsids, showing that neutralization depends on specific interactions of the Fabs with the capsid rather than being a result of simple Fab attachment to a particular site, and the neutralizing and non-neutralizing antibodies must therefore affect the cell infection processes differently. None of the A site Fabs tested here efficiently neutralized the virus.
Blockade of receptor binding is often suggested as a mechanism of viral neutralization, and here we were able to test for this effect using the purified receptor in in vitro assays, and by testing for binding to the receptor on cells. The results indicate that any neutralization differences due to effects on TfR binding would be rather subtle, since all of the Fabs inhibited binding to the purified receptor ectodomain when present in high enough concentrations. However, the highly neutralizing Fabs (E and F) blocked capsid binding to the TfR at the non-saturating concentration of 25 Fab per capsid, and even blocked 80% of the binding at a ratio of 6.25 Fab per capsid. This suggests a mechanism of neutralization that results from a direct effect of small numbers of Fabs on the capsid that blocks TfR binding to the remaining sites on the capsid, perhaps through some allosteric change induced in the capsid. However, in limited studies we have not been able to directly demonstrate any change in the capsid (results not shown). The less efficiently neutralizing or non-neutralizing Fabs also inhibited capsid binding to the feline TfR when added at higher concentrations, indicating that they would not have the same structural effect on the capsids, and that they must leave sufficient binding sites available at lower antibody concentrations for cell binding and endocytosis allowing infection. The TfR is very efficiently taken up by clathrin-mediated endocytosis (Enns, 2002), and even the low affinity binding of the capsid to the canine TfR allows efficient infection of cells (Palermo et al., 2006).
The antibodies against the CPV and FPV capsids recognize only a limited number of structural sites (Hafenstein et al., 2006), suggesting that those sites are particularly efficient at selecting high affinity antibodies and are the result of selection to allow the most efficient replication and transmission in the presence of antibodies. The capsid structures of the parvoviruses infecting vertebrates show a variety of prominent regions at or surrounding the threefold spikes (Chapman, 1996; Kaufmann et al., 2004; McKenna et al., 1999; Padron et al., 2005; Walters et al., 2004; Xie et al., 2002), and epitopes that have been mapped mostly fall on those structures (Costello et al., 1999; Lopez de Turiso et al., 1991; Strassheim et al., 1994; Wobus et al., 2000). Insect parvoviruses are not under antibody selection, and have smoother surfaces (Bruemmer et al., 2005; Simpson et al., 1998), suggesting that the prominent structures benefit the vertebrate viruses when targeted by antibodies, perhaps because the limited number of sites inducing high affinity antibodies allows them to regulate the effects of the antibody binding during infection.
What selects for changes in the viral antigenic structure? Although natural antigenic variants have been seen among the CPV- and FPV-like viruses, most of those changes simultaneously alter TfR receptor binding (Hueffer et al., 2003a; Hueffer et al., 2003b; Palermo et al., 2006), and in this case it appears that antigenic variation is a side-effect of capsid changes that alter receptor binding to mediate host adaptation. FPV and MEV have been in long association with their hosts and are also antigenically conserved, with only a single antigenic variant being identified among MEV isolates due to the change of residue 300 from Ala to Ser (Parrish and Carmichael, 1983; Parrish et al., 1984). The natural antigenic variants of CPV fall within both the A and B sites of the capsid. The change of VP2 residue 93 from Lys to Asn during the emergence of CPV was required for canine TfR binding and canine host range determination, and that also altered antigenic site A (Chang et al. , 1992; Hueffer et al., 2003a; Hueffer et al., 2003b). That difference would not have been subject to selection by preexisting immunity as CPV-2 could not infect cats, so the selection of that site was for host range variation alone. Some combination of the changes between CPV-2 and CPV-2a of VP2 residues 87, 101, 300, and 305 also reduced the affinity of CPV-2a binding of CPV-2a to the feline TfR (Palermo et al., 2006), and allowed the CPV-2a and the more recent strains to infect cats (Truyen et al., 1996). Those changes also altered antigenic site B causing the loss of binding to some antibodies that bound CPV-2 and FPV, and creating the CPV-2a specific antigenic sites (Parrish et al., 1991). That variation also reduced the affinity of the virus for the feline TfR and likely determined the host range for cats, again making it likely that receptor binding was the most important property being selected.
Other antigenic variants have emerged in the CPV-2a background; after 1983 the change of VP2 residue 426 from Asn to Asp was found worldwide, giving the CPV-2b variant (Parrish et al., 1991). That mutation is still seen at various levels among dogs around the world, suggesting that the selection on that site prevents it becoming fixed (Buonavoglia et al., 2000; Costa et al., 2005; Sagazio et al., 1998; Steinel et al., 1998). Since ~2000 viruses containing the VP2 residue 426 to Glu replacement have been seen in various parts of the world, and that variant is increasing in frequency (Martella et al., 2004; Nakamura et al., 2004). Residue 426 is within the TfR binding site, but effects of those mutations on TfR binding have not been described.
Antibody and receptor binding have overlapping effects on the pathogenesis and epidemiology of these viruses. Antibodies in infected and recovered animals protect for life against re-infection, so that the viruses are maintained in nature as a series of acute infections of puppies or kittens which have waning or no maternal immunity. This continual turnover of the virus population allows mutations to be selected within the global populations of hosts over short time periods. Host infection is oronasal, and the virus then circulates systemically to infect cells in many lymphoid tissues and the small intestine, and the virus is vulnerable to both mucosal and plasma antibodies during this process. Ability to infect hosts with low levels of maternal antibodies may be a selected property of these viruses (Elia et al., 2005; Martella et al., 2005; Pollock and Carmichael, 1982; Waner et al., 1996). In addition, although anti-viral antibodies are important for recovery from infection, some viremia continues for a short period in the presence of the developing antibody response (Meunier et al., 1985).
The interactions between viruses, host receptors and antibodies can be complex, even for the simple parvovirus capsids. The extensive overlap between the TfR and antibody binding sites results in many mutations in the capsid surface that affect the binding of both ligands simultaneously. The antibody-capsid interactions of these parvoviruses may share features with other virus-receptor-antibody interactions. The foot and mouth disease virus also shows overlap between the integrin binding sequence and antibody epitopes on the G-H loop of the virus, although the Arg-Gly-Asp binding sequence is quite small and may be conserved while the surrounding epitopes can vary (Domingo et al., 1999; Verdaguer et al., 1999). In other cases the receptor binding and antigenic sites are largely distinct. Some picornaviruses bind their receptors through a lowered region of the capsids (canyon), while most of the antigenic sites are on raised regions (although some antibodies may be able to access the receptor site) (Rossmann et al., 2002; Smith et al., 1996). For the influenza virus hemagglutinin the sialic acids bind in relatively conserved pockets while the antigenic sites are on surface regions which can readily accommodate variation (Skehel and Wiley, 2000; Smith et al., 2004). In HIV the receptor binding sites on the gp120 are protected from antibody by a variety of strategies including overlapping carbohydrate structures and rapid variability, while the conserved chemokine receptor binding site is buried within the structure and not revealed until after CD4 binding (Chen et al., 2005; Kwong et al., 1998). Most viruses of vertebrates are under strong antibody selection and many different solutions are used to allow their success. Since these interactions are key to effective protective antiviral immunity, a better understanding of the processes involved would help in the design of better vaccines in the future.
METHODS AND MATERIALS
Cells and viruses
Crandell Reese Feline Kidney Cells (CRFK) were grown in a 1:1 mixture of McCoy’s 5A and Lebovitz L-15 media with 5% fetal calf serum (FCS). TRVb hamster cells which lack TfR were grown in Hams F-12 medium with 5% FCS, and were transfected with plasmids expressing the feline TfR (TRVb-fTfR) and selected with 400 μg/ml G418 (Hueffer, et al., 2004) . FPV, CPV-2, and CPV-2b virus stocks were prepared from infectious clones transfected into CRFK cells as previously described (Parrish, 1991), and stored at −80°C in aliquots.
Purified Transferrin Receptor
Soluble TfR ectodomain was produced by baculovirus expression in insect cells as previously described (Hueffer, et al., 2004; Palermo, et al., 2006). The dimeric protein was isolated by size exclusion chromatography through a S300 column.
Capsid concentration measurements
Capsid concentrations in virus samples were determined using a capture ELISA. Purified rat anti-CPV (MAb F) bound to an ELISA plate was used to capture virus from the test samples or controls containing known amounts of purified virus. After 1 h the plate was washed with phosphate buffered saline with Tween 20 (PBST). The plates were incubated with a mouse anti-CPV (Mab 8), then with a specific goat anti-mouse HRP-conjugated antibody, and 2,2’-Azinobis(3-ethyl bensthaxoline-6-sulfonic acid) (ABTS) substrate for 30 min.
Anti-CPV and FPV antibodies
Eight different hybridomas were chosen which produced IgG antibodies against CPV or FPV capsids (Table 1)(Parrish and Carmichael, 1983; Parrish, 1982). Hybridomas were grown in 500 ml volumes in gas permeable bags (Nexell, Irving, CA) containing Dulbecco’s minimal essential medium with 5% FCS, then the IgGs were purified using Protein G (GE Healthcare, Piscataway, NJ). Fabs were generated using papain digestion; the Fc portions removed with protein A (GE Healthcare), and the monomeric Fabs were purified by chromatography in a Sephadex G100 column in PBS. Protein concentrations were determined by A280 and by bicinchoninic acid (BCA) assay.
TABLE 1.
Monoclonal antibodies used in these studies, along with the known specificity for particular viruses, and the escape mutants that affect their binding.
| Antibody ID | Source | Epitope recognized | Specificity | Escape mutations (residue in VP2 sequence) |
|---|---|---|---|---|
| 14 | Mouse | Site A | CPV only | 93 N-K, 224 G-R, 224 G-E, 222 H-Y |
| 15 | Mouse | Site B | CPV, FPV | 299 G-E (300 A-D partial) |
| 16 | Mouse | Site B | CPV, FPV | 299 G-E, 300 A-D, 302 N-D |
| 6 | Mouse | Site A | CPV, FPV | 222 H-Y, 224 G-R, 224 G-E |
| 8 | Mouse | Site B | CPV, FPV | 300 A-D, 302 N-D |
| B | Rat | Site A | CPV, FPV | 222 H-Y, 224 G-R, 224 G-E |
| F | Rat | Site B | CPV, FPV | No escape mutant recovered |
| E | Rat | Site B | CPV, FPV | 300 A-D |
Virus neutralization assays
CRFK cells were seeded into 96 well trays at 1.28 x 104 cells per cm2 and incubated overnight. 104 TCID50 of virus and various amounts of IgG or Fab were mixed with DMEM to give a final volume of 0.4 ml. After 1 h at 37°C, each virus was diluted in 10-fold series, and 0.05ml added to replicate wells. After 1 h at 37°C, 0.1 ml of growth medium was added and cells were incubated for 48 hrs. After fixation the infected cells were detected by antibody staining as described (Yuan and Parrish, 2000), and the TCID50 titers calculated.
Affinity of Fabs for CPV
Affinities of Fabs were determined by direct calibration ELISA (Fuchs et al., 1995). Three separate experiments were performed to obtain variables used in determination of affinity. Unless otherwise noted100 ng of capsids and 10 ng of Fabs per well were used, Fabs were diluted in blocking buffer (PBST with 0.5% w/v ovalbumin), and plates washed six times with PBST between inclubations. Fabs were detected with either an anti-mouse or anti-rat HRP-conjugated secondary antibody. Capsids were immobilized to Maxisorp plates (Nunc, Rochester, NY) in carbonate-bicarbonate buffer (pH 9.6) overnight at 4°C. These plates were washed and then blocked with blocking buffer for 2.5 h before use in each experiment.
To determine the rate of Fab-CPV complex formation, Kc, Fabs and capsids were incubated for various times before the unbound Fabs were removed, wells washed and 100 μl of PBST was left in each well until the last time point was completed, when the bound Fabs detected. Non-linear regression analysis estimated the maximum absorbance for each Fab after infinite binding time. The absorbance correlates with the concentration of Fab-CPV complexes by an unknown calibration factor, c. The calibration factor was determined by incubating Fabs with immobilized capsids for varying times, and then serially transferred to more immobilized capsids. The amount of each Fab transferred or retained is determined by the transfer factor, F. After 4 transfers, the plates were washed, bound Fabs were detected, and F and c determined.
The amount of CPV immobilized to the plate was determined by a saturation analysis. Fab concentrations ranging from 166 to 0.04 nM were allowed to interact with capsids for 3.5 hr, plates were washed and Fabs detected. Non linear regression analysis was used to determine the maximum binding that would occur with an infinite concentration of Fabs, and this was used to determine the immobilized CPV concentration by the calibration factor c. Affinities were determined by the equation .
Competition with capsid binding to the feline TfR
The effects of the Fab on capsid binding to the feline TR were tested using in vitro binding assays. Purified feline TfR ectodomain was coated onto Maxisorp black plates (Nunc) in pH 9.6 carbonate buffer at 4°C overnight. After washing the plates were incubated with blocking buffer for 1 h. The Fab fragments were incubated at various concentrations with the CPV-2 empty capsids labeled with Cy2, and then after 1 h the mixtures were added to the wells containing the feline TfR. Control Fabs were prepared from antibodies directed against the capsids of AAV-1 or AAV-5, which did not react with the CPV capsids. After 1 h incubation, the plates were washed and the capsid fluorescence bound determined in a fluorescence reader (Tecan Sapphire, Durham, NC).
Cell binding and uptake
Fluorescence microscopy was used to examine the ability of Fab bound capsids to bind and enter cells. Cy-2 labeled CPV-2 or FPV capsids were incubated with Fabs at ratios between 1:100 to 1:20 for 1 h at 37°C. CRFK cells, or TRVb- cells expressing the feline TfR were washed 3 times with DMEM containing 0.1% BSA, then incubated with Fab-CPV mixtures for 1 h at 37°C. Cells were washer with PBS, fixed in 2% paraformaldehyde (PFA) for 10 min, then washed and visualized under the fluorescent microscope. Images were collected at the same exposure in both phase contrast and fluorescence, and analyzed for virus fluorescence in each cell using Image J (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://rsb.info.nih.gov/ij/, 1997–2006). The area and regions of interest of cells were determined from the phase contrast image, and mean specific fluorescence values for each cell were determined for that same area in the fluorescent channel. Data is the result of at least 25 cells from each treatment, selected randomly using the phase contrast channel.
Acknowledgments
Gail Sullivan and Wendy Weichert provided excellent technical support, and Alex Maltsev helped with data analysis. Supported by grants AI 28385 and AI 33468 from the National Institutes of Health to C.R.P..
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Christian D.S. Nelson, Email: cn38@cornell.edu.
Laura S. Palermo, Email: lmp52@cornell.edu.
Susan L. Hafenstein, Email: shafenst@purdue.edu.
REFERENCES CITED
- Agbandje M, McKenna R, Rossmann MG, Strassheim ML, Parrish CR. Structure determination of feline panleukopenia virus empty particles. Proteins. 1993;16 (4):155–171. doi: 10.1002/prot.340160204. [DOI] [PubMed] [Google Scholar]
- Bailey J, Blankson JN, Wind-Rotolo M, Siliciano RF. Mechanisms of HIV-1 escape from immune responses and antiretroviral drugs. Curr Opin Immunol. 2004;16 (4):470–476. doi: 10.1016/j.coi.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Bruemmer A, Scholari F, Lopez-Ferber M, Conway JF, Hewat EA. Structure of an insect parvovirus (Junonia coenia Densovirus) determined by cryo-electron microscopy. J Mol Biol. 2005;347 (4):791–801. doi: 10.1016/j.jmb.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Buonavoglia C, Martella V, Pratelli A, Tempesta M, Cavalli A, Buonavoglia D, Bozzo G, Elia G, Decaro N, Carmichael L. Evidence for evolution of canine parvovirus type 2 in Italy. J Gen Virol. 2001;82 (12):3021–3025. doi: 10.1099/0022-1317-82-12-3021. [DOI] [PubMed] [Google Scholar]
- Buonavoglia D, Cavalli A, Pratelli A, Martella V, Greco G, Tempesta M, Buonavoglia C. Antigenic analysis of canine parvovirus strains isolated in Italy. New Microbiol. 2000;23 (1):93–96. [PubMed] [Google Scholar]
- Burton DR. Antibodies, viruses and vaccines. Nat Rev Immunol. 2002;2 (9):706–713. doi: 10.1038/nri891. [DOI] [PubMed] [Google Scholar]
- Casal JI, Langeveld JP, Cortes E, Schaaper WW, van Dijk E, Vela C, Kamstrup S, Meloen RH. Peptide vaccine against canine parvovirus: identification of two neutralization subsites in the N terminus of VP2 and optimization of the amino acid sequence. J Virol. 1995;69 (11):7274–7277. doi: 10.1128/jvi.69.11.7274-7277.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SF, Sgro JY, Parrish CR. Multiple amino acids in the capsid structure of canine parvovirus coordinately determine the canine host range and specific antigenic and hemagglutination properties. J Virol. 1992;66 (12):6858–6567. doi: 10.1128/jvi.66.12.6858-6867.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MS. Structural refinement of the DNA-containing capsid of canine parvovirus using RSRef, a resolution-dependent stereochemically restrained real-space refinement method. Acta Crystallogr D Biol Crystallogr. 1996;52 (1):129–142. doi: 10.1107/S0907444995007268. [DOI] [PubMed] [Google Scholar]
- Che Z, Olson NH, Leippe D, Lee WM, Mosser AG, Rueckert RR, Baker TS, Smith TJ. Antibody-mediated neutralization of human rhinovirus 14 explored by means of cryoelectron microscopy and X-ray crystallography of virus-Fab complexes. J Virol. 1998;72 (6):4610–4622. doi: 10.1128/jvi.72.6.4610-4622.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Vogan EM, Gong H, Skehel JJ, Wiley DC, Harrison SC. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature. 2005;433 (7028):834–841. doi: 10.1038/nature03327. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Zak O, Aisen P, Harrison SC, Walz T. Structure of the human transferrin receptor-transferrin complex. Cell. 2004;116 (4):565–576. doi: 10.1016/s0092-8674(04)00130-8. [DOI] [PubMed] [Google Scholar]
- Costa AP, Leite JP, Labarthe NV, Garcia RC. Genomic typing of canine parvovirus circulating in the state of rio de janeiro, Brazil from 1995 to 2001 using polymerase chain reaction assay. Vet Res Commun. 2005;29 (8):735–743. doi: 10.1007/s11259-005-3865-9. [DOI] [PubMed] [Google Scholar]
- Costello F, Steenfos N, Jensen KT, Christensen J, Gottschalck E, Holm A, Aasted B. Epitope mapping of Aleutian mink disease parvovirus virion protein VP1 and 2. Scand J Immunol. 1999;49 (4):347–354. doi: 10.1046/j.1365-3083.1999.00499.x. [DOI] [PubMed] [Google Scholar]
- Domingo E, Verdaguer N, Ochoa WF, Ruiz-Jarabo CM, Sevilla N, Baranowski E, Mateu MG, Fita I. Biochemical and structural studies with neutralizing antibodies raised against foot-and-mouth disease virus. Virus Res. 1999;62 (2):169–175. doi: 10.1016/s0168-1702(99)00042-8. [DOI] [PubMed] [Google Scholar]
- Edwards MJ, Dimmock NJ. A haemagglutinin (HA1)-specific FAb neutralizes influenza A virus by inhibiting fusion activity. J Gen Virol. 2001a;82 (6):1387–1395. doi: 10.1099/0022-1317-82-6-1387. [DOI] [PubMed] [Google Scholar]
- Edwards MJ, Dimmock NJ. Hemagglutinin 1-specific immunoglobulin G and Fab molecules mediate postattachment neutralization of influenza A virus by inhibition of an early fusion event. J Virol. 2001b;75 (21):10208–10218. doi: 10.1128/JVI.75.21.10208-10218.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elia G, Cavalli A, Cirone F, Lorusso E, Camero M, Buonavoglia D, Tempesta M. Antibody levels and protection to canine parvovirus type 2. J Vet Med B Infect Dis Vet Public Health. 2005;52 (7–8):320–322. doi: 10.1111/j.1439-0450.2005.00870.x. [DOI] [PubMed] [Google Scholar]
- Enns CA. The transferrin receptor. In: Templeton DM, editor. Molecular and cellular iron transport. Marcel Dekker; New York: 2002. pp. 71–94. [Google Scholar]
- Fuchs H, Orberger G, Tauber R, Gessner R. Direct calibration ELISA: a rapid method for the simplified determination of association constants of unlabeled biological molecules. J Immunol Methods. 1995;188 (2):197–208. doi: 10.1016/0022-1759(95)00202-2. [DOI] [PubMed] [Google Scholar]
- Gao G, Alvira MR, Somanathan S, Lu Y, Vandenberghe LH, Rux JJ, Calcedo R, Sanmiguel J, Abbas Z, Wilson JM. Adeno-associated viruses undergo substantial evolution in primates during natural infections. Proc Natl Acad Sci U S A. 2003;100 (10):6081–6086. doi: 10.1073/pnas.0937739100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannetti AM, Snow PM, Zak O, Bjorkman PJ. Mechanism for multiple ligand recognition by the human transferrin receptor. PLoS Biol. 2003;1 (3):E51. doi: 10.1371/journal.pbio.0000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Puertas P, Rodriguez F, Oviedo JM, Ramiro-Ibanez F, Ruiz-Gonzalvo F, Alonso C, Escribano JM. Neutralizing antibodies to different proteins of African swine fever virus inhibit both virus attachment and internalization. J Virol. 1996;70 (8):5689–5694. doi: 10.1128/jvi.70.8.5689-5694.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindasamy L, Hueffer K, Parrish CR, Agbandje-McKenna M. Structures of host range-controlling regions of the capsids of canine and feline parvoviruses and mutants. J Virol. 2003;77 (22):12211–12221. doi: 10.1128/JVI.77.22.12211-12221.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafenstein S, Rossmann MG, Palermo LM, Parrish CR. unpublished results. 2006. [Google Scholar]
- Holguin A, Hernandez J, Martinez MA, Mateu MG, Domingo E. Differential restrictions on antigenic variation among antigenic sites of foot-and-mouth disease virus in the absence of antibody selection. J Gen Virol. 1997;78 (3):601–609. doi: 10.1099/0022-1317-78-3-601. [DOI] [PubMed] [Google Scholar]
- Hueffer K, Govindasamy L, Agbandje-McKenna M, Parrish CR. Combinations of two capsid regions controlling canine host range determine canine transferrin receptor binding by canine and feline parvoviruses. J Virol. 2003a;77 (18):10099–10105. doi: 10.1128/JVI.77.18.10099-10105.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hueffer K, Palermo LM, Parrish CR. Parvovirus infection of cells by using variants of the feline transferrin receptor altering clathrin-mediated endocytosis, membrane domain localization, and capsid-binding domains. J Virol. 2004;78 (11):5601–5611. doi: 10.1128/JVI.78.11.5601-5611.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hueffer K, Parker JS, Weichert WS, Geisel RE, Sgro JY, Parrish CR. The natural host range shift and subsequent evolution of canine parvovirus resulted from virus-specific binding to the canine transferrin receptor. J Virol. 2003b;77 (3):1718–1726. doi: 10.1128/JVI.77.3.1718-1726.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann B, Simpson AA, Rossmann MG. The structure of human parvovirus B19. Proc Natl Acad Sci U S A. 2004;101 (32):11628–11633. doi: 10.1073/pnas.0402992101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasse PJ, Sattentau QJ. Occupancy and mechanism in antibody-mediated neutralization of animal viruses. J Gen Virol. 2002;83 (9):2091–2108. doi: 10.1099/0022-1317-83-9-2091. [DOI] [PubMed] [Google Scholar]
- Knossow M, Gaudier M, Douglas A, Barrere B, Bizebard T, Barbey C, Gigant B, Skehel JJ. Mechanism of neutralization of influenza virus infectivity by antibodies. Virology. 2002;302 (2):294–298. doi: 10.1006/viro.2002.1625. [DOI] [PubMed] [Google Scholar]
- Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393 (6686):648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langeveld JP, Casal JI, Cortes E, van de Wetering G, Boshuizen RS, Schaaper WM, Dalsgaard K, Meloen RH. Effective induction of neutralizing antibodies with the amino terminus of VP2 of canine parvovirus as a synthetic peptide. Vaccine. 1994;12 (15):1473–1480. doi: 10.1016/0264-410x(94)90158-9. [DOI] [PubMed] [Google Scholar]
- Langeveld JP, Casal JI, Vela C, Dalsgaard K, Smale SH, Puijk WC, Meloen RH. B-cell epitopes of canine parvovirus: distribution on the primary structure and exposure on the viral surface. J Virol. 1993;67 (2):765–772. doi: 10.1128/jvi.67.2.765-772.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence CM, Ray S, Babyonyshev M, Galluser R, Borhani DW, Harrison SC. Crystal structure of the ectodomain of human transferrin receptor. Science. 1999;286 (5440):779–782. doi: 10.1126/science.286.5440.779. [DOI] [PubMed] [Google Scholar]
- Ledford RM, Patel NR, Demenczuk TM, Watanyar A, Herbertz T, Collett MS, Pevear DC. VP1 sequencing of all human rhinovirus serotypes: insights into genus phylogeny and susceptibility to antiviral capsid-binding compounds. J Virol. 2004;78 (7):3663–3674. doi: 10.1128/JVI.78.7.3663-3674.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebermann H, Mentel R, Bauer U, Pring-Akerblom P, Dolling R, Modrow S, Seidel W. Receptor binding sites and antigenic epitopes on the fiber knob of human adenovirus serotype 3. J Virol. 1998;72 (11):9121–9130. doi: 10.1128/jvi.72.11.9121-9130.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Turiso JA, Cortez E, Ranz A, Garcia J, Sanz A, Vela C, Casal JI. Fine mapping of canine parvovirus B cell epitopes. J Gen Virol. 1991;72 (10):2445–2456. doi: 10.1099/0022-1317-72-10-2445. [DOI] [PubMed] [Google Scholar]
- Ludert JE, Ruiz MC, Hidalgo C, Liprandi F. Antibodies to rotavirus outer capsid glycoprotein VP7 neutralize infectivity by inhibiting virion decapsidation. J Virol. 2002;76 (13):6643–51. doi: 10.1128/JVI.76.13.6643-6651.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martella V, Cavalli A, Decaro N, Elia G, Desario C, Campolo M, Bozzo G, Tarsitano E, Buonavoglia C. Immunogenicity of an intranasally administered modified live canine parvovirus type 2b vaccine in pups with maternally derived antibodies. Clin Diagn Lab Immunol. 2005;12 (10):1243–1245. doi: 10.1128/CDLI.12.10.1243-1245.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martella V, Cavalli A, Pratelli A, Bozzo G, Camero M, Buonavoglia D, Narcisi D, Tempesta M, Buonavoglia C. A canine parvovirus mutant is spreading in Italy. J Clin Microbiol. 2004;42 (3):1333–1336. doi: 10.1128/JCM.42.3.1333-1336.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna R, Olson NH, Chipman PR, Baker TS, Booth TF, Christensen J, Aasted B, Fox JM, Bloom ME, Wolfinbarger JB, Agbandje-McKenna M. Three-dimensional structure of Aleutian mink disease parvovirus: implications for disease pathogenicity. J Virol. 1999;73 (8):6882–6891. doi: 10.1128/jvi.73.8.6882-6891.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier PC, Cooper BJ, Appel MJ, Slauson DO. Pathogenesis of canine parvovirus enteritis: the importance of viremia. Vet Pathol. 1985;22 (1):60–71. doi: 10.1177/030098588502200110. [DOI] [PubMed] [Google Scholar]
- Mochizuki M, Harasawa R, Nakatani H. Antigenic and genomic variabilities among recently prevalent parvoviruses of canine and feline origin in Japan. Vet Microbiol. 1993;38 (1–2):1–10. doi: 10.1016/0378-1135(93)90070-n. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Sakamoto M, Ikeda Y, Sato E, Kawakami K, Miyazawa T, Tohya Y, Takahashi E, Mikami T, Mochizuki M. Pathogenic potential of canine parvovirus types 2a and 2c in domestic cats. Clin Diagn Lab Immunol. 2001;8 (3):663–668. doi: 10.1128/CDLI.8.3.663-668.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Nakamura K, Miyazawa T, Tohya Y, Mochizuki M, Akashi H. Monoclonal antibodies that distinguish antigenic variants of canine parvovirus. Clin Diagn Lab Immunol. 2003;10 (6):1085–1089. doi: 10.1128/CDLI.10.6.1085-1089.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Tohya Y, Miyazawa T, Mochizuki M, Phung HT, Nguyen NH, Huynh LM, Nguyen LT, Nguyen PN, Nguyen PV, Nguyen NP, Akashi H. A novel antigenic variant of canine parvovirus from a Vietnamese dog. Arch Virol. 2004;149 (11):2261–2269. doi: 10.1007/s00705-004-0367-y. [DOI] [PubMed] [Google Scholar]
- Nybakken GE, Oliphant T, Johnson S, Burke S, Diamond MS, Fremont DH. Structural basis of West Nile virus neutralization by a therapeutic antibody. Nature. 2005;437 (7059):764–769. doi: 10.1038/nature03956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osiowy C, Anderson R. Neutralization of respiratory syncytial virus after cell attachment. J Virol. 1995;69 (2):1271–1274. doi: 10.1128/jvi.69.2.1271-1274.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padron E, Bowman V, Kaludov N, Govindasamy L, Levy H, Nick P, McKenna R, Muzyczka N, Chiorini JA, Baker TS, Agbandje-McKenna M. Structure of adeno-associated virus type 4. J Virol. 2005;79 (8):5047–5058. doi: 10.1128/JVI.79.8.5047-5058.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo LM, Hafenstein SL, Parrish CR. Purified feline and canine transferrin receptors reveal complex interactions with the capsids of canine and feline parvoviruses that correspond to their host ranges. J Virol. 2006;80 (17):8482–8492. doi: 10.1128/JVI.00683-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo LM, Hueffer K, Parrish CR. Residues in the apical domain of the feline and canine transferrin receptors control host-specific binding and cell infection of canine and feline parvoviruses. J Virol. 2003;77 (16):8915–8923. doi: 10.1128/JVI.77.16.8915-8923.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JSL, Murphy WJ, Wang D, O'Brien SJ, Parrish CR. Canine and feline parvoviruses can use human or feline transferrin receptors to bind, enter, and infect cells. J Virol. 2001;75 (8):3896–3902. doi: 10.1128/JVI.75.8.3896-3902.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish CR. Mapping specific functions in the capsid structure of canine parvovirus and feline panleukopenia virus using infectious plasmid clones. Virology. 1991;183 (1):195–205. doi: 10.1016/0042-6822(91)90132-u. [DOI] [PubMed] [Google Scholar]
- Parrish CR, Aquadro C, Strassheim ML, Evermann JF, Sgro JY, Mohammed H. Rapid antigenic-type replacement and DNA sequence evolution of canine parvovirus. J Virol. 1991;65 (12):6544–6552. doi: 10.1128/jvi.65.12.6544-6552.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish CR, Carmichael LE. Antigenic structure and variation of canine parvovirus type-2, feline panleukopenia virus, and mink enteritis virus. Virology. 1983;129 (2):401–414. doi: 10.1016/0042-6822(83)90179-4. [DOI] [PubMed] [Google Scholar]
- Parrish CR, Carmichael LE, Antczak DF. Antigenic relationships between canine parvovirus type-2, feline panleukopenia virus and mink enteritis virus using conventional antisera and monoclonal antibodies. Arch Virol. 1982;72 (4):267–278. doi: 10.1007/BF01315223. [DOI] [PubMed] [Google Scholar]
- Parrish CR, Gorham JR, Schwartz TM, Carmichael LE. Characterisation of antigenic variation among mink enteritis virus isolates. Am J Vet Res. 1984;45 (12):2591–2599. [PubMed] [Google Scholar]
- Parrish CR, Have P, Foreyt WJ, Evermann JF, Senda M, Carmichael LE. The global spread and replacement of canine parvovirus strains. J Gen Virol. 1988;69 (5):1111–1116. doi: 10.1099/0022-1317-69-5-1111. [DOI] [PubMed] [Google Scholar]
- Parrish CR, O'Connell PH, Evermann JF, Carmichael LE. Natural variation of canine parvovirus. Science. 1985;230 (4729):1046–1048. doi: 10.1126/science.4059921. [DOI] [PubMed] [Google Scholar]
- Pollock RVH, Carmichael LE. Maternally derived immunity to canine parvovirus infection: transfer, decline and interference with vaccination. J Am Vet Med Assoc. 1982;180 (1):37–42. [PubMed] [Google Scholar]
- Rimmelzwaan GF, Carlson J, UytdeHaag FG, Osterhaus AD. A synthetic peptide derived from the amino acid sequence of canine parvovirus structural proteins which defines a B cell epitope and elicits antiviral antibody in BALB c mice. J Gen Virol. 1990;71 (11):2741–2745. doi: 10.1099/0022-1317-71-11-2741. [DOI] [PubMed] [Google Scholar]
- Rossmann MG, He Y, Kuhn RJ. Picornavirus-receptor interactions. Trends Microbiol. 2002;10 (7):324–331. doi: 10.1016/s0966-842x(02)02383-1. [DOI] [PubMed] [Google Scholar]
- Rota JS, Hummel KB, Rota PA, Bellini WJ. Genetic variability of the glycoprotein genes of current wild-type measles isolates. Virology. 1992;188 (1):135–142. doi: 10.1016/0042-6822(92)90742-8. [DOI] [PubMed] [Google Scholar]
- Rota JS, Wang ZD, Rota PA, Bellini WJ. Comparison of sequences of the H, F, and N coding genes of measles virus vaccine strains. Virus Res. 1994;31 (3):317–330. doi: 10.1016/0168-1702(94)90025-6. [DOI] [PubMed] [Google Scholar]
- Sagazio P, Tempesta M, Buonavoglia D, Cirone F, Buonavoglia C. Antigenic characterization of canine parvovirus strains isolated in Italy. J Virol Methods. 1998;73 (2):197–200. doi: 10.1016/s0166-0934(98)00055-x. [DOI] [PubMed] [Google Scholar]
- Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, Rudd PM, Dwek RA, Stanfield RL, Burton DR, Wilson IA. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science. 2001;293 (5532):1155–1159. doi: 10.1126/science.1061692. [DOI] [PubMed] [Google Scholar]
- Sheshberadaran H, Norrby E. Characterization of epitopes on the measles virus hemagglutinin. Virology. 1986;152 (1):58–65. doi: 10.1016/0042-6822(86)90371-5. [DOI] [PubMed] [Google Scholar]
- Sheshberadaran H, Norrby E, Rammohan KW. Monoclonal antibodies against five structural components of measles virus. II Characterization of five cell lines persistently infected with measles virus. Arch Virol. 1985;83 (3–4):251–268. doi: 10.1007/BF01309921. [DOI] [PubMed] [Google Scholar]
- Simpson AA, Chipman PR, Baker TS, Tijssen P, Rossmann MG. The structure of an insect parvovirus (Galleria mellonella densovirus) at 3.7 Å resolution. Structure. 1998;6 (11):1355–1367. doi: 10.1016/s0969-2126(98)00136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skehel JJ, Wiley DC. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu Rev Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. Mapping the antigenic and genetic evolution of influenza virus. Science. 2004;305 (5682):371–376. doi: 10.1126/science.1097211. [DOI] [PubMed] [Google Scholar]
- Smith TJ, Chase ES, Schmidt TJ, Olson NH, Baker TS. Neutralizing antibody to human rhinovirus 14 penetrates the receptor-binding canyon. Nature. 1996;383 (6598):350–354. doi: 10.1038/383350a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinel A, Venter EH, Van Vuuren M, Parrish CR, Truyen U. Antigenic and genetic analysis of canine parvoviruses in southern Africa. Onderstepoort J Vet Res. 1998;65 (4):239–242. [PubMed] [Google Scholar]
- Strassheim LS, Gruenberg A, Veijalainen P, Sgro JY, Parrish CR. Two dominant neutralizing antigenic determinants of canine parvovirus are found on the threefold spike of the virus capsid. Virology. 1994;198 (1):175–184. doi: 10.1006/viro.1994.1020. [DOI] [PubMed] [Google Scholar]
- Truyen U, Evermann JF, Vieler E, Parrish CR. Evolution of canine parvovirus involved loss and gain of feline host range. Virology. 1996;215 (2):186–189. doi: 10.1006/viro.1996.0021. [DOI] [PubMed] [Google Scholar]
- Truyen U, Gruenberg A, Chang SF, Obermaier B, Veijalainen P, Parrish CR. Evolution of the feline-subgroup parvoviruses and the control of canine host range in vivo. J Virol. 1995;69 (8):4702–4710. doi: 10.1128/jvi.69.8.4702-4710.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truyen U, Parrish CR. Canine and feline host ranges of canine parvovirus and feline panleukopenia virus: distinct host cell tropisms of each virus in vitro and in vivo. J Virol. 1992;66 (9):5399–5408. doi: 10.1128/jvi.66.9.5399-5408.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao J, Chapman MS, Agbandje M, Keller W, Smith K, Wu H, Luo M, Smith TJ, Rossmann MG, Compans RW, Parrish CR. The three-dimensional structure of canine parvovirus and its functional implications. Science. 1991;251 (5000):1456–1464. doi: 10.1126/science.2006420. [DOI] [PubMed] [Google Scholar]
- Varghese R, Mikyas Y, Stewart PL, Ralston R. Postentry neutralization of adenovirus type 5 by an antihexon antibody. J Virol. 2004;78 (22):12320–32. doi: 10.1128/JVI.78.22.12320-12332.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdaguer N, Schoehn G, Ochoa WF, Fita I, Brookes S, King A, Domingo E, Mateu MG, Stuart D, Hewat EA. Flexibility of the major antigenic loop of foot-and-mouth disease virus bound to a Fab fragment of a neutralising antibody: structure and neutralisation. Virology. 1999;255 (2):260–268. doi: 10.1006/viro.1998.9554. [DOI] [PubMed] [Google Scholar]
- Vossen MT, Westerhout EM, Soderberg-Naucler C, Wiertz EJ. Viral immune evasion: a masterpiece of evolution. Immunogenetics. 2002;54 (8):527–542. doi: 10.1007/s00251-002-0493-1. [DOI] [PubMed] [Google Scholar]
- Walters RW, Agbandje-McKenna M, Bowman VD, Moninger TO, Olson NH, Seiler M, Chiorini JA, Baker TS, Zabner J. Structure of adeno-associated virus serotype 5. J Virol. 2004;78 (7):3361–3371. doi: 10.1128/JVI.78.7.3361-3371.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waner T, Naveh A, Wudovsky I, Carmichael LE. Assessment of maternal antibody decay and response to canine parvovirus vaccination using a clinic-based enzyme-linked immunosorbent assay. J Vet Diagn Invest. 1996;8 (4):427–432. doi: 10.1177/104063879600800404. [DOI] [PubMed] [Google Scholar]
- Weichert WS, Parker JS, Wahid ATM, Chang SF, Meier E, Parrish CR. Assaying for structural variation in the parvovirus capsid and its role in infection. Virology. 1998;250 (1):106–117. doi: 10.1006/viro.1998.9352. [DOI] [PubMed] [Google Scholar]
- West AP, Jr, Giannetti AM, Herr AB, Bennett MJ, Nangiana JS, Pierce JR, Weiner LP, Snow PM, Bjorkman PJ. Mutational analysis of the transferrin receptor reveals overlapping HFE and transferrin binding sites. J Mol Biol. 2001;313 (2):385–397. doi: 10.1006/jmbi.2001.5048. [DOI] [PubMed] [Google Scholar]
- Wikoff WR, Wang G, Parrish CR, Cheng RH, Strassheim ML, Baker TS, Rossmann MG. The structure of a neutralized virus: canine parvovirus complexed with neutralizing antibody fragment. Structure. 1994;2 (7):595–607. doi: 10.1016/s0969-2126(00)00062-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobus CE, Hugle-Dorr B, Girod A, Petersen G, Hallek M, Kleinschmidt JA. Monoclonal antibodies against the adeno-associated virus type 2 (AAV-2) capsid: epitope mapping and identification of capsid domains involved in AAV-2-cell interaction and neutralization of AAV-2 infection. J Virol. 2000;74 (19):9281–9293. doi: 10.1128/jvi.74.19.9281-9293.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Bu W, Bhatia S, Hare J, Somasundaram T, Azzi A, Chapman MS. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc Natl Acad Sci U S A. 2002;99(16):10405–10410. doi: 10.1073/pnas.162250899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Q, Chapman MS. Canine parvovirus capsid structure, analyzed at 2.9 Å resolution. J Mol Biol. 1996;264 (3):497–520. doi: 10.1006/jmbi.1996.0657. [DOI] [PubMed] [Google Scholar]
- Yuan W, Parrish CR. Comparison of two single-chain antibodies that neutralize Canine Parvovirus: analysis of an antibody-combining site and mechanisms of neutralization. Virology. 2000;269 (2):471–480. doi: 10.1006/viro.2000.0230. [DOI] [PubMed] [Google Scholar]