

Abstract
Lipopolysaccharides (LPS), otherwise termed ‘endotoxins’, are outer-membrane constituents of Gram-negative bacteria, and play a key role in the pathogenesis of ‘Septic Shock’, a major cause of mortality in the critically ill patient. We had previously defined the pharmacophore necessary for small molecules to specifically bind and neutralize this complex carbohydrate. A series of aryl and aliphatic spermine-sulfonamide analogs were synthesized and tested in a series of binding and cell-based assays in order to probe the effect of lipophilicity on sequestration ability. A strong correlation was indeed found, supporting the hypothesis that endotoxin-neutralizing ability involves a lipophilic or membrane attachment event. The research discussed herein may be useful for the design of additional carbohydrate recognizing molecules and endotoxin-neutralizing drugs.
Lipopolysaccharides (LPS) are the predominant structural components of the outer membrane of Gram-negative bacteria.1,2 Otherwise termed ‘endotoxins’, LPS play a pivotal role in septic shock, a syndrome of systemic toxicity which occurs frequently when the body’s defense mechanisms are compromised.3–6 Gram-negative sepsis is the thirteenth leading cause of overall mortality7 and the number one cause of deaths in the intensive care unit,8 accounting for more than 200,000 fatalities in the US annually.9 The presence of LPS in the systemic circulation causes an uncontrolled activation of the innate immune system10,11 leading to the production of numerous inflammatory mediators, including tumor necrosis factor-α (TNF-α), interleukin-1 β (IL-1β), and interleukin-6 (IL-6),12,13 culminating in the frequently-fatal multiple system organ failure.
The toxic moiety of LPS resides in its glycolipid component called Lipid A,14 which is composed of a hydrophilic, bis-phosphorylated diglucosamine backbone, and a hydrophobic domain of 6 (E. coli) or 7 (Salmonella) acyl chains14 (Fig. 1). The pharmacophore necessary for the neutralization of lipid A15 by small molecules requires two groups protonatable at physiological pH, with an intervening distance of ~ 14 Å, enabling ionic H-bonds between the cationic groups and the lipid A phosphates. In addition, appropriately positioned pendant hydrophobic functionalities are required to further stabilize the resultant complexes via hydrophobic interactions with the polyacyl domain of lipid A (for a recent review, see Ref. 16). In two recent studies of the effect of the hydrocarbon chain length in two series of acylhomospermines, it was shown that C16 is the ideal lipophilic substituent, corresponding to maximal affinity, optimal aqueous solubility (and bioavailability), and neutralization potency.17,18 In building on our earlier work focused upon acylated spermines, this paper examines spermine-sulfonamides composed of various numbers (mono-, di- and tri-substituted) and types of lipophilic groups with a view to correlating such structural modifications on the affinity of binding to LPS and on the potency of abrogating endotoxicity in in vitro neutralization assays. We specifically address several key questions: (i) will spermine-sulfonamides display similar activity to previously studied acylspermines, (ii) what is the relationship between lipophilicity of the substituent hydrophobic groups and binding activity, and (iii) can highly lipophilic aryl groups effectively mimic long hydrocarbon chains?
Figure 1.
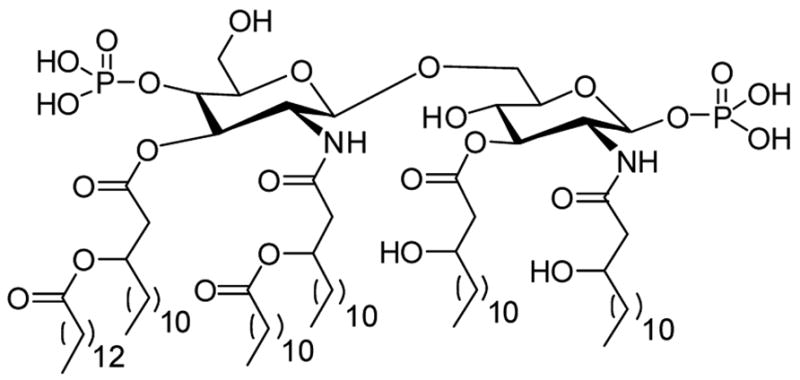
Structure of Lipid A, the toxic principle of lipopolysaccharide.
The analogs described were produced as shown in Scheme 1 (Supplemental Data).
Scheme 1.
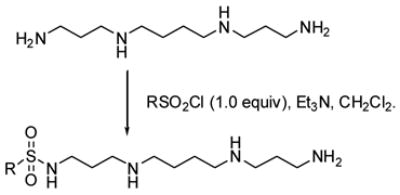
Synthesis of spermine sulfonamides.
The relative binding affinities of the analogs are reported in Table 1 (Bis-substituted analogs were also isolated and showed higher ED50 values, for the sake of brevity, representative data on the mono-substituted compounds are shown) as half-maximal effective displacement of probe (ED50) obtained using a high-throughput fluorescence based displacement assay, employing BODIPY-TR cadaverine (BC).19,20 Polymyxin B (PMB), a decapeptide antibiotic, known to bind and neutralize LPS,21–24 was used as a reference compound.
Table 1.
Binding affinity of mono-substituted spermine sulfonamides.
| Analog | R | Binding ED50 (μM)a |
|---|---|---|
| PMB | --- | 0.31 |
| 1 | CH3(CH2)2CH2- | >10,000 |
| 2 |
|
4877 |
| 3 |
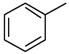
|
2955 |
| 4 |
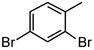
|
2108 |
| 5 |
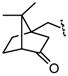
|
1675 |
| 6 |
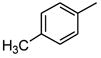
|
884 |
| 7 |
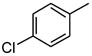
|
158 |
| 8 |
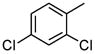
|
56 |
| 9 |
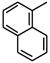
|
34 |
| 10 |
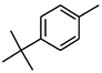
|
25 |
| 11 |
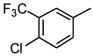
|
21 |
| 12 |
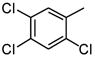
|
19 |
| 13 |
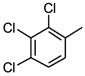
|
9 |
| 14 | CH3(CH2)16CH2- | 6 |
| 15 | CH3(CH2)6CH2- | 5 |
| 16 |
|
4 |
| 17 | CH3(CH2)8CH2- | 3 |
| 18 | CH3(CH2)10CH2- | 3 |
| 19 | CH3(CH2)14CH2- | 2 |
As is evident in Table 1, a clear trend towards higher-affinity binding by those analogs with more lipophilic substituents was observed. An especially instructive series was the chloro-substituted phenyl devivatives: phenyl (3, 2955 μM), 4-monochloro (7, 158 μM), 2,4-dichloro (8, 58 μM), 2,4,5-trichloro (12, 18.7 μM), and 2,3,4-trichloro (13, 9.08 μM).
The correlation between lipophilicity and binding was also observed for the aliphatic monosulfonamides: butyl (1, >10,000 μM), octyl (15, 5.43 μM), decyl (17, 3.7 μM), dodecyl (18, 2.29 μM), hexadecyl (19, 2.34 μM), and octadecyl (14, 6.01 μM). Both series display maximal binding affinity leveling off at a chain length of 16. We analyzed the correlation between experimentally determined binding activity for the monosulfonamide spermine analogs, and their calculated lipophilicity (cLogP). As shown in Figure 2, a distinct correlation between these two parameters was observed, with an R2 value of 0.756 for a first-order exponential fit.
Fig. 2.

Correlation of binding affinity and lipophilicity in spermine monosulfonamides
Inhibition of LPS-induced nitric oxide (NO) production (measured as nitrite) by the sulfonamide analogs in murine J774 cells was performed as published previously.17,25 In parallel we also examined dose-responses in the inhibition of induction of NF-κB (a key transcriptional activator of the innate immune system, leading to uncontrolled cytokine release25,26). NF-κB was quantified using human embryonic kidney 293 cells cotransfected with TLR4 (LPS receptor), CD14 and MD2 (co-receptors), available from InvivoGen, Inc., (HEK-Blue™, San Diego, CA) as described elsewhere.26 A clear relationship between binding affinity and both NO and NF-κB in vitro IC50 values are evident (Fig. 3). While the dynamic range for the BC displacement assay is very large (nM to mM),35 linearity for both the NO and NF-κB assays are limiting, resulting in significant deviations from nonlinearity for IC50 values above 30 μM (1.5 on the log scale). Indeed, piece-wise linear regressions yielded R values of 0.78 and 0.83 for the NO and NF-κB assays, respectively (Fig. 3).
Fig. 3.

Correlation of binding affinity with NF-κB and NO inhibition in vitro of the monosulfonamides. Piece-wise linear regression lines show the experimentally useful range of linearity (≤ 100 μM; solid line).
We have established that NF-κB inhibition is best correlated with a definitive murine model17,25 of endotoxic shock. We therefore sought to compare the potencies of the high affinity long-chain monosulfonamides with PMB. As shown in Fig. 4, several long-chain monosulfonamide compounds compare very favorably with PMB, the dose-response for 19 being virtually indistinguishable from that of PMB (Fig. 4).
Fig. 4.

Comparison of NF-kB inhibition by long-chain sulfonamides and polymyxin B. IC50 values were determined by four-parameter logistic fits.
These results confirm that long-chain aliphatic appendages on spermine-like polyamine scaffolds yield the most potent anti-endotoxin compounds as a consequence of imbuing optimal hydrophobic character. Simple cLogP calculations may thus serve as a useful tool in conjunction with detailed in silico design and docking experiments27 to construct focused libraries. As the last group of mostly unexploited drug targets, complex carbohydrates represent an especially challenging group of biomolecules with which to design specific recognition molecules. The complex interplay of hydrogen-bond complementarity, charge and lipophilicity in interaction with membrane-associated receptors greatly contributes to this challenge. The set of molecules described in this report showcase the role that lipophilicity plays in Lipid A recognition. Given their ease of synthesis, potent LPS-neutralizing effects and activity in a cell-based assay, these spermine sulfonamide analogs may be serve as candidates for animal testing and further preclinical development. A detailed characterization of the endotoxin-sequestering activities of 19 will be reported elsewhere.
Supplementary Material
Supplementary Data: Synthesis and characterization data of 18 and 19.
Acknowledgments
This work was supported from NIH Grant 1U01 AI054785 (S. David).
References
- 1.Lüderitz O, Galanos C, Rietschel ET. PharmacolTher. 1982;15:383–402. doi: 10.1016/0163-7258(81)90051-6. [DOI] [PubMed] [Google Scholar]
- 2.Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zähringer U, Seydel U, Di Padova F, et al. FASEB J. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 3.Hurley JC. ClinInfectDis. 1992;15:840–854. doi: 10.1093/clind/15.5.840. [DOI] [PubMed] [Google Scholar]
- 4.Hurley JC. Drug Saf. 1995;12:183–195. doi: 10.2165/00002018-199512030-00004. [DOI] [PubMed] [Google Scholar]
- 5.Prins JM, van Agtmael MA, Kuijper EJ, van Deventer SJ, Speelman P. JInfectDis. 1995;172:886–891. doi: 10.1093/infdis/172.3.886. [DOI] [PubMed] [Google Scholar]
- 6.Prins JM, Van Deventer SJH, Kuijper EJ, Speelman P. AntimicrobAgents Chemother. 1994;38:1211–1218. doi: 10.1128/aac.38.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gelfand JA, Shapiro L. New Horizons. 1993;1:13–22. [PubMed] [Google Scholar]
- 8.Gasche Y, Pittet D, Sutter P. Outcome and prognostic factors in bacteremic sepsis. In: Sibbald WJ, Vincent JL, editors. Clinical trials for treatment of sepsis. Vol. 95. Springer-Verlag: Berlin; pp. 35–51. [Google Scholar]
- 9.MMWR. 1990;39:31–34. [Google Scholar]
- 10.Ulevitch RJ. ImmunolRes. 2000;21:49–54. [Google Scholar]
- 11.Ulevitch RJ, Tobias P. CurrOpinImmunol. 1999;11:19–23. [Google Scholar]
- 12.Dinarello CA. CurrTopMicrobiolImmunol. 1996;216:133–165. doi: 10.1007/978-3-642-80186-0_7. [DOI] [PubMed] [Google Scholar]
- 13.Michie HR, Manogue KR, Spriggs DR, Revhaug A, O’Dwyer S, Dinarello CA, Cerami A, Wolff SM, Wilmore DW. NEnglJMed. 1988;318:1481–1486. doi: 10.1056/NEJM198806093182301. [DOI] [PubMed] [Google Scholar]
- 14.Raetz CRH, Whitfield C. AnnuRevBiochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.David SA, Mathan VI, Balaram P. JEndotoxinRes. 1995;2:325–336. [Google Scholar]
- 16.David SA. JMolecRecognition. 2001;14:370–387. [Google Scholar]
- 17.Miller KA, Suresh Kumar EVK, Wood SJ, Cromer JR, Datta A, David SA. JMedChem. 2005;48:2589–2599. doi: 10.1021/jm049449j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burns MR, Wood SJ, Miller KA, Nguyen T, Cromer JR, David SA. BioorgMedChem. 2005;13:2523–2536. doi: 10.1016/j.bmc.2005.01.038. [DOI] [PubMed] [Google Scholar]
- 19.Wood SJ, Miller KA, David SA. CombChemHighThroughputScreen. 2004;7:239–249. [Google Scholar]
- 20.Wood SJ, Miller KA, David SA. CombChemHighThroughputScreen. 2004;7:733–743. [Google Scholar]
- 21.Aoki H, Kodama M, Tani T, Hanasawa K. AmJSurg. 1994;167:412–417. doi: 10.1016/0002-9610(94)90126-0. [DOI] [PubMed] [Google Scholar]
- 22.Bucklin SE, Lake P, Logdberg L, Morrison DC. AntimicrobAgents Chemother. 1995;39:1462–1466. doi: 10.1128/aac.39.7.1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mayumi T, Takezawa J, Takahashi H, Kuwayama N, Fukuoka T, Shimizu K, Yamada K, Kondo S, Aono K. Shock. 1999;11:82–86. doi: 10.1097/00024382-199902000-00002. [DOI] [PubMed] [Google Scholar]
- 24.Stokes DC, Shenep JL, Fishman ML, Hidner WK, Bysani GK, Rufus K. JInfectDis. 1989;160:52–57. doi: 10.1093/infdis/160.1.52. [DOI] [PubMed] [Google Scholar]
- 25.David SA, Silverstein R, Amura CR, Kielian T, Morrison DC. AntimicrobAgents Chemother. 1999;43:912–919. doi: 10.1128/aac.43.4.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khownium K, Wood SJ, Miller KA, Balakrishna R, Nguyen TB, Kimbrell MR, Georg GI, David SA. BioorgMedChemLett. 2006;16:1305–1308. doi: 10.1016/j.bmcl.2005.11.059. [DOI] [PubMed] [Google Scholar]
- 27.Guo JX, Wood SJ, David SA, Lushington GH. BioorgMedChemLett. 2006;16:714–717. doi: 10.1016/j.bmcl.2005.10.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data: Synthesis and characterization data of 18 and 19.