

The lectin from the Nigerian legume B. mildbraedii was crystallized in complex with Man(α1-2)Man and data were collected to a resolution of 1.90 Å using synchrotron radiation.
Keywords: Bowringia mildbraedii, legume lectins, carbohydrate recognition
Abstract
The lectin from Bowringia mildbraedii seeds crystallizes in the presence of the disaccharide Man(α1-2)Man. The best crystals grow at 293 K within four weeks after a pre-incubation at 277 K to induce nucleation. A complete data set was collected to a resolution of 1.90 Å using synchrotron radiation. The crystals belong to space group I222, with unit-cell parameters a = 66.06, b = 86.35, c = 91.76 Å, and contain one lectin monomer in the asymmetric unit.
1. Introduction
Lectins are carbohydrate-binding proteins of non-immune origin that specifically recognize carbohydrate structures. They are widespread in all kingdoms of life and mediate a variety of biological processes such as viral, bacterial and parasitic infections, growth and differentiation or cancer metastasis (Taylor & Drickamer, 2003 ▶). Plant lectins (Peumans & Van Damme, 1995 ▶), especially those purified from the leguminous family, are among the best studied group of carbohydrate-binding proteins. Legume lectins have for decades been a paradigm for protein–carbohydrate recognition because of their wide range of specificities for monosaccharides as well as for complex carbohydrates (Sharon & Lis, 1990 ▶).
Since the crystal structure determination of concanavalin A in the early 1970s (Edelman et al., 1972 ▶; Hardman & Ainsworth, 1972 ▶), the crystal structures of about 30 members of the legume lectin family have been determined (http://www.cermav.cnrs.fr/glyco3d/index.php), most of them in complex with one or more carbohydrate ligands. From these studies, it has been established that legume lectins have a conserved monomeric scaffold, but at the same time exhibit a variety of quaternary associations. Their carbohydrate-binding sites consist of four loops (Loris et al., 1998 ▶; Sharma & Surolia, 1997 ▶). Three of these contain a set of (semi-)invariant residues that make up the primary or monosaccharide-binding site (Young & Oomen, 1992 ▶). Carbohydrate specificity is thought to be largely determined by the fourth loop, which is highly variable.
The seeds of the Nigerian legume Bowringia milbraedii contain a Man/Glc-specific lectin (B. milbraedii agglutinin; BMA) that shows its highest affinity for the disaccharide Man(α1-2)Man (Animashaun & Hughes, 1989 ▶; Chawla et al., 1992 ▶). The lectin reacts with human erythrocytes regardless of the blood type and can stimulate human and pig lymphocytes as effectively as concanavalin A or phytohaemagglutinin (Animashaun & Hughes, 1989 ▶). BMA consists of a 29 kDa precursor that is cleaved into an α-chain of 114 amino acids and a β-chain of 116 amino acids. The β-chain contains a single cysteine residue that participates in an inter-β-chain disulfide bond (Chawla et al., 1993 ▶). The protein shows 48% sequence identity with concanavalin A, its closest relative (Fig. 1 ▶). Here, we present the crystallization and preliminary X-ray diffraction analysis of BMA in complex with Man(α1-2)Man.
Figure 1.

Sequence alignment made with ClustalW using default settings (Thompson et al., 1994 ▶) of mannose-specific legume lectins: B. milbraedii (BMA), Canavalia ensiformis concanavalin A (ConA), Phaseolus vulgaris Flt3 receptor-interacting lectin (PvFRIL), L. ochrus isolectin I (LOL-I) and Pterocarpus angolensis seed lectin (PAL). The positions of the β-strands are indicated with arrows. Residues contributing to the monosaccharide-binding site are marked with a black background.
2. Materials and methods
2.1. Purification of BMA by affinity chromatography
B. mildbraedii seeds were a gift from Dr T. Animashaun (National Institute for Medical Research, Mill Hill, London). After initial crushing with a hammer, the seeds were ground in a domestic coffee grinder. The lectin was extracted at 277 K for 24 h in 0.9%(w/v) NaCl (10 ml per gram of ground material). The supernatant obtained after centrifugation at 10 000g for 30 min was applied onto a 30 ml mannose–agarose column (Sigma). After loading, the column was extensively washed with PBS (137 mM NaCl, 3 mM KCl, 8 mM Na2HPO4, 1.75 mM KH2PO4) and subsequently eluted with 0.2 M methyl-α-d-mannopyrannoside in PBS. 3 ml fractions were collected. The peak fractions of the eluant were dialyzed exhaustively against 0.9% NaCl at 277 K to remove the methyl-α-d-mannoside and analyzed for haemagglutinating activity. The active fractions were pooled.
2.2. Gel filtration
Gel-filtration experiments were carried out in 50 mM MES–HCl pH 6.5, 100 mM NaCl, 250 mM glucose on a Superdex 75-HR column using a flow rate of 0.5 ml min−1. 450 µl samples with a protein concentration of about 1 mg ml−1 were loaded onto the column. Thyroglobulin (679 kDa), γ-globulin (158 kDa), ovalbumin (44 kDa), chymotrypsin A (25 kDa), horse myoglobin (17 kDa) and vitamin B12 (1.35 kDa) were used as molecular-weight standards.
2.3. Agglutination assay
One volume of rabbit blood was added to an equal volume of Alsever solution (0.05 g glucose, 0.8 g sodium citrate and 0.42 g NaCl in 100 ml of water) containing heparin (two drops in 5 ml Alsever). The cells were washed three times by centrifugation (450g for 20 min at 277 K) using fresh saline solution [0.9%(w/v) NaCl] each time. After washing, the erythrocyte pellet was resuspended in a total volume of 25 ml of saline solution to render a 4%(v/v) erythrocyte suspension.
Agglutination activity was measured by serially diluting 25 µl of the BMA sample in 25 µl of saline solution in U-shaped 96-well plates (Limbro, Titertek). Finally, 50 µl of the erythrocyte suspension was added to each well. The plates were incubated at room temperature for 1–2 h. The haemagglutination activity was estimated visually and expressed as the agglutination titre (the highest dilution that still showed agglutination).
2.4. Crystallization
For all crystallization experiments, BMA was dialyzed against 150 mM NaCl and concentrated to 14 mg ml−1. Crystallization conditions were screened by the hanging-drop vapour-diffusion method using Hampton Research Crystal Screen and Crystal Screen II (Hampton Research, Riverside, CA, USA; Jancarik & Kim, 1991 ▶) at 293 K. Drops consisting of 3 µl BMA solution and 3 µl precipitant were equilibrated against 1 ml reservoir solution. To optimize the conditions for crystallization, protein concentration, pH, incubation temperature and precipitant concentration were varied. We attempted to crystallize ligand-free protein as well as co-crystallization with methyl-α-d-mannopyrannoside, Man(α1-2)Man, Man(α1-3)Man, Man(α1-3)[Man(α1-6)]Man, sucrose, trehalose and turanose. Different incubation times at 277 K followed by transfer to a final temperature of 293 K were also assayed for optimization.
2.5. Data collection
X-ray data were collected at 100 K on EMBL beamline X11 of the DESY synchrotron, Hamburg, Germany. The crystals were soaked in a cryoprotectant solution containing 0.1 M MES pH 6.5 and 32.5% PEG 20 000 and frozen directly in the cryostream. Data collection was carried out using a MAR CCD detector and using synchrotron radiation at a wavelength of 0.8123 Å. The detector was placed at a distance of 165 mm. In total, 195 frames were recorded, each covering 0.7° of rotation. The data were indexed and integrated with DENZO, merged with SCALEPACK (Otwinowski & Minor, 1997 ▶) and converted to structure-factor amplitudes using the CCP4 program TRUNCATE (Collaborative Computational Project, Number 4, 1994 ▶).
3. Results and discussion
BMA purified using affinity chromatography behaves mostly as a dimer (and a small fraction of tetramer) as assessed by analytical gel filtration (Fig. 2 ▶ a). The minimum protein concentration required for hemagglutination was approximately 1 µg ml−1, corresponding to a dilution of 215. The protein shows as two bands on SDS–PAGE (Fig. 2 ▶ b), corresponding to the reported α- and β-chains (Chawla et al., 1993 ▶). Proteolytic processing of a precursor chain is common in the legume lectin family. In addition to removal of the N-terminal signal peptide, several legume lectins have ragged C-terminal ends (Young et al., 1995 ▶). Others consist of tetramers that contain two full-length and two C-terminal truncated subunits (Roberts et al., 1982 ▶; Hamelryck et al., 1999 ▶). Most lectins from members of the Vicieae tribe are proteolytically cleaved into a 6000 kDa α-chain and a 20 000 kDa β-chain (Hemperly & Cunningham, 1983 ▶) at a site distinct from the proteolytic cleavage observed for BMA (Chawla et al., 1993 ▶). The most elaborate post-translational processing described to date is undoubtedly found in concanavalin A, in which first a 15-amino-acid glycopeptide is removed, followed by partial ligation of the original N- and C-termini to produce a circularly permuted protein (Carrington et al., 1985 ▶). Some legume lectins from the Diocleinae subtribe, including concanavalin A, have been found to show a pH-dependent dimer–tetramer equilibrium (Bouckaert et al., 2000 ▶; Wah et al., 2001 ▶). For others, including the seed lectin from Dioclea grandiflora, the quaternary state is not affected over a wide range of pH values. Gel-filtration experiments of BMA at different pH values indicate that the oligomerization state of BMA is not affected by this parameter (data not shown).
Figure 2.

Purification of BMA. (a) Analytical gel-filtration profile of BMA on a Superdex 75-HR column. The weights of the molecular-weight standards are indicated. (b) 12% SDS–PAGE under reducing conditions showing the purified lectin in lane 1 and a molecular-weight marker in lane 2. The molecular weights of the marker proteins are indicated in kDa.
Microcrystals of ligand-free BMA were obtained under three conditions: (i) 0.2 M magnesium acetate, 0.1 M sodium cacodylate pH 6.5, 20% PEG 8000, (ii) 0.2 M ammonium sulfate, 0.1 M sodium acetate pH 4.6, 25% PEG 4000 and (iii) 0.1 M MES pH 6.5, 12% PEG 20 000 (Fig. 3 ▶ a). All three conditions were subjected to optimization by varying temperature, PEG concentration and pH and by the addition of a variety of carbohydrate ligands.
Figure 3.

Crystals of B. mildbraedii agglutinin. (a) Original microcrystals grown from 0.1 M MES pH 6.5, 12% PEG 20 000. (b) Crystals of BMA in the presense of trehalose. (c) Crystals of BMA in the presence of Man(α1-2)Man before optimization. (d) Typical crystals of the BMA–Man(α1-2)Man complex after optimization. The scale bar in each panel corresponds to 0.2 mm.
This eventually led to macrocrystals in condition (iii) when the ligands trehalose or Man(α1-2)Man were added and the crystals were grown at 277 K (Figs. 3 ▶ b and 3 ▶ c). No macrocrystals were obtained in the absence of the sugar nor with any other sugar tried. Trehalose and Man(α1-2)Man cocrystals are different in shape. The trehalose complex formed small oval-shaped crystals (Fig. 3 ▶ b), while the Man(α1-2)Man complex crystallized as needles (Fig. 3 ▶ c). The best crystals for the Man(α1-2)Man co-crystals were obtained using 15% PEG 20 000 and 0.1 M MES pH 6.7. However, nucleation was erratic. Consistent nucleation could be achieved by setting up the crystallization experiments at 277 K and after two weeks transferring them to 293 K. After another two weeks of incubation at 293 K, the large crystals shown in Fig. 3 ▶(d) appeared consistently. These crystals belong to space group I222 or I212121, with unit-cell parameters a = 66.60, b = 86.35, c = 91.76 Å, and diffract to 1.9 Å on beamline X11 of the DESY synchrotron, Hamburg, Germany (Fig. 4 ▶). A full data set from a BMA crystal was collected to a resolution of 1.90 Å. The statistics of the data collections are given in Table 1 ▶. Assuming the presence of one monomer in the asymmetric unit gives a Matthews coefficient of 2.7 Å3 Da−1 (Matthews, 1968 ▶), corresponding to a solvent content of 53.4%. Crystal symmetry can generate a dimer or a tetramer in space group I222, but not in space group I212121.
Figure 4.

Diffraction pattern of BMA–Man(α1-2)Man crystals. The pattern is clean and extends to 1.90 Å resolution. The crystal was exposed at 100 K after soaking in 32.5% PEG 20 000 for cryoprotection. The rotation angle used was 0.7°, the crystal-to-detector distance was 165 mm and the wavelength was 0.8123 Å. The inset shows a detail of the diffraction pattern with spots visible near the edge of the detector.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Detector | MAR CCD |
| Beamline | X11 |
| Data-collection temperature (K) | 100 |
| Wavelength (Å) | 0.8123 |
| Unit-cell parameters (Å) | a = 66.60, b = 86.35, c = 91.76 |
| Space group | I222 |
| Content of the AU | 1 monomer |
| Resolution range (Å) | 15.0–1.90 (1.97–1.90) |
| No. of observed reflections | 99945 (7476) |
| No. of unique reflections | 18493 (1699) |
| Rmerge† | 0.095 (0.310) |
| Completeness (%) | 87.3 (80.9) |
| I/σ(I) | 13.2 (4.1) |
| Redundancy | 5.4 (4.4) |
R
merge = 
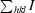 .
.
Crystals of the Man(α1-2)Man complex of a similar shape and size as described above also grow overnight at 277 K when raising the pH of the crystallization buffer to 8.7 (with a corresponding change of buffer to 0.1 M Tris instead of 0.1 M MES). However, these crystals do not diffract. Similarly, crystals grown in the presence of trehalose (pH 6.5) instead of Man(α1-2)Man also do not diffract.
BMA belongs to the Man/Glc-specific group of legume lectins, several members of which have been studied in detail. Concanavalin A and its close relatives are the best studied members both from the viewpoint of structure as well as from that of thermodynamics (Dam & Brewer, 2002 ▶). Concanavalin A is most specific for the trisaccharide Man(α1-3)[Man(α1-6)]Man (Dam et al., 1998 ▶; Naismith & Field, 1996 ▶; Rozwarski et al., 1998 ▶; Williams et al., 1992 ▶) and the pentasaccharide GlcNAc(β1-2)Man(α1-3)[GlcNAc(β1-2)Man(α1-6)]Man (Moothoo & Naismith, 1998 ▶). The lectin from Pterocarpus angolensis binds the same pentasaccharide, but using a different binding mode (our unpublished results). Lectins belonging to the Vicieae tribe such as that from Lathyrus ochrus, on the other hand, require the presence of a fucose attached to the Asn-linked GlcNAc residue of biantennary complex-type glycans for high-affinity binding (Debray et al., 1981 ▶; Kornfeld et al., 1981 ▶; Bourne et al., 1994 ▶). Other Man/Glc-binding legume lectins such as FRIL do not seem to possess any additional subsites flanking the monosaccharide-binding site (Hamelryck et al., 2000 ▶; Mo et al., 1999 ▶). BMA, which is most specific for Man(α1-2)Man, thus belongs to yet another subgroup. It will therefore be of interest to learn how oligosaccharide specificity is achieved by this lectin. In addition, the quaternary structure of the protein will be of interest. Currently, four different types of dimers (Audette et al., 2000 ▶; Delbaere et al., 1993 ▶; Einspahr et al., 1986 ▶; Hamelryck et al., 1999 ▶; Loris et al., 2004 ▶; Manoj et al., 2000 ▶; Prabu et al., 1998 ▶; Shaanan et al., 1991 ▶) and four different types of tetramers (Banerjee et al., 1994 ▶; Dessen et al., 1995 ▶; Hardman & Ainsworth, 1972 ▶; Edelman et al., 1972 ▶; Hamelryck et al., 1996 ▶; Lescar et al., 2002 ▶; Tempel et al., 2002 ▶) have been identified. The quaternary associations add an extra layer of specificity to the recognition of multivalent carbohydrates (Brewer et al., 2002 ▶; Sacchettini et al., 2001 ▶) and are governed by amino-acid substitutions at the lectin surface (Brinda et al., 2004 ▶; Srinivas et al., 2001 ▶) but not by differential glycosylation (Kulkarni et al., 2004 ▶) as was once assumed (Shaanan et al., 1991 ▶).
Acknowledgments
We thank Theresa Animashaun for kindly providing seeds of B. milbraedii. The authors acknowledge the use of synchrotron beamtime at the EMBL beamlines at the DORIS storage ring (Hamburg, Germany) and the ESRF (Grenoble, France). This work was funded by the Vlaams Interuniversitair Instituut voor Biotechnologie (VIB), the Onderzoeksfonds of the Vrije Universiteit Brussel (OZR) and the Fonds voor Wetenschappelijk Onderzoek Vlaanderen (FWO).
References
- Animashaun, T. & Hughes, R. C. (1989). J. Biol. Chem.264, 4657–4663. [PubMed] [Google Scholar]
- Audette, G. F., Vandonselaar, M. & Delbaere, L. T. (2000). J. Mol. Biol.304, 423–433. [DOI] [PubMed] [Google Scholar]
- Banerjee, R., Mande, S. C., Ganesh, V., Das, K., Dhanaraj, V., Mahanta, S. K., Suguna, K., Surolia, A. & Vijayan, M. (1994). Proc. Natl Acad. Sci. USA, 91, 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert, J., Loris, R. & Wyns, L. (2000). Acta Cryst. D56, 1569–1576. [DOI] [PubMed] [Google Scholar]
- Bourne, Y., Mazurier, J., Legrand, D., Rouge, P., Montreuil, J., Spik, G. & Cambillau, C. (1994). Structure, 2, 209–219. [DOI] [PubMed] [Google Scholar]
- Brewer, C. F., Miceli, M. C. & Baum, L. G. (2002). Curr. Opin. Struct. Biol.12, 616–623. [DOI] [PubMed] [Google Scholar]
- Brinda, K. V., Mitra, N., Surolia, A. & Vishveshwara, S. (2004). Protein Sci.13, 1735–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington, D. M., Auffret, A. & Hanke, D. E. (1985). Nature (London), 313, 64–67. [DOI] [PubMed] [Google Scholar]
- Chawla, D., Animashaun, T. & Hughes, R. C. (1992). FEBS Lett.298, 291–196. [DOI] [PubMed] [Google Scholar]
- Chawla, D., Animashaun, T., Hughes, R. C., Harris, A. & Aitken, A. (1993). Biochim. Biophys. Acta, 1202, 38–46. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Dam, T. K. & Brewer, C. F. (2002). Chem. Rev.102, 387–429. [DOI] [PubMed] [Google Scholar]
- Dam, T. K., Cavada, B. S., Grangeiro, T. B., Santos, C. F., de Sousa, F. A., Oscarson, S. & Brewer, C. F. (1998). J. Biol. Chem.273, 12082–12088. [DOI] [PubMed] [Google Scholar]
- Debray, H., Decout, D., Strecker, G., Spik, G. & Montreuil, J. (1981). Eur. J. Biochem.117, 41–55. [DOI] [PubMed] [Google Scholar]
- Delbaere, L. T., Vandonselaar, M., Prasad, L., Quail, J. W., Wilson, K. S. & Dauter, Z. (1993). J. Mol. Biol.230, 950–965. [DOI] [PubMed] [Google Scholar]
- Dessen, A., Gupta, D., Sabesan, S., Brewer, C. F. & Sacchettini, J. C. (1995). Biochemistry, 34, 4933–4942. [DOI] [PubMed] [Google Scholar]
- Edelman, G. M., Cunningham, B. A., Reeke, G. N. Jr, Becker, J. W., Waxdal, M. J. & Wang, J. L. (1972). Proc. Natl Acad. Sci. USA, 69, 2580–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einspahr, H., Parks, E. H., Suguna, K., Subramanian, E. & Suddath, F. L. (1986). J. Biol. Chem.261, 16518–16527. [PubMed] [Google Scholar]
- Hamelryck, T. W., Dao-Thi, M.-H., Poortmans, F., Chrispeels, M. J., Wyns, L. & Loris, R. (1996). J. Biol. Chem.271, 20479–20485. [DOI] [PubMed] [Google Scholar]
- Hamelryck, T. W., Loris, R., Bouckaert, J., Dao-Thi, M.-H., Strecker, G., Imberty, A., Fernandez, E., Wyns, L. & Etzler, M. E. (1999). J. Mol. Biol.286, 1161–1177. [DOI] [PubMed] [Google Scholar]
- Hamelryck, T. W., Moore, J. G., Chrispeels, M. J., Loris, R. & Wyns, L. (2000). J. Mol. Biol.299, 875–883. [DOI] [PubMed] [Google Scholar]
- Hardman, K. D. & Ainsworth, C. F. (1972). Biochemistry, 11, 4910–4919. [DOI] [PubMed] [Google Scholar]
- Hemperly, J. J. & Cunningham, B. A. (1983). Trends Biochem. Sci.8, 100–102.
- Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst.24, 409–411. [Google Scholar]
- Kornfeld, K., Teirman, M. L. & Kornfeld, R. (1981). J. Biol. Chem.256, 6633–6640. [PubMed] [Google Scholar]
- Kulkarni, K. A., Srinivasta, A., Mitra, N., Surolia, A., Vijayan, M. & Suguna, K. (2004). Proteins, 56, 821–827. [DOI] [PubMed] [Google Scholar]
- Lescar, J., Loris, R., Mitchell, E., Gautier, C., Chazalet, V., Cox, V., Wyns, L., Perez, S., Breton, C. & Imberty, A. (2002). J. Biol. Chem.277, 6608–6614. [DOI] [PubMed] [Google Scholar]
- Loris, R., Hamelryck, T., Bouckaert, J. & Wyns, L. (1998). Biochim. Biophys. Acta, 1383, 9–36. [DOI] [PubMed] [Google Scholar]
- Loris, R., Van Walle, I., De Greve, H., Beeckmans, S., Deboeck, F., Wyns, L. & Bouckaert, J. (2004). J. Mol. Biol.335, 1227–1240. [DOI] [PubMed] [Google Scholar]
- Manoj, N., Srinivas, V. R., Surolia, A., Vijayan, M. & Suguna, K. (2000). J. Mol. Biol.302, 1129–1137. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Mo, H., Meah, Y., Moore, J. G. & Goldstein, I. J. (1999). Glycobiology, 9, 173–179. [DOI] [PubMed] [Google Scholar]
- Moothoo, D. N. & Naismith, J. H. (1998). Glycobiology, 8, 173–181. [DOI] [PubMed] [Google Scholar]
- Naismith, J. H. & Field, R. A. (1996). J. Biol. Chem.271, 972–976. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Peumans, W. J. & Van Damme, E. J. M. (1995). Plant Physiol.109, 347–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabu, M. M., Sankaranaraynan, R., Puri, K. D., Sharma, V., Surolia, A., Vijayan, M. & Suguna, K. (1998). J. Mol. Biol.276, 787–796. [DOI] [PubMed] [Google Scholar]
- Roberts, D. D., Etzler, M. E. & Goldstein, I. J. (1982). J. Biol. Chem.257, 9198–9204. [PubMed] [Google Scholar]
- Rozwarski, D. A., Swami, B. M., Brewer, C. F. & Sacchettini, J. C. (1998). J. Biol. Chem.273, 32818–32825. [DOI] [PubMed] [Google Scholar]
- Sacchettini, J. C., Baum, L. G. & Brewer, C. F. (2001). Biochemistry, 40, 3009–3015. [DOI] [PubMed] [Google Scholar]
- Shaanan, B., Lis, H. & Sharon, N. (1991). Science, 254, 862–866. [DOI] [PubMed] [Google Scholar]
- Sharma, V. & Surolia, A. (1997). J. Mol. Biol.267, 433–445. [DOI] [PubMed] [Google Scholar]
- Sharon, N. & Lis, H. (1990). FASEB J.4, 3198–3208. [DOI] [PubMed] [Google Scholar]
- Srinivas, V. R., Reddy, G. B., Ahmad, N., Swaminathan, C. P., Mitra, N. & Surolia, A. (2001). Biochim. Biophys. Acta, 1527, 102–111. [DOI] [PubMed] [Google Scholar]
- Taylor, M. E. & Drickamer, K. (2003). Introduction to Glycobiology. Oxford University Press.
- Tempel, W., Tschampel, S. & Woods, R. J. (2002). J. Biol. Chem.277, 6615–6621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). Nucleic Acids Res.22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wah, D. A., Romero, A., Gallego del Sol, F., Cavada, B. S., Ramos, M. V., Grangeiro, T. B., Sampaio, A. H. & Calvete, J. J. (2001). J. Mol. Biol.310, 885–894. [DOI] [PubMed] [Google Scholar]
- Williams, B. A., Chervenak, M. C. & Toone, E. J. (1992). J. Biol. Chem.267, 22907–22911. [PubMed] [Google Scholar]
- Young, N. M. & Oomen, R. P. (1992). J. Mol. Biol.228, 924–934. [DOI] [PubMed] [Google Scholar]
- Young, N. M., Watson, D. C., Yaguchi, M., Adar, R., Arango, R., Rodriguez-Arango, E., Sharon, N., Blay, P. K. & Thibault, P. (1995). J. Biol. Chem.270, 2563–2570. [DOI] [PubMed] [Google Scholar]