

Three structures of a putative RNA 5-methyluridine methyltransferase from T. thermophilus, including its complex with S-adenosyl-l-homocysteine, are presented. The structures reveal the mode of cofactor binding, architecture of the putative active site, and the presence of a deep cleft adjacent to the active site that may bind RNA.
Keywords: PUA domain, RNA-modification enzyme, 5-methyluridine methyltransferase, S-adenosyl-l-homocysteine
Abstract
The Thermus thermophilus hypothetical protein TTHA1280 belongs to a family of predicted S-adenosyl-l-methionine (AdoMet) dependent RNA methyltransferases (MTases) present in many bacterial and archaeal species. Inspection of amino-acid sequence motifs common to class I Rossmann-fold-like MTases suggested a specific role as an RNA 5-methyluridine MTase. Selenomethionine (SeMet) labelled and native versions of the protein were expressed, purified and crystallized. Two crystal forms of the SeMet-labelled apoprotein were obtained: SeMet-ApoI and SeMet-ApoII. Cocrystallization of the native protein with S-adenosyl-l-homocysteine (AdoHcy) yielded a third crystal form, Native-AdoHcy. The SeMet-ApoI structure was solved by the multiple anomalous dispersion method and refined at 2.55 Å resolution. The SeMet-ApoII and Native-AdoHcy structures were solved by molecular replacement and refined at 1.80 and 2.60 Å, respectively. TTHA1280 formed a homodimer in the crystals and in solution. Each subunit folds into a three-domain structure composed of a small N-terminal PUA domain, a central α/β-domain and a C-terminal Rossmann-fold-like MTase domain. The three domains form an overall clamp-like shape, with the putative active site facing a deep cleft. The architecture of the active site is consistent with specific recognition of uridine and catalysis of methyl transfer to the 5-carbon position. The cleft is suitable in size and charge distribution for binding single-stranded RNA.
1. Introduction
Post-transcriptional modification of RNA nucleotides occurs in all three kingdoms of life. Approximately 100 distinct types of modifications have been identified to date (Rozenski et al., 1999 ▶; http://medstat.med.utah.edu/RNAmods). Modified nucleotides are most prevalent in tRNA and rRNA, but are also present in various other cellular RNAs. Although the knowledge of modifications that occur is considerable, less is known about the biological function of the modified nucleotides and the enzymes that catalyze the modifications. One common type of nucleotide modification is methylation, which can occur at O, N or C atoms. In a recent bioinformatics survey of proteins involved in RNA metabolism, Anantharaman et al. (2002 ▶) identified a family of proteins conserved in the archaeal and bacterial kingdoms that are predicted to be RNA methyltransferases (MTases). Proteins of this family, as typified by the hypothetical protein MJ1653 from Methanococcus jannaschii, are characterized by an N-terminal RNA-binding PUA domain and a C-terminal S-adenosyl-l-methionine (AdoMet) dependent MTase catalytic domain of the class I Rossmann-fold-like type.
The Thermus thermophilus genome encodes a hypothetical protein, designated TTHA1280, which is a member of the MJ1653 family of proteins. Proteins of this family share several regions of conserved residues, many of which correspond to sequence motifs characteristic of the Rossmann-fold-like MTases (Fig. 1 ▶). The Rossmann-fold-like MTase domain is composed of a central seven-stranded β-sheet (strand order 3-2-1-4-5-7-6) surrounded by three α-helices on each side (reviewed in Fauman et al., 1999 ▶; Schubert et al., 2003 ▶). Motifs I and II define the cofactor-binding site and are located at the C-terminal ends of the first and second strands, respectively. Motifs IV and VI are located at the C-terminal ends of the fourth and fifth strands, respectively, and typically contain residues involved in catalysis and substrate recognition. The MJ1653 family of proteins contain the conserved sequence DPPXF, where X is any amino acid, in motif IV and the conserved sequence SCS in motif VI (Fig. 1 ▶). Nucleic acid MTases that catalyze methyl transfer to the 5-carbon position of a pyrimidine base utilize a nucleophilic cysteine residue in their catalytic mechanism (Kealey et al., 1994 ▶). Recent biochemical and structural studies have defined specific cysteine residues within motifs IV and VI that distinguish DNA 5-methylcytidine (m5C), RNA m5C and RNA 5-methyluridine (m5U) MTases (Liu & Santi, 2000 ▶; Foster et al., 2003 ▶; Lee et al., 2004 ▶). DNA m5C MTases utilize an invariant cysteine residue in motif IV as the catalytic nucleophile, whereas RNA m5C and m5U MTases utilize an invariant cysteine residue in motif VI. RNA m5C MTases also contain a second conserved cysteine residue in motif IV, but its precise role in the catalytic mechanism is unclear. The presence of a conserved cysteine in motif VI (Cys326 in TTHA1280) and the absence of a cysteine in motif IV suggest that the MJ1653 family of proteins are RNA m5U MTases.
Figure 1.

Amino-acid sequence alignment of selected MJ1653 family proteins from bacterial and archaeal species. The secondary-structure elements of TTHA1280 are shown above the sequence. Identical residues are highlighted with a red background and similar residues are shown in red text with a white background. The putative catalytic cysteine residue is indicated with a black star, residues that form hydrogen bonds or hydrophobic contacts with the AdoHcy ligand are indicated with blue triangles and residues with possible roles in substrate base recognition (as described in text) are indicated with green triangles. The alignment was performed with CLUSTALW (Thompson et al., 1994 ▶) and the figure was generated with ESPript (Gouet et al., 1999 ▶). The bacterial species are Thermus thermophilus (TTHA), Escherichia coli (EC) and Aquifex aeolicus (AQ). The archaeal species are Thermococcus kodakaraensis (TK) and Methanococcus jannaschii (MJ).
The catalytic mechanism of RNA m5U MTases is well characterized (Kealey et al., 1994 ▶; Lee et al., 2005 ▶; Fig. 2 ▶). The motif VI cysteine thiolate of the enzyme attacks the C6 position of the base, forming a covalent Michael adduct and activating C5 for electrophilic substitution. The methyl group of AdoMet is transferred to C5 with inversion of configuration and trans to the Cys—S—C6 thioether linkage. General base-mediated abstraction of the proton at C5 allows β-elimination of the enzyme and release of the methylated base.
Figure 2.

Catalytic mechanism of AdoMet-dependent RNA m5U MTases (adapted from Kealey et al., 1994 ▶).
What are possible RNA substrates for a family of predicted RNA m5U MTases? m5U modifications have been identified in tRNA of archaea and bacteria and in 23S rRNA and tmRNA of bacteria (Rozenski et al., 1999 ▶; http://medstat.med.utah.edu/RNAmods). The proteins responsible for three of the four known m5U modifications in Escherichia coli, at position U54 of tRNAs and positions U747 and U1939 of 23S rRNA, have been identified. It has been suggested that methylation of U341 in tmRNA might be catalyzed by the same enzyme that methylates U54 of tRNAs (Felden et al., 1998 ▶). It seems plausible that additional m5U modifications remain to be characterized.
Here, we present the crystal structures of the TTHA1280 apoprotein and its complex with S-adenosyl-l-homocysteine (AdoHcy) and compare the structures with the recently determined structures of E. coli RumA, a 23S rRNA m5U MTase (Lee et al., 2004 ▶, 2005 ▶). We show that TTHA1280 and RumA share similar active-site architectures and we suggest that TTHA1280 probably also catalyzes methyl transfer to the 5-carbon position of uridine. We suggest residues that could position uridine at the active site and we show that each subunit of the dimer contains a deep cleft adjacent to the active site that appears to be well suited for binding single-stranded RNA.
2. Materials and methods
2.1. Cloning, expression, purification and analytical ultracentrifugation
The gene encoding TTHA1280 was PCR amplified from T. thermophilus HB8 genomic DNA and cloned into the pET11a expression vector (Novagen). The native protein was expressed in E. coli strain Rosetta (DE3). TTHA1280 was purified by denaturation of heat-labile E. coli proteins at 343 K for 15 min, followed by a series of column chromatography steps using an AKTA Explorer 10S system (Amersham Bioscience). The course of the purification was monitored by SDS–PAGE. The column steps consisted of HiTrap Q HP, Resource ISO, HiTrap Heparin, Superdex 75HR and a final HiTrap Heparin step. The selenomethionine (SeMet) substituted TTHA1280 was expressed in E. coli strain B834 (DE3) and purified in the same manner. Sedimentation-equilibrium analytical ultracentrifugation experiments were carried out as previously described (Handa et al., 2003 ▶) using a Beckman Coulter Proteome Lab XL-1 protein characterization system.
2.2. Crystallization
The purified proteins were concentrated to ∼8.8 mg ml−1 in storage buffer containing 20 mM Tris–HCl pH 8.0, 150 mM NaCl and 1 mM DTT. Hanging drops containing 1–2 µl of the protein in storage buffer were mixed with an equal volume of crystallization reagent and equilibrated over 0.5 ml of reservoir solution at 293 K. Two initial crystallization conditions were identified for the SeMet-labelled protein using the Index Screen from Hampton Research (reagent Nos. 18 and 58). The initial conditions were refined by optimizing the reagent composition and screening additives. The final reservoir solution for crystal form I contained 35% polypropylene glycol (PPG) P400, 0.1 M sodium acetate pH 5.2, 3% 1,6-hexanediol and 10 mM l-cysteine. The final reservoir solution for crystal form II contained 1 M sodium potassium phosphate pH 7.0–7.6 and 3% sucrose. Plate-shaped crystals appeared within 1 d for crystal form I and within 3 d for crystal form II. Microseeding permitted single three-dimensional crystals free of visual defects to be obtained within 1–2 d after seeding for both forms. Crystal form III was obtained by cocrystallizing the native protein with S-adenosyl-l-homocysteine (AdoHcy; Sigma). AdoHcy was added to the native protein in storage buffer to a final concentration of 1 mM before setting up hanging drops with the form I reservoir solution. The space group, unit-cell parameters and diffraction limit for the three crystal forms are listed in Table 1 ▶. Hereafter, the three crystal forms will be referred to as SeMet-ApoI for crystal form I, SeMet-ApoII for crystal form II and Native-AdoHcy for crystal form III.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution bin. NA, not applicable.
| Crystal form | SeMet-ApoI | SeMet-ApoII | Native-AdoHcy | ||
|---|---|---|---|---|---|
| Data collection | |||||
| Space group | C2 | P1 | C2 | ||
| Unit-cell parameters | |||||
| a () | 252.7 | 77.6 | 137.8 | ||
| b () | 46.0 | 82.3 | 46.1 | ||
| c () | 139.6 | 79.6 | 134.1 | ||
| () | 90 | 96.3 | 90 | ||
| () | 100.1 | 90.1 | 109.9 | ||
| () | 90 | 103.1 | 90 | ||
| Resolution range () | 45.832.55 (2.642.55) | 39.551.80 (1.861.80) | 43.42.60 (2.692.60) | ||
| Wavelength () | 0.9791 (peak) | 0.9794 (edge) | 0.9640 (remote) | 0.9640 | 1.00 |
| No. of observations | 180956 | 144136 | 172049 | 476979 | 91791 |
| Unique reflections† | 97119 | 86580 | 96545 | 328489 | 24665 |
| Completeness (%) | 97.7 (91.5) | 86.1 (80.7) | 96.1 (88.3) | 93.7 (86.5) | 99.7 (99.2) |
| I/(I) | 10.6 (2.5) | 11.3 (2.1) | 11.8 (2.3) | 7.4 (2.4) | 22.1 (3.0) |
| R merge ‡ (%) | 4.8 (25.3) | 4.6 (29.7) | 4.8 (26.8) | 8.3 (22.6) | 5.1 (34.8) |
| MAD phasing | |||||
| FOM after SOLVE | 0.32 | NA | NA | ||
| FOM after RESOLVE | 0.61 | NA | NA | ||
| Refinement | |||||
| Resolution range | 45.832.55 | 39.551.80 | 43.42.60 | ||
| No. of reflections (working/test) | 88648/4493 (remote data) | 297649/15049 | 22605/1146 | ||
| R cryst § (%)/R free ¶ (%) | 22.3/27.9 | 20.3/22.9 | 25.8/29.7 | ||
| B value from Wilson plot (2) | 40.2 | 11.0 | 59.3 | ||
| Mean B value (2) | 53.8 | 20.4 | 74.9 | ||
| Protein molecules per ASU | 4 | 4 | 2 | ||
| No. of protein atoms | 12139 | 12172 | 6041 | ||
| No. of water atoms | 38 | 1105 | 32 | ||
| No. of heterogen atoms | 8 (1 hexane-1,6-diol) | 44 (8 PO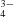 , 4K+) , 4K+) |
62 (2AdoHcy, 1glycerol, 1 acetate) | ||
| R.m.s bond-length deviation () | 0.007 | 0.005 | 0.008 | ||
| R.m.s. bond-angle deviation () | 1.30 | 1.40 | 1.30 | ||
| Ramachandran plot, residues in†† (%) | |||||
| Most favored | 86.6 | 89.8 | 82.2 | ||
| Additional allowed | 12.8 | 9.6 | 16.3 | ||
| Generously allowed | 0.6 | 0.6 | 1.5 | ||
| Disallowed | 0 | 0 | 0 | ||
Values are before merging of Friedel pairs for SeMet-ApoI and SeMet-ApoII.
R
merge = 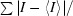
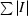 , where I is the intensity measurement for a given reflection and I is the average intensity for multiple measurements of the reflection.
, where I is the intensity measurement for a given reflection and I is the average intensity for multiple measurements of the reflection.
R
cryst = 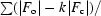
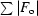 .
.
R free was calculated using a randomly selected 5% subset of the total reflections that was omitted from the refinement.
As defined in PROCHECK (Laskowski et al., 1993 ▶).
2.3. Data collection and processing
SeMet-ApoI and Native-AdoHcy crystals were transferred directly into cryoprotectant composed of 28% PPG P400, 4% glycerol, 0.1 M sodium acetate pH 5.2, 3% 1,6-hexanediol and 1 mM l-cysteine and flash-frozen in a gaseous liquid-nitrogen stream. SeMet-ApoII crystals were dialyzed overnight into cryoprotectant consisting of 2.6 M sodium potassium phosphate pH 7.6 and 10.5% sucrose and frozen as above. Diffraction data were collected at beamline BL26B1 of the SPring-8 synchrotron facility (Hyogo, Japan) using a Jupiter CCD detector. A three-wavelength MAD data set (peak, edge, high remote) was collected from a single SeMet-ApoI crystal and single-wavelength data sets were collected from single SeMet-ApoII and Native-AdoHcy crystals. The data sets were indexed, integrated and scaled with the HKL2000 package (Otwinowski & Minor, 1997 ▶). The data-collection statistics are listed in Table 1 ▶.
2.4. Structure determination and refinement
The SeMet-ApoI structure was solved by the MAD method (Hendrickson & Ogata, 1997 ▶). 15 of 16 expected selenium positions were automatically located and refined using the SOLVE package (Terwilliger & Berendzen, 1999 ▶). The Se substructure and phases from SOLVE were input into RESOLVE for automated density modification (Terwilliger, 2000 ▶) and model building (Terwilliger, 2003 ▶). The RESOLVE model-building feature built 77% of the total residues (43% with side chains). The model was completed by iterative cycles of manual model building in O (Jones et al., 1991 ▶) and refinement against the remote data set in CNS v.1.1 (Brünger et al., 1998 ▶) with simulated annealing, energy minimization and grouped B-factor refinement. NCS restraints were applied to the main chain of the four molecules in the asymmetric unit (ASU), except for regions where the molecules differed.
The SeMet-ApoII and Native-AdoHcy structures were solved by molecular replacement (MR). The solution for the SeMet-ApoII crystal form was obtained with MOLREP v.7.3 (Vagin & Teplyakov, 1997 ▶) and utilized a protomer from the SeMet-ApoI structure as the search model. The anomalous scattering phases were not included. The solution for the Native-AdoHcy crystal form was obtained with CNS v.1.1 (Brünger et al., 1998 ▶) and utilized a protomer from the SeMet-ApoII structure as the search model. Rigid-body refinement of the SeMet-ApoII MR solution in CNS yielded an R free of 42.5%, while the Native-AdoHcy MR solution yielded an R free of 38.2%. F o − F c electron-density maps calculated with the Native-AdoHcy MR phases and contoured at 3σ revealed unambiguous density for two AdoHcy molecules, one bound to each of the two protein molecules in the ASU. The SeMet-ApoII and Native-AdoHcy models were completed by iterative cycles of manual rebuilding in O and refinement in CNS using simulated annealing, energy minimization and restrained individual B-factor refinement. NCS restraints were initially applied on the main chain of the four molecules in the ASU of the SeMet-ApoII structure, but were removed in the final stages of refinement. For the Native-AdoHcy structure, NCS restraints were applied on the two protein molecules, except where they differed, and on the two AdoHcy molecules. Grouped occupancy refinement of the AdoHcy molecules did not yield occupancies significantly different than one nor a lower R free value; therefore, in the final model their occupancies were not refined. The progress of each refinement was monitored by the R free value based on 5% of the reflections. The MAD phasing and refinement statistics are listed in Table 1 ▶.
3. Results and discussion
3.1. Quality of the models
Two structures of the SeMet-labelled TTHA1280 apoprotein in different crystal forms and one structure of the native protein bound to AdoHcy were solved and refined as described in §2. The SeMet-ApoI structure was refined at 2.55 Å resolution to an R factor of 22.3% and an R free of 27.9%. The ASU contained four protein molecules arranged as one AB dimer with non-crystallographic twofold symmetry and single C and D molecules positioned about distinct crystallographic twofold axes such that the CC and DD dimer dyad axes coincide with the crystallographic axis. The final SeMet-ApoI model contains all amino-acid residues (1–382) for each chain, other than residues 189–193 of chain A, which were disordered. The SeMet-ApoII structure was refined at 1.80 Å resolution to an R factor of 20.3% and an R free of 22.9%. The four molecules in the ASU were arranged as two dimers (AB and CD) and the final model contains all amino-acid residues for each chain. The Native-AdoHcy structure was refined at 2.60 Å resolution to an R factor of 25.8% and an R free of 29.7%. The ASU contained two protein molecules, each bound to one AdoHcy molecule, and positioned about a distinct crystallographic twofold axis such that the AA and BB dimer dyad axes coincide with the crystallographic axis. The final model contains all amino-acid residues other than 187–192 of chain A, which were disordered. The stereochemical quality of the models is good as verified by PROCHECK (Laskowski et al., 1993 ▶; Table 1 ▶). The ten distinct subunits from the three crystal forms superimpose with an average Cα r.m.s.d. of 0.66 Å (based on 45 pairwise combinations). In the following sections, we describe the TTHA1280 structure based on the AB dimer of the SeMet-ApoII model and chain A of the Native-AdoHcy model because these subunits exhibit the best geometry and the lowest B factors.
3.2. Overall structure of the TTHA1280 apoprotein
TTHA1280 crystallized as a homodimer in three distinct crystal forms and each form exhibited the same dimer interface. Sedimentation-equilibrium analytical ultracentrifugation experiments indicated a molecular weight of ∼87 kDa in solution, corresponding to the dimer (data not shown). These results strongly suggest that the biological unit of TTHA1280 is the homodimer. Ribbon diagrams of the protomer and dimer structures of the apoprotein are shown in Fig. 3 ▶. The TTHA1280 protomer is composed of three α/β-domains: N-terminal (residues 1–65), central (residues 66–194) and C-terminal (residues 195–382) (Fig. 3 ▶ a). The domains are arranged to give an overall clamp-like shape, with the N-terminal and central domains forming one half of the clamp and the C-terminal domain forming the other half. The most striking feature of the structure is the presence of a deep cleft at the centre of the clamp. The dimensions of a single subunit are approximately 60 × 45 × 30 Å.
Figure 3.

Overall structure of the TTHA1280 apoprotein. (a) Ribbon diagram of a single subunit of the apoprotein presented in stereo with secondary-structure elements labelled. The N-terminal, central and C-terminal domains are colored cyan, lime and yellow, respectively. The putative catalytic Cys326 is shown in stick representation and coloured red. (b) Ribbon diagram of the apoprotein homodimer viewed down the non-crystallographic twofold axis. (c) Molecular surface of a single subunit of the apoprotein coloured by amino-acid sequence conservation. A semi-transparent surface is superimposed on a ribbon diagram oriented as in (a). The strictly conserved residues shown in Fig. 1 ▶ are coloured cyan, the similar residues are coloured green and the unconserved residues are coloured white. The square highlights the putative active site. This and all subsequent figures were generated with PyMOL (DeLano, 2002 ▶).
The N-terminal domain is the smallest of the three domains and consists of a six-stranded mixed β-sheet (strand order 3-1-4-5-6-2) with a short α-helix inserted between strands 1 and 2. A DALI search (Holm & Sander, 1993 ▶) for similar folds in the PDB confirmed that the N-terminal domain is a PUA domain. PUA domains were named based on their presence in pseudouridine synthase (TruB) and archaeosine tRNA guanine transglycosylase (ArcTGT), two enzymes that modify ribonucleotides in tRNA (Aravind & Koonin, 1999 ▶). The PUA domains of TruB and ArcTGT superimpose on the TTHA1280 PUA domain with an r.m.s.d. of their Cα positions of 2.1 and 1.9 Å, respectively.
The central domain consists of three α-helices and two distinct β-sheets, large and small, with their respective strands oriented almost perpendicular to each other. The large mixed β-sheet contains five strands in the order 7-8-9-10-11. Two long kinked α-helices flank one side of the larger sheet, while the other side opens to the central cleft. A short α-helix is inserted between strands 10 and 11. A small antiparallel β-sheet (strands 12 and 13) links the large sheet with the C-terminal domain. The small sheet and the loop after strand 13 form the bottom of the central cleft. A DALI search for similar folds yielded low similarity (Z score 2.8 and Cα r.m.s.d. 3.2 Å) to the central domain of E. coli RumA, a 23S rRNA m5U MTase. However, visual comparison of the two structures indicates that the two central domains are more similar than the DALI Z score suggests (§3.3).
The C-terminal domain of TTHA1280 is the largest of the three domains. It forms the classic class I Rossmann-fold-like MTase catalytic domain that is present in the majority of AdoMet-dependent MTase enzymes (reviewed in Fauman et al., 1999 ▶; Schubert et al., 2003 ▶). A DALI search confirmed the structural similarity of the C-terminal domain to many class I MTase catalytic domains; most notably those of E. coli Fmu, a 16S rRNA m5C MTase (Z score 15.0 and Cα r.m.s.d. 3.0 Å) and E. coli RumA (Z score 13.9 and Cα r.m.s.d. 2.3 Å). Although these domains are structurally similar, their amino-acid sequences do not exhibit significant similarity. The sequence motifs I, II, IV and VI are located close together in the three-dimensional structure at the C-terminal ends of β-strands 14, 15, 17 and 18, respectively. This region faces the central cleft and probably forms the active site, with the putative catalytic nucleophile, Cys326, pointing towards the putative cofactor-binding site.
In the homodimer, the subunits are positioned with the clefts on opposite sides of the dimer, open to the solvent (Fig. 3 ▶ b). All of the subunit interactions involve the surface of the protein on the side opposite the cleft and active site. Presumably, dimer formation is not needed for catalytic competence. Dimerization might function to generate an intersubunit substrate-binding surface large enough to accommodate a complex RNA structure. The subunit interactions form a relatively small contact surface that occludes ∼2600 Å2 of accessible surface area or ∼7.6% of the total. Contacts are made between the two C-terminal domains and between the N-terminal and central domains of one subunit and the C-terminal domain of the opposing subunit. The contacts involve the C-terminus of α1, α10 and β19 and the loops connecting α1 and β2, β7 and β8, and β19 and β20. The dimer interface is predominantly hydrophobic, but hydrogen bonds are also involved. A primary interaction between the two C-terminal domains involves the packing of Leu366 from one subunit into a hydrophobic patch composed of Val339, Ala340, Ala343, Leu350 and Phe379 from the opposing subunit. Similarly, Leu349 packs against a hydrophobic patch in the opposing subunit that is formed at the interface of the three domains by the aliphatic portion of the Arg16 side chain, Leu18, Phe62, Leu104 and Leu365. The main-chain carbonyl of Val364 and the amide of Leu366 form intersubunit hydrogen bonds with the main-chain amide of Val352 and the carbonyl of Leu350, respectively.
The residues involved in dimer formation are not conserved among the proteins shown in Fig. 1 ▶, suggesting that other MJ1653 family members do not necessarily form dimers. The residues that are strictly conserved or similar among the proteins cluster around the putative active site, the bottom and sides of the cleft, the interfaces between the three domains within a subunit or at structurally important positions such as the hydrophobic core of the molecule or turns (Fig. 3 ▶ c).
3.3. Comparison with E. coli RumA
E. coli RumA is an RNA m5U MTase specific for position U1939 of 23S rRNA (Agarwalla et al., 2002 ▶). TTHA1280 and RumA do not exhibit significant amino-acid sequence similarity, although they share certain conserved amino acids in their C-terminal domains; most notably an aspartate and proline in motif IV and a cysteine in motif VI. Nonetheless, they share a similar three-domain structure that includes a small N-terminal RNA-binding domain, similar central domains and a common C-terminal MTase catalytic domain (Fig. 4 ▶). RumA differs in that it is monomeric, it contains a 4Fe–4S cluster and additional secondary-structure elements not present in TTHA1280 and its N-terminal domain forms an all-β OB fold (Lee et al., 2004 ▶). Interestingly, the positions of β7 and α2 are similar in the two central domains, but the overall topology of these domains differs owing to the presence of extra secondary-structure elements in RumA (Fig. 4 ▶). The position of the RumA N-terminal domain with respect to the rest of the molecule is different to that observed in TTHA1280, resulting in a wider shallower RNA-binding groove as opposed to a deep cleft.
Figure 4.

Comparison of TTHA1280 and E. coli RumA. A single TTHA1280 subunit is shown on the left, next to the RumA monomer (PDB code 1uwv). The proteins are positioned with their C-terminal domains (yellow) in the same orientation. The RumA N-terminal domain is coloured red and secondary-structure elements present in RumA, but not in TTHA1280, are coloured grey. The portion of the RumA central domain that is equivalent to the TTHA1280 central domain is colored lime for elements that exhibit the same topology as in TTHA1280 and pink for elements with topology different to that in TTHA1280. The RumA 4Fe–4S cluster is shown in stick representation. Selected secondary-structure elements are labelled as in Fig. 3 ▶(a) for TTHA1280 or as in Lee et al. (2004 ▶) for RumA. The squares highlight the putative (TTHA1280) and experimentally confirmed (RumA) active sites.
3.4. Cofactor binding and architecture of the putative active site
The native TTHA1280 protein was cocrystallized with the byproduct of the MTase reaction, S-adenosyl-l-homocysteine (AdoHcy), and the structure was determined at 2.60 Å resolution. Despite the moderate resolution, the electron density for the AdoHcy molecules was unambiguous. Only negligible differences in some side-chain conformations were observed between the apoprotein and ligand-bound structures. AdoHcy binds at the C-terminal end of β14, in an orientation similar to that observed in other class I AdoMet-dependent MTase enzymes (reviewed in Schubert et al., 2003 ▶; Fig. 5 ▶ a). Conserved residues in motifs I, II and IV form most of the binding site. Phe217 makes an edge-to-face hydrophobic stacking interaction with the adenine base moiety. Asn265 Oδ1 accepts a hydrogen bond from N6 of the adenine ring and the carboxylate of Asp238 accepts two hydrogen bonds from the 2′- and 3′-hydroxyl groups of the ribose sugar moiety. The amino group of AdoHcy is a hydrogen-bond donor to Asp286 Oδ2 and the main-chain carbonyl of Phe217. The carboxylate of AdoHcy accepts a hydrogen bond from the guanidino group of Arg202. The positive and negative charges of the AdoHcy amino and carboxylate groups are neutralized by their interactions with Asp286 and Arg202, respectively.
Figure 5.

AdoHcy binding and comparison of the TTHA1280 and RumA active sites. (a) Stereoview of the putative TTHA1280 active site from the 2.6 Å resolution Native-AdoHcy structure. The protein is shown as a ribbon diagram with selected side chains and the AdoHcy ligand displayed as sticks. Atoms are coloured red for oxygen, blue for nitrogen, orange for sulfur, yellow for protein C atoms and green for AdoHcy C atoms. The red dashes indicate hydrogen bonds between the protein and AdoHcy. The simulated-annealing composite omit 2F o − F c electron-density map is shown as a blue mesh contoured at 1σ and displayed within 2 Å of the ligand. (b) Stereoview of the RumA active site from the RumA–RNA–AdoHcy ternary complex (PDB code 2bh2). For clarity, only the uracil base of the RNA substrate is shown. The base is methylated at the C5 position and has an F atom in place of the C5 hydrogen, preventing β-elimination of the enzyme (Lee et al., 2005 ▶). Uracil C atoms are coloured cyan and the F atom is coloured magenta.
The architecture of the TTHA1280 active site is similar to that of the RumA active site in many respects, although notable differences exist. The position of the motif VI cysteine (Cys326 in TTHA1280; Cys389 in RumA) in relation to the AdoHcy cofactor is very similar in the two proteins and both proteins use the motif IV aspartate (Asp286 in TTHA1280; Asp363 in RumA) to help position the cofactor (Figs. 5 ▶ a and 5 ▶ b; Lee et al., 2005 ▶). RumA orients the target uridine in the active site by forming hydrogen bonds between the Gln265 side chain and N3 and O4 of the base (Fig. 5 ▶ b). Although there is no Gln265 equivalent in TTHA1280, Asp197 and Gln198, two residues conserved among the MJ1653 family of proteins, are good candidates for residues that could orient uridine in the active site through hydrogen bonds with N3 and O4 of the base. The most notable difference between the two active sites concerns the general base. In RumA, Glu424 abstracts the proton from C5 after methyl transfer occurs (Fig. 5 ▶ b; Lee et al., 2005 ▶). The equivalent position in TTHA1280 is occupied by a leucine (Leu374) that is conserved among the MJ1653 family of proteins (Fig. 1 ▶). Another residue, perhaps Asp286, or an activated water molecule might provide the necessary general base function for TTHA1280. Lastly, product release is thought to be facilitated by the steric clash that would occur between the methyl group of the product and the backbone carbonyl of a conserved proline residue in motif IV (Pro364 in RumA; Lee et al., 2005 ▶). The position of TTHA1280 Pro287 is consistent with such a mechanism (Fig. 5 ▶ a). While the above analysis does not prove that TTHA1280 catalyzes methyl transfer to the 5-carbon position of uridine, the similar active-site architectures of TTHA1280 and RumA suggests that they share a core catalytic function.
3.5. The central cleft and RNA binding
The cleft varies in width from approximately 11 to 18 Å and is roughly 15 Å deep as viewed in Figs. 3 ▶(a) and 3 ▶(c). The inner edges of the cleft exhibit a predominantly positive electrostatic surface potential. In the dimer, this positively charged surface extends upward from the cleft to include the PUA domain, the N-terminus of α9 and a portion of the C-terminal domain from the opposing subunit (Fig. 6 ▶). In contrast, the opposite side of the dimer contains a relatively even distribution of positive and negative regions. The presence of two large continuous stretches of positively charged surface suggests that the dimer could accommodate the simultaneous binding of two RNA substrates that each make contacts with the cleft and PUA domain of one subunit and the C-terminal domain of the opposing subunit. The dimensions, charge distribution and shape of the cleft appear well suited for binding single-stranded RNA, presumably through recognition of the sugar–phosphate backbone.
Figure 6.

Electrostatic surface potential of the TTHA1280 apoprotein homodimer. The left image shows the same view as in Fig. 3 ▶(b). Positively charged regions are coloured blue, negatively charged regions red and neutral regions white. The colour ramp is from −8 to +8kT, where k is the Boltzmann constant and T is the absolute temperature. The surface potential was calculated with APBS (Baker et al., 2001 ▶) assuming a solvent of 150 mM NaCl. The molecular surface was generated with PyMOL (DeLano, 2002 ▶).
Recent structural studies of RNA-modification enzymes in complex with tRNA and rRNA substrates have revealed a trend of major rearrangements in RNA structure occurring upon binding the enzyme (Ishitani et al., 2003 ▶; Lee et al., 2005 ▶). This highlights the difficulty of predicting a specific RNA substrate based on the size and shape of the cleft. The presence of the PUA domain makes it tempting to speculate that the substrate is a tRNA molecule; however, relatively few PUA domains have been experimentally characterized. PUA domains may also bind RNAs other than tRNA. The ArcTGT PUA domain binds to the acceptor stem of tRNA to help position the target nucleotide in the tRNA D-arm at the active site of the catalytic domain, which is located far away from the PUA domain (Ishitani et al., 2003 ▶). The TTHA1280 PUA domain may play a similar role in positioning the RNA substrate in relation to the cleft and active site. The PUA domains are the least conserved regions within the MJ1653 family (Fig. 1 ▶), suggesting that the specific RNA-binding mechanism may differ among members of the family, although they may all bind the same class of RNA, whether it be tRNA, rRNA or another RNA.
4. Conclusions
The MJ1653 family of proteins is widespread in the archaeal and bacterial kingdoms. Such a broad evolutionary distribution indicates an important role for this family of proteins in RNA metabolism. The structures of TTHA1280 presented here support the amino-acid sequence-based predictions that the MJ1653 family of proteins catalyze methyl transfer to the 5-carbon position of uridine. Specifically, the structures provide a detailed view of cofactor binding and the putative active site and suggest that the motif VI Cys326 is a catalytic nucleophile and that Asp197 and Gln198 might be involved in base recognition. Genetic and biochemical experiments aimed at proving the catalytic activity and mechanism and determining the specific RNA substrates of the MJ1653 family of proteins will benefit from the foundation laid here by the TTHA1280 crystal structures.
Supplementary Material
PDB reference: SeMet-ApoI, 1wxw, r1wxwsf
PDB reference: SeMet-ApoII, 1wxx, r1wxxsf
PDB reference: Native-AdoHcy, 2cww, r2cwwsf
Acknowledgments
We thank Hongfei Wang for collecting the Native-AdoHcy data set and Mutsuko Kukimoto-Niino and Ryogo Akasaka for conducting the analytical ultracentrifugation experiments. We also thank Masami Nishida, Ayako Tatsuguchi and Takaho Terada for protein preparation. This work was supported by the RIKEN Structural Genomics/Proteomics Initiative (RSGI), the National Project on Protein Structural and Functional Analysis, Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- Agarwalla, S., Kealey, J. T., Santi, D. V. & Stroud, R. M. (2002). J. Biol. Chem. 277, 8835–8840. [DOI] [PubMed] [Google Scholar]
- Anantharaman, V., Koonin, E. V. & Aravind, L. (2002). Nucleic Acids Res. 30, 1427–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind, L. & Koonin, E. V. (1999). J. Mol. Evol. 48, 291–302. [DOI] [PubMed] [Google Scholar]
- Baker, N. A., Sept, D., Joseph, S., Holst, M. J. & McCammon, J. A. (2001). Proc. Natl Acad. Sci. USA, 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J.-S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T. & Warren, G. L. (1998). Acta Cryst. D54, 905–921. [DOI] [PubMed] [Google Scholar]
- DeLano, W. L. (2002). The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA.
- Fauman, E. B., Blumenthal, R. M. & Cheng, X. (1999). S-Adenosylmethionine-Dependent Methyltransferases: Structures and Functions, edited by X. Cheng & R. M. Blumenthal, pp. 1–38. Singapore: World Scientific Publishing.
- Felden, B., Hanawa, K., Atkins, J. F., Himeno, H., Muto, A., Gesteland, R. F., McCloskey, J. A. & Crain, P. F. (1998). EMBO J. 17, 3188–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster, P. G., Nunes, C. R., Greene, P., Moustakas, D. & Stroud, R. M. (2003). Structure, 11, 1609–1620. [DOI] [PubMed] [Google Scholar]
- Gouet, P., Courcelle, E., Stuart, D. I. & Metoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed] [Google Scholar]
- Handa, N., Terada, T., Kamewari, Y., Hamana, H., Tame, J. R. H., Park, S.-Y., Kinoshita, K., Ota, M., Nakamura, H., Kuramitsu, S., Shirouzu, M. & Yokoyama, S. (2003). Protein Sci. 12, 1621–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrickson, W. A. & Ogata, C. M. (1997). Methods Enzymol. 276, 494–523. [DOI] [PubMed]
- Holm, L. & Sander, C. (1993). J. Mol. Biol. 233, 123–138. [DOI] [PubMed] [Google Scholar]
- Ishitani, R., Nureki, O., Nameki, N., Okada, N., Nishimura, S. & Yokoyama, S. (2003). Cell, 113, 383–394. [DOI] [PubMed] [Google Scholar]
- Jones, T. A., Zou, J. Y., Cowan, S. W. & Kjeldgaard, M. (1991). Acta Cryst. A47, 110–119. [DOI] [PubMed] [Google Scholar]
- Kealey, J. T., Gu, X. & Santi, D. V. (1994). Biochimie, 76, 1133–1142. [DOI] [PubMed] [Google Scholar]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291. [Google Scholar]
- Lee, T. T., Agarwalla, S. & Stroud, R. M. (2004). Structure, 12, 397–407. [DOI] [PubMed] [Google Scholar]
- Lee, T. T., Agarwalla, S. & Stroud, R. M. (2005). Cell, 120, 599–611. [DOI] [PubMed] [Google Scholar]
- Liu, Y. & Santi, D. V. (2000). Proc. Natl Acad. Sci. USA, 97, 8263–8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Rozenski, J., Crain, P. F. & McCloskey, J. A. (1999). Nucleic Acids Res. 27, 196–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert, H. L., Blumenthal, R. M. & Cheng, X. (2003). Trends Biochem. Sci. 28, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T. C. (2000). Acta Cryst. D56, 965–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger, T. C. (2003). Acta Cryst. D59, 34–44.
- Terwilliger, T. C. & Berendzen, J. (1999). Acta Cryst. D55, 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). Nucleic Acids Res. 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin, A. & Teplyakov, A. (1997). J. Appl. Cryst. 30, 1022–1025. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: SeMet-ApoI, 1wxw, r1wxwsf
PDB reference: SeMet-ApoII, 1wxx, r1wxxsf
PDB reference: Native-AdoHcy, 2cww, r2cwwsf