

The CcdA C-terminal domain was crystallized in complex with CcdB in two crystal forms that diffract to beyond 2.0 Å resolution.
Keywords: plasmid addiction, toxin–antitoxin systems
Abstract
CcdA and CcdB are the antidote and toxin of the ccd addiction module of Escherichia coli plasmid F. The CcdA C-terminal domain (CcdAC36; 36 amino acids) was crystallized in complex with CcdB (dimer of 2 × 101 amino acids) in three different crystal forms, two of which diffract to high resolution. Form II belongs to space group P212121, with unit-cell parameters a = 37.6, b = 60.5, c = 83.8 Å and diffracts to 1.8 Å resolution. Form III belongs to space group P21, with unit-cell parameters a = 41.0, b = 37.9, c = 69.6 Å, β = 96.9°, and diffracts to 1.9 Å resolution.
1. Introduction
Bacterial toxin–antitoxin systems (TA systems) have recently attracted much attention because of their crucial role in bacterial physiology (Gerdes et al., 2005 ▶; Lewis, 2005 ▶) and because of their potential for the development of novel antibacterial drugs (Engelberg-Kulka et al., 2004 ▶). TA systems are operons that consist of a gene encoding a toxin preceded by a gene encoding the antidote or antitoxin. In general, the toxin is a stable long-lived protein, while the antitoxin is only marginally stable and prone to proteolysis. The antitoxin reversibly inactivates the toxin by forming a non-covalent complex with it. Upon activation of the system, the antidote is quickly degraded, liberating the toxin. Two classes of toxins can be distinguished based upon their targets. Members of the CcdB and ParE families (Bernard & Couturier, 1992 ▶; Jiang et al., 2002 ▶) inhibit gyrase, while other families such as RelE and MazF mediate the cleavage of mRNA bound to the ribosome (Christensen et al., 2003 ▶; Pedersen et al., 2003 ▶).
The physiological function of TA systems is highly debated. Initially, TA systems were discovered on low copy-number plasmids and phages, where they were found to be essential for the maintenance of these extrachromosomal elements in the absence of external selective pressure (for a review, see Engelberg-Kulka & Glaser, 1999 ▶). Cells that become cured from the plasmid stop growing. Thus, TA systems can be seen as a type of selfish DNA that parasitizes on a host. Recent surveys of bacterial genomes have led to the detection of a large number of chromosome-residing TA systems that can be classified into seven distinct families (Pandey & Gerdes, 2005 ▶; Anantharaman & Aravind, 2003 ▶). Some authors have argued that these TA systems mediate altruistic cell death under conditions of nutrient stress (Amitai et al., 2004 ▶; Engelberg-Kulka & Hazan, 2003 ▶; Aizenman et al., 1996 ▶). Conversely, it has been proposed that TA systems function as stress-induced regulators that do not kill the cell but rather bring it reversibly into a state of physiological stasis (Gerdes et al., 2005 ▶; Christensen et al., 2001 ▶). Finally, evidence is accumulating for a role in the emergence of persistor cells that are responsible for multidrug tolerance in biofilms (Lewis, 2005 ▶).
The ccd system is the earliest described TA system (Miki, Chang et al., 1984 ▶; Miki, Yoshioka et al., 1984 ▶) as well as one of the best characterized. It ensures the maintenance of the F-plasmid in Escherichia coli populations and is thus the archetype of a plasmid addiction system (Jaffe et al., 1985 ▶; Gerdes et al., 1986 ▶). Crystal structures of the toxin CcdB and its complex with a relevant fragment of gyrase are available (Loris et al., 1999 ▶; Dao-Thi et al., 2005 ▶). Structural information on the antidote CcdA remains elusive. CcdA has a low thermodynamic stability (Dao-Thi et al., 2000 ▶) and is degraded by the Lon protease (Van Melderen et al., 1994 ▶, 1996 ▶). Genetic studies indicate that CcdA is a two-domain protein consisting of a globular N-terminal dimerization and DNA-binding domain followed by a C-terminal domain that binds CcdB (Bernard & Couturier, 1991 ▶; Salmon et al., 1994 ▶). The latter may be intrinsically unstructured in the absence of CcdB. As both CcdA and CcdB are dimers and are bivalent for each other, a series of complexes with different stoichiometries can be formed, ultimately leading to insoluble chains of alternating CcdA and CcdB dimers (Dao-Thi et al., 2002 ▶). The latter phenomenon has hampered crystallization of a CcdA–CcdB complex. Here, we report the crystallization of the monomeric C-terminal half of CcdA (CcdAC36; 36 amino acids; MW 4366 Da) in complex with CcdB (101 amino acids; monomeric MW 11 706 Da). The structure of this CcdB–CcdAC36 complex will shed further light on the mechanism of antidote action and on the evolutionary relationships between different TA families.
2. Material and methods
2.1. Purification of CcdB
E. coli strain CSH50 with the CcdB permissive mutation R462C in the GyrA gene, harbouring plasmid pULB2250 (a pKK223-3 derivative with the ccdB gene under control of the tac promotor; Bahassi et al., 1999 ▶), was grown at 310 K in a Luria–Bertani broth (LB) culture supplemented with 100 µg ml−1 ampicillin and 50 µg ml−1 streptomycin. The culture was induced with 0.5 mM IPTG at an OD600nm of approximately 1. Cells were harvested by centrifugation at 277 K (15 min at 4000g) and suspended in cold 50 mM Tris–HCl pH 7.8, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 0.1 mg ml−1 AEBSF and 1 µg ml−1 leupeptin. Cells were broken at 277 K in a French Press (103 MPa in a 20K cell; Spectronic Instruments, Rochester, NY, USA) and cell debris were removed by centrifugation for 30 min at 12 000g. Ammonium sulfate (30%) was added to the supernatant and it was kept for 1 h at 277 K. Precipitated proteins were removed by centrifugation (30 min at 12 000g). The supernatant was brought to 80% ammonium sulfate and was kept for 1 h at 277 K. The protein pellet (30 min at 12 000g) was dissolved in 50 mM Tris–HCl pH 8.2 and dialyzed against the same buffer in a 3.5 kDa cutoff dialysis tubing (Spectrapor, Houston, TX, USA) to lower the conductivity to 0.5 mS cm−1. The dialyzed pool was loaded onto a Poros20 HQ anion-exchange column (Perseptive Biosystems, Cambridge, MA, USA) equilibrated in 50 mM Tris–HCl pH 8.2 and operated at 300 ml h−1. After a five-column-volume wash, the column was eluted with a 20-column-volume linear gradient to 0.5 M NaCl in the same buffer. CcdB eluted at 190 mM NaCl in the gradient. The CcdB-containing fractions were pooled, diluted until an OD280nm of 0.05 was reached and ammonium sulfate was slowly added to a final concentration of 1.5 M. This pool was loaded onto Isopropyl Resource15 (Amersham Pharmacia Biotech, Uppsala, Sweden) equilibrated in 20 mM Tris–HCl pH 8.0, 1.5 M ammonium sulfate and operated at 300 ml h−1. After a five-column-volume wash, the column was eluted with a 20-column-volume linear gradient to 20 mM Tris–HCl pH 8.0. CcdB eluted at 1 M ammonium sulfate. The CcdB-containing fractions were pooled and concentrated on a 3 kDa cutoff low-protein-binding membrane (Millipore) in a concentrator cell under nitrogen pressure. The final purification and buffer change run was performed on a Superdex75 PG (16/60; Amersham Bioscience, Uppsala, Sweden) size-exclusion column in 50 mM Tris–HCl pH 8.0, 150 mM NaCl. CcdB was thereupon concentrated to 10 mg ml−1 (assuming a 1:1 stoichiometry for the complex). The purified protein shows essentially as a single band of the correct molecular weight on SDS–PAGE (Fig. 1 ▶ a).
Figure 1.

(a) 10% SDS–PAGE of redissolved crystals. Lane 1, form I crystals; lane 2, form III crystals; lane 3, form II crystals; lane M, molecular-weight markers (kDa); lane 4, purified CcdB; lane 5, CcdAC36. (b) Form I crystals of the complex between CcdB and CcdAC36. The scale bar represents 0.1 mm. (c) Crystals of form II. (d) Crystals of form III.
2.2. Preparation of the CcdB–CcdAC36 complex
The C-terminal half of CcdA from E. coli plasmid TP181, CcdAC36 (amino-acid sequence NH2-RRLRPERWKVANQEGMAEVARFIEMNGSFADENRDW-COOH), was synthesized by solid-phase synthesis by Alta Bioscience, University of Birmingham, UK. The peptide was claimed to be at least 88% pure by the vendor and showed as a single band on 10% SDS–PAGE (Fig. 1 ▶ a). It is stable in water upon storage at 253 K, but degradation was detected upon prolonged incubation at room temperature, similar to observations on full-length F-plasmid CcdA (data not shown). The CcdB–CcdAC36 complex was prepared by slowly mixing equal volumes of CcdB (10 mg ml−1 in 50 mM Tris–HCl pH 8.0, 150 mM NaCl) and CcdAC36 (5 mg ml−1 dissolved in water) to obtain an approximately 1:1.5 ratio. This mixture was subsequently subjected to gel filtration on a Superdex-75 column (in 50 mM Tris–HCl pH 8.0, 150 mM NaCl). The pure complex (extinction coefficient 27 880 M −1 cm−1 at 280 nm calculated for a 1:1 complex) was concentrated to 10.0 mg ml−1 in the same buffer.
2.3. Crystallization
Initial screening of crystallization conditions was carried out with the Hampton Research Crystal Screen, Crystal Screen II and Natrix kits, the Stura MD1-20 Footprint Screen and the DeCODE Genetics Wizard I and Wizard II screens using the hanging-drop vapour-diffusion method. Drops consisting of 1.5 µl protein solution (10 mg ml−1 in 50 mM Tris–HCl pH 8.0, 150 mM NaCl) and 1.5 µl precipitant solution were equilibrated against 0.5 ml precipitant solution. Further optimization of crystal growth was performed using repetitive seeding. The initial microcrystalline precipitate was used for streak-seeding to obtain a first generation of crystals. In each case, 1 µl of microcrystalline precipitate was diluted in 20 µl precipitant solution and vortexed. A cat whisker was passed through the seed solution and subsequently passed through the fresh crystallization drop. Further improvement was obtained using repetitive microseeding. For these experiments, a stock solution of seeds was prepared by crushing a few crystals of the previous generation with a needle in 20 µl precipitant solution followed by vortexing. This seed stock was diluted serially in precipitant solution, each time by a factor of 20 (highest dilution factor 160 000). 0.1 µl of each dilution was added to different crystallization drops (1.5 µl of protein solution and 1.5 µl of precipitant solution, pre-equilibrated overnight) to grow the next generation of crystals. To further increase crystal size and quality, precipitant concentration, protein concentration and pH were also varied during seeding optimization experiments.
2.4. Crystal characterization and data collection
Data collection for form II crystals was performed at EMBL beamline X11 of the DESY synchrotron (Hamburg, Germany). The wavelength was 0.8125 Å and the MAR CCD detector was placed at a distance of 100 mm. 200 frames of 1.0° rotation were collected using an exposure time of 10 s per frame. Data for form III crystals were collected on beamline ID14-2 of the ESRF synchrotron (Grenoble, France). Here, the wavelength was 0.934 Å and the detector distance was 150 mm. 720 frames of 0.5° were collected using an exposure time of 3 s per frame. All data were indexed and integrated with DENZO, scaled with SCALEPACK (Otwinowski & Minor, 1997 ▶) and converted to structure-factor amplitudes using the CCP4 program TRUNCATE (Collaborative Computational Project, Number 4, 1994 ▶).
3. Results and discussion
Initial screening for crystals of the CcdB–CcdAC36 complex produced microcrystalline aggregates in three conditions. Optimization combined with repetitive seeding eventually resulted in macrocrystals, which we designate as forms I, II and III.
Crystals of form I were grown by microseeding in 1.0 M ammonium phosphate, 100 mM Tris–HCl pH 8.5. They are bipyramidal in shape, with their largest dimension reaching 200 µm (Fig. 1 ▶ b). These crystals do not diffract when exposed to synchrotron X-ray radiation.
Crystals of form II were obtained after repetitive microseeding in 200 mM calcium acetate, 100 mM MES pH 6.0, 20%(w/v) PEG 8000. They appear as clusters of small blocks (maximum dimension 200 µm; Fig. 1 ▶ c) which can be separated with the Hampton microtools. These crystals belong to space group P212121, with unit-cell parameters a = 37.6, b = 60.5, c = 83.8 Å, and diffract to 1.8 Å resolution on beamline X11 of the DESY synchrotron (Table 1 ▶). For data collection, the crystals were frozen directly in the cold stream without any need for an additional cryoprotectant.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Form II | Form III | |
|---|---|---|
| Resolution (Å) | 15.0–1.8 (1.94–1.80) | 15.0–1.9 (1.97–1.90) |
| Completeness (%) | 99.9 | 93.8 |
| 〈I/σ(I)〉 | 12.7 (4.0) | 12.5 (3.5) |
| No. of measured reflections | 103726 (9670) | 89162 (3792) |
| No. of unique reflections | 21626 (2149) | 17171 (998) |
| No. of reflections >3σ | 15617 (906) | 12961 (354) |
| Rmerge† | 0.075 (0.524) | 0.096 (0.442) |
R
merge = 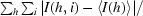
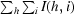 .
.
Crystals of form III were grown, again by seeding, in 200 mM magnesium chloride, 100 mM Tris–HCl pH 8.5, 30%(w/v) PEG 4000. They form clusters of needles with a largest dimension of 200 µm (Fig. 1 ▶ d). They belong to space group P21, with unit-cell parameters a = 41.0, b = 37.9, c = 69.6 Å, β = 96.9°, and diffract to 1.9 Å resolution on beamline ID14-1 of the ESRF (Table 1 ▶). For data collection, a single crystal was isolated using the Hampton microtools and flash-frozen in the cryostream without addition of further cryoprotectant.
The observed unit cells are different from those observed for free CcdB (Steyaert et al., 1993 ▶; Dao-Thi et al., 1998 ▶). The crystal packing for both crystal forms II and III appears to be tight (Matthews, 1968 ▶), with calculated Matthews coefficient (V M) values of 1.7 and 1.9 Å3 Da−1, respectively, for a 2:1 complex. For a 2:2 complex, these values would be 1.5 and 1.7 Å3 Da−1. Given the small size of the crystals, form III in particular, and the observation that they diffract beyond 2 Å resolution, tight packing and low solvent content are not unexpected (Kantardjieff & Rupp, 2003 ▶). As an additional check crystals of each form were washed and subjected to SDS–PAGE to confirm the presence of both CcdB and CcdAC36 in the crystals. As seen in Fig. 1 ▶(a), both proteins are present, although the exact stoichiometry will have to await structure determination.
A number of properties of CcdA may have contributed to our failure to crystallize full-length CcdA or its complex with CcdB. CcdA has a low thermodynamic stability (Dao-Thi et al., 2000 ▶). A low stability does not in itself necessarily preclude crystallization, as has been shown by studies on point mutants of T4 lysozyme and RNase T1 that are only marginally stable (Matthews, 1993 ▶; De Vos et al., 2001 ▶). However, a low thermodynamic stability implies that the protein spends a larger fraction of the time in a (partly) unfolded state and is thus more prone to attack by possible proteases. CcdA is indeed very protease-sensitive. Purified samples of CcdA (and other addiction antidotes such as MazE or Phd) degrade when stored unfrozen, which may be a consequence of trace contaminations with a protease (N. De Jonge, M.-H. Dao-Thi & R. Loris, unpublished results). However, the use of additional purification steps does not seem to influence this degradation, leading to the intriguing possibility of a form of autoproteolysis, something which has not yet been experimentally tested. In this respect, it is remarkable that the CcdAC36 seems to have degraded somewhat in the form I and form II crystals. A third culprit is the intrinsically unfolded nature of the C-terminal domain of CcdA. In the case of MazE, this problem could be solved by cocrystallization with either a MazE-specific camel antibody VHH domain (Loris et al., 2003 ▶) or with its toxin MazF (Kamada et al., 2003 ▶). Both strategies failed in the case of CcdA. Attempts to co-crystallize full-length CcdA and CcdB lead to irreversible precipitation as a consequence of the bivalency of both proteins (Dao-Thi et al., 2002 ▶). Here, by using only the monomeric C-terminal half of CcdA, CcdAC36, we were able to prepare a soluble CcdB–CcdA36 complex that was succesfully crystallized. The structure of this complex will provide insight into CcdA–CcdB specificity as well as into the proposed evolutionary relationship between the ccd-type and maz-type TA systems.
Acknowledgments
This work was supported by grants from OZR-VUB, VIB and FWO-Vlaanderen. NDJ received financial support from IWT. The authors acknowledge the use of EMBL beamlines BW7A (DESY, Hamburg, Germany) and ID14-2 (ESRF, Grenoble, France). We thank Kris Pauwels and Inge Van Molle for critically reading the manuscript.
References
- Aizenman, E., Engelberg-Kulka, H. & Glaser, G. (1996). Proc. Natl Acad. Sci. USA, 93, 6059–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amitai, S., Yassin, Y. & Engelberg-Kulka, H. (2004). J. Bacteriol.186, 8295–8300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman, V. & Aravind, L. (2003). Genome Biol.4, R81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahassi, E. M., O’Dea, M. H., Allali, N., Messens, J., Gellert, M. & Couturier, M. (1999). J. Biol. Chem.274, 10936–10944. [DOI] [PubMed] [Google Scholar]
- Bernard, P. & Couturier, M. (1991). Mol. Gen. Genet.226, 297–304. [DOI] [PubMed] [Google Scholar]
- Bernard, P. & Couturier, M. (1992). J. Mol. Biol.226, 735–745. [DOI] [PubMed] [Google Scholar]
- Christensen, S. K., Mikkelsen, M., Pedersen, K. & Gerdes, K. (2001). Proc. Natl Acad. Sci. USA, 98, 14328–14333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen, S. K., Pedersen, K., Hansen, F. G. & Gerdes, K. (2003). J. Mol. Biol.332, 809–819. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763. [Google Scholar]
- Dao-Thi, M.-H., Charlier, D., Loris, R., Maes, D., Messens, J., Wyns, L. & Backmann, J. (2002). J. Biol. Chem.277, 3733–3742. [DOI] [PubMed] [Google Scholar]
- Dao-Thi, M.-H., Messens, J., Wyns, L. & Backmann, J. (2000). J. Mol. Biol.299, 1373–1386. [DOI] [PubMed] [Google Scholar]
- Dao-Thi, M.-H., Van Melderen, L., De Genst, E., Afif, H., Buts, L., Wyns, L. & Loris, R. (2005). J. Mol. Biol.348, 1091–1102. [DOI] [PubMed] [Google Scholar]
- Dao-Thi, M.-H., Wyns, L., Poortmans, F., Bahassi, E. M., Couturier, M. & Loris, R. (1998). Acta Cryst. D54, 975–981. [DOI] [PubMed] [Google Scholar]
- De Vos, S., Backmann, J., Prevost, M., Steyaert, J. & Loris, R. (2001). Biochemistry, 40, 10140–10149. [DOI] [PubMed] [Google Scholar]
- Engelberg-Kulka, H. & Glaser, G. (1999). Annu. Rev. Microbiol.53, 43–70. [DOI] [PubMed] [Google Scholar]
- Engelberg-Kulka, H. & Hazan, R. (2003). Science, 301, 467–468. [DOI] [PubMed] [Google Scholar]
- Engelberg-Kulka, H., Sat, B., Reches, M., Amitai, S. & Hazan, R. (2004). Trends Microbiol.12, 66–71. [DOI] [PubMed] [Google Scholar]
- Gerdes, K., Christensen, S. K. & Lobner-Olesen, A. (2005). Nature Rev. Microbiol.3, 371–382. [DOI] [PubMed]
- Gerdes, K., Rasmussen, P. B. & Molin, S. (1986). Proc. Natl Acad. Sci. USA, 83, 3116–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe, A., Ogura, T. & Hiraga, S. (1985). J. Bacteriol.163, 841–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y., Pogliano, J., Helinski, D. R. & Konieczny, I. (2002). Mol. Microbiol.44, 971–979. [DOI] [PubMed] [Google Scholar]
- Kamada, K., Hanaoka, F. & Burley, S. K. (2003). Mol. Cell, 11, 875–884. [DOI] [PubMed] [Google Scholar]
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci.12, 1865–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, K. (2005). Biochemistry (Moscow), 70, 267–274.
- Loris, R., Dao-Thi, M.-H., Bahassi, E. M., Van Melderen, L., Poortmans, F., Liddington, R., Couturier, M. & Wyns, L. (1999). J. Mol. Biol.285, 1667–1677. [DOI] [PubMed] [Google Scholar]
- Loris, R., Marianovsky, I., Lah, J., Laeremans, T., Engelberg-Kulka, H., Glaser, G., Muyldermans, S. & Wyns, L. (2003). J. Biol. Chem.278, 28252–28257. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed] [Google Scholar]
- Matthews, B. W. (1993). Annu. Rev. Biochem.62, 139–160. [DOI] [PubMed] [Google Scholar]
- Miki, T., Chang, Z. T. & Horiuchi, T. (1984). J. Mol. Biol.174, 627–646. [DOI] [PubMed] [Google Scholar]
- Miki, T., Yoshioka, K. & Horiuchi, T. (1984). J. Mol. Biol.174, 605–625. [DOI] [PubMed] [Google Scholar]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Pandey, D. P. & Gerdes, K. (2005). Nucleic Acids Res.33, 966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen, K., Zavialov, A. V., Pavlov, M. Y., Elf, J., Gerdes, K. & Ehrenberg, M. (2003). Cell, 112, 131–140. [DOI] [PubMed] [Google Scholar]
- Salmon, M. A., Van Melderen, L., Bernard, P. & Couturier, M. (1994). Mol. Gen. Genet.244, 530–538. [DOI] [PubMed] [Google Scholar]
- Steyaert, J., Van Melderen, L., Bernard, P., Dao-Thi, M.-H., Loris, R., Wyns, L. & Couturier, M. (1993). J. Mol. Biol.231, 513–515. [DOI] [PubMed] [Google Scholar]
- Van Melderen, L., Bernard, P. & Couturier, M. (1994). Mol. Microbiol.11, 1151–1157. [DOI] [PubMed] [Google Scholar]
- Van Melderen, L., Dao-Thi, M.-H., Lecchi, P., Gottesman, S., Couturier, M. & Maurizi, M. R. (1996). J. Biol. Chem.271, 27730–27738. [DOI] [PubMed] [Google Scholar]