

Summary
AnsA is the cytoplasmic asparaginase from Escherichia coli involved in intracellular asparagine utilization. Analytical ultracentifugation and X-ray crystallography reveal that AnsA forms a tetrameric structure as a dimer of two intimate dimers. Kinetic analysis of the enzyme reveals that AnsA is positively cooperative, displaying a sigmoidal substrate dependence curve with an [S]0.5 of 1 mM L-asparagine and a Hill coefficient (nH) of 2.6. Binding of L-asparagine to an allosteric site was observed in the crystal structure concomitant with a reorganization of the quarternary structure, relative to the apo enzyme. The carboxyl group of the bound asparagine makes salt bridges and hydrogen bonds to Arg240, while the Nδ2 nitrogen interacts with Thr162. Mutation of Arg240 to Ala increases the [S]0.5 to 5.9 mM, presumably by reducing the affinity of the site for L-asparagine, although the enzyme retains cooperativity. Mutation of Thr162 to Ala results in an active enzyme with no cooperativity. Transmission of the signal from the allosteric site to the active site appears to involve subtle interactions at the dimer-dimer interface and relocation of Gln118 into the vicinity of the active site to position the probable catalytic water molecule. These data define the structural basis for the cooperative regulation of the intracellular asparaginase that is required for proper functioning within the cell.
Keywords: enzyme mechanisms, kinetics, positive cooperativity, allostery, leukemia
Introduction
Asparaginases are widely distributed in nature from bacteria to mammals and play a central role in amino acid metabolism and utilization. Asparagine is hydrolyzed to aspartate by the action of an asparaginase (EC 3.5.1.1), and the aspartate is then transaminated to oxaloacetate, an intermediate in the tricarboxylic acid cycle. Aspartate is also converted into fumarate during the urea cycle. Thus, asparaginases are important in maintaining nitrogen balance and the levels of amino acids within cells.
The model Gram negative bacterium, Escherichia coli, possesses two L-asparaginases: the cytoplasmic type I form encoded by ansA and the periplasmic type II form encoded by ansB.1,2 Type I asparaginase is constitutively expressed and required for the growth of the bacteria on asparagine as the sole nitrogen source.1,3–6 In contrast, the type II asparaginases are primarily expressed under anaerobic conditions with nutrient starvation, and their expression is tightly regulated by cyclic AMP and the oxygen-sensing regulator of fumarate and nitrate reduction (FNR) protein.1,4,6,7 Consistent with their different metabolic roles, the two enzyme types display very different affinities for asparagine. The type I enzyme has an apparent KM for asparagine of 3.5 mM, as befits an enzyme involved in catabolysis of amino acids.6 Type II enzymes, which scavenge nitrogen from the environment, display a KM for the substrate in the μM range.1 The first crystal structure of a type I enzyme, from the archaea Pyrococcus horikoshii, was recently reported,8 while the structures of type II enzymes have been extensively studied.9–12 Given the high degree of sequence similarity between the type I and type II enzymes (Figure 1), it is not surprising that the overall fold of both classes is highly conserved. However, the type I protein crystallized as a dimer rather than the common tetrameric architecture of the type II proteins, consistent with an earlier report in which the E. coli AnsA appeared to behave as a dimer by gel filtration chromatography.13
Figure 1. Sequence alignment of selected asparaginases.

The alignment and secondary structure locations are with respect to the E. coli type I (AnsA) enzyme described in this paper. The linker region that joins the N- and C-terminal domains of the structure is indicated. Regions of sequence identity and sequence similarity are shown in blue boxes and blue text, respectively. The conserved tyrosine with a putative catalytic role is indicated by black arrowheads (residue 24 in ecAnsA). The sequences of three intracellular AnsA enzymes and three extracellular AnsB enzymes are shown; ec, E. coli; ph, Pyrococcus horikoshii; sc, Saccharomyces cerevisiae; er, Erwinia chrysanthemi. For clarity, the signal sequences at the N-termini of AnsB enzymes are not shown, and an extended N-terminal region (amino acids 1–46) in scAnsA has been removed. The figure was generated with ESPript 2.2.57
Type II asparaginases are used in the treatment of acute lymphoblastic leukemia (ALL).14,15 As such, the type II enzymes from E. coli,11,16–24 Erwinia chrysanthemi,10,24–28 Wolinella succinogenes,9 Pseudomonas strain 7A29–31 and others12,32 have been extensively studied at the structural and mechanistic level. In contrast, the type I E. coli AnsA protein does not have therapeutic activity either in vitro or in vivo,2 and the reasons for this lack of activity are not clearly understood. Although the short half-life of AnsA in plasma (ca. 15 min) may explain its lack of activity in animal models, it does not explain its lack of activity in cell culture. Most likely, it is the biochemical properties of the enzyme that are responsible,2 and these could include the observed high KM for asparagine, the substrate specificity or potential allosteric kinetics. It is important to understand the structural and kinetic properties that distinguish potentially therapeutically useful asparaginases from those that are not because the current therapeutics have a number of practical and medical drawbacks that limit their use.33–36 Therefore, we have conducted an in-depth structure-function analysis of E. coli AnsA to aid our understanding of type I asparaginases and to identify the crucial differences between the non-therapeutic and the therapeutic asparaginases typified by E. coli AnsB. Our data, from both crystal structure analysis and analytical ultracentrifugation, unequivocally demonstrate that E. coli AnsA is a tetramer. In addition, structural, kinetic and mutagenesis data reveal that AnsA exhibits strong positive cooperativity through the action of a second, allosteric, L-asparagine binding site that modulates the architecture of the tetramer and the active site environment.
Results
Structural Analysis of Apo AnsA
His-tagged AnsA produced crystals (APOM) in space group P21 that diffracted to 1.89 Å and contained four monomers in the asymmetric unit (ASU). The structure was determined by molecular replacement, and the crystallographic and refinement details are shown in Table 1. The structure reveals a tetrameric architecture that corresponds to the ASU.
Table 1.
Data Refinement and Statistics
| Apo AnsA | Complexed
|
||
|---|---|---|---|
| Native AnsA | AnsA[T162A] | ||
| APOM | CNATM | CT162A | |
| Data Collectiona | |||
| Space group | P21 | P21 | P21 |
| Cell dimensions | |||
| a, b, c (Å) | 57.8, 151.6, 87.1 | 90.3, 89.8, 93.1 | 90.5, 89.8, 93.3 |
| α, β, γ (°) | 90.0, 88.6, 90.0 | 90.0, 117.0, 90.0 | 90.0, 117.1, 90.0 |
| Beamline | ID22, SER-CAT | ID22, SER-CAT | BM22, SER-CAT |
| Resolution range(Å) | 50.0-1.88 (1.93-1.88) | 50.0-1.90 (1.97-1.90) | 50.0-1.82 (1.89-1.82) |
| Wavelength(Å) | 1.01259 | 1.01259 | 0.97923 |
| No. of reflections | |||
| Measured | 468,621 | 338,886 | 377,415 |
| Unique | 116,888 | 95,017 | 119,400 |
| Rsym(%)b | 8.0 (31.0) | 8.4 (24.6) | 8.0 (29.8) |
| Completeness(%) | 96.2 (85.8) | 90.6 (81.3) | 99.7 (98.8) |
| Redundancy | 4.0 (3.6) | 3.6 (3.2) | 3.2 (2.5) |
| I/σ(I) | 18.2 (4.4) | 17.9 (5.0) | 15.7 (2.4) |
| Refinement | |||
| Resolution range(Å) | 50.0-1.89 | 50.0-1.90 | 50.0-1.82 |
| No. of reflection(F>0σ(F)) | 115,353 | 89,743 | 115,053 |
| Rwork(%)c | 18.4 | 22.3 | 20.9 |
| Rfree(%)d | 20.7 | 26.4 | 22.6 |
| No. of atoms | |||
| Protein | 10,200 | 9,589 | 10,013 |
| Ligand/ion | 72 | 159 | 136 |
| Water | 561 | 575 | 515 |
| Average B factors(Å2) | |||
| Protein | 27 | 20 | 26 |
| Ligand | 39 | 15 | 22 |
| Water | 37 | 24 | 32 |
| Root mean square deviations from ideal values | |||
| Bond lengths(Å) | 0.007 | 0.013 | 0.005 |
| Bond angles(degrees) | 1.5 | 1.4 | 1.4 |
| Residues from Ramachandran plot | |||
| Most favored(%) | 92.2 | 92.1 | 91.3 |
| Additional allowed(%) | 7.4 | 7.4 | 7.9 |
| Generously allowed(%) | 0.4 | 0.5 | 0.7 |
| Contents of structure e | |||
| Chain A | 2–280, 287–337 | 3–17, 40–279, 288–336 | 3–19, 25–280, 286–336 |
| Chain B | 3–280, 287–337 | −4–13, 28–280, 288–337 | −5–19, 28–280, 287–337 |
| Chain C | 2–280, 286–337 | 3–13, 34–280, 288–336 | 3–18, 25–280, 286–337 |
| Chain D | 1–280, 287–337 | −5–13, 30–279, 288–336 | −5–19, 25–280, 288–337 |
| PDB code | 2P2D | 2P2N | 2HIM |
Highest resolution shell is shown in parenthesis.
Rsym = Σ|(I−<I>)|IΣ(I), where I is the observed intensity.
Rwork = Σ||Fobs|−|Fcalc|| / Σ|Fobs|, where Fobs and Fcalc are observed and calculated structure factors.
Rfree is the R value obtained for a test set of reflections consisting of randomly selected 5% subset of the data set excluded from refinement.
Residues with negative numbers derive from the 6 × His tag.
Structure of the Monomer
The overall structure of the monomer is very similar to those of the related asparaginases, and includes the unusual the left-handed crossover structural motif.9–11 The molecule comprises two α/β domains that are connected by an extended but structured linker region (Figure 1 and 2(a)). The N-terminal and C-terminal domains span residues 1-190 and 211-338, respectively, and the linker region encompasses the intervening residues 191-210. The larger N-terminal domain is constructed around a 5-stranded parallel β-pleated sheet arranged in the order β2 (residues 47-54), β1 (4-12), β3 (84-90), β4 (111-116) and β5 (146-150), and the sheet is extended by three short anti-parallel β-strands β6 (154-157), β7 (180-183) and β8 (187-190) that associate with β5. The central β-sheet is flanked by four α-helices, helices α1 (29-38) and α4 (127-142) on one side and helices α2 (65-79) and α3 (93-104) on the other. Two features to note in the N-terminal domain are a proline-mediated kink at the end of helix α1 at residue 39, and an extended β-ribbon structure (160-174) in the long loop that connects β6 and β7. The smaller C-terminal domain is constructed around a central 4-stranded parallel β-sheet arranged in the order β9 (211-217), β10 (235-241), β11 (266-270) and β12 (294-297). This β-sheet is flanked by five α-helices, α5 (223-231), α6 (251-263) and α7 (287-293) on one side and α8 (302-314) and α9 (319-327) on the other.
Figure 2. Overall structure of E. coli AnsA.
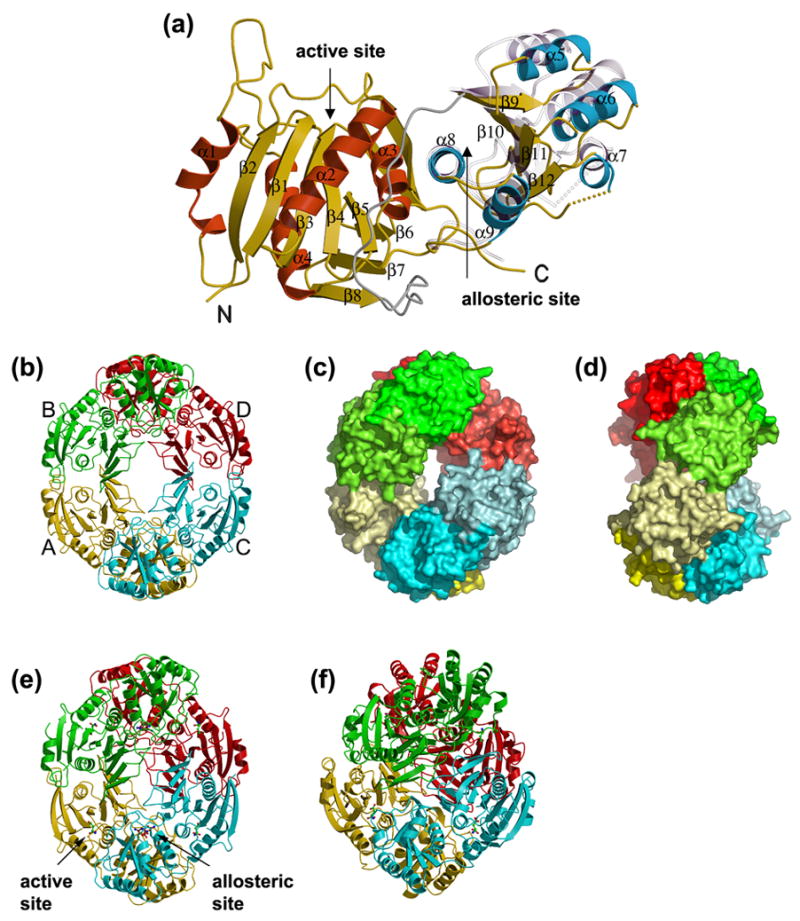
(a) A ribbon representation of the apo AnsA monomer showing the secondary structure elements. The N-terminal domain α-helices are shown in red, the C-terminal domain α-helices are blue and the β-strands are in yellow in both domains of the molecule. The linker region is shown in grey. Shown semi-transparently is the position of the C-terminal domain relative to the fixed N-terminal domain in the structure of AnsA complexed with L-asparagine. Note that the movement is a rotation around helix α8 at the domain interface. (b–d) The tetrameric structure of apo AnsA with monomers A, B, C and D (labeled in b) in yellow, green, cyan and red, respectively. (b) A ribbon representation; (c) a surface representation showing the donut shape with the hole in the center; (d) a 90° rotation of (c) showing the tight (top) and loose (front, middle) interfaces. In (b) and (c), the N- and C-terminal domains are shaded differently for clarity. (e) The tetrameric structure of AnsA complexed with L-asparagine in a ribbon representation showing aspartate (green) at the four active sites and asparagine (brown) at the four allosteric sites (one of each site is indicated). Compared to the apo structure (b), the tetramer is more compact and the central hole is smaller. (f) The tetrameric structure of E. coli AnsB in ribbon representation in which the central hole is absent due to a more orthogonal packing of tight dimers.
Structure of the Tetramer
Four identical monomers associate into a tetrameric structure that can more accurately be described as a dimer of dimers. Thus, two monomers (A and C, and B and D in our nomenclature) interact via a tight interface to create a crescent shaped dimer, and two dimers pack more loosely to create a “doughnut” with a hole in the center (Figure 2(b), 2(c) and 2(d)).
The tight dimer interface is mostly formed by interactions between C-terminal domains. Figure 3(a) shows a close up view of the interactions. At the surface, helices 5 and 5′ (from the opposite monomers) interact in an anti-parallel fashion, and prominent reciprocal interactions include a hydrogen bond between the Nδ2 nitrogen of Asn228 and the backbone carbonyl oxygen of Gln232′, aromatic packing of Phe229 and Phe229′, and a hydrophobic interaction between Val225 and Val234′. Further towards the center of the molecule, strands β9 and β9′ associate to create an 8-stranded β-sheet that spans the dimer. Specific reciprocal interactions in this region of the interface include hydrogen bonds between the hydroxyl of Tyr218 and the carbonyl oxygen of Ile212′, and the hydroxyl of Tyr96 and the Oε1 oxygen of Gln272′, and stacking interactions between the guanidinium groups of Arg240 and Arg240′. Finally, Met274 forms a tight hydrophobic interaction with its dimeric counterpart across the interface. Interactions between the N- and N′-terminal domains across the tight dimer interface do not exist, but there are some significant interactions between the N- and C′-terminal domains that involve the β-ribbon (160-174) which reaches across the interface. Notably, the Nδ1 nitrogen of His165 forms a hydrogen bond with Oγ oxygen of Ser275′ and the carbonyl oxygen of Ala164 forms a hydrogen bond to the backbone amide of Met274′.
Figure 3. Monomer-monomer interfaces in the AnsA tetramer.
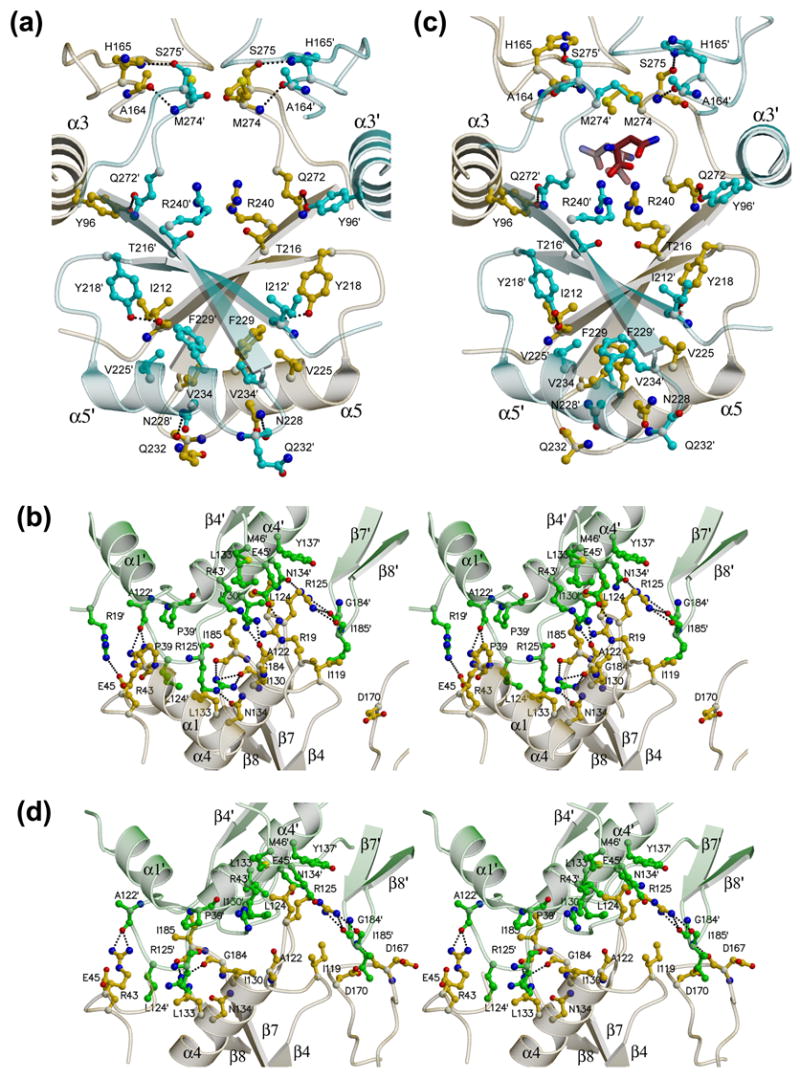
Panels (a) and (c) show the intimate dimer (A/C) interface in the apo structure and the asparagine-bound structure, respectively. Panels (b) and (d) show stereoviews of the loose dimer (A/B) interface in the apo structure and the asparagine-bound structure, respectively. Monomer A is yellow, B is green and C is in cyan, and the two allosteric asparagine molecules in (c) have red carbon atoms. Details are provided in the text.
The dimer-dimer interface that completes the tetramer is exclusively mediated by interacting N-terminal domains. A close up view of the interactions is shown in Figure 3(b). The interface is centered on the N-terminal halves of helices α4 and α4′ that interact in an anti-parallel fashion. Flanking these are the loops between strands β7 and β8, and β4 and α4, and their dimeric counterparts. There is also a van der Waals interaction between Pro39 and Pro39′ that kinks the end of helix α1. A notable feature of the interface is that three arginine side chains reach across and make specific interactions. Arg19 forms a salt bridge with Glu45′, Arg43 forms a hydrogen bond to the carbonyl oxygen of Ala122′, and Arg125 makes three hydrogen bonds to the Oδ1 oxygen of Asn134′ and the carbonyl oxygens of Gly184′ and Ile185′. In addition, the interface involves a number of hydrophobic interactions between Ile130 and Ile130′, Ile119 and Ile185′, and Leu124 within a pocket formed by the side chains of Arg43′, Glu45′, Met46′, Leu133′ and Tyr137′.
Identification of an allosteric site
AnsA was pre-mixed with asparagine to form a complex and then subjected to crystallization trials. High quality crystals (CNATM) in space group P21 were obtained that diffracted to 1.90 Å resolution and structural analysis revealed a tetramer in the ASU. Crystallographic and refinement details are shown in Table 1. Clear electron density was located in the active sites of all four monomers, and this will be described in detail later.
An unexpected feature of the complex is the presence of 4 tightly bound asparagine molecules at equivalent locations within the tetramer distant from the active site (Figure 4). This suggested that asparagine is not only the substrate of the enzyme but also acts as an allosteric effector molecule. This is supported by a small but significant reorganization and compaction of the quaternary structure that maintains the 222 symmetry (Figure 2(e)). The putative allosteric site is at the N-terminus of helix α8 and close to the tight dimer interface (Figure 4(a)). Within the dimer, the two allosteric sites are less than 7.5 Å apart (as measured between the alpha carbons of the bound asparagines). The mode of binding within each monomer is such that the asparagine connects the N-terminus of α8 (residues 302 and 303) to surrounding elements of the structure. Specifically, the carboxyl group forms salt bridges and hydrogen bonds with the paired arginines 240 and 240′ at the dimer interface, the amino group donates two hydrogen bonds to the carbonyl oxygens of Cys273 and Thr271, the amido nitrogen Nδ2 donates two hydrogen bonds to the Oγ1 oxygen of Thr162 and the Oε2 oxygen of Glu303, and the Oδ1 oxygen receives a hydrogen bond from the backbone amide nitrogen of Val302 (Figure 4(b)). The bound asparagine slightly rotates α8 relative to the apo structure, and this movement results in a rotation of the C-terminal domain relative to the N-terminal domain (Figure 2(a)), and a slight compaction of the tetramer (Figure 2(e)). The axis of helix α8 is the hinge about which the movement occurs.
Figure 4. The AnsA allosteric asparagine binding pocket.
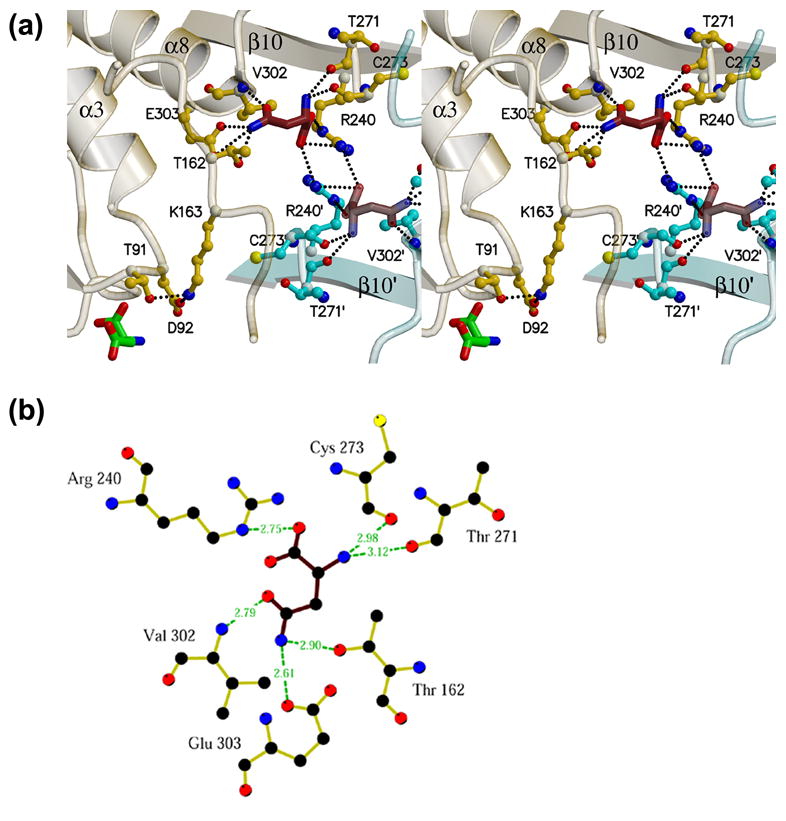
(a) The pocket is at the tight dimer interface (between monomers A and C in this view) at the N-terminus of helix α8 and spanning dyad-related arginines 240 and 240′. Details of the interaction are provided in the text. Elements of monomer A and C are shown in yellow and cyan, respectively. Note that two dyad-related pockets are visible in this view, as well as the active site aspartate in monomer A adjacent to Thr91. Note also that Thr162 and Lys163 connect the allosteric and active sites within one monomer. (b) Diagrammatic representation of the hydrogen bonding interactions of asparagine (brown bonds) in the allosteric site (yellow bonds). Figure was generated with Ligplot.58
Inspection of the two monomer-monomer interfaces within the tetramer reveals the changes that are necessary to accommodate this rotation of the C-terminal domain. At the tight monomer-monomer (A/C) interface (Figure 3(c)), there is an adjustment of helices α5 and α5′ at the periphery where the domain rotation generates the largest displacement of over 3 Å. The movement of the helices is such that Asn228 and Asn228′ can no longer form interfacial hydrogen bonds but pack against each other in a van der Waals interaction. In addition, the paired Phe229 and Phe229′ no longer interact and Phe229 now packs with Val225′. At the dimer-dimer interface (A/B) (Figure 3(d)), the N-terminal domain moves towards its partner by some 4.5 Å, and helices α4 and α4′ twist slightly with respect to each other (compare Figure 3(b) and 3(d)). Also, the loop connecting strand β1 and helix α1 becomes disordered, as well as varying parts of the N-terminal region of helix α1 (residues 18-39, 14-27, 14-33 and 14-29, in monomers A, B, C and D, respectively). Despite the domain movements, the interfacial residue-residue interactions within the dimer-dimer interface are largely unaffected because the loop which contains the key residues Ala122, Leu124 and Arg125 moves to maintain the contacts. Notable changes include the loss of the salt bridge between Arg19 and Glu45′ that may contribute to the loss of order around the former residue, and the loss of the hydrogen bond between Arg125 and Asn134′.
Further Insights into AnsA Allostery
Analytical Ultracentrifugation
The crystallographic evidence for allostery in AnsA is largely based on the proposed tetrameric architecture of the enzyme. During purification, the wild type AnsA protein eluted from the S200 gel filtration column with a retention time consistent with a tetrameric structure, and not as a dimer as reported previously.13 To date, all of the type II asparaginase crystal structures have revealed a homotetrameric quaternary organization with 222 symmetry,9–11 and it is not surprising that the type I enzyme is also a tetramer. However, as shown in Figure 2(f) for E. coli AnsB (3ECA), the tetrameric arrangements are significantly different for the two enzymes. In AnsB, the crescent shaped dimers pack orthogonally such that the concave surfaces mesh tightly in contrast with the doughnut-like architecture of AnsA. In addition, the recently-determined structure of the type I enzyme from P. horikoshii (PhAnsA) suggests a homodimer.8 These differences are consistent with the observed tenuous nature of the dimer-dimer interaction, but it was crucial to the interpretation of the allosteric movements to accurately and definitively establish the multimeric state of AnsA in solution.
Sedimentation equilibrium experiments yielded a value of 154,402 Da (Figure 5(a)), close to the predicted value of 157,160 Da (4 × 39,290) for tetrameric His-tagged AnsA protein. Sedimentation velocity experiments (Figure 5(b)) in addition to sedimentation coefficients also provide information on the shape of a protein. The c(s) analysis show that AnsA oligomerizes into a tetramer with an s-value of 4.59 S at 4 °C (at standard conditions of 20 °C and water as a solvent, s20,w, is 7.60 S) and a best-fit weight-average frictional ratio (f/f0) of 1.37. A spherical compact protein molecule would have a ratio of 1.2 and thus 1.37 indicates a slight deviation from this shape that is consistent with the crystallographically observed doughnut. Molar mass (M) information was extracted from the sedimentation velocity data and yielded an M-value of 159,949 Da, very close to the theoretical value of the tetramer.
Figure 5. Analytical Ultracentrifugation of AnsA.
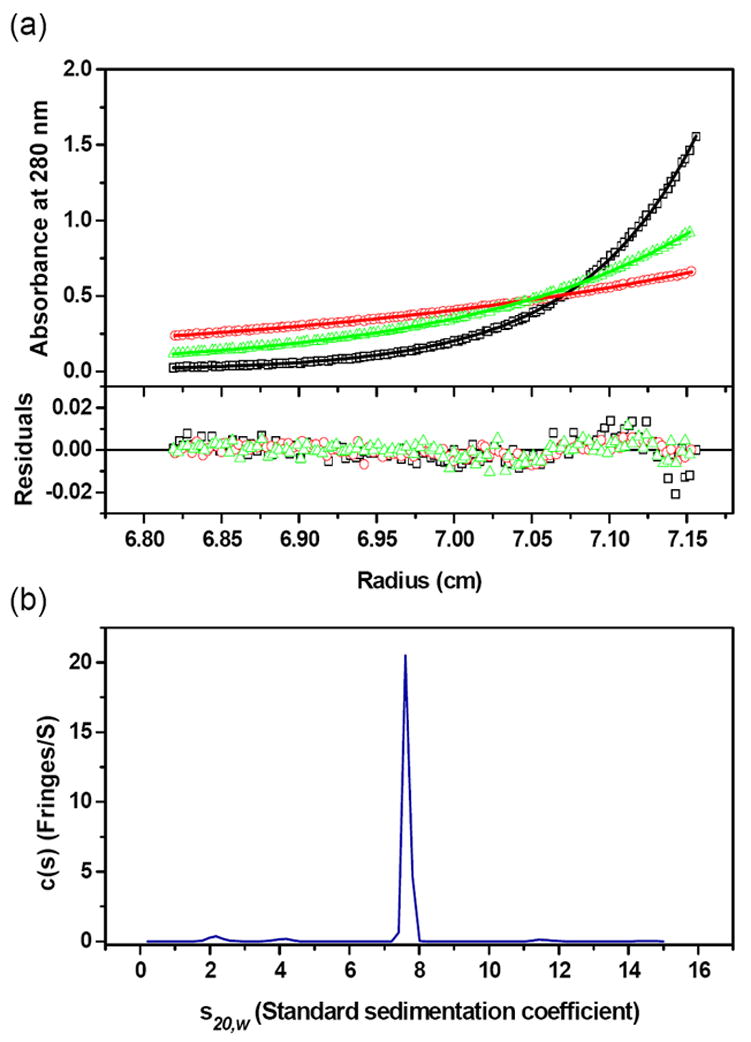
(a) Absorbance scans at 280 nm at equilibrium are plotted versus the distance from the axis of rotation for AnsA. Protein samples were centrifuged at 4 °C for at least 48 h at 5,000 (red circle), 7,000 (green triangle), and 10,000 (black square) rpm. The solid lines represent the global nonlinear least squares best-fit of all the data to a single molecular species. For clarity only the sample with a loading protein concentration of 0.5 mg/ml is shown. Residuals of the fit at all rotor speeds are also shown and the r.m.s. deviation is 0.005 absorbance units. (b) The s-values of the c(s) distribution were converted to standard condition s20,w-values (20 °C, water as solvent), and plotted as shown. The r.m.s. deviation is 0.010 fringes. Experiments were conducted at a loading protein concentration of 1.1 mg/ml at 4 °C and at a rotor speed of 50,000 rpm. The s20,w-value of AnsA is 7.60 S.
Kinetics and Mutagenesis
Wild-type AnsA was assayed for activity towards L-asparagine. At 20 mM substrate, the specific activity was 173 nmoles/min/μg and the Vmax was 16.6 nmoles ammonia formed per minute (Table 2). As substrate concentration was reduced, it was observed that the enzyme was not following Michaelis-Menten kinetics. A Hill plot (not shown) of the resulting velocities indicated that the enzyme exhibited strong positive cooperativity, with a Hill constant (nH) of 2.6. The concentration of substrate required for half maximal velocity ([S]0.5) was 1.2 mM.
Table 2.
Kinetic properties of the AnsA wild type and mutants.
| Protein | Vmax (nmoles·min−1) | S0.5 (mM) | nH |
|---|---|---|---|
| Wild type | 16.6 | 1.2 | 2.6 |
| R240A | 41.6 | 5.9 | 4.2 |
| T162A | 29.4 | 4.6 | 1.0 |
| D170Q | 70 | 1.2 | 2.8 |
| Q118D | ND | ||
| T14A | ND | ||
| T14V | ND | ||
| T91A | ND | ||
| T91V | ND | ||
| S61Q | ND |
Values from a single representative experiment are shown. ND =None detected.
The putative allosteric effector asparagines at the tight dimer interface interact intimately with the paired Arg240 and Arg240′, and this residue was mutated to Ala to potentially reduce the binding affinity at the allosteric site. The AnsA[R240A] protein had a similar specific activity to the wild type at 20 mM (saturating) asparagine, but was less active at lower substrate concentrations, with an [S]0.5 of 5.9 mM (Table 2). The nH for this protein (4.2) was higher than the wild type. These data support the importance of asparagine binding at the allosteric site for the activation of AnsA catalysis. The asparagine also interacts with the side chain of Thr162, and this residue is directly adjacent to the active site Lys163 (Figure 4(a)). The AnsA[T162A] mutant was prepared to investigate whether Thr162 acts as a conduit to transfer information from the allosteric site to the active site. This protein had a similar specific activity to the wild type protein at saturating substrate concentrations, but displayed very different kinetic properties. Instead of exhibiting cooperativity, it followed Michaelis-Menten kinetics, and gave a straight line when analyzed by double reciprocal plot (not shown). From these data, a KM of 4.6 mM for L-asparagine was obtained (Table 2). A Hill plot confirmed lack of cooperativity, with an nH of 1.0. Crystals were grown of the AnsA[T162A]-asparagine complex that were very similar to those of wild type AnsA-asparagine (Table 1), and the structure revealed four asparagine molecules docked at the allosteric effector site making identical interactions as observed in the wild-type complex. In addition, the two structures are practically identical and display the same global modifications to the quaternary structure that represents the active conformation of the enzyme.
Insights into the AnsA Active Site
The Apo Structure
The active site locale of type II and, more recently, type I, asparaginases is well documented and features two conserved threonine side chains that mediate sequential ‘ping-pong’ nucleophilic attacks during amidohydrolysis.8,10,16,18,19,26,37 The active site of apo AnsA is shown in Figure 6(a), and the two key threonines correspond to Thr14 in the extended loop between β1 and α1, and Thr91 in the loop between β3 and α3. The substrate binding pocket is occupied by electron density that we interpret as a molecule of glycerol. A number of alternative interpretations were tested, but glycerol refined the best and glycerol molecules were located elsewhere in the structure. To confirm the roles of the two threonines in the catalytic mechanism of AnsA we constructed four mutants: AnsA[T14A], AnsA[T14V], AnsA[T91A] and AnsA[T91V]. The proteins were all expressed to high level, purified and assayed for asparaginase activity. All four mutants had no measurable activity in our assay, indicating a level of activity at least 5,000-fold lower than the wild-type protein (Table 2).
Figure 6. Stereoviews of the AnsA active site.
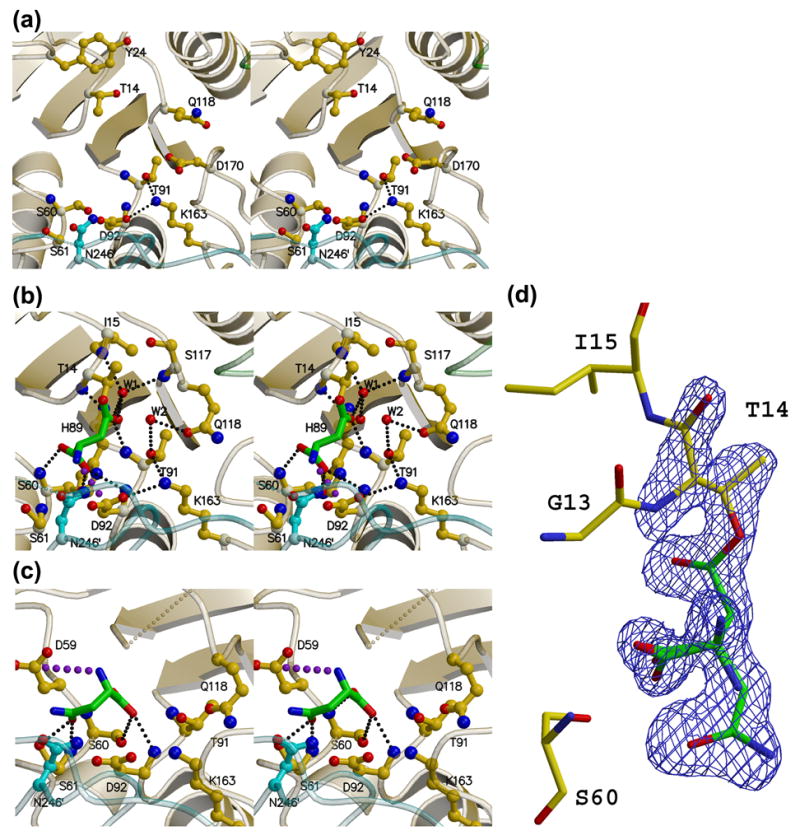
(a) The apo enzyme (APOM). (b) The AnsA-asparagine complex showing the covalently attached product aspartate (green) at the active site. Water molecule W2 hydrogen bonded to Thr91 and Gln118 is ideally positioned to act as the nucleophile that will release the product (green). (c) The AnsA-asparagine complex showing asparagine (green) bound in the non-productive alternate conformation. In each panel, monomer A is yellow, B is green and C is cyan. Details of these interactions are provided in the text, but the key active site residues are Thr14, Thr91, Lys163 and Asp92. Note how Gln118 changes conformation as the tetramer is compacted in (b) versus (a). Note also the pseudo mirror symmetry in the active site that accommodates the two alternate binding modes in (b) and (c). Large purple dots indicate a salt bridge, while small, light gray dots indicate missing structural elements. (d) Electron density observed in the active site that was interpreted as overlapped aspartate and asparagine in the non-productive alternate conformation. The Fo-Fc simulated annealing omit map is displayed at a contour level of 4σ (blue).
The AnsA-Asparagine Complex
Electron density was present at all four active sites in co-crystals of both wild type AnsA and AnsA[T162A] complexed with substrate. However, it was consistently too large for either asparagine (substrate) or aspartate (product) and was interpreted as a mixture of a productive conformation of aspartate and an unproductive, alternative conformation of asparagine (see below). In the productive mode, substrate had been converted into product. In monomer A of the wild type and all four monomers of the mutant, there was clear evidence in the electron density of covalent attachment of the product to the active site with the Cγ carbon of aspartate connected to the Oγ1 oxygen of Thr14. In the other three wild type monomers, the reaction appears to have gone to completion, and no such covalent linkage was seen. Non-covalently and covalently bound aspartates both reside in the active site in a nearly identical manner, and reveal a number of key interactions with conserved active site residues. Covalent attachment is shown in Figure 6(b) and adds two hydrogen bonds between the product and the backbone amides of Thr14 and Ile15 that are now visible in the active site loop. The former donates a hydrogen bond to the free side chain oxygen, and the latter donates a hydrogen bond to the ordered W1 water molecule that is anchored by additional hydrogen bonds to the backbone carbonyl oxygen and amide nitrogen of His89 and Ser117, respectively, where the C-termini of strands β3 and β4 begin to diverge. The amino group of aspartate is presumably charged because it forms a salt bridge with Asp92. One of the two carboxyl oxygen atoms receives a hydrogen bond from the backbone amide of Ser60, and the second oxygen receives hydrogen bonds from the Oγ oxygen of Ser60 and the backbone amide of Asp92. The side chain carboxyl group of the bound aspartate only interacts with the active site via one of the oxygens; it receives a hydrogen bond from the backbone amide nitrogen of Thr91 and a second hydrogen bond from water molecule W1. Finally, the side chains of Asp92, Lys163, Thr91 and Gln118 form a hydrogen bonded pathway that involves a second ordered water molecule (W2) located 3.0 Å from the carbonyl C atom (Cγ) of the ligand in the wild type complex, and 3.3 Å away in CT162A.
Overall, there are minimal structural changes within the monomer in the AsnA-asparagine complex, but there is a significant reorganization in the active site locale at the A/B/C interface. As seen by comparing Figure 7(a) and 7(b) (also visible in Figure 6), this region of the tetramer undergoes a compaction that alters the conformation of the adjacent extended β-ribbon structure (160-174) in the long loop that connects β6 and β7. This change in conformation is caused by the movement of the loop connecting β7′ and β8′ in the adjacent monomer, and the resulting close interaction with Ile185′ leads to a reorientation of Phe169 and Asp170. Residues 160-174 are close to the active site and the space occupied by Asp170 in the apo structure directly adjacent to Thr91 and Lys163 becomes occupied by Gln118 in the AsnA-asparagine complex. This structural change at the active site is a direct result of the putative allosteric movements in the quaternary structure elicited by the asparagine bound at the allosteric site and it was of interest to investigate whether they could be replicated by mutagenesis. The key event is for Asp170 and Gln118 to switch positions during the conformational change. Replacement of Asp170 with a Gln in the mutant AnsA[D170Q] resulted in a protein with kinetic parameters virtually indistinguishable from the wild type enzyme (Table 2). On the other hand, changing Gln118 to an Asp resulted in an enzyme, AnsA[Q118D], with no detectable asparaginase activity. Taken together, these data suggest that the correct positioning of Gln118 is required for enzyme function and it is not the presence of Asp170 per se that is switching the enzyme off.
Figure 7. Relative positioning of Gln118 and Asp170 at the AnsA active site.
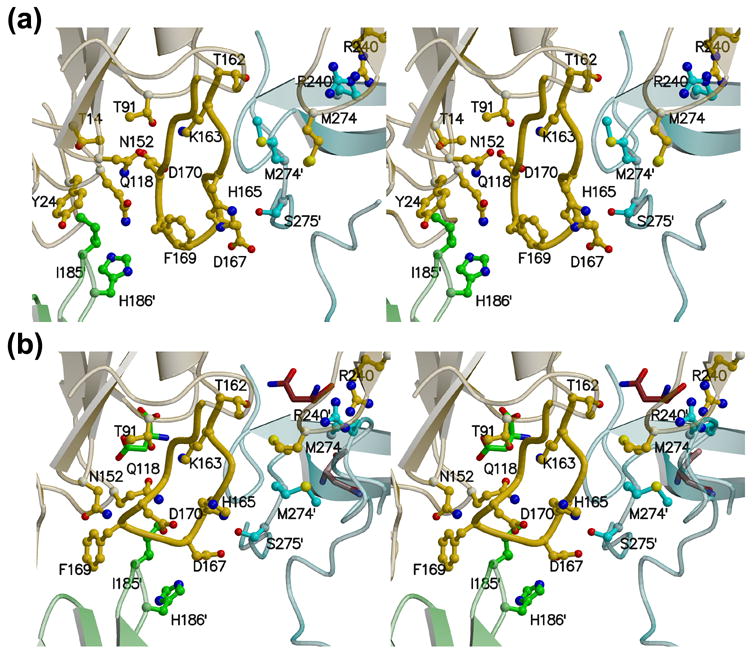
During the allosteric switch upon binding asparagine, the two loops containing Gln118 and Asp170 adjacent to the active site change conformation. As a result, Gln118 occupies the space adjacent to Thr91 and Lys163 that was previously occupied by Asp170 in the apo structure. (a) Stereoview of the apo active site. (b) Stereoview of the AnsA-asparagine active site. In each panel, monomer A is yellow, B is green and C is cyan. Note how the active site locale is at the convergence of three subunits. Details are provided in the text.
Substrate Specificity
Asparaginases from different organisms display varying activities towards L-glutamine and this presumably reflects the differential abilities of their active sites to accommodate the additional carbon atom on the side chain.27,38 This feature has therapeutic consequences,33 and one of our goals is to understand this ancillary activity at the structural level. It has been noted that the optimal length of the substrate is tightly constrained by the binding pocket.27,38 The amido group at one end must lie precisely between the two catalytic threonines for hydrolysis to occur. At the opposite end, the carboxyl group is involved in interactions with the conserved Ser60, while the amino group interacts with Asp92 (Figure 6(b)). Consistent with this, bound glutamine is forced to bow in the active site, and glutaminase activity correlates with one particular residue that dictates whether or not space is available for this to occur.27,38 In our AnsA structure, this residue is Asn246′ that reaches across from the adjacent monomer (Figure 6). Enzymes with low glutaminase activity such as E. coli AnsB have an asparagine residue at this position, whereas enzymes with high activity such as Erwinia AnsB have the smaller serine at this location (Figure 1). This observation is supported by the fact that E. coli AnsA had no measurable activity towards L-glutamine at the concentrations of substrate and enzyme tested (not shown), indicating it is at least 5,000-fold less active towards this substrate than asparagine under these conditions. Interestingly, PhAnsA has an atypical glycine at this location and would be predicted to have substantial glutaminase activity.
Alternate Substrate-Binding Conformation
An unexpected feature of the active sites of both wild type AnsA and AnsA[T162A] complexed with substrate was an alternate substrate binding orientation. In both structures, the electron density was interpreted and successfully refined as an approximately 50% occupancy of aspartate in the productive binding mode (Figure 6(b)) and 50% occupancy of asparagine in a non-productive orientation (Figure 6(c)). Considering the shape of the density (Figure 6(d)) and the fact that citrate was present in the crystallization and cryoprotectant buffers, a molecule of citrate was a possible alternative interpretation. However, four lines of evidence argue against this. First, although our resolution is insufficient to make unequivocal distinctions between oxygen and nitrogen atoms, the side chain electron density is clearly asymmetric and consistent with the with larger oxygen and smaller nitrogen of asparagine and not the symmetric carboxylate group of citrate. Second, evidence in the electron density of a covalent attachment to Thr14 was clearly observed in one monomer of the wild type protein and in all four monomers of the AnsA[T162A] mutant. Citrate, having carboxylic acid groups at either end, would not be expected to react with a nucleophilic threonine, and thus would not be expected to form a covalent adduct with the protein. Third, electron density representing either just the active or the alternate conformation could be seen in the crystal structures (obtained in the same citrate buffer) of AnsA[T14V] and AnsA[T14A] mutants complexed with substrate (data not shown). Thus both modes of binding are observable independently of each other. Fourth, the enzyme was about 2-fold more active in a citrate buffer at pH 6.0 than in a bis-Tris buffer at the same pH (data not shown). If citrate bound tightly to the active site, it would be expected to act as an inhibitor of the enzyme, but this did not appear to be the case.
The relationship between the two modes is an approximate 180° rotation about the Cα-C bond such that the α-carboxyl groups almost exactly superimpose and the side chains point in opposite directions. Structurally, this second binding mode is possible for three reasons: there is an approximate mirror symmetry in the active site that allows the carboxyl and amino groups to engage in similar interactions; there is a pocket in AnsA that can accommodate the side chain in the second orientation; and there are hydrogen bond acceptors and donors around the pocket to interact with the repositioned side chain. For steric reasons, only one binding mode is possible in one active site at any given time. Details of the second binding mode are shown in Figure 6(c) and the mirror symmetry is apparent by comparing it with Figure 6(b). The carboxyl group can still form hydrogen bonds with the backbone amides of Ser60 and Asp92 and the Oγ oxygen of Ser60, and the amino group forms a salt bridge with Asp59 across the pseudo mirror plane from Asp92. Within the side chain pocket, the Oδ1 oxygen forms hydrogen bonds with the backbone amide nitrogen and Oγ oxygen of Ser61, and the Nδ2 nitrogen can potentially interact with the exposed side chain of Asn246′ across the tight dimer interface, although the rotamer orientation of the latter is not suitable in our crystal structures.
Discussion
AnsA is an Allosteric Enzyme
Although there have been many studies on the structure and mechanism of asparaginases, this is the first to explore the allosteric nature of the type I asparaginase from E. coli. Our structural, kinetic, mutagenesis and hydrodynamic results are consistent with the conclusion that the activity of E. coli AnsA depends on the binding of asparagine to four allosteric sites that are distinct from the catalytic active site. Binding of asparagine to the allosteric site activates a cooperative conformational switch from an inactive to an active form at around 1 mM asparagine. An earlier report suggested that the type I E. coli protein exhibits Michaelis-Menten kinetics with a KM of 3.5 mM,6 but our kinetic data clearly show that the enzyme exhibits positive cooperativity with a Hill coefficient of 2.6. The crystal structure of the type I archaeal enzyme from P. horikoshii indicates it exists as a dimer and earlier reports concluded that E. coli AnsA is a dimer.8,13 However, AnsA crystallizes as a tetramer and there is little doubt from our analytical ultracentrifugation experiments that AnsA is a tetramer in solution. Like the other tetrameric asparaginases, AnsA can be described as a dimer of intimate dimers.
The structural characterization of the allosteric binding pocket reveals how it controls the conformational change that activates the quaternary structure. The conformational change in the monomers that elicits the compaction of the tetramer arises from a hinge-like motion between the N- and C-terminal domains around the axis of helix α8 at the domain interface. This movement is allowed based on normal mode analysis, which predicts that rotation around the hinge represents a low energy flexing of the monomer.39 We suggest that the binding of the allosteric asparagines favors the active conformation by anchoring one end of the α–helix to the surrounding protein.
The importance of the allosteric site to catalysis is supported by the removal of the key residue Arg240. The AnsA[R240A] mutant is more highly cooperative than wild-type (nH shifted from 2.6 to 4.2), and the concentration of substrate required for half-maximal velocity increases to 5.9 mM. These kinetics are explained by the fact that loss of Arg240 decreases the affinity of the allosteric site for asparagine, resulting in a higher asparagine concentration being required to switch on the enzyme. Type II asparaginases are clinically useful agents against certain forms of cancer, and the E. coli and Erwinia AnsB proteins are front line drugs in the treatment of childhood leukemia.14,15 These enzymes function by depleting extracellular asparagine that is required for the growth of these tumors. In contrast, the type I proteins tested to date have failed to exhibit anti-cancer activity. Our studies on the allostery of the type I asparaginase illustrate why this class of asparaginases fail to efficiently deplete asparagines and suggest that the search for alternate forms of therapeutic proteins should avoid the intracellular allosteric asparaginases.
Our study is not the first to report allostery in asparaginases. The yeast Saccharomyces cerevisiae has cytoplasmic and extracellular asparaginases.40 The intracellular protein exhibits a low affinity for substrate and sigmoidal substrate-activity dependence, with half maximal velocity at 1 mM asparagine. In contrast, the extracellular protein exhibits no cooperativity and has a KM of about 10 μM. It has been suggested that the gene for the cytoplamsic yeast protein arose by duplication of the type II (extracellular) gene in the yeast lineage since, by sequence analysis, both yeast proteins appear to be type II asparaginases (Figure 1).40–42 Therefore it would seem that the regulation for both the intracellular E. coli AnsA and the intracellular yeast asparaginase evolved convergently due to the need for tight regulation on this critical enzyme in asparagine catabolism. In contrast, the type II asparaginases are involved in nitrogen scavenging and exist in a permanent state of high activity to efficiently hydrolyze asparagine in the environment. The observation that asparaginase II inhibits protein synthesis in cell free extracts indicates that intracellular expression of the type II activity is toxic.43,44
The Active Site
The basic asparaginase catalytic mechanism is established and supported by previous crystallographic data. Amidohydrolysis proceeds by two sequential nucleophilic attacks by the paired active site threonines.10,16,18,19,26,37 The first attacking threonine releases ammonia and generates a covalent enzyme-product intermediate. During the second attack, the opposing threonine activates a water molecule for product release. An early question concerning the mechanism was which of the two threonines performs the first half-reaction. This arose because the first catalytic threonine is located in a flexible loop with no obvious means of activation, and it was considered more likely that the second, rigid, threonine would be a better candidate for the first step.26 However, kinetic and mutagenesis data together with the direct visualization of the covalent enzyme-product intermediate confirm that Thr14 in the flexible loop performs the first nucleophilic attack.19 We also trapped the covalent enzyme intermediate in the wild type and AnsA[T162A]-asparagine complexes, and this intermediate does indeed reside on Thr14 in AnsA. The initial nucleophilic attack by Thr14 would be facilitated by an oxyanion hole to accept the transient build up of negative charge on the Oδ1 oxygen. In our covalent intermediates, the Oδ1 oxygen occupies a pocket that includes a water molecule (W1) and the backbone amides of Thr14 and Thr91. This structure identifies the oxyanion hole, consistent with other structures.19
Important unresolved questions about the active site are the two mechanisms that activate nucleophic attack by the two threonines. A number of studies suggest that the role of the adjacent conserved tyrosine (Tyr24 in AnsA) is to abstract the proton from the theonine Oγ1 oxygen, but then the question is: how is the tyrosine activated? Both residues are part of the same flexible loop, and no structures to date have visualized this loop in a catalytically competent conformation that shows the activation mechanism. This is also a deficiency in our AnsA structures where the active site loop is disordered or in a catalytically incompetent conformation. One suggestion based on the structure of the Pseudomonas enzyme is that a conserved glutamic acid from the adjacent monomer participates in an activating Thr-Tyr-Glu triad.31 This idea does not receive support from our structures or from mutagenesis experiments.26 The mechanism by which Thr91 is activated is less problematical, although specific details are still lacking. In all known asparaginase structures, this threonine is associated with a highly conserved Ser-Asp-Lys triad that corresponds to residues Ser60, Asp92 and Lys163 in AnsA. The threonine Oγ1 oxygen is adjacent to the Nζ nitrogen of the lysine which acts as a general base. We hypothesized that the location of Lys163 is a key factor in the allosteric activation, and in the AnsA[T162A] mutant, the loss of the hydrogen bond from asparagine to Thr162 in the allosteric site would lead to a displacement of the adjacent Lys163 and disruption of the triad. Although no difference in the position of Lys163 was observed in any of our structures, we may not have trapped the appropriate intermediate form. AnsA[T162A] does lose positive cooperativity, and thus Thr162 clearly plays a role in communicating information between the allosteric and active sites.
Trapping of the covalent enzyme-product intermediate in all four monomers of AnsA[T162A] but in just one of the wild type AnsA monomers suggests that the role of allostery is to accelerate the second nucleophilic attack for product release and regeneration of the active site. Clues as to how this occurs come from the allosteric conformational change in the loop that connects β6 and β7 (residues 160 to 174) adjacent to the active site, and the presence of a strategically located water molecule (W2 in Figure 6) in the complexed active sites. The conformational change spatially replaces Glu170 with Gln118 directly adjacent to the Ser-Asp-Lys triad, and mutagenesis reveals that the Gln118 is absolutely required for activity. The water molecule W2 is ideally positioned to be activated by the Oγ1 of Thr91 and to attack the asparagine-enzyme intermediate to release product. W2 is hydrogen bonded to Gln118, which in turn participates in the catalytic triad hydrogen bonding arrangement. Thus, the allosteric movement is required to establish a pocket that binds and activates a nucleophilic water molecule. One caveat to this scenario is that Gln118 is replaced by a methionine in type II asparaginases, including the intracellular yeast asparaginase (Figure 1). However, in the known type II structures, the β6-β7 loop adopts the structure found in activated AnsA. It is notable that the active site and the β6-β7 loop are located within the quaternary structure in the one position where three monomers converge (monomers A, B and C; Figure 7), and are appropriately positioned to ‘sense’ the compaction that occurs upon the binding of asparagine at the allosteric site. The loop between β10 and α6 from the neighboring monomer within the tight dimer (monomer C) flanks one side of the active site, and the loop between β7 and β8 from the loosely associated monomer (monomer B) flanks the other side. Finally, the region of the active site loop that contains Tyr24 is not visible in our complexed structures. This suggests that the loop becomes disordered upon completion of the first half reaction, and it is not obvious why the product remains covalently bound at the active site in our structure.
Alternate Substrate-Binding Conformation
The physiological relevance, if any, of the alternate substrate binding mode at the active site is unknown, but it would be predicted to lower the efficiency of the enzyme by reducing the number of substrate molecules that can bind in the correct configuration for hydrolysis. This feature has not been reported for any of the E. coli AnsB structures, but the corresponding region of the AnsB active site does reveal three potentially pertinent differences. Firstly, type II asparaginases contain a conserved glutamate (283 in E. coli AnsB) that reaches across the dimer interface to make additional interactions with the bound substrate. This residue has been implicated in substrate specificity.27 There is no equivalent residue in type I asparaginases and its absence in AnsA would partly compromise the correct binding of substrate. Secondly, in AnsB, Ser61 is replaced by a glutamine that reduces the size of the pocket. We tested this by making the AnsA[S61Q] mutant, but the structure (not shown) revealed that the glutamine side chain adopts a different rotamer than is observed in AnsB and the alternate substrate conformation can still be accommodated. Surprisingly, AnsA[S61Q] was found to be inactive, and the structure revealed no obvious reasons for this. It may well be that the S61Q mutation actually stabilizes rather than destabilizes the alternate conformation. Finally, AnsB does not have an equivalent residue to Asp59 that forms a salt bridge with the amino group of the substrate in the alternate conformation, and this may be the more important residue for this mode of binding.
Comparison to Pyrococcus horikoshii asparaginase
The only other structure of a type I asparaginase that has been determined is PhAnsA from the archaeon P. horikoshii.8 Nothing is known about the cellular localization or biochemical properties of the PhAnsA. The monomeric structure is very similar to that of E. coli AnsA, and the sequences share sufficient identity (37%) to classify the enzyme as a type I asparaginase (Figure 1). The major difference is that PhAnsA appears to exist as a dimer rather than a tetramer, and an analysis of the putative dimer-dimer interface compared to the type II enzymes supported the crystallographic result.8 Our tetrameric type I structure allows us to re-analyze the putative dimer-dimer interface more accurately, and there is indeed no evidence of a similar interface in PhAnsA. Thus, none of the key interfacial residues shown in Figure 3(b) are conserved, and a superposition of the two type I structures reveals that this region deviates the most although the secondary structure is largely conserved.
It was suggested that a rigid gate in PhAnsA composed of a β-turn (residues 12-30) stabilized by ionic interactions with the carboxy-terminal domain of the other subunit closes the active site and leads to the low affinity of these proteins for their substrate.8 Extracellular asparaginases have a flexible loop in this region, and a high affinity for substrate.8,21 The E. coli AnsA does not have a rigid β-turn in this region. Further, residues missing (unstructured) from our AnsA structure (280–286) are analogous to those involved in the stabilizing interactions in PhAnsA. Thus the gate in the E. coli AnsA enzyme can be considered open and the reasons for the high KM of type I asparaginases cannot be explained by this hypothesis.
The dimer structure might also suggest that PhAnsA would not be allosteric because the changes in the quaternary structure of E. coli AnsA involve movements of the entire tetramer. When the two enzymes are structurally superimposed, the PhAnsA apo enzyme closely matches the AnsA-asparagine conformation rather than the apo conformation suggesting that PhAnsA is in the active state. Further, PhAnsA has a Met in place of Gln118, as do the AnsB-like asparaginases (Figure 1). We therefore predict that PhAnsA is not allosteric, and this conclusion is supported by two additional observations. First, we have suggested that the conformation of the active site loop in AnsA (residues 160-174) that connects β6 and β7 reflects whether the enzyme is ‘on’ or ‘off’, and the corresponding loop in PhAnsA is in the ‘on’ conformation. Second, although there is a putative allosteric binding pocket in PhAnsA at the N-terminus of helix α8, it is crucially different from that of AnsA. The paired arginines that bind two asparagines across the tight dimer interface are replaced with glutamates (residue 230), and the actual pocket is largely occupied by a lysine residue (292). Interestingly, this lysine is located at the N-terminus of helix α8, and the Nζ nitrogen exactly coincides with the amine of the allosteric asparagine in AnsA and interacts in a similar way with two adjacent backbone carbonyl oxygens. Thus, the lysine residue appears to stabilize the active conformation of PhAnsA in the same way that the allosteric asparagine does in activated AnsA. PhAnsA might therefore not have the same biochemical properties or cellular localization as E. coli AnsA.
Methods
Construction of His-AnsA expression vector
The ansA gene was cloned by the polymerase chain reaction from the K12 strain of E. coli, InvαF’, using the primers AnsA For and Rev synthesized in the Hartwell Center for Biotechnology at St Jude (for a list of oligonucleotides used in this study, please contact R.J.H.). The 1087 bp fragment thus obtained was ligated into pCR4Blunt Topo (Invitrogen) according to the manufacturer’s instructions and transformed into Top10 cells. A clone with the correct sequence was identified, the DNA digested with the restriction enzymes NdeI and BamHI and the ansA-containing fragment ligated into similarly digested pET-15b (Novagen). A correct clone was isolated and the plasmid designated pET-AnsA.
Mutagenesis of AnsA
The QuikChange II mutagenesis kit (Stratagene) was used for mutagenesis, with pET-AnsA as the template. For each mutation, a complementary pair of forward and reverse oligos were designed according to the manufacturer’s recommendations. Several clones were picked for each experiment and the entire region was sequenced. A single plasmid containing only the desired mutation was then selected and used for His-tagged protein expression and purification as described below for the wild-type.
Protein Purification
The pET-AnsA plasmid was transformed into E. coli BL21(DE3) (Novagen) and an overnight culture grown in LB media with 100 μg/ml ampicillin at 37 °C. This culture was used to inoculate 2 L of the same media, and grown to an optical density of 0.6 at 600 nm at 37 °C with shaking at 250 rpm. IPTG was then added to 1 mM final concentration, and growth continued for 3 h. Cells were harvested by centrifugation. The pellet was resuspended in MCAC 0 buffer45 (50 mM Tris-Cl, pH 7,9, 500 mM NaCl) and lyzed by passage through a microfluidizer. The cleared lysate was then applied to a 5 ml His-Trap column (GE Healthcare) previously charged with nickel and equilibrated with MCAC 0. His-tagged AnsA was eluted with a linear gradient of imidazole in MCAC 0 and applied to a S200 26/60 size exclusion column equilibrated with 50 mM Tris-Cl pH 7.5, 1 mM DTT, 1 mM EDTA, 150 mM NaCl. Protein was concentrated to 20 mg/ml, flash frozen and stored at −80 °C for crystallography, or stored at −20 °C with 50% (v/v) glycerol for enzyme assays.
Crystallization and Data collection
All crystals of AnsA in the absence and presence of L-asparagine were grown using the hanging-drop vapor-diffusion method at 18 °C. AnsA in the absence of L-asparagine produced a monoclinic crystal (APOM) in a few days by mixing 2 μl of protein solution (20 mg/ml in 25 mM Tris-Cl, pH 7.5, 0.5 mM DTT, 0.5 mM EDTA, and 75 mM NaCl) with 2 μl of reservoir solution containing 0.1 M MES, pH 6.0 and 22.5% MPD. For data collection, the APOM crystal was transferred into cryoprotectant solution containing 0.1 M MES, pH 6.0, 0.5 mM DTT, 50 mM NaCl, 22.5 % MPD and 34 % glycerol, and flash-frozen using liquid nitrogen. For co-crystallization of AnsA and L-asparagine, the complex was first formed by dissolving L-asparagine in the protein solution, removing the excess undissolved material by centrifugation and setting up the drops. The native AnsA complex produced monoclinic crystals (CNATM) and were grown by mixing 2 μl of complex solution with 2 μl of reservoir solution containing 0.1 M citric acid/trisodium citrate, pH 4.0 and 1.0 M sodium chloride. For data collection, the CNATM crystal was transferred into 0.1 M citric acid, pH 4.0, 1.0 M sodium chloride, 0.5 mM L-aspartic acid, 0.5 mM DTT and 50 % ethylene glycol as cryoprotectant, and flash-frozen using liquid nitrogen. The same procedures were used to grow and flash-freeze complex crystals with the AnsA[T162A] point mutant (CT162A) that was isomorphous with CNATM. Diffraction data sets from all crystals were collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22-ID and 22-BM beamlines at the Advanced Photon Source (APS), Argonne National Laboratory. All data sets were collected at 100 K and processed using HKL2000.46 Details of the crystals and the data collection statistics are summarized in Table 1.
Structure Determination
The structure of the apo protein (APOM) was solved by molecular replacement (MR) using the dimer of the type I L-asparaginase from Pyrococcus horikoshii, PhA (PDB entry 1WLS; Ref 8) as a search model. To create the model, an amino acid sequence alignment was generated and all non-identical residues in the search model were replaced by alanine. Rotation and translation functions were calculated using the program MOLREP47 with data in the resolution range of 30 - 3.0 Å, and this revealed two dimers in the asymmetric unit. Rigid body refinement and conjugate gradient minimization refinement produced a model with Rwork and Rfree values of 43.7 % and of 45.2 %, respectively at 30 - 2 Å resolution. The model was improved by iterative cycles of refinement and model building using CNS, Xtalview and Refmac5,48,49,50 and water and glycerol molecules were identified and incorporated. The active site contained strong electron density peaks that could be interpreted as glycerol molecules, phosphate ions, or sulfate ions. However, they were eventually refined as glycerol molecules which are consistent with the crystallization and cryoprotectant conditions. The final model had Rwork and Rfree values of 18.4 % and of 20.7 %, respectively at 50 – 1.89 Å resolution. The CNATM structure was determined by molecular replacement using MOLREP, with the data in resolution range of 30 - 3.0 Å and a monomer of the APOM molecule as the search model. A dimer corresponding to the ‘intimate dimer’ was located by the first rotation and translation search, and a second run with this dimer as the search model identified the complete tetramer in the asymmetric unit. An initial rigid body refinement gave Rwork and Rfree values of 46.4% and of 46.5%, respectively at 30 – 3 Å resolution. Subsequent iterative cycles of refinement using CNS, Xtalview and Refmac5 produced the final model that contained four L-aspartate molecules, eight L-asparagine molecules, four chloride ions, several ethylene glycol molecules, and ordered water molecules. The final Rwork and Rfree values were 22.3 % and of 26.4 %, respectively for data between 50.0 – 1.9 Å resolution. The CT162A structure was directly refined using the isomorphous CNATM structure. The final Rwork and Rfree values were 20.9 % and 22.6 %, respectively, for data between 50.0 – 1.82 Å resolution. The refinement statistics for all three structures are given in Table 1. To check for model bias during refinement, simulated annealing composite omit maps were calculated using CNS, and PROCHECK was used to validate the final structures.51
Analytical Ultracentifugation
Experiments were carried out in a ProteomeLab XL-I analytical ultracentrifuge with an eight-hole Beckman An-50 Ti rotor and cells containing either sapphire or quartz windows and charcoal-filled Epon double-sector centerpieces (Beckman Coulter, Fullerton, CA). The density and viscosity of the buffer (50 mM Tris-Cl pH 7.4, 150 mM NaCl, 1 mM EDTA) at 4 and 20 °C were calculated from its composition and the molar extinction coefficient at 280 nm, partial specific volume at 4 and 20 °C and molecular weight of the protein were calculated based on its amino acid composition using the software SEDNTERP.52 All samples were dialysed against the ultracentrifugation buffer and the dialysate was used as an optical reference. Sedimentation equilibrium was attained at 48 h at a rotor temperature of 4 °C at increasing speeds of 5,000, 7,000 and 10,000 rpm, using the long column technique.53 Protein at concentrations of 0.16 and 0.5 mg/ml (175 μl) was loaded into double-sector centrepieces and absorbance distributions recorded at 280 nm in 0.001 cm radial intervals with 20 replicates for each point. Global least squares modelling was performed at multiple concentrations and rotor speeds with the software SEDPHAT (www.analyticalultracentrifugation.com) using the single-species model. For the sedimentation velocity experiments the loading volume of 400 μl was identical for the reference and sample chambers of the double-sector centrepiece. Following an overnight temperature equilibration at 4°C at rest the rotor was accelerated to 50,000 rpm and refractive index profiles recorded in 1 min intervals with the Rayleigh interference optical system. Interference fringe displacement profiles were analysed with the software SEDFIT (www.analyticalultracentrifugation.com) using a model for continuous sedimentation coefficient distribution c(s) with deconvolution of diffusional effects.54,55 The sedimentation coefficient distribution c(s) was calculated with maximum entropy regularization at a confidence level of p = 0.7 and at a resolution of sedimentation coefficients of n = 100. The positions of the meniscus and bottom, as well as time-invariant and radial noises, were fitted. The s-value of the protein was determined by integration of the main peak of c(s). The molecular weight distribution, c(M), was calculated using the values determined from the c(s) distribution. The best-fit weight-average frictional ratio (f/f0) was determined from the c(s) analysis, and a single value was assumed.54 The s-values of the c(s) data were converted to standard conditions (20 °C, water as solvent) with the program SEDNTERP52 and plotted.
Enzyme Assays
A stopped assay which used Nessler’s reagent to measure the amount of ammonia released was employed.56 Typically, a reaction contained 25 mM Tris-Cl pH 8.6, 0.25 to 40 mM L-asparagine (or L-glutamine) and between 1 ng and 1 μg of His-AnsA (wild type or mutant) in a final volume of 250 μl. Reactions were incubated for 30 minutes at 37 °C, then stopped by the addition of 50 μl of ice-cold 1.5 M trichloroacetic acid (TCA) and placed in an ice bath. After a brief centrifugation, 250 μl of the supernatant was transferred to a semi-micro cuvette to which 200 μl Nessler’s reagent (Aldrich) and 550 μl water had already been added. The optical density was read at 436 nm. For kinetic experiments, 1.5 ml assay were established with approximately 20 ng of AnsA. 250 μl aliquots were removed at timed intervals and placed into tubes containing 50 μl of 1.5 M TCA in an ice bath and then treated as above. The linear portion of the reaction was used. A standard curve was generated with ammonium sulfate standards. Control reactions lacking either enzyme or substrate were included with each experiment, and subtracted from the data.
Acknowledgments
The technical assistance of the Kelly Akin and Dana Roeber of the St Jude Protein Production Facility is greatly appreciated. This work was supported by the National Institutes of Health Grants AI070721 and GM34496, Cancer Center (CORE) Support Grant CA21765 and the American Lebanese and Syrian Associated Charities.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Cedar H, Schwartz JH. Localization of the two L-asparaginases in anaerobically grown Escherichia coli. J Biol Chem. 1967;242:3753–3755. [PubMed] [Google Scholar]
- 2.Schwartz JH, Reeves JY, Broome JD. Two L-asparaginases from E. coli and their action against tumors. Proc Natl Acad Sci U S A. 1966;56:1516–1519. doi: 10.1073/pnas.56.5.1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spring KJ, Jerlstrom PG, Burns DM, Beacham IR. L-asparaginase genes in Escherichia coli: isolation of mutants and characterization of the ansA gene and its protein product. J Bacteriol. 1986;166:135–142. doi: 10.1128/jb.166.1.135-142.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cedar H, Schwartz JH. Production of L-asparaginase II by Escherichia coli. J Bacteriol. 1968;96:2043–2048. doi: 10.1128/jb.96.6.2043-2048.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casale TD, Sollitti P, Chesney RH. Cytoplasmic L-asparaginase: isolation of a defective strain and mapping of ansA. J Bacteriol. 1983;154:513–515. doi: 10.1128/jb.154.1.513-515.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Willis RC, Woolfolk CA. Asparagine utilization in Escherichia coli. J Bacteriol. 1974;118:231–241. doi: 10.1128/jb.118.1.231-241.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jennings MP, Beacham IR. Analysis of the Escherichia coli gene encoding L-asparaginase II, ansB, and its regulation by cyclic AMP receptor and FNR proteins. J Bacteriol. 1990;172:1491–1498. doi: 10.1128/jb.172.3.1491-1498.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao M, Yasutake Y, Morita H, Tanaka I. Structure of the type I L-asparaginase from the hyperthermophilic archaeon Pyrococcus horikoshii at 2.16 Å resolution. Acta Crystallogr D Biol Crystallogr. 2005;61:294–301. doi: 10.1107/S0907444904032950. [DOI] [PubMed] [Google Scholar]
- 9.Lubkowski J, Palm GJ, Gilliland GL, Derst C, Rohm KH, Wlodawer A. Crystal structure and amino acid sequence of Wolinella succinogenes L-asparaginase. Eur J Biochem. 1996;241:201–207. doi: 10.1111/j.1432-1033.1996.0201t.x. [DOI] [PubMed] [Google Scholar]
- 10.Miller M, Rao JK, Wlodawer A, Gribskov MR. A left-handed crossover involved in amidohydrolase catalysis. Crystal structure of Erwinia chrysanthemi L-asparaginase with bound L-aspartate. FEBS Lett. 1993;328:275–279. doi: 10.1016/0014-5793(93)80943-o. [DOI] [PubMed] [Google Scholar]
- 11.Swain AL, Jaskolski M, Housset D, Rao JK, Wlodawer A. Crystal structure of Escherichia coli L-asparaginase, an enzyme used in cancer therapy. Proc Natl Acad Sci U S A. 1993;90:1474–1478. doi: 10.1073/pnas.90.4.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka S, Robinson EA, Appella E, Miller M, Ammon HL, Roberts J, Weber IT, Wlodawer A. Structures of amidohydrolases. Amino acid sequence of a glutaminase-asparaginase from Acinetobacter glutaminasificans and preliminary crystallographic data for an asparaginase from Erwinia chrysanthemi. J Biol Chem. 1988;263:8583–8591. [PubMed] [Google Scholar]
- 13.Jerlstrom PG, Bezjak DA, Jennings MP, Beacham IR. Structure and expression in Escherichia coli K-12 of the L-asparaginase I-encoding ansA gene and its flanking regions. Gene. 1989;78:37–46. doi: 10.1016/0378-1119(89)90312-0. [DOI] [PubMed] [Google Scholar]
- 14.Pui CH, Evans WE. Acute lymphoblastic leukemia. N Engl J Med. 1998;339:605–615. doi: 10.1056/NEJM199808273390907. [DOI] [PubMed] [Google Scholar]
- 15.Pui CH, Campana D, Evans WE. Childhood acute lymphoblastic leukaemia - current status and future perspectives. The Lancet Oncology. 2001;2:597–607. doi: 10.1016/S1470-2045(01)00516-2. [DOI] [PubMed] [Google Scholar]
- 16.Harms E, Wehner A, Aung HP, Rohm KH. A catalytic role for threonine-12 of E. coli asparaginase II as established by site-directed mutagenesis. FEBS Lett. 1991;285:55–58. doi: 10.1016/0014-5793(91)80723-g. [DOI] [PubMed] [Google Scholar]
- 17.Derst C, Henseling J, Rohm KH. Probing the role of threonine and serine residues of E. coli asparaginase II by site-specific mutagenesis. Protein Eng. 1992;5:785–789. doi: 10.1093/protein/5.8.785. [DOI] [PubMed] [Google Scholar]
- 18.Wehner A, Harms E, Jennings MP, Beacham IR, Derst C, Bast P, Rohm KH. Site-specific mutagenesis of Escherichia coli asparaginase II. None of the three histidine residues is required for catalysis. Eur J Biochem. 1992;208:475–480. doi: 10.1111/j.1432-1033.1992.tb17210.x. [DOI] [PubMed] [Google Scholar]
- 19.Palm GJ, Lubkowski J, Derst C, Schleper S, Rohm KH, Wlodawer A. A covalently bound catalytic intermediate in Escherichia coli asparaginase: crystal structure of a Thr-89-Val mutant. FEBS Lett. 1996;390:211–216. doi: 10.1016/0014-5793(96)00660-6. [DOI] [PubMed] [Google Scholar]
- 20.Kozak M, Jaskolski M. Crystallization and preliminary crystallographic studies of a new crystal form of Escherichia coli L-asparaginase II (Ser58Ala mutant) Acta Crystallogr D Biol Crystallogr. 2000;56:509–511. doi: 10.1107/s0907444900000081. [DOI] [PubMed] [Google Scholar]
- 21.Aung HP, Bocola M, Schleper S, Rohm KH. Dynamics of a mobile loop at the active site of Escherichia coli asparaginase. Biochim Biophys Acta. 2000;1481:349–359. doi: 10.1016/s0167-4838(00)00179-5. [DOI] [PubMed] [Google Scholar]
- 22.Kozak M, Jurga S. A comparison between the crystal and solution structures of Escherichia coli asparaginase II. Acta Biochim Pol. 2002;49:509–513. [PubMed] [Google Scholar]
- 23.Sanches M, Barbosa JA, de Oliveira RT, Abrahao NJ, Polikarpov I. Structural comparison of Escherichia coli L-asparaginase in two monoclinic space groups. Acta Crystallogr D Biol Crystallogr. 2003;59:416–422. doi: 10.1107/s0907444902021200. [DOI] [PubMed] [Google Scholar]
- 24.Jaskolski M, Kozak M, Lubkowski J, Palm G, Wlodawer A. Structures of two highly homologous bacterial L-asparaginases: a case of enantiomorphic space groups. Acta Crystallogr D Biol Crystallogr. 2001;57:369–377. doi: 10.1107/s0907444900020175. [DOI] [PubMed] [Google Scholar]
- 25.Howard JB, Carpenter FH. L-asparaginase from Erwinia carotovora. Substrate specificity and enzymatic properties. J Biol Chem. 1972;247:1020–1030. [PubMed] [Google Scholar]
- 26.Aghaiypour K, Wlodawer A, Lubkowski J. Do bacterial L-asparaginases utilize a catalytic triad Thr-Tyr-Glu? Biochim Biophys Acta. 2001;1550:117–128. doi: 10.1016/s0167-4838(01)00270-9. [DOI] [PubMed] [Google Scholar]
- 27.Aghaiypour K, Wlodawer A, Lubkowski J. Structural basis for the activity and substrate specificity of Erwinia chrysanthemi L-asparaginase. Biochemistry. 2001;40:5655–5664. doi: 10.1021/bi0029595. [DOI] [PubMed] [Google Scholar]
- 28.Lubkowski J, Dauter M, Aghaiypour K, Wlodawer A, Dauter Z. Atomic resolution structure of Erwinia chrysanthemi L-asparaginase. Acta Crystallogr D Biol Crystallogr. 2003;59:84–92. doi: 10.1107/s0907444902019443. [DOI] [PubMed] [Google Scholar]
- 29.Lubkowski J, Wlodawer A, Ammon HL, Copeland TD, Swain AL. Structural characterization of Pseudomonas 7A glutaminase-asparaginase. Biochemistry. 1994;33:10257–10265. doi: 10.1021/bi00200a005. [DOI] [PubMed] [Google Scholar]
- 30.Jakob CG, Lewinski K, LaCount MW, Roberts J, Lebioda L. Ion binding induces closed conformation in Pseudomonas 7A glutaminase-asparaginase (PGA): crystal structure of the PGA-SO42−-NH4+ complex at 1.7 Å resolution. Biochemistry. 1997;36:923–931. doi: 10.1021/bi961979x. [DOI] [PubMed] [Google Scholar]
- 31.Ortlund E, LaCount MW, Lewinski K, Lebioda L. Reactions of Pseudomonas 7A glutaminase-asparaginase with diazo analogues of glutamine and asparagine result in unexpected covalent inhibitions and suggests an unusual catalytic triad Thr-Tyr-Glu. Biochemistry. 2000;39:1199–1204. doi: 10.1021/bi991797d. [DOI] [PubMed] [Google Scholar]
- 32.Lubkowski J, Wlodawer A, Housset D, Weber IT, Ammon HL, Murphy KC, Swain AL. Refined crystal structure of Acinetobacter glutaminasificans glutaminase-asparaginase. Acta Crystallogr D Biol Crystallogr. 1994;50:826–832. doi: 10.1107/S0907444994003446. [DOI] [PubMed] [Google Scholar]
- 33.Reinert RB, Oberle LM, Wek SA, Bunpo P, Wang XP, Mileva I, Goodwin LO, Aldrich CJ, Durden DL, McNurlan MA, Wek RC, Anthony TG. Role of Glutamine Depletion in Directing Tissue-specific Nutrient Stress Responses to L-Asparaginase. J Biol Chem. 2006;281:31222–31233. doi: 10.1074/jbc.M604511200. [DOI] [PubMed] [Google Scholar]
- 34.Hernandez-Espinosa D, Minano A, Martinez C, Perez-Ceballos E, Heras I, Fuster JL, Vicente V, Corral J. L-asparaginase-induced antithrombin type I deficiency: implications for conformational diseases. Am J Pathol. 2006;169:142–153. doi: 10.2353/ajpath.2006.051238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evans WE, Tsiatis A, Rivera G, Murphy SB, Dahl GV, Denison M, Crom WR, Barker LF, Mauer AM. Anaphylactoid reactions to Escherichia coli and Erwinia asparaginase in children with leukemia and lymphoma. Cancer. 1982;49:1378–1383. doi: 10.1002/1097-0142(19820401)49:7<1378::aid-cncr2820490713>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 36.Duval M, Suciu S, Ferster A, Rialland X, Nelken B, Lutz P, Benoit Y, Robert A, Manel AM, Vilmer E, Otten J, Philippe N. Comparison of Escherichia coli-asparaginase with Erwinia-asparaginase in the treatment of childhood lymphoid malignancies: results of a randomized European Organisation for Research and Treatment of Cancer-Children’s Leukemia Group phase 3 trial. Blood. 2002;99:2734–2739. doi: 10.1182/blood.v99.8.2734. [DOI] [PubMed] [Google Scholar]
- 37.Ehrman M, Cedar H, Schwartz JH. L-asparaginase II of Escherichia coli. Studies on the enzymatic mechanism of action. J Biol Chem. 1971;246:88–94. [PubMed] [Google Scholar]
- 38.Derst C, Henseling J, Rohm KH. Engineering the substrate specificity of Escherichia coli asparaginase. II Selective reduction of glutaminase activity by amino acid replacements at position 248. Protein Sci. 2000;9:2009–2017. doi: 10.1110/ps.9.10.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hollup SM, Salensminde G, Reuter N. WEBnm@: a web application for normal mode analyses of proteins. BMC Bioinformatics. 2005;6:52. doi: 10.1186/1471-2105-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dunlop PC, Meyer GM, Ban D, Roon RJ. Characterization of two forms of asparaginase in Saccharomyces cerevisiae. J Biol Chem. 1978;253:1297–1304. [PubMed] [Google Scholar]
- 41.Bonthron DT, Jaskolski M. Why a “benign” mutation kills enzyme activity. Structure-based analysis of the A176V mutant of Saccharomyces cerevisiae L-asparaginase I. Acta Biochim Pol. 1997;44:491–504. [PubMed] [Google Scholar]
- 42.Borek D, Jaskolski M. Sequence analysis of enzymes with asparaginase activity. Acta Biochim Pol. 2001;48:893–902. [PubMed] [Google Scholar]
- 43.Arnstein HR, Barwick CW, Lange JD, Thomas HD. Control of protein synthesis by amino acid supply. The effect of asparagine deprivation on the translation of messenger RNA in reticulocyte lysates. FEBS Lett. 1986;194:146–150. doi: 10.1016/0014-5793(86)80066-7. [DOI] [PubMed] [Google Scholar]
- 44.Iwamaru Y, Miyake M, Arii J, Tanabe Y, Noda M. An inhibitory factor for cell-free protein synthesis from Salmonella enteritidis exhibits cytopathic activity against Chinese hamster ovary cells. Microb Pathog. 2001;31:283–293. doi: 10.1006/mpat.2001.0471. [DOI] [PubMed] [Google Scholar]
- 45.Petty KJ. Metal-chelate affinity chromatography. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Strugh K, editors. Current Protocols in Molecular Biology. John Wiley & Sons, Inc.; 1994. pp. 10.11.8–10.11.22. [DOI] [PubMed] [Google Scholar]
- 46.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 47.Vagin A, Teplyakov A. An approach to multi-copy search in molecular replacement. Acta Crystallogr D Biol Crystallogr. 2000;56:1622–1624. doi: 10.1107/s0907444900013780. [DOI] [PubMed] [Google Scholar]
- 48.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 49.McRee DE. A visual protein crystallographic software system for X11/Xview. J Molecular Graphics. 1992;10:44–46. [Google Scholar]
- 50.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Method Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 51.Laskowski RA, McArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the quality of protein structures. J Appl Crystallogr. 1993;26:282–291. [Google Scholar]
- 52.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. In: Analytical Centrifugation in Biochemistry and Polymer Science. Harding SE, Rowe AJ, Horton JC, editors. The Royal Society of Chemistry; Cambridge: 1992. pp. 90–125. [Google Scholar]
- 53.Vistica J, Dam J, Balbo A, Yikilmaz E, Mariuzza RA, Rouault TA, Schuck P. Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal Biochem. 2004;326:234–256. doi: 10.1016/j.ab.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 54.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schuck P, Perugini MA, Gonzales NR, Howlett GJ, Schubert D. Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophys J. 2002;82:1096–1111. doi: 10.1016/S0006-3495(02)75469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wriston JC., Jr Asparaginase. Methods Enzymol. 1985;113:608–618. doi: 10.1016/s0076-6879(85)13082-x. [DOI] [PubMed] [Google Scholar]
- 57.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 58.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Prot Eng. 1995;8:127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]