

Abstract
Cell migration, a highly complex physiological phenomenon that requires the co-ordinated and tightly regulated function of several proteins, is mediated by a number of signalling pathways. Elucidation of the molecular mechanisms of cell migration impacts our comprehension of numerous cell functions, ranging from development and immune surveillance to angiogenesis and metastasis. The scaffold protein IQGAP1, which binds multiple proteins and regulates their functions, promotes cell motility. Many of the IQGAP1 binding proteins have been implicated in cell migration. In this study, we employed a multifaceted strategy to identify proteins that contribute to IQGAP1-stimulated cell migration. Using specific IQGAP1 point mutant constructs, an interaction with actin was shown to be essential for IQGAP1 to increase cell migration. In contrast, eliminating the binding of Ca2+/calmodulin, but not Ca2+-free calmodulin, augmented the ability of IQGAP1 to stimulate cell migration. Consistent with these findings, selective inhibition of calmodulin function at the plasma membrane with a specific peptide inhibitor enhanced cell migration mediated by IQGAP1. Interestingly, immunofluorescence staining and confocal microscopy suggest that localization of Cdc42 at the leading edge is not necessary for maximal migration of epithelial cells. Coupled with the observations that Cdc42 and Rac1 contribute to IQGAP1-stimulated cell migration, these data suggest that IQGAP1 serves as a junction to integrate multiple signalling molecules to facilitate cell migration.
Keywords: IQGAP1, calmodulin, scaffold, cell migration, actin
INTRODUCTION
Migration is a fundamental characteristic of cells that is necessary for normal embryonic development, protection against infection, tissue development and tissue repair [1, 2]. Numerous proteins and signalling pathways interact to orchestrate directed cell movement. The molecular mechanisms underlying cell migration are highly complex, but published studies from many laboratories have enhanced our comprehension of this process. Based on our current understanding, cell migration is a cyclic process [1]. A migration-promoting agent induces cell polarization and the extension of protrusions in the direction of migration. Adhesion to the extracellular matrix or adjacent cells stabilizes the protrusions and serves as sites of traction for migration. The adhesions at the rear of the cell are detached, allowing the cell to move forward. The cytoskeleton, particularly actin and microtubules, participates with molecular motors to move the cells forward [2]. Multiple proteins are involved in the steps of cell migration. These include the Rho family of GTPases (RhoA, Rac1 and Cdc42), phosphoinositide 3-kinases, atypical protein kinase C, the WASP/WAVE family and Src family kinases [1].
As outlined above, the cytoskeleton is an integral component of cell motility. IQGAP1 is an important regulator of the cytoskeleton [3]. IQGAP1 modulates actin function both by directly binding to F-actin [4, 5] and indirectly via Cdc42 and Rac1 [6–9]. In addition, IQGAP1 appears to capture growing microtubule ends by binding CLIP-170 in a process regulated by Cdc42 and Rac1 [10]. Consistent with its role as a modulator of cytoskeletal function, IQGAP1 contributes to cell motility. Mataraza et al. [11] were the first to document that IQGAP1 promotes cell motility. They showed that overexpression of IQGAP1 enhances cell migration, while specific knockdown of endogenous IQGAP1 by small inhibitory RNA (siRNA) significantly reduces cell motility [11]. A number of other investigators subsequently validated a role for IQGAP1 in cell motility [12–14].
IQGAP1 binds to multiple proteins, including calmodulin, actin, Cdc42, Rac1, E-cadherin, β-catenin, adenomatous polyposis coli (APC) and components of the mitogen-activated protein (MAP) kinase pathway (specifically MEK and ERK) [15–18]. Initial investigation revealed that the interaction of IQGAP1 with Cdc42 and Rac1 contributes to its effects on cell migration [11]. Interestingly, IQGAP1 is necessary for Cdc42 to increase epithelial cell motility [11], implying a bi-directional communication between IQGAP1 and Cdc42/Rac1 in cell migration. Notwithstanding these findings, several other binding partners of IQGAP1, including actin, ERK, Ca2+, calmodulin and microtubules, have been implicated in cell migration [1, 19]. Therefore, in this study we investigated the molecular mechanisms by which IQGAP1 promotes cell motility. Our data reveal that proteins in addition to Cdc42 and Rac1 participate in IQGAP1-stimulated cell motility. We document that an interaction of IQGAP1 with actin is necessary and also show that calmodulin influences this function of IQGAP1. Collectively these findings provide insight into the complex mechanism of cell migration.
EXPERIMENTAL PROCEDURES
Reagents
Cell culture reagents were from Invitrogen (Grand Island, NY). The anti-IQGAP1 polyclonal and monoclonal antibodies [4, 17] and anti-calmodulin monoclonal antibody [20] have been previously characterized. Anti-Myc monoclonal antibody (9E10.2) was manufactured by Maine Biotechnology. Antibodies were purchased as follows: monoclonal anti-Cdc42 from Transduction Laboratories (Franklin Lakes, NJ), anti-green fluorescent protein antibody (for Western blotting) from Clontech (Mountain View, CA) and horseradish peroxidase-conjugated secondary antibodies from GE Healthcare (Piscataway, NJ). Enhanced chemiluminescence (ECL) reagents were from Millipore (Billerica, MA). All other reagents were of standard analytical grade.
Plasmid Constructs
Myc-tagged human IQGAP1 in pcDNA3 vector [4] was used. The construction of IQGAP1·apoCaM(−) and IQGAP1·CaM(−), point mutant constructs that lack binding to apocalmodulin and calmodulin, respectively, has been described previously [21]. IQGAP1G75Q was generated by site-directed mutagenesis using Strategene Quick Change site-directed mutagenesis method. Thermal cycling was performed with 5’-GAGGGGCTTAGGAATCAGGTCTACCTTGCCAA-3’ and 5’-TTGGCAAGGTAGACCTGATTCCTAAGCCCCTC-3’ (mutated residues are underlined) as primer pair, pSCRIPTII-IQN as template and pfu DNA polymerase. These changes result in replacement of Gly-75 with G1n. The template was digested with Dpn1 and transformed into DH5α. After the sequence was confirmed by DNA sequencing, the IQN in pcDNA3-Myc-IQGAP1 was replaced with the IQN segment containing the G75Q mutant. Full-length IQGAP1 was tagged at the N-terminus with tandem enhanced green fluorescent protein (GFP) as described [22]. GFP-tagged versions of IQGAP1·apoCaM(−) and IQGAP1·CaM(−) were prepared by replacing a 2.1 Kb Pac1-Af1II fragment in wild type pEGFP-IQGAP1 with the same fragment from pcDNA3-IQGAP1·apoCaM(−). or pcDNA3-IQGAP1·CaM(−). The sequence of the constructs was confirmed by both restriction mapping and DNA sequencing.
Cell Culture and Transfection
MCF-7 human breast epithelial cells (from American Type Culture Collection) were maintained in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS). MCF-7 cells which stably overexpress either pcDNA3 (MCF/V) or pcDNA3-myc-IQGAP1 (MCF/I) have been described previously; MCF/I cells have 3-fold more IQGAP1 than MCF/V cells [8]. Transient transfections were performed using FuGENE 6 (Roche Molecular Biochemicals) as previously described [23]. Five micrograms of the relevant plasmid was transfected into MCF-7 cells. After 48 h incubation, cells were lysed and equal amounts of protein lysate were resolved by SDS-PAGE and Western blotting. Blots were probed with the antibodies indicated in the figures. IQGAP1 siRNA was engineered by oligonucleotide hybridisation as previously described [11], cloned into vector mU6pro and transfected into MCF-7 cells using FuGENE 6.
A synthetic gene that encodes the calmodulin-binding sequence of myosin-light chain kinase [24, 25] (kindly provided by Marcia Kaetzel and John Dedman, University of Cincinnati) was used. The construct contains four tandem calmodulin-binding peptide (CaMBP) repeats. The CaMBP was tagged with a yellow-green variant of enhanced green fluorescent protein (EYFP) and inserted into plasmids that selectively target it to two subcellular domains, the nucleus or the plasma membrane [25]. The correct subcellular localization of these CaMBPs has been verified by confocal microscopy [25]. The EYFP-containing plasmid was used as control.
Western Blotting and Immunoprecipitation
Confluent 100 mm plates of MCF-7 cells were wounded multiple times as described [11]. At different times after wounding (see Fig. 1), cells were lysed in buffer A (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EGTA, 1% Triton X-100 and protease inhibitors). Equal amounts of protein were subjected to SDS-PAGE and blots were probed with anti-Cdc42 or anti-IQGAP1 monoclonal antibodies, followed by horseradish peroxidase-conjugated goat anti-mouse antibodies and developed with ECL. For immunoprecipitation, equal amounts of protein lysate were incubated with anti-IQGAP1 polyclonal antibodies for 3 h at 4 °C. Anti-IQGAP1 immune complexes were collected for 2 h with 40 µl Protein A-Sepharose (Amersham Pharmacia Biotech), washed five times with buffer A and Western blotting for IQGAP1 and Cdc42 was carried out as outlined above.
Fig. 1.
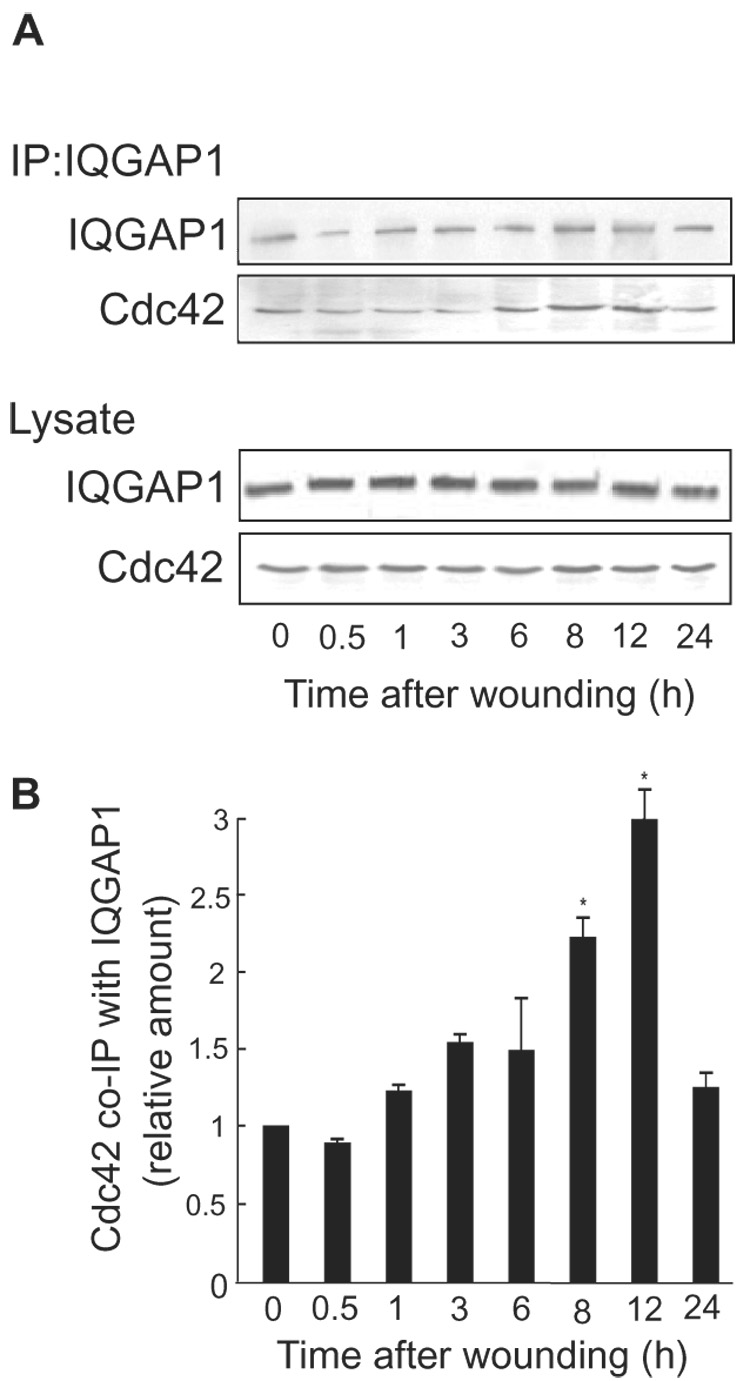
Increase in co-immunoprecipitation of Cdc42 with IQGAP1 over wounding timecourse. A, confluent 100 mm plates of MCF-7 cells were wounded multiple times and lysed at the indicated times after wounding. Equal amounts of protein were subjected to SDS-PAGE and blots were probed with anti-IQGAP1 and anti-Cdc42 antibodies (Lysate), followed by horseradish peroxidase-conjugated goat anti-mouse antibodies and developed with ECL. Equal amounts of protein were also immunoprecipitated (IP) with anti-IQGAP1 antibodies. Complexes were collected with Protein A-Sepharose, resolved by SDS-PAGE, and Western blots were probed with anti-IQGAP1 and anti-Cdc42 antibodies. Data are representative of 3 independent experiments. B, the relative amount of Cdc42 that co-immunoprecipitated with IQGAP1 was quantified by densitometry and corrected for the amount of IQGAP1 immunoprecipitated. Data, expressed relative to the amount of Cdc42 at time 0, represent the means ± S.E. (n=3). *, significantly different from time 0 (p<0.05).
Migration Assays
Migration assays were performed essentially as previously described [11]. Briefly, Transwells with 8 micron pores were placed in 12-well tissue culture plates (Corning, Inc.) and the underside of the membranes was coated with human collagen I (Collaborative Biomedical Products) at 37 °C. Collagen was removed after 18 h and replaced with DMEM. Cells were trypsinized, washed once in DMEM and counted with a hemacytometer. Cells were resuspended at a concentration of 200 000/ml in DMEM containing 5 mg/ml BSA and 1.5 ml of this suspension was added to the top chamber of the Transwell, while 1.5 ml of DMEM was added to the lower chamber. After 16 h, assays were terminated by wiping the top of the Transwell membrane with a cotton swab to remove non-migratory cells. Membranes were rinsed with PBS, then fixed and stained with DiffQuick (Baxter Scientific Products) and mounted on glass slides. Quantification of cells was performed by counting four microscope fields per well using a 10X objective (at least 600 cells were counted for each Transwell membrane). All assays were done in triplicate.
Actin Binding Assay
In vitro actin binding assays were performed essentially as previously described [26]. GST fusion proteins were expressed in Escherichia coli and isolated with glutathione-Sepharose as described [4]. GST-IQGAP1, GST-IQGAP1G75Q, and GST (as a negative control) were bound to glutathione-Sepharose beads and incubated for 15 min with 10 µM F-actin in phosphate-buffered saline (PBS) containing 1 mM MgCl2 and 1 mM DTT at 4 °C. Unbound actin was removed by centrifugation for 5 s at 13,000 × g. Under these conditions virtually none of the F-actin was in the pellet in the absence of IQGAP1 on the beads (see Fig. 3A). Bound actin was visualized by Coomassie blue staining.
Fig. 3.
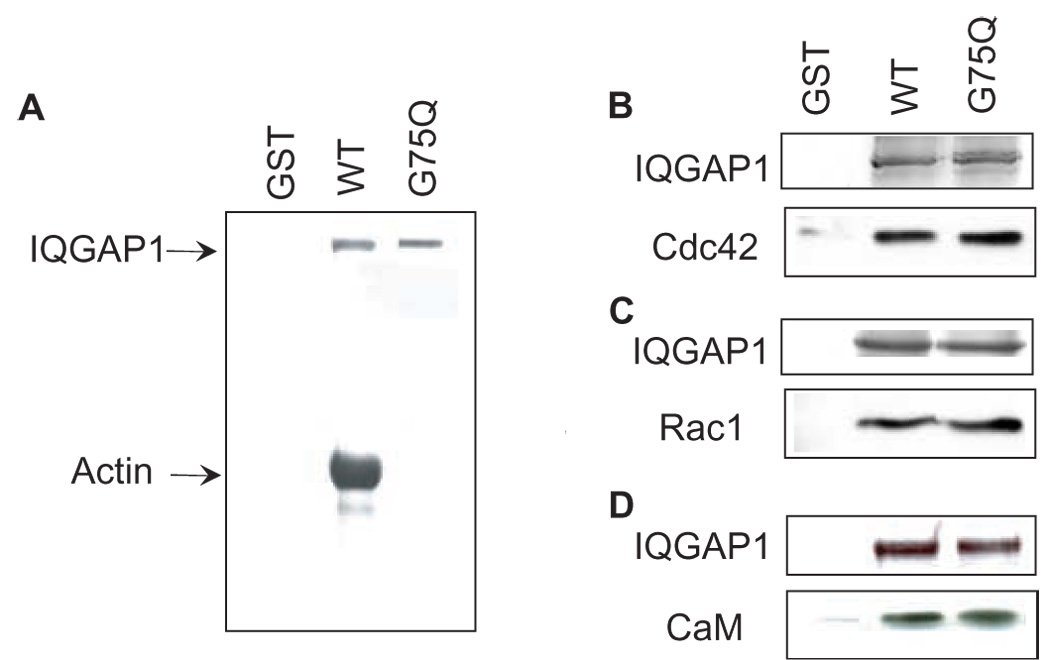
IQGAP1G75Q does not bind F-actin. A, GST, wild type (WT) GST-IQGAP1 or GST-IQGAP1G75Q (G75Q) bound to glutathione-Sepharose beads was incubated for 15 min with F-actin. Samples were washed, resolved by SDS-PAGE and bound actin was visualized by Coomassie blue staining. Data are representative of three independent experimental determinations. B–D, equal amounts of GST, wild type GST-IQGAP1 or GST-IQGAP1G75Q bound to glutathione-Sepharose were incubated with 1.5 mg of protein lysate from MCF-7 cells. After washing, the bound proteins were resolved by SDS-PAGE. The gel was cut into two pieces; the top half was stained with Coomassie blue to visualize the GST-IQGAP1 constructs. The bottom half was transferred to polyvinylidene difluoride membrane and probed for Cdc42 (B), Rac1 (C) or calmodulin (CaM) (D). Representative experiments of three independent determinations are shown.
GST Pull-down Assays
Equal amounts of GST, GST-IQGAP1 (wild type) or GST-IQGAP1G75Q bound to glutathione-Sepharose beads were incubated with 1.5 mg protein lysate from MCF-7 cells. After 3 h at 4 °C, samples were sedimented by centrifugation and washed 5 times with buffer A. Samples were resolved by SDS-PAGE and the gel was cut into two pieces. The top half of the gel was stained with Coomassie blue to visualize the GST-tagged IQGAP1 constructs. The bottom half of the gel was transferred to polyvinylidene difluoride membranes. The blots were probed with antibodies to Cdc42, Rac1 or calmodulin. Separate analyses were performed for each protein.
Wound Healing Assay
The wound healing assay was performed as described [11]. MCF-7 cells were plated onto glass coverslips and transiently transfected with GFP-tagged wild type IQGAP1, IQGAP1·apoCaM(−) or IQGAP1·CaM(−). Cells were allowed to grow until confluent, then the monolayer was scraped with a pipette tip and washed to remove dislodged cells. After 8 h, cells were processed for immunocytochemistry.
Immunofluorescence Staining and Confocal Microscopy
Immunocytochemistry was performed essentially as described [11, 27] with a few modifications. Briefly, cells were washed twice with phosphate-buffered saline (PBS) and incubated in 4% paraformaldehyde-PBS for 20 min at 4 °C. After washing twice in PBS, cells were permeabilized in 0.2% Triton X-100 with 3% BSA in PBS for 1 h at 22 °C. Polyclonal anti-Cdc42 and monoclonal anti-GFP antibodies (both from Santa Cruz Biotechnology) were added in 0.2% Triton/1% BSA for 16 h at 4 °C, followed by Alexa-Fluor 488-labelled anti-rabbit and Alexa-Fluor 543-labelled anti-mouse IgG secondary antibodies (Molecular Probes), and Alexa-Fluor 633-conjugated phalloidin (Invitrogen) (to visualize actin) for 1 h. The anti-GFP antibodies were used to enhance the intensity of the GFP signal.
The stained cells were analyzed using a Zeiss LSM 510 confocal microscope and analyzed with Zeiss LSM software. The number of migrating cells which had the relevant GFP-tagged IQGAP1 construct, Cdc42 or actin at the leading edge was quantified.
Miscellaneous
Protein concentrations were measured with the DC protein asay (Bio-Rad). Densitometry of ECL signals was performed with Un-scan-it software (Silk Scientific Corp.). Statistical analyses were assessed by Student’s t test with InStat software (GraphPad Software, Inc.).
RESULTS
Cell migration promotes the interaction of Cdc42 with IQGAP1
The time-dependent interaction between IQGAP1 and Cdc42 in migrating cells was examined by co-immunoprecipitation. Confluent plates of MCF-7 cells, wounded multiple times, were lysed at different time intervals after wounding. Equal amounts of protein lysate were immunoprecipitated with anti-IQGAP1 antibodies. This revealed that the amount of Cdc42 that co-immunoprecipitated with IQGAP1 increased in a time-dependent manner, with a maximum increase of 2.99 ± 0.2-fold (mean ± S.E., n=3) seen at 12 h (Fig. 1A). Binding of Cdc42 to IQGAP1 returned virtually to baseline levels by 24 h, after the wound had closed. The total amounts of Cdc42 and IQGAP1 in the cells did not change (Fig. 1). These data reveal a significantly enhanced association between IQGAP1 and Cdc42 in migrating cells and provide additional evidence to support our prior observation [11] that the interaction between Cdc42 and IQGAP1 contributes to cell migration.
IQGAP1 targets in addition to Cdc42 and Rac1 participate in IQGAP1-mediated cell migration
We previously documented that IQGAP1 promotes cell migration in a Cdc42- and Rac1-dependent manner [11]. In order to ascertain whether this interaction is sufficient for IQGAP1 to increase cell migration, we examined the effect on cell migration of a mutant IQGAP1. By deleting 24 amino acids from the Cdc42- and Rac1-binding region of IQGAP1, we produced a construct (termed IQGAP1ΔMK24) that specifically lacks binding to Cdc42 and Rac1 [28]. Transient overexpression of wild-type IQGAP1 significantly enhanced (by 2.49 ± 0.09-fold) migration of MCF-7 cells through Transwell pores (Fig. 2). Conversely, transient knockdown of IQGAP1 by siRNA reduced MCF-7 cell migration. These data are consistent with our prior observations (obtained with MCF-7 cells in which IQGAP1 was stably overexpressed or knocked down) that increasing and decreasing intracellular IQGAP1 concentrations increase and decrease, respectively, cell motility [11]. Transient transfection of MCF-7 cells with IQGAP1ΔMK24 increased cell migration, but substantially less than that produced by wild type IQGAP1 (Fig. 2). Western blotting of cell lysates verified that IQGAP1ΔMK24 and wild-type IQGAP1 were expressed at similar levels (Fig. 2C) and that siRNA reduced endogenous IQGAP1 (data not shown). These data indicate that proteins other than Cdc42 and Rac1 contribute to IQGAP1-stimulated cell migration.
Fig. 2.
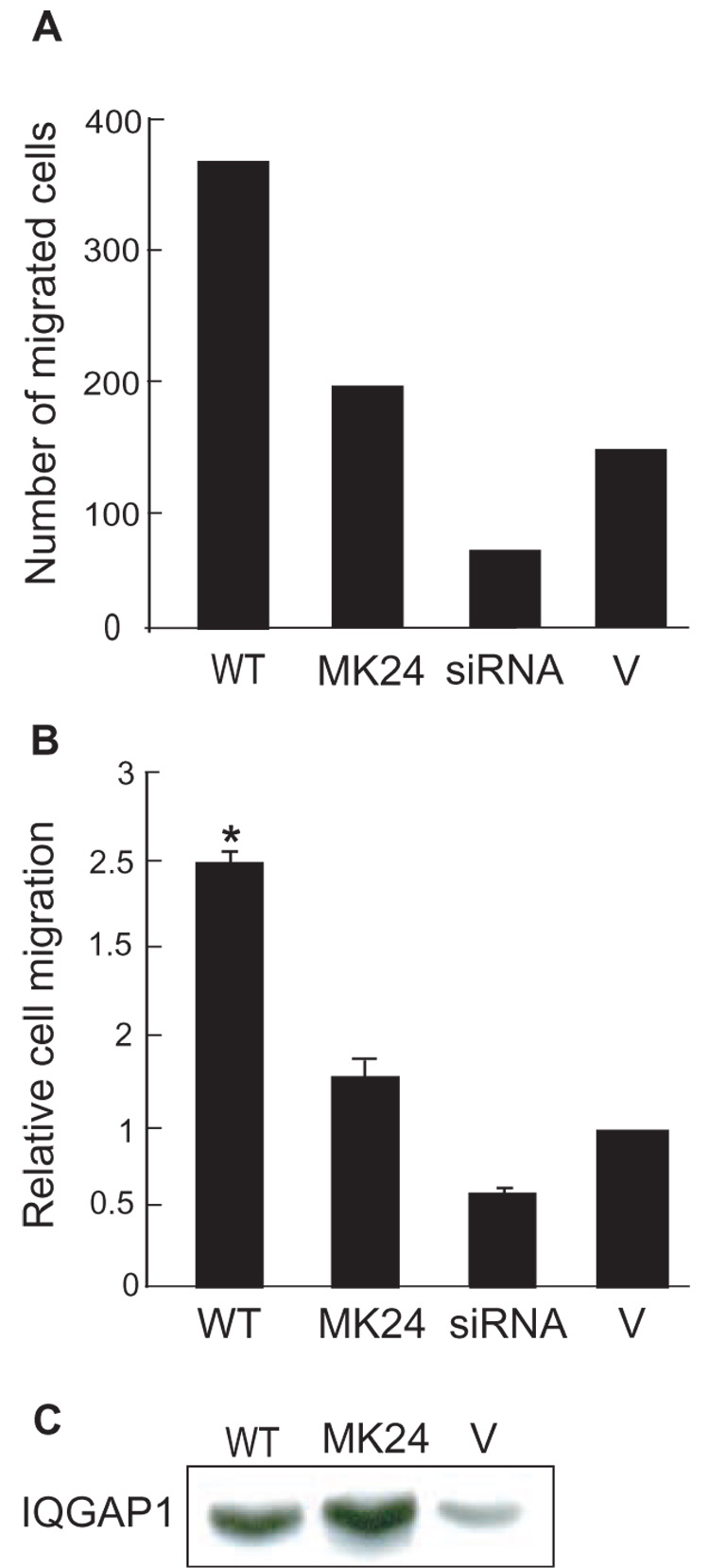
Effect of IQGAP1ΔMK24 on cell migration. Migration through Transwell membranes was performed with MCF-7 cells transiently transfected with wild type (WT) IQGAP1, IQGAP1ΔMK24 (MK24), IQGAP1 siRNA (siRNA) or vector (V). Migration was quantified by counting fields of migratory cells under a light microscope. Four fields per well were counted and all assays were done in triplicate. A, a representative experiment is shown to indicate the relative number of cells migrating. B, data, expressed relative to migration of vector-transfected cells, represent the means ± S.E. from 3 independent experimental determinations. *, significantly different from vector-transfected cells (p<0.001). C, a representative Western blot to show overexpression of IQGAP1 proteins. Equal amounts of protein lysate from MCF-7 cells transfected with wild type IQGAP1, IQGAP1ΔMK24 or vector were resolved by SDS-PAGE and Western blots were probed with anti-IQGAP1 antibody.
Binding to actin is necessary for IQGAP1 to promote cell motility
Actin, a target of IQGAP1, is required for cell migration [29]. To determine whether an interaction with actin is necessary for IQGAP1 to promote cell migration, we generated a point mutant IQGAP1 construct that specifically lacks binding to actin. Gly75 in the actin-binding calponin homology domain was mutated to Gln. The construct is termed IQGAP1G75Q. In vitro analysis with purified proteins revealed that F-actin bound to GST-IQGAP1 (Fig. 3A). In contrast, GST-IQGAP1G75Q was unable to pull down F-actin (Fig. 3A). Binding was specific for IQGAP1 as F-actin did not interact with GST alone. Because IQGAP1G75Q has replacement of only a single amino acid, it is unlikely that binding to proteins other than actin will be altered. Nevertheless, it is important to establish this premise prior to conducting assays in intact cells. Therefore, we incubated GST-IQGAP1G75Q and GST-IQGAP1 with cell lysates and compared the relative amounts of proteins that bound. These data reveal that binding of Cdc42, Rac1 and calmodulin to IQGAP1G75Q and to wild type IQGAP1 are essentially the same (Fig. 3B–D), verifying that the point mutation does not interfere with these interactions.
The requirement of actin for IQGAP1 to enhance cell motility was assessed by transiently transfecting IQGAP1G75Q into MCF-7 cells. Although expressed to essentially the same level as the wild type protein (Fig. 4B), IQGAP1G75Q failed to increase the ability of MCF-7 cells to migrate through a Transwell membrane (Fig. 4A). These data document that an interaction with actin is necessary for IQGAP1 to promote cell motility.
Fig. 4.
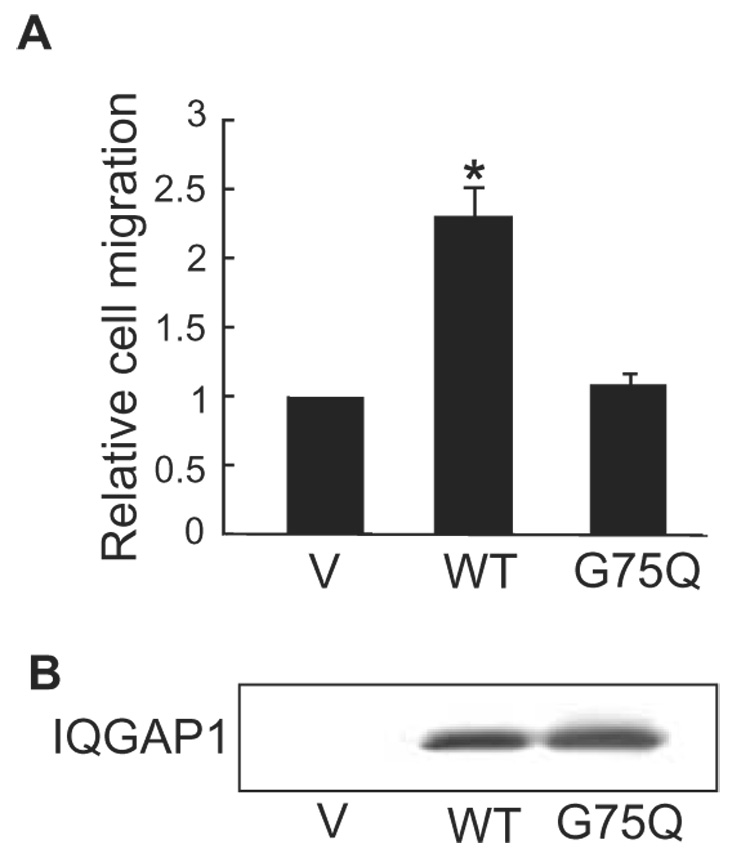
IQGAP1G75Q is unable to promote cell migration. A, Transwell migration assays were performed with MCF-7 cells transiently transfected with vector (V), wild type (WT) IQGAP1, or IQGAP1G75Q (G75Q). Migration was quantified by counting fields of migratory cells under a light microscope. Data, expressed relative to migration of vector-transfected cells, represent the means ± S.E. (n=3). *, significantly different from vector-transfected cells (p<0.01). B, a representative Western blot derived from cells transfected as described for panel A. Equal amounts of protein were resolved and probed with anti-myc antibody (all IQGAP1 constructs are myc-tagged).
Effect of calmodulin on IQGAP1-stimulated cell migration
Ca2+ and calmodulin have been reported to participate in cell migration [30–32]. Moreover, in the presence of Ca2+, calmodulin abrogates the binding of IQGAP1 to Cdc42 [7]. Therefore, we examined the effect of calmodulin on IQGAP1-stimulated cell migration. Analysis was performed with two point-mutant IQGAP1 constructs we developed: IQGAP1·CaM(−), which is unable to bind calmodulin, and IQGAP1·apoCaM(−), which lacks binding to apocalmodulin (Ca2+-free calmodulin) but binds Ca2+/calmodulin [21]. Replacement of basic charged arginine residues in each of the four IQ motifs (and glutamine in the second IQ motif) yields a construct to which apocalmodulin does not bind. To eliminate all calmodulin binding, selected hydrophobic residues and the amino acid immediately proximal to the glutamine (the “I” of the IQ) were replaced in each IQ motif. These constructs have been described and characterized in detail previously [21]. Overexpression of IQGAP1·apoCaM(−) enhanced cell migration by essentially the same magnitude as that produced by wild type IQGAP1 (Fig. 5A). [Note: The vector and wild type IQGAP1 data presented here are the same as those shown in Fig. 4A as the analysis of IQGAP1·CaM(−), IQGAP1·apoCaM(−) and IQGAP1G75Q were done in parallel. The vector and wild type data are reproduced here to facilitate interpretation.] Transient transfection of IQGAP1·CaM(−) increased cell migration by 3.8 ± 0.34-fold, significantly more than that produced by wild type IQGAP1 (Fig. 5A). All IQGAP1 constructs were expressed at similar levels (Fig. 5B). These findings suggest that binding of Ca2+/calmodulin attenuates IQGAP1-stimulated cell migration, while apocalmodulin has no effect.
Fig. 5.
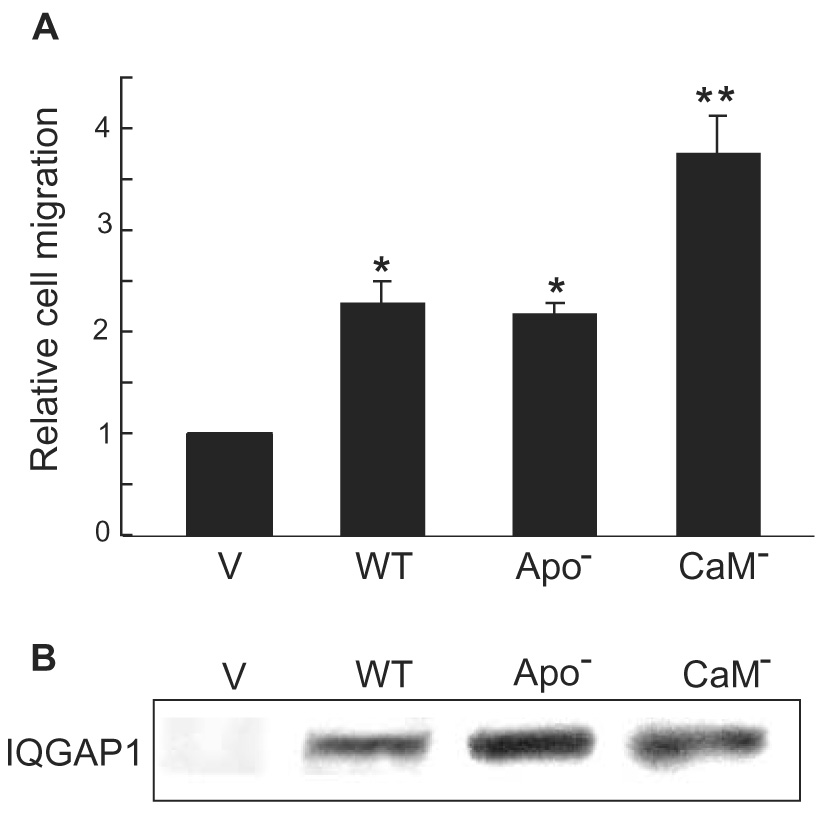
IQGAP1·CaM(−) further increases IQGAP1-stimulated cell migration. A, Transwell migration assays were performed with MCF-7 cells transiently transfected with empty pcDNA3 vector (V), wild type (WT) IQGAP1, IQGAP1·apoCaM(−) (Apo−) or IQGAP1·CaM(−) (CaM−). Migration was quantified by counting fields of migratory cells under a light microscope. Data, expressed relative to migration of vector-transfected cells, represent the means ± S.E. (n=3). [Note: The vector and wild type IQGAP1 data presented here are the same as those shown in Fig. 4A as the analysis of IQGAP1·CaM(−), IQGAP1·apoCaM(−) and IQGAP1G75Q were done in parallel. The vector and wild type data are reproduced here to facilitate interpretation.] *, significantly different from vector-transfected cells (p<0.01). **, significantly different from vector-transfected cells (p<0.001). B, equal amounts of protein lysate from MCF-7 cells transiently transfected as described for panel A were processed by SDS-PAGE and Western blotting. Blots were probed with anti-myc antibody.
The possible role of calmodulin in IQGAP1-stimulated cell migration was also examined by a complementary strategy. A peptide corresponding to the calmodulin-binding domain of myosin-light chain kinase specifically inhibits calmodulin function in cells [24, 25]. By selectively targeting this construct to the nucleus or plasma membrane, we are able to inhibit calmodulin function in discrete subcellular domains [25]. To determine the effect on migration stimulated by IQGAP1, we used cells that stably overexpress IQGAP1. Termed MCF/I, these cells have 3-fold more IQGAP1 than the control, vector-transfected MCF/V cells [11, 17, 33, 34]. As previously documented [11], the motility of MCF/I cells is greater than that of MCF/V cells (Fig. 6A). Inhibition of calmodulin in the nucleus did not alter the ability of IQGAP1 overexpression to augment cell migration. In contrast, when calmodulin was selectively inhibited at the membrane, the ability of IQGAP1 to increase cell migration was significantly enhanced (Fig. 6A). The membrane-targeted peptide did not substantially alter MCF/V cell migration, suggesting that the effect in MCF/I cells is due to an effect of calmodulin on IQGAP1. The lack of effect of the nuclear-targeted peptide further supports this hypothesis as IQGAP1 is not seen in the nucleus. This effect is not due to increased expression of the membrane-targeted calmodulin-binding peptide as Western blotting reveals that it is expressed at a lower level than the nuclear-targeted peptide (Fig. 6B).
Fig. 6.
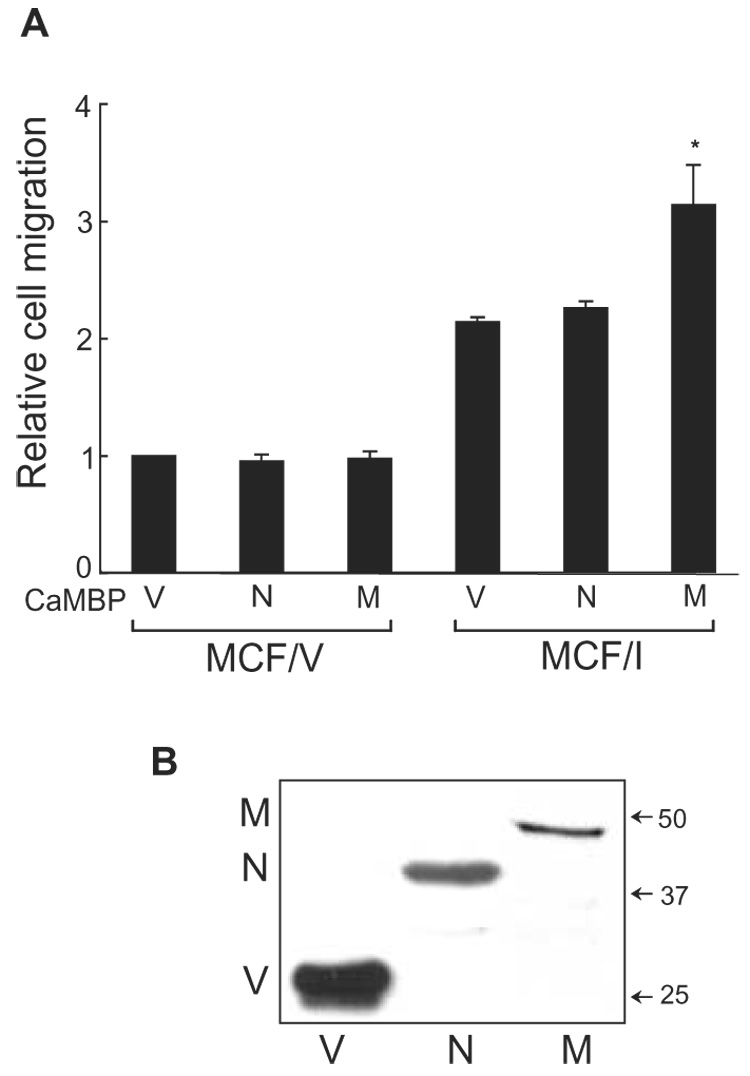
Inhibition of calmodulin augments IQGAP1-stimulated cell migration. A, migration assays were performed using MCF/V and MCF/I cells transiently transfected with calmodulin-binding peptide (CaMBP), targeted to the nucleus (N) or targeted to the membrane (M). The empty vector (V) was transfected as control. Migration was quantified by counting fields of migratory cells under a light microscope. Data, expressed relative to migration of MCF/V cells transfected with vector, represent the means ± S.E. (n=3). *, significantly different from MCF/I cells transfected with vector (p<0.05). B, representative Western blot. Cells were transiently transfected with empty pEYFP vector (V) or YFP-tagged CaMBP targeted to the nucleus (N) or the membrane (M). After 48 h, equal amounts of protein lysate were resolved by SDS-PAGE and probed for YFP. The positions of migration of molecular weight markers are indicated on the right. Note that the membrane-targeting sequence slightly reduces the migration of CaMBP.
Examination of proteins at the leading edge of migrating cells
We employed the widely-used strategy of wound healing to examine the localization of selected proteins at the leading edge of migrating cells. MCF-7 cells were transfected with GFP-tagged IQGAP1 constructs and confluent plates of cells were wounded. Migrating cells were fixed 8 h later and analyzed by immunocytochemistry and confocal microscopy. GFP-tagged wild type IQGAP1 localizes at the leading edge of migrating MCF-7 cells (Fig. 7A). This pattern is essentially identical to that we previously observed with endogenous IQGAP1 [11]. Both Cdc42 and actin co-localize with the wild type IQGAP1 at the leading edge (Fig. 7A). Consistent with the effects produced on cell migration (see Fig. 5A), transfection of GFP-tagged IQGAP1·apoCaM(−) yielded virtually identical results. IQGAP1·apoCaM(−) accumulates at the leading edge of migrating cells where it co-localizes with Cdc42 and actin (Fig. 6). Quantification of the percentage of migrating cells which have IQGAP1, Cdc42 and actin at the leading edge reveals that the patterns for wild type IQGAP1 and IQGAP1·apoCaM(−) are the same (Fig. 7B). The IQGAP1 constructs are at the leading edge in virtually all migrating cells. In contrast, IQGAP1·CaM(−) is at the leading edge in ~50% of the migrating cells (Fig. 7). Similarly, the number of motile cells transfected with IQGAP1·CaM(−) which have Cdc42 at the leading edge is significantly reduced by 50%. The subcellular location of actin is not changed significantly by IQGAP1·CaM(−) (Fig. 7). These data indicate that IQGAP1·CaM(−) has altered subcellular location in migrating cells and suggest that the IQGAP1 construct changes the localization of Cdc42.
Fig. 7.
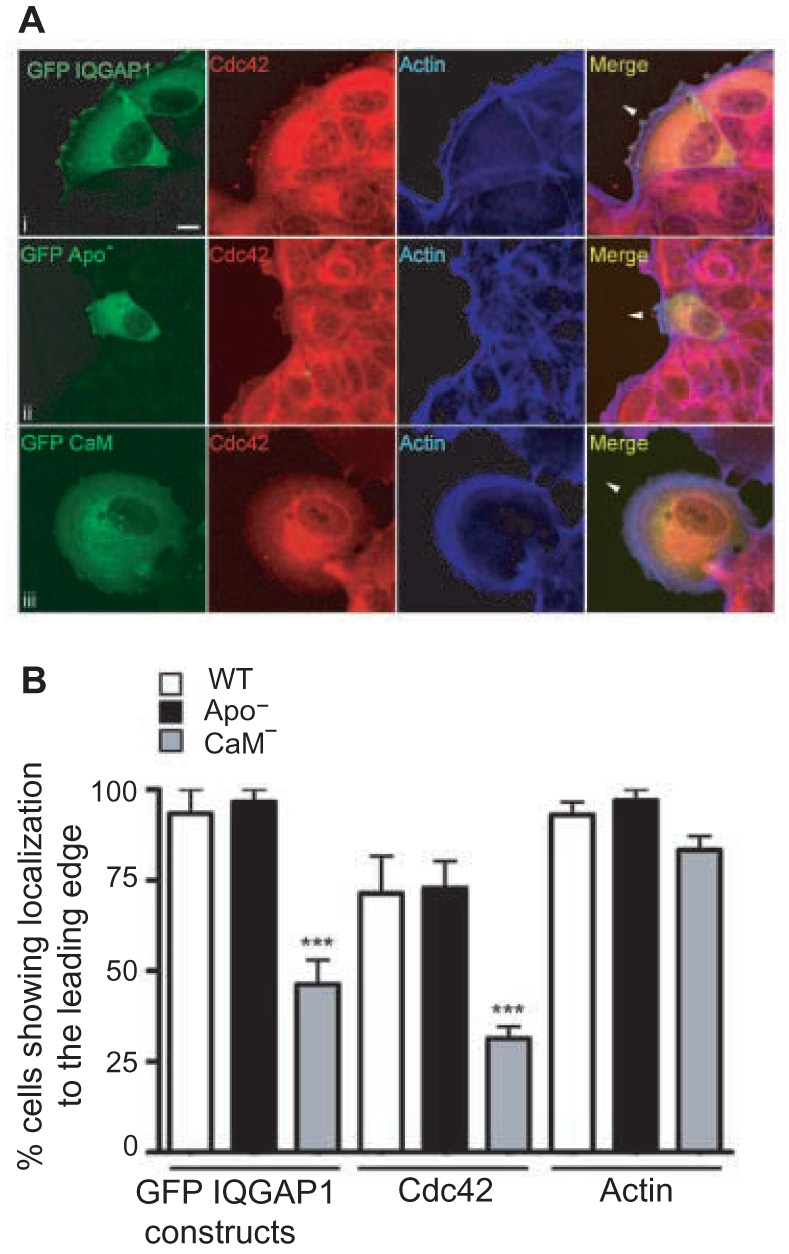
IQGAP1·CaM(−) displays reduced localization to the leading edge of migrating cells. A, MCF-7 cells were transfected with wild-type GFP-IQGAP1, GFP-IQGAP1·apoCaM(−) (GFP Apo−) or GFP-IQGAP1·CaM(−) (GFP CaM−), and subjected to the wound healing assay as described under Experimental Procedures. After 8 h, cells were fixed and processed for immunocytochemistry. Cells were stained for GFP (green), Cdc42 (red) and actin (blue). Merge represents a composite of all three channels. Arrowheads indicate the direction of cell movement. Representative images of two independent experiments, each performed in quadruplicate, are shown. Scale bar, 10 µm. B, the number of migrating cells in which the GFP-tagged IQGAP1 constructs [wild type (WT) IQGAP1 (clear bars), IQGAP1·apoCaM(−) (Apo−, black bars) or IQGAP1·CaM(−) (CaM−, grey bars)], Cdc42 or actin are localized to the leading edge were quantified. Data are expressed as a percentage of total cells counted and represent the means ± S.E.M. (n=50). * p<0.001 compared to cells transfected with GFP-tagged wild type IQGAP1.
DISCUSSION
Accumulating evidence reveals that IQGAP1 is an integral component of the molecular machinery that mediates cell migration. This is not surprising as IQGAP1 is a significant modulator of cytoskeletal architecture across a wide spectrum of organisms, ranging from yeast to Xenopus to mammals [15]. IQGAP1 influences numerous cell functions, including actin polymerization, microtubule function, cell-cell adhesion and neurite outgrowth [3, 15, 16, 35]. These effects are mediated by a direct interaction between IQGAP1 and multiple-binding proteins [3, 16]. Many of these proteins participate in cell migration, and we therefore investigated the role of selected targets in IQGAP1-mediated cell motility.
We observed that the association of Cdc42 with IQGAP1 increases when MCF-7 cells migrate into a wound, and remains high while the cells are migrating. The interaction returns to basal levels after the wound has healed and cell migration has ceased. Previously, we demonstrated that dominant negative constructs of Cdc42 or Rac1 prevented the enhanced motility produced by IQGAP1 [11]. These findings suggest that Cdc42 and Rac1 are components of IQGAP1-stimulated cell motility. Nevertheless, the data presented in this study reveal that binding partners of IQGAP1 other than Cdc42 and Rac1 also have a role. A mutant IQGAP1 construct that specifically lacks binding to Cdc42 and Rac1 [28] is able to increase cell motility, albeit less than that produced by wild type IQGAP1. Not surprisingly, the interaction of IQGAP1 with actin is essential for IQGAP1 to promote cell motility. Elimination of actin binding by replacing Gly75 of IQGAP1 with Gln completely abrogated the ability of IQGAP1 to enhance cell migration. These data do not conflict with the findings seen with Cdc42 and Rac1. Spatially controlled polymerization of actin is at the origin of cell motility [29]. Actin polymerization is required for cells to extend protrusions in the direction of cell migration [1]. The Rho GTPases, Rac1 and Cdc42, are required for formation of lamellipodia and filopodia. Several targets of Rac1 and Cdc42 mediate actin polymerization. IQGAP1 has been shown to increase cross-linking of F-actin in vitro, an effect that is augmented by active Cdc42 and Rac1 [36]. One could thus envisage that, as we previously speculated [3, 4], IQGAP1 serves to couple active Cdc42 (and Rac1) to the actin cytoskeleton, thereby enabling the Rho GTPases to induce cell migration.
Intracellular Ca2+ regulates several processes necessary for cell migration [31]. There is also compelling evidence that calmodulin participates in cell motility [19, 30, 32, 37]. We previously documented that Ca2+/calmodulin attenuates the binding of IQGAP1 to Cdc42; no effect was observed with Ca2+-free calmodulin [7]. One would thus anticipate that reducing the association between Ca2+/calmodulin and IQGAP1 would promote the interaction of IQGAP1 with Cdc42, thereby enhancing cell migration. In this model, apocalmodulin, which does not interfere with the binding of Cdc42 to IQGAP1 [7], would have no effect. This hypothesis was confirmed by the analyses with the point mutant IQGAP1 constructs that selectively lack binding to calmodulin. Transfection of IQGAP1·apoCaM(−), which cannot bind apocalmodulin but binds normally to Ca2+/calmodulin [21], increases cell migration by essentially the same magnitude as wild type IQGAP1. In contrast, transfection of IQGAP1·CaM(−), a point mutant IQGAP1 construct that fails to interact with Ca2+/calmodulin [21], augments cell migration substantially more than wild type IQGAP1.
Further insight into the molecular interactions among Cdc42, calmodulin and IQGAP1 in cell migration was obtained by examining protein localization. In virtually all migrating cells, both wild type IQGAP1 and IQGAP1·apoCaM(−) accumulate at the leading edge, where they co-localize with Cdc42 and actin. A different pattern is observed with IQGAP1·CaM(−). IQGAP1·CaM(−) was detected at the leading edge of only 50% of migrating cells. Importantly, expression of IQGAP1·CaM(−) prevented Cdc42, but not actin, from accumulating at the leading edge of cells migrating into the wound. These data imply that IQGAP1·CaM(−) augments cell migration by preventing Cdc42 from moving to the leading edge in a large proportion of cells or by inducing the translocation of Cdc42 from the front of the migrating cells to another subcellular region. The findings are congruent with our prior observations that IQGAP1 influences the subcellular localization of Cdc42 [8]. Overexpression of wild type IQGAP1 significantly increased the amount of Cdc42 in the membrane fraction of cells, while the dominant negative IQGAP1ΔGRD prevented bradykinin from inducing translocation of Cdc42 to the membrane.
How Rho, Rac1 and Cdc42 function in signalling during cell migration remains incompletely understood. While there is agreement that activated Rac1 is required at the leading edge of migrating cells, the location of Cdc42 is less well defined [1]. Differences among cell types, coupled with the participation of Cdc42 in cell-cell adhesion [38], further complicate the issue. For example, Cdc42 is not required for wound closure by Madin-Darby canine kidney (MDCK) cells [39]. In addition to inducing the formation of filopodia, Cdc42 regulates cell polarity during movement [40]. This is achieved by both restricting where lamellipodia form and by localizing the microtubule organizing center and Golgi apparatus in front of the nucleus [1, 41]. Microtubule reorganization appears to be more important for the migration of slow-moving cells [1], such as MCF-7 cells, than fast-moving cells. Polarity is required for a cell to migrate. Thus, IQGAP1·CaM(−) may promote motility of MCF-7 cells by localizing Cdc42 in front of the nucleus, thereby enhancing cell polarity. Further studies are required to test this hypothesis, but it is supported by the demonstration that IQGAP1 contributes to polarity of fibroblasts by establishing a polarized microtubule array [10]. Regardless of the mechanism, our data suggest that localization of Cdc42 at the leading edge is not necessary for maximal migration of MCF-7 epithelial cells.
Additional support for a regulatory role for calmodulin in IQGAP1-mediated cell migration is provided by a complementary strategy. We previously speculated that IQGAP1 may influence cell motility by increasing the levels of active Cdc42 (and Rac1) in migratory cells [11]. If this hypothesis is correct, blocking the interaction of calmodulin with IQGAP1 at cell membranes should enhance the interaction of IQGAP1 with Cdc42 and augment IQGAP1-stimulated cell migration. Consistent with this model, a calmodulin-binding peptide that specifically and selectively inhibits calmodulin function at membranes [25] significantly increased cell migration promoted by IQGAP1. In contrast, when the peptide was targeted to the nucleus, which inhibits nuclear calmodulin function [25], IQGAP1-stimulated cell migration was not altered. Collectively these data reveal that Ca2+/calmodulin signalling provides an additional level to fine tune the ability of IQGAP1 to increase cell migration.
Many of the proteins that bind IQGAP1 have been implicated in cell migration. Testing every one of these is clearly beyond the scope of the current study. Increased cell migration and invasion are fundamental prerequisites for metastasis of tumour cells, which is the major cause of mortality of solid tumours [42]. A steadily increasing body of evidence implicates IQGAP1 in metastasis. For example, Richard Hynes’s group demonstrated that IQGAP1 was one of 32 only genes (out of ~10 500 analyzed) that were upregulated in metastatic melanoma [43]. The insight provided by the data presented in this work substantially enhances our comprehension of the molecular mechanisms by which IQGAP1 contributes to cell migration (and metastasis). Elucidation of these pathways is necessary to develop novel therapeutic agents to treat metastatic disease.
ACKNOWLEDGEMENTS
We thank Robert Krikorian for expertly preparing the manuscript. Confocal analysis was performed at the imaging facility of the Harvard Center for Neurodegeneration and Repair.
This work was supported by grants from the National Institutes of Health (to D.B.S).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Science. 2003;302(5651):1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 2.Pollard TD. Nature. 2003;422(6933):741–745. doi: 10.1038/nature01598. [DOI] [PubMed] [Google Scholar]
- 3.Briggs MW, Sacks DB. FEBS Lett. 2003;542(1–3):7–11. doi: 10.1016/s0014-5793(03)00333-8. [DOI] [PubMed] [Google Scholar]
- 4.Ho YD, Joyal JL, Li Z, Sacks DB. J. Biol. Chem. 1999;274(1):464–470. doi: 10.1074/jbc.274.1.464. [DOI] [PubMed] [Google Scholar]
- 5.Mateer SC, McDaniel AE, Nicolas V, Habermacher GM, Lin MJ, Cromer DA, King ME, Bloom GS. J. Biol. Chem. 2002;277(14):12324–12333. doi: 10.1074/jbc.M109535200. [DOI] [PubMed] [Google Scholar]
- 6.Hart MJ, Callow MG, Souza B, Polakis P. EMBO J. 1996;15(12):2997–3005. [PMC free article] [PubMed] [Google Scholar]
- 7.Joyal JL, Annan RS, Ho YD, Huddleston ME, Carr SA, Hart MJ, Sacks DB. J. Biol. Chem. 1997;272(24):15419–15425. doi: 10.1074/jbc.272.24.15419. [DOI] [PubMed] [Google Scholar]
- 8.Swart-Mataraza JM, Li Z, Sacks DB. J. Biol. Chem. 2002;277:24753–24763. doi: 10.1074/jbc.M111165200. [DOI] [PubMed] [Google Scholar]
- 9.Erickson JW, Cerione RA, Hart MJ. J. Biol. Chem. 1997;272:24443–24447. doi: 10.1074/jbc.272.39.24443. [DOI] [PubMed] [Google Scholar]
- 10.Fukata M, Watanabe T, Noritake J, Nakagawa M, Yamaga M, Kuroda S, Matsuura Y, Iwamatsu A, Perez F, Kaibuchi K. Cell. 2002;109(7):873–885. doi: 10.1016/s0092-8674(02)00800-0. [DOI] [PubMed] [Google Scholar]
- 11.Mataraza JM, Briggs MW, Li Z, Entwistle A, Ridley AJ, Sacks DB. J. Biol. Chem. 2003;278(42):41237–41245. doi: 10.1074/jbc.M304838200. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe T, Wang S, Noritake J, Sato K, Fukata M, Takefuji M, Nakagawa M, Izumi N, Akiyama T, Kaibuchi K. Dev. Cell. 2004;7(6):871–883. doi: 10.1016/j.devcel.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 13.Yamaoka-Tojo M, Ushio-Fukai M, Hilenski L, Dikalov SI, Chen YE, Tojo T, Fukai T, Fujimoto M, Patrushev NA, Wang N, Kontos CD, Bloom GS, Alexander RW. Circ. Res. 2004;95(3):276–283. doi: 10.1161/01.RES.0000136522.58649.60. [DOI] [PubMed] [Google Scholar]
- 14.Kholmanskikh SS, Koeller HB, Wynshaw-Boris A, Gomez T, Letourneau PC, Ross ME. Nat. Neurosci. 2006;9(1):50–57. doi: 10.1038/nn1619. [DOI] [PubMed] [Google Scholar]
- 15.Briggs MW, Sacks DB. EMBO reports. 2003;4:571–574. doi: 10.1038/sj.embor.embor867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown MD, Sacks DB. Trends Cell Biol. 2006;16:242–249. doi: 10.1016/j.tcb.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 17.Roy M, Li Z, Sacks DB. Mol. Cell. Biol. 2005;25:9740–9752. doi: 10.1128/MCB.25.18.7940-7952.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mateer SC, Wang N, Bloom GS. Cell Motil. Cytoskeleton. 2003;55(3):147–155. doi: 10.1002/cm.10118. [DOI] [PubMed] [Google Scholar]
- 19.Pettit EJ, Fay FS. Physiol. Rev. 1998;78(4):949–967. doi: 10.1152/physrev.1998.78.4.949. [DOI] [PubMed] [Google Scholar]
- 20.Sacks DB, Porter SE, Ladenson JH, McDonald JM. Anal Biochem. 1991;194(2):369–377. doi: 10.1016/0003-2697(91)90243-m. [DOI] [PubMed] [Google Scholar]
- 21.Li Z, Sacks DB. J. Biol. Chem. 2003;278:4347–4352. doi: 10.1074/jbc.M208579200. [DOI] [PubMed] [Google Scholar]
- 22.Ren JG, Li Z, Crimmins DL, Sacks DB. J. Biol. Chem. 2005;280:34548–34557. doi: 10.1074/jbc.M507321200. [DOI] [PubMed] [Google Scholar]
- 23.Li Z, Kim SH, Higgins JM, Brenner MB, Sacks DB. J. Biol. Chem. 1999;274(53):37885–37892. doi: 10.1074/jbc.274.53.37885. [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Campos B, Jamieson GA, Jr., Kaetzel MA, Dedman JR. J. Biol. Chem. 1995;270(51):30245–30248. doi: 10.1074/jbc.270.51.30245. [DOI] [PubMed] [Google Scholar]
- 25.Li L, Li Z, Sacks DB. J. Biol. Chem. 2003;278(2):1195–1200. doi: 10.1074/jbc.M210708200. [DOI] [PubMed] [Google Scholar]
- 26.Buss F, Kendrick-Jones J, Lionne C, Knight AE, Cote GP, Paul Luzio J. J. Cell Biol. 1998;143(6):1535–1545. doi: 10.1083/jcb.143.6.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim SH, Li Z, Sacks DB. J. Biol. Chem. 2000;275(47):36999–37005. doi: 10.1074/jbc.M003430200. [DOI] [PubMed] [Google Scholar]
- 28.Mataraza JM, Briggs MW, Li Z, Frank R, Sacks DB. Biochem. Biophys. Res. Commun. 2003;305(2):315–321. doi: 10.1016/s0006-291x(03)00759-9. [DOI] [PubMed] [Google Scholar]
- 29.Pantaloni D, Le Clainche C, Carlier MF. Science. 2001;292(5521):1502–1506. doi: 10.1126/science.1059975. [DOI] [PubMed] [Google Scholar]
- 30.Walker JW, Gilbert SH, Drummond RM, Yamada M, Sreekumar R, Carraway RE, Ikebe M, Fay FS. Proc. Natl. Acad. Sci. U.S.A. 1998;95(4):1568–1573. doi: 10.1073/pnas.95.4.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee J, Ishihara A, Oxford G, Johnson B, Jacobson K. Nature. 1999;400(6742):382–386. doi: 10.1038/22578. [DOI] [PubMed] [Google Scholar]
- 32.Giuliano KA, Taylor DL. Curr. Opin. Cell Biol. 1995;7(1):4–12. doi: 10.1016/0955-0674(95)80038-7. [DOI] [PubMed] [Google Scholar]
- 33.Briggs MW, Li Z, Sacks DB. J. Biol. Chem. 2002;277(9):7453–7465. doi: 10.1074/jbc.M104315200. [DOI] [PubMed] [Google Scholar]
- 34.Roy M, Li Z, Sacks DB. J. Biol. Chem. 2004;279:17329–17337. doi: 10.1074/jbc.M308405200. [DOI] [PubMed] [Google Scholar]
- 35.Li Z, McNulty DE, Marler KJM, Lim L, Hall C, Annan RS, Sacks DB. J. Biol. Chem. 2005;280:13871–13878. doi: 10.1074/jbc.M413482200. [DOI] [PubMed] [Google Scholar]
- 36.Fukata M, Kuroda S, Fujii K, Nakamura T, Shoji I, Matsuura Y, Okawa K, Iwamatsu A, Kikuchi A, Kaibuchi K. J. Biol. Chem. 1997;272(47):29579–29583. doi: 10.1074/jbc.272.47.29579. [DOI] [PubMed] [Google Scholar]
- 37.Lawson MA, Maxfield FR. Nature. 1995;377(6544):75–79. doi: 10.1038/377075a0. [DOI] [PubMed] [Google Scholar]
- 38.Casanova JE. Am. J. Physiol. Gastrointest. Liver Physiol. 2002;283(5):G1015–G1019. doi: 10.1152/ajpgi.00255.2002. [DOI] [PubMed] [Google Scholar]
- 39.Fenteany G, Janmey PA, Stossel TP. Curr. Biol. 2000;10(14):831–838. doi: 10.1016/s0960-9822(00)00579-0. [DOI] [PubMed] [Google Scholar]
- 40.Nobes CD, Hall A. J. Cell Biol. 1999;144(6):1235–1244. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Etienne-Manneville S, Hall A. Nature. 2002;420(6916):629–635. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 42.Condeelis J, Segall JE. Nat. Rev. Cancer. 2003;3(12):921–930. doi: 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- 43.Clark EA, Golub TR, Lander ES, Hynes RO. Nature. 2000;406(6795):532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]