

Abstract
Dihydroxyacetone phosphate (DHAP) dependent aldolases have been widely used for organic synthesis. The major drawback of DHAP-dependent aldolases is their strict donor substrate specificity toward DHAP, which is expensive and unstable. Here we report the development of an in vivo selection system for the directed evolution of the DHAP dependent aldolase, L-rhamnulose-1-phosphate aldolase (RhaD), to alter its donor substrate specificity from DHAP to dihydroxyacetone (DHA). We also report preliminary results on mutants that were discovered with this screen. A strain deficient in the L-rhamnose metabolic pathway in E. coli (ΔrhaDAB, DE3) was constructed and used as a selection host strain. Co-expression of L-rhamnose isomerase (rhaA) and rhaD in the selection host did not restore its growth on minimal plate supplemented with L-rhamnose as a sole carbon source, because of the lack of L-rhamnulose kinase (RhaB) activity and the inability of WT RhaD aldolase to use unphosphorylated L-rhamnulose as a substrate. Use of this selection host and co-expression vector system gives us an in vivo selection for the desired mutant RhaD which can cleave unphosphorylated L-rhamnulose and allow the mutant to grow in the minimal media. An error-prone PCR (ep-PCR) library of rhaD gene on the co-expression vector was constructed and introduced into the rha- mutant, and survivors were selected in minimal media with L-rhamnose (MMRha media). An initial round of screening gave mutants allowing the selection strain to grow on MMRha plates. This in vivo selection system allows rapid screening of mutated aldolases that can utilize dihydroxyacetone as a donor substrate.
1. Introduction
Aldolases have been attractive and widely used catalysts for organic synthesis due to their ability to catalyze the formation of new carbon-carbon bonds with high regio- and stereo- selectivities. Dihydroxyacetone phosphate (DHAP) dependent aldolases catalyze aldol condensation with DHAP as a nucleophilic donor substrate, and have been used successfully for syntheses of many interesting unnatural monosaccharides and related compounds.1–6 The unnatural monosaccharide L-Fructose has many potential applications such as non-caloric sweetener, inhibition of various glycosidases, or as a useful chiral building block for pharmaceutical intermediates.7, 8 Several chemical 9–11 or chemoenzymatic methods 12–14 have been reported for the syntheses of L-fructose. Recently, we reported a one-pot synthesis of L-fructose from DHA and DL-glyceraldehyde using L-rhamnulose 1-phosphate aldolase (RhaD) and borate buffer. 15
The major drawback of DHAP dependent aldolases is their strict donor substrate specificity toward DHAP. Wild type DHAP dependent aldolases do not accept DHA as a substrate. The crystal structure of RhaD in binary complex with a DHAP analogue shows the phosphate moiety forms strong hydrogen bonds with four residues in the active site, which is the major source of binding affinity between the aldolase and DHAP. 16 The requirement for DHAP as a donor substrate limits practical synthetic application because of the high cost of DHAP and its instability. However, several chemical 17–19 or chemo-enzymatic 20–24 syntheses of DHAP have been described. In many cases, phospholyrated substrates or products are unstable and difficult to manipulate, and the phosphate moiety of the product usually needs to be removed to obtain the desired product. Thus aldolases utilizing dihydroxyacetone (without phosphate) as the donor substrate is desired, and our strategy is to alter the donor substrate specificity of RhaD by means of directed evolution to be capable of accepting DHA (Fig. 1).
Fig. 1.
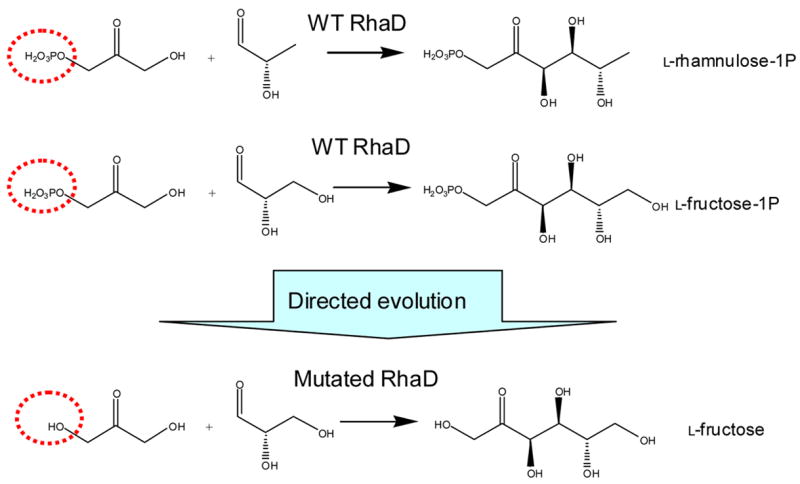
Directed evolution of RhaD to alter its donor substrate specificity
Directed evolution 25 has been successfully employed to improve the properties of biocatalysts by altering activity, substrate specificity, enantioselectivity, or enzyme stability. 26–29 Aldolases have also been subjected to directed evolution to alter their substrate specificity or enantioselectivity, 30 invert the stereoselectivity, 31, 32 or improve the stability. 33, 34 Directed evolution can be divided into two steps. The first is construction of mutated libraries by error-prone PCR, DNA shuffling, saturation mutagenesis, or other methods.35 The second step is screening/selection for the desired mutants. In vitro screening is a commonly used strategy. However, each individual mutated enzyme is assayed separately for the desired activities. Therefore the number of mutated enzymes that can be tested may be limited in the absence of automation systems, or making the process of screening a very large library is very time consuming. In vivo selection is an alternative method to allow high throughput screening for desired enzyme activity. 36–40 In the in vivo selection strategy, the desired enzymatic activities should be linked to the survival of host strains to allow an activity-based assay evaluating large library sizes of up to 107 to 1010. In vivo selection systems for the directed evolution of a pyruvate-dependent aldolase, 39 and an acetaldehyde-dependent aldolase, 40 have been reported. However there has been no report describing in vivo selection for DHAP-dependent aldolases.
Here we report the development of an in vivo selection system for the directed evolution of RhaD to alter its donor substrate specificity from DHAP to DHA. Use of an E. coli deletion mutant of L-rhamnose metabolic genes as a selection host gives us an in vivo selection for the desired mutated RhaD which can cleave unphosphorylated L-rhamnulose, allowing rapid screening of mutated aldolases utilizing dihydroxyacetone as a donor substrate.
2. Results and Discussion
Development of in vivo selection system in E. coli
Our strategy for the in vivo selection involved the metabolic engineering of the endogenous E. coli L-rhamnose pathway as shown in Fig. 2. 41 L-Rhamnose, which is actively incorporated into E. coli cells, is first isomerized to the corresponding ketose, L-rhamnulose, by L-rhamnose isomerase (RhaA). Next, L-rhamnulose is phosphorylated by rhamnulose kinase (RhaB) to give the natural substrate of RhaD, L-rhamnulose-1-phosphate. RhaD splits rhamnulose-1-P by a retroaldol reaction into two three-carbon carbohydrates, DHAP and L-lactaldehyde, which are then metabolized through glycolysis or the TCA cycle. Since the wild type RhaD can not cleave L-rhamnulose, the disruption of the rhamnulose kinase gene (rhaB-) leads to inability to grow on minimal media supplemented with L-rhamnose as a carbon source. Use of the disruptant as a selection host allowed us to screen for mutated RhaD which are capable of accepting unphosphorylated L-rhamnulose by selecting the survivors in MMRha.
Fig. 2.
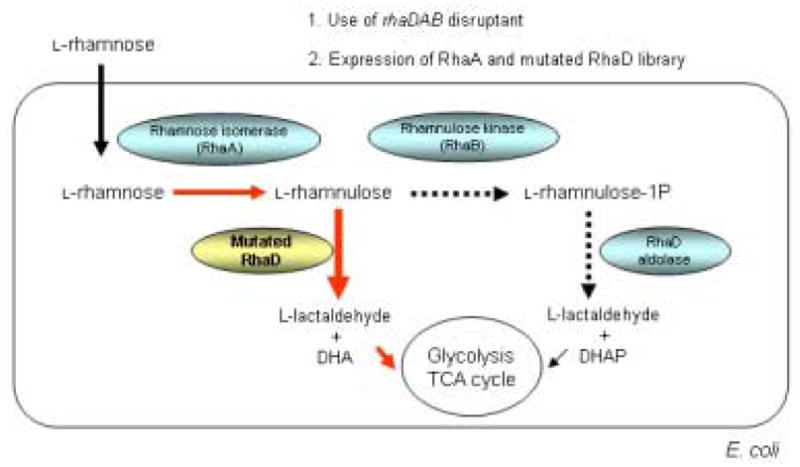
Metabolic engineering of E. coli rhamnose pathway for in vivo selection development
As an initial trial, we disrupted the rhamnulose kinase gene (rhaB) of E. coli BL21 (DE3) by homologous recombination, and the resulting disruptant showed significantly slower growth on MMRha plate than the wild type strain. However, background growth of the disruptants on MMRha due to the appearance of revertants, and a lowered expression level of rhamnose isomerase probably because of disorder of the rha operon caused by the gene disruption, made the rhaB- disruptant a less than ideal selection host, yielding a high rate of false positive clones. Therefore, we changed the host strain to E. coli BW25113 which lacks all of the rhaBAD genes, and RhaA and RhaD were co-expressed on a plasmid. In order to facilitate the T7 expression system in this strain, λDE3 lysogene was introduced into BW25113 to afford BW25113 (DE3). E. coli BW25113 (DE3) was confirmed not to grow on MMRha plate after one week incubation. Thus this strain can be used as a selection host strain without appearance of revertants. Next, RhaA and RhaD were co-expressed in E. coli harboring pETDRhaAD (Fig. 3). Both the RhaA and RhaD were overexpressed in the soluble fraction (Fig. 4, Table 1), allowing us to utilize commercially available L-rhamnose as a sole carbon source. This was desirable because L-rhamnulose is not commercially available.
Fig. 3.
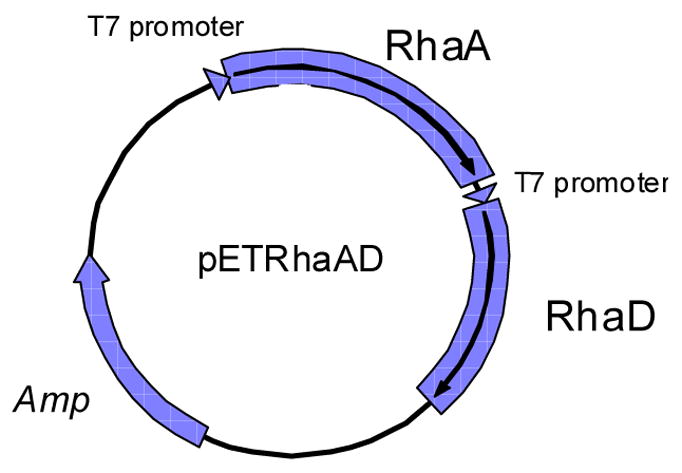
Co-expression vector of rhaA and rhaD.
Fig. 4.
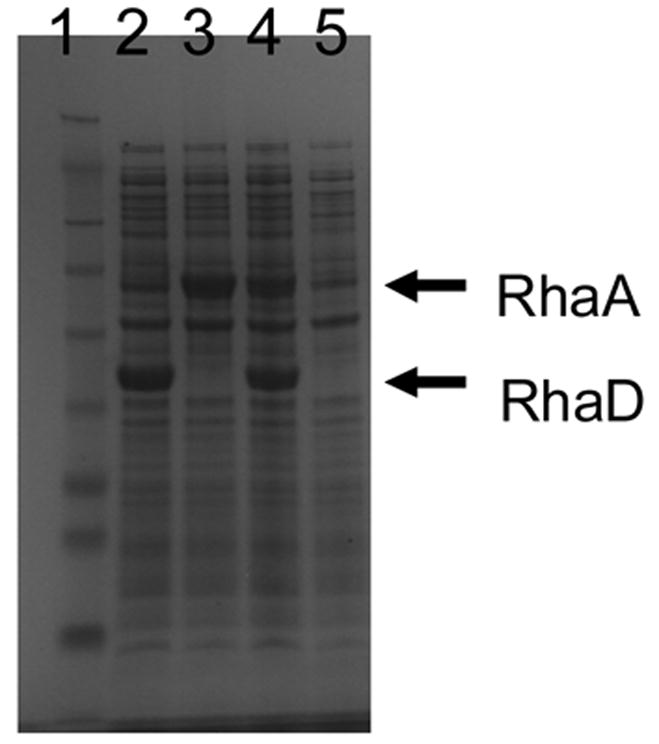
Co-expression of RhaA and RhaD in E. coli BW25113 (DE3). Lane 1, MW marker; 2, pETDRhaD; 3, pETDRhaA; 4, pETDRhaAD; 5, pETDuet (control).
Table 1.
Co-expression of RhaA and RhaD in E. coli BW25113 (DE3).
| plasmids | RhaD activity * (U/mg) | RhaA activity ** (U/mg) |
|---|---|---|
| pETRhaAD | 0.90 | 14 |
| pETRhaA | N.D. | 66 |
| pETRhaD | 1.3 | N.D. |
| pET | N.D. | N.D. |
5 mM L-rhamnulose-1-P as a substrate
50 mM L-rhamnose as a substrate
N.D., Not detected
In vivo selection for mutated RhaD using MMRha liquid media
An ep-PCR library of rhaD gene was constructed on the co-expression vector pETDRhaAD, and introduced into the selection strain. E. coli BW25113 (DE3) harboring the wild type pETDRhaAD can not grow in the MMRha media. In contrast, transformants from the ep-PCR library grew in MMRha media after 3 days incubation, suggesting that some mutants within the ep-PCR library allowed the selection host to survive in MMRha. In order to enrich the fast-growing cells which may possess the desired RhaD mutants, broth was seeded into a fresh MMRha media and cultivated again. The enrichment was performed twice and plasmids were extracted from each round of enrichment and named as selected library 1 and 2 (see Materials and Methods). Next, the effectiveness of the selection and enrichment experiments was assessed by transformation of fresh selection E. coli cells with either the non-mutated control DNA or the selected library DNA. As a result, transformants with selected library 2 grew first, and transformants with selected library 1 grew on the next day, while strains harboring the non-mutated control plasmid pETDRhaAD did not grow after one week cultivation (Fig. 5). These results suggested that the selected library DNA restored the growth of the rhaBAD disruptant in MMRha media and the desired mutants could be concentrated through the enrichment process.
Fig. 5.
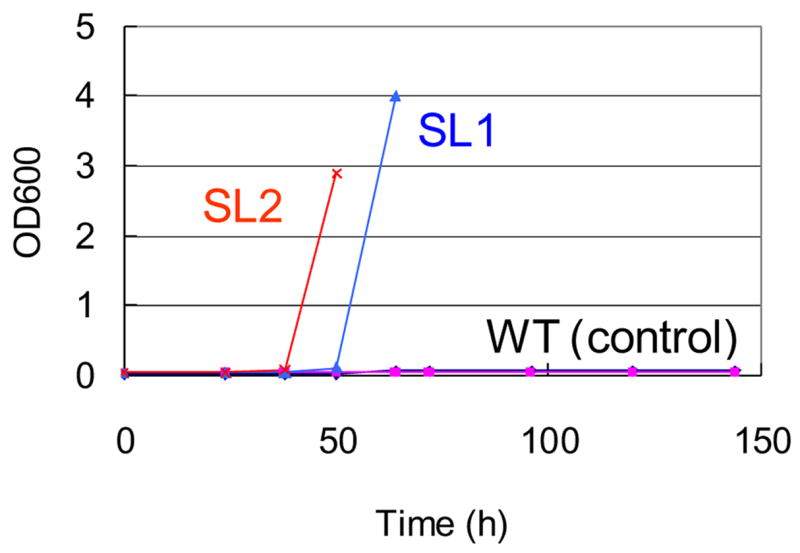
Growth of E. coli BW25113 (DE3) harboring the selected library and the wild type plasmid pETDRhaAD in minimal media containing L-rhamnose as a sole carbon source. SL1, selected library 1; SL2, selected library 2; WT (control), pETDRhaAD.
In vitro assay for L-rhamnulose aldolase activity
The selected library 2 plasmids were introduced into the selection host again, and survivors on MMRha plates were selected. Transformants with the selected library 2 plasmids appeared after 3 days incubation, while transformants with the control plasmid (pETDuet) did not grow on the MMRha, even after 7 days incubation, suggesting that in vivo selection also works on MMRha plates.
Next, each individual colony was picked, cultivated in 96 well plates containing LB media supplemented with 10 μM IPTG to allow the over-expression of RhaA and mutated RhaD. L-Rhamnulose aldolase activity was evaluated using cell-free extracts as enzyme sources. Co-expression of RhaA with mutated RhaD allowed us to use L-rhamnose as the starting substrate for this assay, instead of L-rhamnulose which is not commercially available. Formation of L-rhamnulose from L-rhamnose under the assay conditions has been confirmed by the detection of ketose formation using the cysteine-carbazole method 42 (see Materials and Methods). In the initial round of in vitro screening, several colonies showed L-rhamnulose aldolase activity. In order to exclude the possibility of mutations on the genomic DNA of the E. coli host strain, plasmids were extracted from these colonies, and the selection host strain was transformed again by each mutated plasmid. The re-transformants also grew on the MMRha plate (Fig. 6), confirming that the restoration of growth is derived from their plasmids. The RhaD genes of these mutated plasmids were sequenced, showing both plasmids possessed the same two amino acid substitutions (C142Y, T158S) in the protein sequence (Table 2, Fig. 7). Characterization of these mutants and further rounds of screening are in progress.
Fig. 6.

In vivo selection on a minimal plate containing L-rhamnose as a sole carbon source.
Table 2.
Mutations in the evolved RhaD. Mutant 10 harbored a silent mutation1.
| Mutants | amino acid substitution (nucleotide substitution) |
|---|---|
| Mutant 9 | C142Y (TGC→TAC), T158S (ACC→AGC) |
| Mutant 10 | C142Y (TGC→TAC), T158S (ACC→AGC), V10 (GTC→GTG) 1 |
Fig. 7.
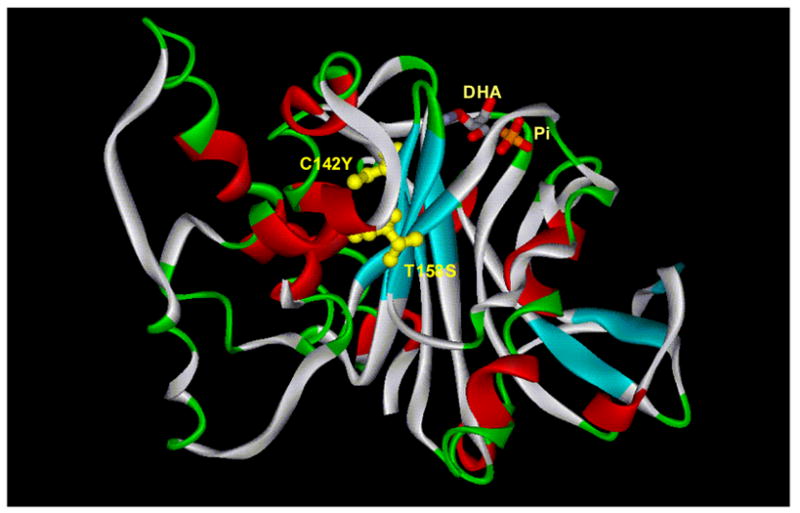
Mutation sites in the evolved RhaD mapped onto the crystal structure of WT RhaD. 16 Phosphate and dihydroxyacetone bound to RhaD are present in the DHAP binding site. Mutations observed in the evolved RhaD are shown in yellow. Both mutation sites are remote from the DHAP binding site.
3. Conclusions
In this paper, we report the development of an in vivo selection system for the directed evolution of a DHAP-dependent aldolase, RhaD. This cell-survival based selection enabled us to screen large library of mutated enzymes. Further work on evolution of RhaD is in progress to develop a DHA-dependent aldolase, and to understand how we can alter the donor substrate specificity of aldolases in general. Although altering the donor substrate specificity of aldolases is desirable for the advancement of these enzymes as biocatalysts for organic synthesis, there has been no report thus far describing the alteration of donor substrate specificity of aldolases by directed evolution. The selection strategy demonstrated here could be applied for directed evolution of other DHAP-dependent aldolases. Moreover, we are currently applying this selection host for the directed evolution of another aldolase, D-fructose-6-phosphate aldolase, to invert its stereoselectivity (from D-fructose configuration to L-fructose configuration), which is another important challenge in the directed evolution of aldolases.
4. Materials and Methods
Strains and culture media
E. coli BW25113 (rhaDAB-) was obtained from the E. coli genetic stock center in Yale university. λDE3 lysogene was introduced into this strain using T7 (DE3) lysogenization kit (Novagen) The E. coli BW25113 (DE3) strain was used as a selection host strain. Rhamnose minimal media (MMRha) contained 1X M9 salt (Sigma), 0.5% L-rhamnose, 0.5 g/L MgSO4-7H2O, 0.01 g/L CaCl2-2H2O, 0.01 g/L FeSO4-7H2O, 0.001 g/L ZnSO4-7H2O, 0.004 g/L MnCl2-4H2O, 0.005 g/L thiamine-HCl, 0.01 g/L Ca D-pantothenic acid, 30 μg/L biotin. For the expression of RhaA and RhaD, 100 μg/L carbenicillin and 0.01 mM IPTG were added, and 1.5% Bacto agar was used for agar plates. MacConkey agar plates with L-rhamnose (40 g/L MacConkey base (Difco), 10 g/L L-rhamnose, and 1.5 % agar) was used for the plate-based in vivo selection.
Construction of co-expression vector pETRhaAD
The rhaA gene was amplified from chromosomal DNA of E. coli W3110 by PCR with following two primers; RhaA5Nco, 5′-ggcgccatggccactcaactggaacagg-3′, and RhaA3Hind, 5′-ccgaagcttttacccgcggcgactcaaaatttc-3′. The amplified fragment was cloned into the NcoI and HindIII site of pETDuet vector (Novagene) to give pETDRhaA. The rhaD gene was also amplified from chromosomal DNA of E. coli W3110 by PCR with following the two primers; RhaD5Nde, 5′-cgcgcatatgcaaaacattactcagtcctgg-3′, and RhaD3Xho, 5′-cggctcgagttacagcgccagcgcactggcgag-3′. The amplified 650 bp fragment was cloned into the NdeI and XhoI site of pETDuet vector to give pETDRhaD, or cloned into the same sites of pETDRhaA to afford the co-expression vector pETDRhaAD. E. coli BL21 (DE3) was transformed by pETDRhaAD, pETDRhaA, pETDRhaD or pETDuet, and transformants were cultivated in LB medium containing 50 μg/ml carbenicillin, 10 μM IPTG at 37°C for 16 h. The recombinant RhaA and RhaD were expressed in the soluble fraction, and appeared as major bands on SDS-PAGE.
Construction of ep-PCR library
The template DNA fragment of the rhaD gene was amplified from pETDRhaAD using primers Duet5, 5′-agttaagtataagaaggagatatacatatg-3′, and Duet3, cgcagcagcggtttctttaccagactcgagtta-3′. Ep-PCR was performed using GeneMorph II random mutagenesis kit (Stratagene) with primers Duet5 and Duet3, following the manual. Typically, 300 ng template DNA was used for 50 μl PCR reaction to introduce 1–3 mutations per entire coding region of rhaD gene except for ATG encoding the first Met. The ep-PCR fragments were ligated into the NdeI/XhoI site of pETDRhaA and ElectroTen-Blue Electroporation competent cells (Strategene) were transformed after ethanol precipitation, and transformants were selected on LB-carbenicillin plates. Approximately 1.2 × 108 colonies were collected from the plates and cultivated in 500 ml LB-carbenicillin media for 4h. Plasmids were extracted from the harvested cells and used as the ep-PCR library.
In vivo selection in MMRha media
Electro-competent cells of BW25113 (DE3) were prepared from early exponential culture (OD600=0.4). Cells harvested from 500 ml culture broth were washed with water (2 times) and 10% glycerol, and suspended in 0.5 ml 10% glycerol. Fifty microliters of the electrocells were transformed by 1 μg of the library plasmid, and after expression and saline wash, cultivated in 500 ml MMRha media containing 1% L-rhamnose as a sole carbon source at 37°C with shaking. A total of 1.4 × 106 transformants were added. After 72 h cultivation, OD600 reached 2.1 and plasmids were extracted from 100 ml culture, producing selected library 1. In order to enrich the desired mutants, 0.5 ml of the broth was added into 50 ml (1% seed) fresh MMRha media containing 0.5% L-rhamnose, and cultivated for 24 h (OD600 reached 3.0). Again 100 μl of the broth was added into 50 ml (0.2% seed) fresh MMRha media and cultivated for 22 h (OD600 reached to 5.5). Plasmids were extracted from the harvested cells, producing selected library 2.
In vivo selection on MMRha plates
E. coli BW25113 (DE3) was transformed by 1 μg of Selected Library 2 or pETDuet, to give 1.3 × 108 and 1.4X 108 cfu respectively, and plated onto MMRha plates containing 100 μg/L carbenicillin and 0.01 mM IPTG. After 48 h incubation at 37°C, over 104 colonies appeared with the selected library 2, while no colony was observed with the control plasmid pETDuet. Colonies were picked from selected library 2 plates onto LB-carbenicillin plates, and subjected to the in vitro assay for rhamnulose aldolase activity.
Expression and purification of DHA kinase
For an enzyme-coupled assay of DHA, His-tagged DHA kinase was prepared as follows. The dhaK gene was amplified from chromosomal DNA of Citrobacter braakii ATCC 6750 (formerly named as C. freundii) by PCR with the following two primers; pETDhaK-SNde, 5′-ggcccatatgtctcaattcttttttaaccaacgc-3′, and pETDhaK-ASXho, 5′-cgcctcgaggcccagctcactctccgctagcgctttaaacacc-3′. The amplified 1.6 kbp fragment was cloned into NdeI and XhoI site of pET26 vector (Novagene) to give pETDhaK. E. coli BL21 (DE3) was transformed by pETDhak, and transformants were cultivated in LB medium containing 50 μg/ml kanamycin at 37°C overnight. One milliliter of the broth was seeded into 100 ml of LB-kanamycin medium, and cultivated at 30°C. ITPG (final concentration of 1 mM) was added when OD600 reached to 0.5–0.7, and cultivated for another 4 h. The recombinant DhaK was expressed in the soluble fraction, and appeared as a major band on SDS-PAGE. His-tagged DhaK was purified by Ni2+ affinity column chromatography. Cells were suspended in Binding buffer (50 mM Hepes-NaOH (pH 8.0), 500 mM NaCl, 20 mM imidazole, and 1 mM β-mercaptoethanol), and disrupted by sonication. After removing the debris by centrifuge, the soluble fraction was applied onto a Ni2+ affinity column, washed with binding buffer, and His-tagged DhaK was eluted with Elution buffer (Binding buffer containing 250 mM imidazole. The elutant was collected and dialyzed against buffer containing 10 mM Hepes-NaOH (pH 7.5), 100 mM NaCl, and 2 mM β-mercaptoethanol.
DhaK activity was determined spectrophotometrically at 30°C by monitoring the decrease of NADH absorbance at 340 nm under the following conditions. 50 mM Tris-HCl (pH 8.0), 1 mM DHA, 1 mM ATP, 0.25 mM NADH, 1 mM dithiothieitol, 2.5 mM MgCl2, 6.4 U/ml, α-glycerophosphate dehydrogenase (Sigma), 11 U/ml triosephosphate isomerase (Sigma).
In vitro screening for L-rhamnulose aldolase activity using enzyme-coupled assay
Each colony was inoculated into 1 ml LB media containing 100 μg/ml carbenicillin and 0.01 mM IPTG in 96 well plates, and cultivated for 16 h. Cells were harvested by centrifuge and supernatants was discarded carefully by decantation. Cells were suspended in 400 μl lysis buffer containing 50 mM Tris-HCl (pH 8.0), 50 mM KCl, and 0.5 mg/ml lysozyme. The suspensions were frozen with liquid nitrogen and incubated at room temperature for 20 min followed by incubation at 37°C for another 20 min. After sonication at 4°C for 20 min and centrifuge, the supernatants were used as cell-free extract. For the aldoase reaction, 10 μl of cell-free extracts were transferred to 96 well plates and 90 μl of reaction mixture (50 mM Tris-HCl (pH 8.0), 100 mM L-rhamnose, 6.4 U/ml, α-glycerophosphate dehydrogenase (Sigma), 11 U/ml triosephosphate isomerase (Sigma), 0.75 U/ml DhaK, 0.5 mM NADH, 1 mM ATP, and 5 mM MgCl2, at final concentrations respectively) was dispensed. Absorbance at 340 nm was determined by plate reader, and plates were incubated at 37°C for 3h. Control reactions were performed without L-rhamnose, and L-rhamnulose aldolase activities were calculated based on the difference of the absorbance.
RhaD activity assays
Retroaldol activity for L-rhamnulose 1-phosphate was determined spectrophotometrically at 37°C by monitoring the decrease of an absorbance at 340 nm under the following conditions. 50 mM Tris-HCl (pH 7.5), 50 mM KCl, L-rhamnulose 1-phosphate, 0.25 mM NADH, 6.4 U/ml, α-glycerophosphate dehydrogenase (Sigma), 11 U/ml triosephosphate isomerase (Sigma).
RhaA activity assays
The activity of the isomerase was assayed using L-rhamnose as substrate and measuring the formation of L-rhamnulose by the cysteine-carbazol method.42 The reaction mixture contained 50 mM Tris-HC1 buffer (pH 7.5), 0.1 mM MnCl2, 50 mM L-rhamnose and the enzyme in a total volume of 100 μl. The mixture was incubated at 37 °C, and reactions were quenched by the addition of 10 μl 10% trichloroacetic acid. L-Rhamnulose concentrations were determined by the cysteine-carbazol method using D-fructose as a ketose standard.
Acknowledgments
We thank the NIH for financial support (GM044154). M.S. was supported by Ajinomoto Co. Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Samland AK, Sprenger GA. Appl Microbiol Biotechnol. 2006;71:253–264. doi: 10.1007/s00253-006-0422-6. [DOI] [PubMed] [Google Scholar]
- 2.Gijsen HJM, Qiao L, Fitz W, Wong C-H. Chem Rev. 1996;96:443–473. doi: 10.1021/cr950031q. [DOI] [PubMed] [Google Scholar]
- 3.Takayama S, McGarvey GJ, Wong C-H. Annu Rev Microbiol. 1997;51:285–310. doi: 10.1146/annurev.micro.51.1.285. [DOI] [PubMed] [Google Scholar]
- 4.Fessner W-D, Badia J, Eyrisch O, Schneider A, Sinerius G. Tetrahedron Lett. 1992;33:5231–5234. [Google Scholar]
- 5.Fessner W-D, Sinerius G, Schneider A, Dreyer M, Schulz GE, Badia J, Aguilar J. Angew Chem Int Ed Engl. 1991;30:555–558. [Google Scholar]
- 6.Schoevaart R, van Rantwijk F, Sheldon RA. J Org Chem. 2000;65:6940–6943. doi: 10.1021/jo000492y. [DOI] [PubMed] [Google Scholar]
- 7.Levin GV, Zehner LR, Saunders JP, Beadle JR. Am J Clin Nutr. 1995;62:1161–1168. doi: 10.1093/ajcn/62.5.1161S. [DOI] [PubMed] [Google Scholar]
- 8.Muniruzzaman S, Pan YT, Zeng Z, Atkins B, Izumori K, Elbein AD. Glycobiology. 1996;6:795–803. doi: 10.1093/glycob/6.8.795. [DOI] [PubMed] [Google Scholar]
- 9.Matsumoto T, Enomoto T, Kurosaki T. J Chem Soc, Chem Commun. 1992;8:610–611. [Google Scholar]
- 10.Cheng CC, Whistler RL. Carbohydr Res. 1988;175:265–271. [Google Scholar]
- 11.Gizaw Y, BeMiller JN. Carbohydr Res. 1995;266:81–85. [Google Scholar]
- 12.Dhawale MR, Szarek WA, Hay GW, Kropinski AM. Carbohydr Res. 1986;155:262–265. [Google Scholar]
- 13.Itoh H, Izumori K. J Ferment Bioeng. 1996;81:351–353. [Google Scholar]
- 14.Henderson I, Sharpless KB, Wong C-H. J Am Chem Soc. 1994;116:558–561. [Google Scholar]
- 15.Sugiyama M, Hong Z, Whalen LJ, Greenberg WA, Wong C-H. Adv Synth Catal. 2006;348:2555–2559. [Google Scholar]
- 16.Kroemer M, Merkel I, Schulz GE. Biochemistry. 2003;42:10560–10568. doi: 10.1021/bi0349266. [DOI] [PubMed] [Google Scholar]
- 17.Jung SH, Jeong J-H, Miller P, Wong C-H. J Org Chem. 1994;59:7182–7184. [Google Scholar]
- 18.Charmantray F, El Blidi L, Gefflaut F, Hecquet L, Bolte J, Lemaire M. J Org Chem. 2004;69:9310–9312. doi: 10.1021/jo048697k. [DOI] [PubMed] [Google Scholar]
- 19.Meyer O, Rohmer M, Grosdemange-Billiard C. Tetrahedron Lett. 2004;45:7921–7923. [Google Scholar]
- 20.Wong C-H, Whitesides GM. J Org Chem. 1983;48:3199–3205. [Google Scholar]
- 21.Fessner W-D, Sinerius G. Angew Chem Int Ed Engl. 1994;33:209–212. [Google Scholar]
- 22.Fessner WD, Walter C. Angew Chem, Int Ed Engl. 1992;31:614–616. [Google Scholar]
- 23.Charmantray F, Dellis P, Samreth S, Hecquet L. Tetrahedron Lett. 2006;47:3261–3263. [Google Scholar]
- 24.van Herk T, Hartog AF, Schoemaker HE, Wever R. J Org Chem. 2006;71:6244–6247. doi: 10.1021/jo060644a. [DOI] [PubMed] [Google Scholar]
- 25.Stemmer WPC. Nature. 1994;370:389–391. doi: 10.1038/370389a0. [DOI] [PubMed] [Google Scholar]
- 26.Bloom JD, Meyer MM, Meinhold P, Otey CR, MacMillan D, Arnold FH. Curr Opin Struc Biol. 2005;15:447–452. doi: 10.1016/j.sbi.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 27.Jaeger KE, Eggert T. Curr Opin Biotechnol. 2004;15:305–313. doi: 10.1016/j.copbio.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 28.Reetz MT. Angew Chem, Int Ed. 2002;41:1335–1338. doi: 10.1002/1521-3773(20020415)41:8<1335::aid-anie1335>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 29.Taylor SV, Kast P, Hilvert D. Angew Chem Int Ed. 2001;40:3310–3335. doi: 10.1002/1521-3773(20010917)40:18<3310::aid-anie3310>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 30.Wada M, Hsu C-C, Franke D, Mitchell M, Heine A, Wilson I, Wong C-H. Bioorg Med Chem. 2003;11:2091–2098. doi: 10.1016/s0968-0896(03)00052-x. [DOI] [PubMed] [Google Scholar]
- 31.Williams GJ, Domann S, Nelson A, Berry A. Proc Natl Acad Sci. 2003;100:3143–3148. doi: 10.1073/pnas.0635924100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsu C-C, Hong Z, Wada M, Franke D, Wong C-H. Proc Natl Acad Sci. 2005;102:9122–9126. doi: 10.1073/pnas.0504033102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hao J, Berry A. Protein Engineering Design & Selection. 2004;17:689–697. doi: 10.1093/protein/gzh081. [DOI] [PubMed] [Google Scholar]
- 34.Jennewein1 B, Schurmann M, Wolberg M, Hilker I, Luiten R, Wubbolts M, Mink D. Biotechnol J. 2006;1:537–548. doi: 10.1002/biot.200600020. [DOI] [PubMed] [Google Scholar]
- 35.Franke D, Hsu C-C, Wong C-H. Methods Enzymol. 2004;388:224–238. doi: 10.1016/S0076-6879(04)88020-0. [DOI] [PubMed] [Google Scholar]
- 36.MacBeath G, Shang X, Hilvert D. Science. 1998;279:1958–1961. doi: 10.1126/science.279.5358.1958. [DOI] [PubMed] [Google Scholar]
- 37.Magliery TJ, Anderson JC, Schultz PG. J Mol Biol. 2001;307:755–769. doi: 10.1006/jmbi.2001.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schmidt DMZ, Mundorff EC, Dojka M, Bermudez E, Ness JE, Govindarajan S, Babbitt PC, Minshull J, Gerlt JA. Biochemistry. 2003;42:8387–8393. doi: 10.1021/bi034769a. [DOI] [PubMed] [Google Scholar]
- 39.Griffiths JS, Cheriyan M, Corbell JB, Pocivavsek L, Fierke CA, Toone EJ. Bioorg Med Chem. 2004;12:4067–4074. doi: 10.1016/j.bmc.2004.05.034. [DOI] [PubMed] [Google Scholar]
- 40.DeSantis G, Liu J, Clark DP, Heine A, Wilson IA, Wong C-H. Bioorg Med Chem. 2003;11:43–52. doi: 10.1016/s0968-0896(02)00429-7. [DOI] [PubMed] [Google Scholar]
- 41.Moralejo P, Egan SM, Hidalgo E, Aguilar J. J Bacteriol. 1993;175:5585–5594. doi: 10.1128/jb.175.17.5585-5594.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garcia-Junceda E, Shen G, Alajarin R, Wong C-H. Bioorg Med Chem. 1995;3:1349–1355. doi: 10.1016/0968-0896(95)00119-2. [DOI] [PubMed] [Google Scholar]