

Abstract
Phenylketonuria (PKU) is a common genetic disorder in humans that arises from deficient activity of phenylalanine hydroxylase (PAH), which catalyzes the conversion of phenylalanine to tyrosine. There is a resultant hyperphenylalanemia with subsequent impairment in cognitive abilities, executive functions and motor coordination. The neuropathogenesis of the disease has not been completely elucidated, however, oxidative stress is considered to be a key feature of the disease process. Hyperphenylalanemia also adversely affects monoaminergic metabolism in the brain. For this reason we chose to evaluate the nigrostriatum of Pahenu2 mice, to determine if alterations of monoamine metabolism resulted in morphologic nigrostriatal pathology. Furthermore, we believe that recent developments in adeno-associated virus (AAV)-based vectors have greatly increased the potential for long-term gene therapy and may be a viable alternative to dietary treatment for this metabolic disorder. In this study we identified neurodegenerative changes with regenerative responses in the nigrostriatum of Pahenu2 mice that are consistent with oxidative injury and occurred as early as 4 weeks of age. These neuropathologic changes were reversed following portal vein delivery of a recombinant adeno-associated virus-mouse phenylalanine hydroxylase-woodchuck hepatitis virus post-transcriptional response element (rAAV-mPAH-WPRE) vector to Pahenu2 mice and corresponded to rapid reduction of serum Phe levels.
Keywords: Phenylketonuria, Oxidative stress, Monoamines, Gene therapy, Adeno-associated virus
1. Introduction
Phenylketonuria (PKU) is a disorder of amino acid metabolism caused by a deficiency of hepatic phenylalanine hydroxylase (PAH) activity. Under these conditions, phenylalanine (Phe) is unable to be converted to tyrosine (Tyr), which results in millimolar concentrations of serum Phe. The increased Phe is subsequently metabolized to the organic acid ketones phenylpyruvate, phenylacetate, and phenylactate. The disease manifests itself primarily as a neurologic disorder, and if untreated, can result in microcephaly, mental retardation, epilepsy, and progression of motor disorders in the second or third decade. The treatment is difficult to implement and consists of lifetime dietary restriction of phenylalanine (Krause et al., 1985; Scriver, 1995; Smith and Wolff, 1974).
The etiology of the neurologic disease process in PKU patients has not been fully elucidated but is thought to occur secondarily to increased concentrations of phenylalanine in the blood. One factor that is considered to play a role in the neuropathogenesis of the disease is oxidative stress. The role of oxidative damage in the neuropathology of organic acidurias is presented in a review by Wajner et al. (2004) who discusses considerable evidence in which organic acid accumulation associated with various metabolic disorders will induce free radical and reactive species (RS) generation and will also decrease antioxidant defenses in tissues. These reactive species can inhibit various complexes of the respiratory chain and critical enzymes necessary for energy production resulting in metabolic failure, cell injury and cell death (Castro et al., 1994, 1998; Radi et al., 2002; Wyse et al., 2001). Free radicals also provoke cell injury and death directly by breaking cell lipid membranes, destroying proteins, and oxidizing DNA (Halliwell, 2001; Halliwell and Gutteridge, 1989a). In some of these disorders, the toxic metabolites were thought to be organic acids such as the phenylketones in PKU (Huttenlocher, 2000).
Hyperphenylalanemia (HPA) of PKU also results in the disruption of amino acid transport and metabolic pathways involving any of the aromatic amino acids, and protein synthesis. Elevation of serum Phe inhibits enzymes important in the conversion of tyrosine and tryptophan to their respective neurotransmitter derivatives, affecting catecholamine metabolism in the brain (Kaufman, 1999; Puglisi-Allegra et al., 2000). For this reason, we chose to evaluate the nigrostriatum of Pahenu2 mice for pathologic changes that may occur secondary to monoaminergic metabolic alterations.
Additionally, we also wish to illustrate that, while it is clear that even though dietary restriction can be achieved in well-controlled patients; PKU still remains a significant cause of serious birth defects. A number of studies suggest that these controlled patients still suffer some cognitive loss and clear neurologic stress (Diamond et al., 1997; Levy, 1999; Pietz et al., 1997, 1998; Pietz, 1998). Thus while dietary therapy for PKU can alleviate many of the most deleterious effects of the disease, it is not completely successful from a clinical standpoint. We believe that recent developments in gene therapy, particularly in adeno-associated virus (AAV)-based vectors, have greatly increased the potential for long-term expression of the enzyme, and may hold a true cure for this metabolic disorder. In this study we identify neurodegenerative changes with regenerative responses in the nigrostriatum of Pahenu2 mice ranging in age from 4 weeks to 8 months old, which are reversed with portal vein delivery of a rAAV-mPAH-WPRE vector.
2. Results
2.1. Cytoplasmic vacuolar degeneration and paucity of neuronal cell bodies occurs in dopaminergic neurons of the substantia nigra in PKU mice early as 4 weeks of age
By comparing the entire substantia nigra/ventral tegmental area (SN/VTA) series in 4-week- and 8-month-old PKU and wild-type mice with tyrosine hydroxylase (TH) immunohistochemistry, it was evident there was a clear reduction of dopaminergic cell bodies within these regions in PKU mice. Fig. 1 illustrates the comparison of 4-week (top row) to 8-month (bottom row) mice and shows the PKU mice in the right column have a reduction in TH-expressing neuronal cell bodies compared to their wild-type counterparts in the left column. Fig. 2 examines the substantia nigra pars compacta (SNPc) in 3 different adult animals of both genotoypes and illustrates a reduction in cell bodies as well as cytoplasmic vacuolar degeneration in the PKU animals in the right column. Dopaminergic neuronal cell bodies of the ventral tegmental area were relatively spared as seen in Fig. 1.
Fig. 1.

Tyrosine hydroxylase immunoreactivity and cell body density are decreased in the SN/VTA of PKU mice (B=4 weeks; D=8 months) compared to wild-type mice (A=4 weeks; C=8 months). There is neuropil vacuolation in the VTA of the 8-month PKU mouse (D, arrow). Tyrosine hydroxylase immunoperoxidase method; scale bar=200 μm.
Fig. 2.

Reduction in dopaminergic cell body density is more pronounced in the substantia nigra pars compacta in 3 adult PKU mice (D, E, F) than 3 adult wild-type mice (A, B, C). Cytoplasmic vacuolation is apparent in the PKU mice. Tyrosine hydroxylase immunoperoxidase method; scale bar=25 μm.
To objectively assess the average cell-occupied area in the SNPc for each mouse, we obtained surface plots of each section (Fig. 3), which combines the total volume of each cell per slide in pixels. Line profile comparisons of the cellular aspect of the SNPc to the acellular background gave us the cell-occupied area, which we then used to calculate the mean area/ SNPc for each mouse and genotype. In PKU mice as young as 4 weeks of age, there were significantly less TH-positive cells compared to their wild-type counterparts (Fig. 3). As both genotypes aged, both the wild-type and PKU mice exhibited a reduction in average cell area, but 8-month PKU mice were significantly more affected than 8-month-old wild-type mice (data not shown). There was also a statistically significant difference in average cell area between 4-week and 8-month PKU mice; however, there was no statistical difference between 4-week- and 8-month-old wild-type mice (data not shown). Surface plots of the SNPc with line profiling evaluation (Fig. 3E) demonstrated a significantly marked reduction in the average cell area of all PKU mice compared to wild-type mice used in this study.
Fig. 3.

Average cell area of the SNPc is decreased in PKU mice. Surface plots of the SNPc of 4-week-old PKU mice (B, D) demonstrate a reduction in average cell area when compared to wild-type mice (A, C) of the same age. Comparison of all PKU mice to wild-type mice used in this study shows a significant reduction of TH-immunopositive cells in the SNPc (E). P<0.05. Tyrosine hydroxylase immunoperoxidase method; scale bar=100 μm.
The most striking abnormality occurred in dopaminergic neuronal cell bodies of the SNPc of PKU mice and consisted of intracytoplasmic vacuolation and mild spongiform change of the surrounding neuropil (Fig. 4). These intracytoplasmic vacuoles were round and multiple, 1–5 μm in diameter, sharp bordered and clear. In the cytoplasm of some neurons, there were clusters of smaller vacuoles, approximately 1 μm in diameter, and gave the neuron a cloudy, fragile, less delineated appearance. No changes were observed in the nuclei. Additionally, there was moderate spongiform or vacuolar change within the neuropil surrounding the affected neuronal cell bodies.
Fig. 4.

Neurodegenerative changes in dopaminergic neurons of the SN/VTA are evident as early as 4 weeks of age in PKU mice. Cytoplasmic vacuolation (arrows) within dopaminergic neurons in the SN/VTA of PKU mice is evident at 4 weeks (B) and 8 months of age in PKU mice (D) compared to wild-type mice (A=4 weeks; C=8 months). Tyrosine hydroxylase immunoperoxidase method; scale bar=25 μm.
Neuronophagia was a prominent feature of the SN/VTA (Fig. 5), which was previously reported by our laboratory (Embury et al., 2005). The SN/VTA was infiltrated by cd11b macrophages (small arrows) that phagocytized the degenerating neuronal cell bodies (large arrows). This phagocytosis resulted in overall reduction of cell bodies and damage to the adjacent neuropil resulting in spongiform change (Fig. 1). Macrophages frequently exhibited TH immunopositivity as a result of phagocytosis of dopaminergic neurons.
Fig. 5.

Neuronophagia by infiltrative microglial/macrophages (small arrows) of dopaminergic neurons that have undergone vacuolar degeneration (large arrows) in the SN/VTA region of a 4-week-old PKU mouse. Note shredding and vacuolation of surrounding neuropil. Tyrosine hydroxylase immunoperoxidase method; scale bar=10 μm.
2.2. Striatal evaluation of PKU mice reveals a deficit of nerve terminal TH immunopositivity and reduction in monoamine metabolite concentration
Tyrosine hydroxylase immunohistochemistry revealed a marked reduction in staining intensity within the striata of 4-week- and 8-month-old PKU mice compared to wild-type mice of similar ages (Fig. 6). The delineation of TH immunoreactivity between the striatal (Str) and cortical (Cx) regions was severely diminished in the PKU mice. Cross-sections of TH-immunopositive axons were not readily appreciated in the PKU mice. There was roughly an 84% difference in pixel intensity between the 4-week-old PKU mice and wild-type mice, and an 81% reduction in intensity between the 8-month-old PKU and wild-type mice.
Fig. 6.

TH immunohistochemical staining in coronal sections passing through the striatal region of untreated wild-type and PKU mice. Note the severe reduction of TH immunoreactivity of striatal nerve terminals in 4-week-old (B) and 8-month-old (D) PKU mice when compared to 4-week-old (A) and 8-month-old (C) wild-type mice. STR, striatum; CX, cortex. Tyrosine hydroxylase immunoperoxidase method; scale bar=25 μm.
Striatal monoamine concentration was assessed in 4-week-old PKU and wild-type mice using high performance liquid chromatography (HPLC) analysis (Fig. 7). There was no difference in dopamine (DA), serotonin (5-hydroxytryptamine, 5-HT) and homovanillic acid (HVA) concentrations between PKU and wild-type mice. However, we found concentrations of 5-hydroxyindoleacetic acid (5-HIAA), 3,4-dihydroxy-phenylacetic acid (DOPAC) and 5-HIAA/5-HT ratios to be significantly reduced in PKU mice. The DOPAC/DA and HVA/DA ratios were not significantly affected.
Fig. 7.

Striatal DOPAC (A), 5-HIAA (B), concentrations and the 5-HIAA/5-HT ratio (C) are significantly reduced in 4 wk old PKU mice. ANOVA; P<0.05, n=5/group.
2.3. Presence of striatal nestin and GFAP-expressing cells in PKU mice
Growing evidence suggests that astroglial cells may be involved in regulating neuronal activities as well as modulating survival of nigrostriatal dopamine neurons. In a 1-methyl-4 phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model for Parkinson's disease (PD) Chen et al. (2004) reported the presence of brain-derived neurotrophic factor (BDNF) and nestin-expressing astroglial cells in the neostriatum. We chose to assess the striatum of PKU mice for similar changes and did observe immunopositive nestin and glial fibrillary acid protein (GFAP) cells scattered throughout the striata of 4-week- and 8-month-old PKU mice (Fig. 8). These cells were relatively numerous (approximately 2–3 cells/40× field) and occasionally closely associated with blood vessels. The nestin-expressing cells sporadically displayed occasional “foot processes” that wrapped around the perimeter of the blood vessel. These scattered cells were 5–10 μm in diameter with round, to unipolar (fusiform or “tadpole”), to bipolar to tripolar processes indicative of immature neuronal morphology. These cells had cytoplasmic nestin and GFAP antigen but BDNF expression was not detected in these cells. The nestin-expressing cells were not present in the SN/VTA of 4-week- and 8-month-old PKU or wild-type mice. There were few (1/(3) 40× fields) randomly scattered astrocytes that were 2–4 μm in diameter and had a “starburst” morphology that expressed BDNF and GFAP antigen, however, there was no difference in the number of these astrocytes between PKU and wild-type mice.
Fig. 8.

Nestin (left column) and GFAP (right column) immunoreactive cells are present in coronal striatal sections of adult PKU mice. Numerous, randomly scattered nestin (A)- and GFAP (D)-expressing cells occur in the striatum of PKU mice. These cells are 5 to 10 μm long and both have similar morphology with unipolar (B, E) to tripolar processes (not shown) and are not present in wild-type mice (C, F). Nestin, GFAP immunoperoxidase method; scale bar=25 μm (A, C, D, F); scale bar=10 μm (B, E).
2.4. Correction of hyperphenylalanemia is associated with alleviation of neuronal injury and macrophage influx in gene therapy treated mice
Five male PKU and three control mice were treated with various doses of the rAAV2-mPAH-WPRE vector. Two weeks following the surgery, serum Phe was reduced to clinically normal levels in male mice, ranging from approximately 0.1 to 0.4 mM (Fig. 9) and remained at these levels for up to 24 weeks (Laipis et al., unpublished). Moreover, the gene therapy led to marked improvement in weight and coat color in these mice. Fig. 9 illustrates that reduction of serum phenylalanine by varying concentrations of vector ranging from 1.3–6.7×1010 resulted in alleviation of the macrophage infiltrate in the SN/VTA regions of PKU mice that were previously reported by our laboratory (Embury et al., 2005). The black bars represent the presence of macrophage infiltrate in the SN/VTA, while the white bars indicate no macrophage infiltrate.
Fig. 9.
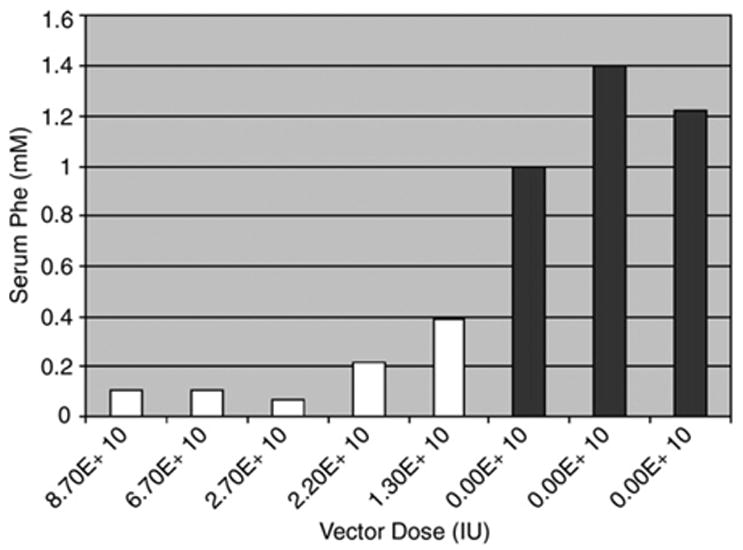
Serum Phe levels are reduced in PKU mice following gene therapy. Average serum Phe levels were calculated beginning 2 weeks after treatment until the end of the experiment at 24+ weeks. LRS control mice (vector dose of 0) were injected with lactated ringers only and doses received are in IU. Black bars denote the presence of macrophage infiltration in the SN/VTA and white bars indicate their absence. Reduction of serum phenylalanine to near normal levels is associated with the disappearance of cellular infiltrate.
TH immunohistochemistry in Fig. 10 reveals the cytoplasmic vacuolar degeneration present in PKU mice (B) is no longer present following administration of the vector (C) and results in a similar morphologic appearance to wild-type mice (A). The presence of these cells is further illustrated in Fig. 11 and shows the macrophage infiltrate of the SN/VTA in PKU mice (B, E) is eliminated following administration of the rAAV-mPAH-WPRE vector (C, F) and is similar to the wild-type animal (A, D). Further immunohistochemical studies confirmed these CD11b and iNOS-expressing macrophages that occurred in PKU mice were not evident in wild-type mice or rAAV-mPAH-WPRE treated mice (data not shown).
Fig. 10.
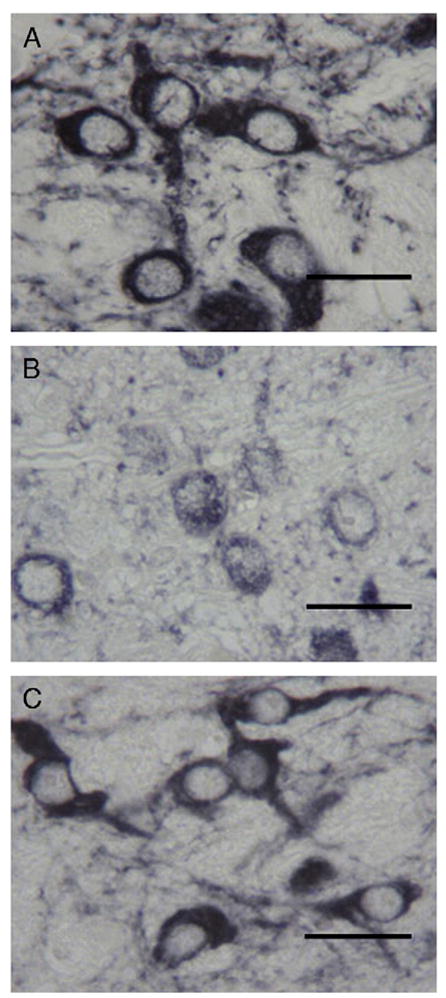
Tyrosine hydroxylase immunoreactivity reveals vacuolar degeneration of dopaminergic neurons in PKU mice (B) is alleviated following administration of rAAV-mPAH-WPRE vector to PKU mice (C). The normal morphology is restored and similar to wild-type mice (A). Tyrosine hydroxylase immunoperoxidase method; scale bar=10 μm.
Fig. 11.
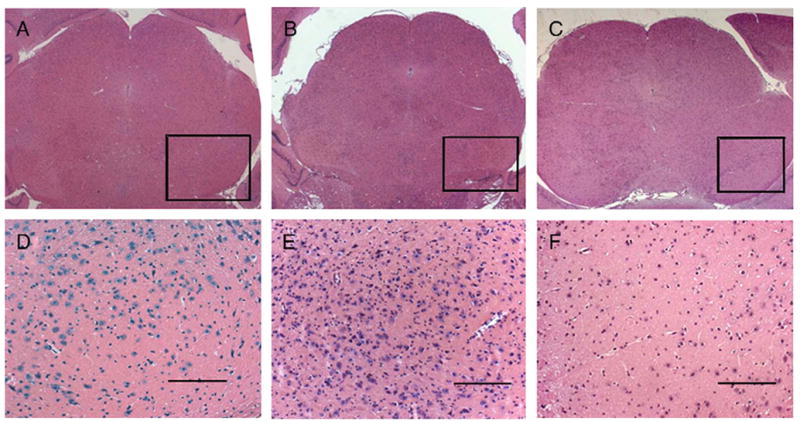
Sections from the mesencephalon of an untreated Pahenu2 mouse (B, E) compared to wild-type BTBR (A, D) and an rAAV-mPAH-WPRE treated male (C, F) that received a dose of 1.3×1010 IU through portal vein administration. There is a marked microglial infiltrate that is most pronounced in the pars reticulata within the homozygous recessive mouse (E), and not apparent in wild-type or vector treated animals. Hematoxylin and eosin, scale bar=50 μm.
3. Discussion
The results of this study provide evidence of reversible neurodegenerative injury to dopaminergic neurons of the SN/VTA in Pahenu2 mice. We offer light microscopic confirmation of somatic injury to the neuron that includes cytoplasmic vacuolar degeneration and subsequent neuronophagia by infiltrative macrophages with resultant spongiform change of the surrounding neuropil. The nuclei were unaffected which supports the diagnosis of reversible cell injury. Distal axonal projections to the striatum were also adversely affected and characterized by a marked reduction in TH immunopositivity and reduction in DOPAC and 5-HIAA concentrations. There is evidence of attempts at regenerative responses in the striatum by infiltration of nestin and GFAP-expressing cells with an immature neuronal morphology. These adverse changes in the nigrostriatum are reversed by reduction of serum Phe levels through rAAV-mPAH-WPRE gene therapy administration.
Ercal et al. (2002) provided experimental evidence of oxidative damage in Pahenu2 mice that included increased concentration of the lipid peroxidation by-product malon-dialdehyde in brain tissue, decreased glutathione/glutathione disulfide also in brain and increased catalase and glucose-6-phosphate dehydrogenase in erythrocytes of these animals. Other studies indicating the role of oxidative stress in PKU include human studies by Artuch et al. (2001, 2004); Colome et al. (2003); and Sierra et al. (1998). Sirtori et al. (2005) demonstrated the involvement of oxidative stress in human PKU patients through increased levels of thiobarbituric acid reactive species (indicative of lipid peroxidation) and decreased total antioxidant reactivity, suggestive of deficient capacity to handle increases in reactive species. Martinez-Cruz et al. (2002) determined that in an experimentally induced rat model of maternal PKU, oxidative stress parameters were increased and adversely influenced morphologic and biochemical development in rat pup brain and cerebellum.
The neuronal vacuolation in the SN/VTA region of these PKU mice is similar to those changes described in a murine transgenic model of human amyotrophic lateral sclerosis (Dal Canto and Gurney, 1994) which express a mutant Cu, Zn superoxide dismutase (SOD). The lesions described for the murine mutant superoxide dismutase transgenic were localized to the large motor neurons of the spinal anterior horns, and appeared as collections of small, tightly packed cytoplasmic vacuoles, which often occupied the entire neuronal cell body. No changes were evident in the nuclei. In older animals, vacuoles were localized both inside neuronal cell bodies and in the surrounding neuropil. Ultrastructural evaluation of these vacuoles were localized to the rough endoplasmic reticulum with dispersion of the ribosomes, and dilation of cisternae resulting in vacuole formation. The second structure involved vacuole formation in mitochondria. Dal Canto and Gurney (1994) speculated these changes in the SOD mutant could be due to an accumulation of superoxide with subsequent hydroxyl radical formation. Additionally, formation of peroxynitrite (OONO−) from superoxide ( ) and nitric oxide (NO−) may have been factor in the neuronal and neuropil vacuolation.
A report of four 3- to 8-month-old Rottweiler puppies describes intracytoplasmic neuronal vacuolation of the cerebellar roof nuclei and nuclei of the extrapyramidal system, as well as neurons in both dorsal nerve root ganglia, myenteric plexus and other ganglia of or the autonomic nervous system (Kortz et al., 1997). The histologic features of the neuronal vacuolation reported in those puppies are similar to those found in the SN/VTA of PKU mice. Ultrastructural evaluation of the vacuoles in the Rottweiler puppies included distention and dilation of both the Golgi apparatus and the smooth and rough endoplasmic reticulum. Cummings and de (1988) reported a litter of three miniature poodle puppies with vacuolar degenerative changes diffusely throughout the cerebral cortex and cerebellar nuclei. Cortical and cerebellar neurons were distended by pale cytoplasmic vacuoles and was referable ultrastructurally to marked dilation of endoplasmic reticulum.
Numerous studies have provided experimental evidence demonstrating oxidative stress in human PKU patients and animal models (Artuch et al., 2001, 2004; Colome et al., 2003; Ercal et al., 2002; Martinez-Cruz et al., 2002; Sierra et al., 1998; Sirtori et al., 2005). Our laboratory previously demonstrated increased inducible nitric oxide synthase production by infiltrative cd11b macrophage cells in the mesencephalon and hypothalamus of PKU mice (Embury et al., 2005), providing further evidence of oxidative processes occurring in these regions.
The effects of free radical damage can be wide-ranging and include lipid peroxidation of cytosolic and mitochondrial membranes, as well as oxidative modifications in cellular proteins and DNA. According to Kumar et al. (2005), reversible cell injury is defined as the ability to compensate for various derangements in cell membrane integrity or generation of ATP and if the injurious stimulus abates, will return to normalcy. Cellular injury can be detected by light microscope evaluation. On microscopic examination, small clear vacuoles may be seen within the cytoplasm, and these represent distended and pinched off segments of endoplasmic reticulum. This pattern of nonlethal injury is called hydropic change or vacuolar degeneration. The ultrastructural changes of reversible cell injury include dilation of the endoplasmic reticulum with detachment and disaggregation of polysomes as well as plasma membrane alterations, and mitochondrial changes. The light microscope changes in VTA/SN dopaminergic neurons of the PKU mice in this study are consistent with these definitions. Ultrastructural studies on the brains of these mice are pending.
Ercal et al. (2002) provides speculation regarding possible initiating events of oxidative stress in PKU brains that include but are not limited to: (1) increased concentrations of Phe in the PKU brain may be metabolized by tyrosine hydroxylase into dihydroxyphenylalanine. Subsequent oxidation of this molecule can produce superoxide ( ) and hydrogen peroxide (H2O2) through a complicated series of reactions (Halliwell and Gutteridge, 1989b). (2) Increased phenylalanine was shown to inhibit Na+-K+ ATPase rat synaptosomes resulting in electron leakage from the inner mitochondrial membrane thereby increasing free radical concentration (Dwivedy and Shah, 1982). (3) Alternative pathways of phenylalanine metabolism may induce free radical formation (Swaiman and Wu, 1984).
In this study we also established reduced TH immunoreactivity and monoamine discrepancies in the striata of Pahenu2 mice which could possibly be attributed to anterograde degeneration through cell body injury in the SN/VTA region, resulting in loss of the distal part of the axon. Dopamine is deaminated and dehydrogenated to form 3,4-dihydroxy-phenylacetic acid (DOPAC) by monoamine oxidase (MAO-A) and aldehyde dehydrogenase. 5-HT is similarly metabolized to 5-HIAA by MAO and aldehyde dehydrogenase. MAO-A predominates in neuronal tissue and is located in striatal synaptic nerve terminals. We surmise the decreased concentrations of striatal 5-HIAA, DOPAC and 5-HIAA/5-HT ratios in the 4-week-old PKU mice may be attributed to decreased monoamine oxidase levels caused by a deterioration in synaptic nerve terminals of the striatum. It will be important to evaluate older animals' striatal monoamine concentrations to determine if the reduction is more severe and generalized possibly affecting all monoaminergic metabolites.
Models of rotenone toxicity that include intranigral infusion of the neurotoxin results in anterograde degeneration of SN neurons over a period of months (Saravanan et al., 2005). Rotenone specifically affects the electron transport chain causing increased levels of oxygen free radicals resulting in oxidative stress, culminating in severe damage of dopaminergic neurons. Similarities between this rotenone model, as well as the 1-methyl-4 phenyl-1,2,3,6-tetrahydropyridine (MPTP) rat and mouse models of Parkinson's disease in terms of reactive oxygen species generation, and the neurodegenerative changes observed in these PKU mice are evident. The neurodegenerative changes in the PKU mouse model could prove useful in the elucidation of pathogenesis of other neurodegenerative diseases such as Parkinson's disease (PD).
In a MPTP mouse model for Parkinson's disease, Chen et al. (2004) identified up-regulation of nestin-expressing cells that were also immunoreactive for GFAP and BDNF in the striatum. Chen speculated these cells might play important roles in the protection of nigrostriatal dopamine neurons in the pathogenesis of Parkinson's disease. We chose to examine the striata of Pahenu2 mice to determine if similar changes were evident and observed developmentally immature cells that were immunoreactive for nestin and GFAP and frequently located near blood vessels. There was a clear morphologic progression of these cells from small and round, to unipolar or tadpole-shaped, to bi- and tripolar immature neurons all which showed cytoplasmic immunoreactivity for nestin and GFAP. There was no evidence of BDNF expression in these immature cells in our hands. We speculate these immature cells with spherical to unipolar to bipolar morphology represent immature neurites that may play a role in regenerative responses to nigrostriatal damage.
Finally, portal vein administration of rAAV-mPAH-WPRE vector to Pahenu2 mice resulted in rapid and long-term reduction of serum Phe. Evaluation of the dopaminergic neurons in the SN/VTA of these treated mice had a normal morphology and showed no evidence of neurodegenerative changes described in this study. Also the cd11b iNOS positive macrophages described in Embury et al. (2005) were no longer present following AAV gene therapy. We propose that by lowering serum Phe through gene therapy administration, oxidative damage is halted and normal dopaminergic cell body morphology is restored, indicating these neurodegenerative changes are reversible. These results reveal that the rAAV-mPAH-WPRE vector was useful in simultaneous reduction of serum phenylalanine levels and diminished presence of cd11b-inos positive microglia in dopaminergic regions of male PKU mice. We redesigned the rAAV vector to contain the Woodchuck hepatitis virus post-transcriptional regulatory element 3' of the mPAH cDNA (rAAV-mPAH-WPRE) to establish increased efficacy of response in female PKU mice which are more severely affected by the disease. Inclusion of the WPRE element has been shown to increase expression of transgenes 8- to 10-fold in vitro and 2- to 4-fold in vivo (Paterna et al., 2000; Schambach et al., 2000). In transient cell transfections, our rAAV-mPAH-WPRE vector is 2- to 4-fold more effective than the rAAV-mPAH vector (Laipis et al., unpublished). Unfortunately however, we have observed a marked increased incidence of systemic neoplastic disease in these animals associated with the use of WPRE and do not recommend its use for traditional gene therapy (Embury et al., 2005, American Society of Gene Therapy, St. Louis, MO).
To summarize, we propose the following schematic diagram may be useful in illustrating a possible explanation for the neuropathogenesis of the disease (Fig. 12).
Fig. 12.
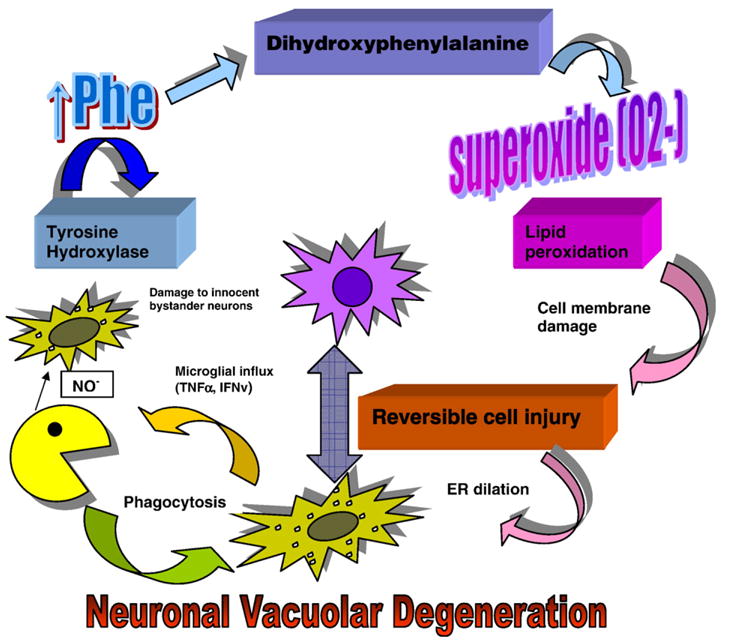
Proposed schematic diagram of possible neuropathogenic mechanisms occurring in hyperphenylalanemic PKU animals (see text).
In PKU, hyperphenylalanemia results in excessive brain Phe accumulation. There is subsequent formation of dihydroxyphenylalanine by tyrosine hydroxylase resulting in free radical formation which injures dopaminergic cell bodies. This may occur through free radical damage ensuing in lipid peroxidation and endoplasmic reticulum (ER) dilation causing cytoplasmic vacuolation, cell damage and some cell death. It is possible there is a release of cytokines such as tumor necrosis factor and gamma-interferon by dead and dying cells to induce signaling influx of the macrophages. We have previously established that there is increased iNOS expression by the infiltrating macrophages resulting in increased NO production. It is possible the NO may have a neuroprotective effect, but there may also be cellular damage to innocent bystander neurons. The NO may result in disruption of ER and further vacuolar degeneration of innocent bystanders. Anterograde nigrostriatal degeneration caused by damaged cell bodies culminate in striatal nestin–GFAP-expressing neurite formation as a regenerative response. These proposed mechanisms are alleviated by reduction of serum Phe by gene therapy.
4. Experimental procedures
4.1. The Pahenu2 mouse model
The Pahenu2 mouse model was initially created through ethylnitrosurea mutagenesis of BTBR mice (Shedlovsky et al., 1993). The mutation is a T to C transition changing Phe 263 to Ser, and incidentally creates a new Alw261 restriction site in exon 7 of the PAH gene on chromosome 10 (McDonald and Charlton, 1997). The Pahenu2 mutation is confirmed by polymerase chain reaction (PCR) amplification of an exon 7 genomic fragment followed by digestion with Alw26I.
4.2. Animal use, surgical procedures and tissue collection
Animal procedures were completed according to the guidelines for animal care at the University of Florida (Gainesville, FL). All mice selected for gene therapy experiments were between 10 and 14 weeks old. Approximately 0.3 ml of vector solution was delivered with a 29-g needle directly into the portal vein following isoflurane anesthesia and ventral mid-line exposure of the vein. Hemostasis was achieved with cotton tipped applicators. 4–0 silk and staples were used to close the incision. Four adult Pahenu2 males received rAAV2-mPAH-WPRE vector doses ranging from 1.3×1010 to 7×1010 infectious units (IU).
A minimum of 3 to 6 homozygous and wild-type animals of both sexes were used for morphologic and immunohistochemical evaluation of the nigrostriatum along with the gene therapy-treated animals. Mice were anesthetized with sodium pentobarbital (80 mg/kg, IP), and intracardially perfused with 4% paraformaldehyde. Brains were removed from the skull following 24 h fixation in 4% paraformaldehyde, transferred to 70% ethanol and routinely processed and embedded in paraffin wax. Each section was 4 μm thick and collected at 40-μm intervals through the entire brain, and alternately stained with hematoxylin and eosin (H&E). Adjacent unstained sections were retained for immunohistochemical studies. Anatomical regions of interest were verified with Paxinos and Franklin's The Mouse Brain In Stereotaxic Coordinates brain atlas (Franklin and Paxinos, 1997).
4.3. Immunohistochemistry
The following antibodies were used for immunostaining individual brain sections: rat anti-mouse cd11b (Serotec, Raleigh NC), anti-inducible nitric oxide synthase (iNOS), rabbit polyclonal (ABR, Golden CO), mouse anti-tyrosine hydroxylase (TH) (Chemicon, Temecula, CA), mouse anti-nestin (Chemicon), mouse anti-human glial fibrillary acidic protein (GFAP) (Serotec), and rabbit anti pro-brain-derived neurotrophic factor (BDNF) (Chemicon). Digest-All 2 (Zymed, San Francisco, CA) was used for antigen retrieval. All tissues were incubated for 12–15 min in 3% hydrogen peroxidase (H202) to quench endogenous peroxidase activity and blocked using the Streptavidin/Biotin Blocking Kit (Vector Laboratories, Burlingame, CA). Tissues were incubated at 4 °C for 24 h at with appropriate primary antibody of dilutions ranging from 1:50–1:200. Appropriate secondary biotinylated antibody (1:100–1:200) (Vector Laboratories) containing 1% normal serum was applied, followed by incubation with Vectastain-Elite ABC (Vector Laboratories) conjugate and visualized with appropriate chromagen (Vector Laboratories). 3,3'-Diaminobenzidine tetrachloride (DAB) with nickel was used for visualization of tyrosine hydroxylase. Sections were examined with bright-field microscopy using a Nikon Labophot-2 microscope. Microscopic images were captured with a QCLR3 3.3 million pixel QColor 3 Olympus digital camera linked to QCapture Image Pro Plus 5.1 image analysis software. For final image output, all images were processed using Adobe Photoshop CS software.
4.4. Immunohistochemistry data analysis
Sections were collected every 40 μm throughout the entire anterioposterior extent of the substantia nigra. Evaluation of TH-immunopositive neurons in the ventral mesencephalon was carried out by visualizing the entire SNPc in one field at 10× magnification. Both the right and left sides were used. Using QCapture Plus Image analysis software, a surface plot was obtained of each image. This enables the volume and intensity of each cell to be combined to obtain the sum cell area and relative percentage of cell occupied space in the field. The line profile function was then used to obtain a plot of the average area and intensity of the image in pixels. The reference option was selected to establish a baseline background value and the difference was calculated between the background and the actual cell field. Each slide was used to calculate a total average cell field per substantia nigra per mouse. Statistical significance between different genotypes of mice was compared using one-way analysis of variance (ANOVA). Measurement of intensity of TH-immunopositive terminals in the striatum was calculated in a similar manner using QCapture Plus image analysis software.
4.5. Determination of biogenic amine levels
PKU and wild-type mice of varying ages were sacrificed by decapitation. The rostral aspect of the left and right striata were collected and frozen on dry ice until further processing for the analyses of biogenic amine levels. The remaining tissue was retained in 4% paraformaldehyde for striatal immunohistochemical evaluation. The tissues were sonicated in ice-cold 0.2 M HClO4 (20% W/V) that contained 100 ng/ml of an internal standard (3,4-dihydroxybenzylamine). After separation by a brief centrifugation, 150 μl of the supernatant was passed through a 0.2 μm Nylon-66 filter and 25 μl of the filtrate was injected on to a HPLC column coupled to an electrochemical detector as previously described (Ali et al., 1994; Zhang et al., 2004). The concentrations of DA, DOPAC, and HVA were calculated using a three-point standard curve generated by determining the known amounts of internal standards.
4.6. Statistical analysis
The data were statistically evaluated for significance by one-way analysis of variance (ANOVA). Results are given as mean± SEM values. Values of P≤0.05 were considered significant.
4.7. Recombinant viruses
The rAAV-mPAH vector was derived from p43CB-AAT (Song et al., 2001) by replacement of the AAT cDNA with the mPAH cDNA. The WPRE vector was constructed by inserting the WPRE and bovine growth hormone polyadenylation signal between the NotI and MfeI restriction sites replacing the original SV40 polyA signal. Recombinant virus production was performed as described previously (Zolotukhin et al., 2002). Briefly, vector DNA was co-transfected into HEK-293 cells with the pDG plasmid, which contains AAV's rep and cap genes along with the required adenovirus genes. After 48 h a cell pellet was obtained, freeze-thawed, and the lysate was separated on an iodixonal step-gradient. The virus was purified on a heparin affinity column, and after concentration the virus was titered by quantitative competitive PCR and infectious center assay.
Acknowledgments
Supported in part by the National Institutes of Health (NCRR K01 RR019979; DK58327,) and March of Dimes (6-FY02-155).
Abbreviations
- AAV
adeno-associated virus
- BDNF
brain-derived neurotrophic factor
- CX
cortex
- DA
dopamine
- DAB
3,3'-diaminobenzidine tetrachloride
- DOPAC
3,4-dihydroxy-phenylacetic acid
- GFAP
glial fibrillary acidic protein
- H202
hydrogen peroxidase
- 5-HIAA
5-hydroxyindoleacetic acid
- 5-HT
5-hydroxytryptamine (serotonin)
- HPA
hyperphenylalanemia
- HVA
homovanillic acid
- INOS
inducible nitric oxide synthase
- MPTP
1-methyl-4 phenyl-1,2,3,6-tetrahydropyridine
- PAH
phenylalanine hydroxylase
- PD
Parkinson's disease
- PKU
phenylketonuria
- PHE
phenylalanine
- rAAV-mPAH-WPRE
recombinant adeno-associated virus-mouse phenylalanine hydroxylase-woodchuck hepatitis virus post transcriptional response element
- RS
reactive species
- SN
substantia nigra
- SNPc
substantia nigra pars compacta
- STR
striatal
- TH
tyrosine hydroxylase
- TYR
tyrosine
- VTA
ventral tegmental area
References
- Ali SF, Newport GD, Holson RR, Slikker W, Jr, Bowyer JF. Low environmental temperatures or pharmacologic agents that produce hypothermia decrease methamphetamine neurotoxicity in mice. Brain Res. 1994;658:33–38. doi: 10.1016/s0006-8993(09)90007-5. [DOI] [PubMed] [Google Scholar]
- Artuch R, Colome C, Vilaseca MA, Sierra C, Cambra FJ, Lambruschini N, Campistol J. Plasma phenylalanine is associated with decreased serum ubiquinone-10 concentrations in phenylketonuria. J Inherit Metab Dis. 2001;24:359–366. doi: 10.1023/a:1010500502275. [DOI] [PubMed] [Google Scholar]
- Artuch R, Colome C, Sierra C, Brandi N, Lambruschini N, Campistol J, Ugarte D, Vilaseca MA. A longitudinal study of antioxidant status in phenylketonuric patients. Clin Biochem. 2004;37:198–203. doi: 10.1016/j.clinbiochem.2003.10.017. [DOI] [PubMed] [Google Scholar]
- Castro L, Rodriguez M, Radi R. Aconitase is readily inactivated by peroxynitrite, but not by its precursor, nitric oxide. J Biol Chem. 1994;269:29409–29415. [PubMed] [Google Scholar]
- Castro LA, Robalinho RL, Cayota A, Meneghini R, Radi R. Nitric oxide and peroxynitrite-dependent aconitase inactivation and iron-regulatory protein-1 activation in mammalian fibroblasts. Arch Biochem Biophys. 1998;359:215–224. doi: 10.1006/abbi.1998.0898. [DOI] [PubMed] [Google Scholar]
- Chen LW, Hu HJ, Liu HL, Yung KK, Chan YS. Identification of brain-derived neurotrophic factor in nestin-expressing astroglial cells in the neostriatum of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice. Neuroscience. 2004;126:941–953. doi: 10.1016/j.neuroscience.2004.04.020. [DOI] [PubMed] [Google Scholar]
- Colome C, Artuch R, Sierra C, Brandi N, Lambruschini N, Campistol J, Vilaseca MA. Plasma thiols and their determinants in phenylketonuria. Eur J Clin Nutr. 2003;57:964–968. doi: 10.1038/sj.ejcn.1601631. [DOI] [PubMed] [Google Scholar]
- Cummings JF, de LA. A study of cerebellar and cerebral cortical degeneration in miniature poodle pups with emphasis on the ultrastructure of Purkinje cell changes. Acta Neuropathol (Berl) 1988;75:261–271. doi: 10.1007/BF00690534. [DOI] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol. 1994;145:1271–1279. [PMC free article] [PubMed] [Google Scholar]
- Diamond A, Prevor MB, Callender G, Druin DP. Prefrontal cortex cognitive deficits in children treated early and continuously for PKU. Monogr Soc Res Child Dev. 1997;62:i–208. [PubMed] [Google Scholar]
- Dwivedy AK, Shah SN. Effects of phenylalanine and its deaminated metabolites on Na+, K+-ATPase activity in synaptosomes from rat brain. Neurochem Res. 1982;7:717–725. doi: 10.1007/BF00965524. [DOI] [PubMed] [Google Scholar]
- Embury JE, Reep RR, Laipis PJ. Pathologic and immunohistochemical findings in hypothalamic and mesencephalic regions in the pah(enu2) mouse model for phenylketonuria. Pediatr Res. 2005;58:283–287. doi: 10.1203/01.PDR.0000170000.78670.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercal N, ykin-Burns N, Gurer-Orhan H, McDonald JD. Oxidative stress in a phenylketonuria animal model. Free Radic Biol Med. 2002;32:906–911. doi: 10.1016/s0891-5849(02)00781-5. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. Academic Press; San Diego: 1997. [Google Scholar]
- Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging. 2001;18:685–716. doi: 10.2165/00002512-200118090-00004. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. Clarendon Press; Oxford: 1989a. [Google Scholar]
- Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. Clarendon Press; Oxford: 1989b. [Google Scholar]
- Huttenlocher PR. The neuropathology of phenylketonuria: human and animal studies. Eur J Pediatr. 2000;159 (Suppl 2):S102–S106. doi: 10.1007/pl00014371. [DOI] [PubMed] [Google Scholar]
- Kaufman S. A model of human phenylalanine metabolism in normal subjects and in phenylketonuric patients. Proc Natl Acad Sci U S A. 1999;96:3160–3164. doi: 10.1073/pnas.96.6.3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortz GD, Meier WA, Higgins RJ, French RA, McKiernan BC, Fatzer R, Zachary JF. Neuronal vacuolation and spinocerebellar degeneration in young Rottweiler dogs. Vet Pathol. 1997;34:296–302. doi: 10.1177/030098589703400405. [DOI] [PubMed] [Google Scholar]
- Krause W, Halminski M, McDonald L, Dembure P, Salvo R, Freides D, Elsas L. Biochemical and neuropsychological effects of elevated plasma phenylalanine in patients with treated phenylketonuria. A model for the study of phenylalanine and brain function in man. J Clin Invest. 1985;75:40–48. doi: 10.1172/JCI111695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Abbas AK, Fausto N, Robbins SL, Cotran RS. Robbins and Cotran Pathologic Basis of Disease. Elsevier Saunders; Philadelphia: 2005. [Google Scholar]
- Levy HL. Phenylketonuria: old disease, new approach to treatment. Proc Natl Acad Sci U S A. 1999;96:1811–1813. doi: 10.1073/pnas.96.5.1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Cruz F, Pozo D, Osuna C, Espinar A, Marchante C, Guerrero JM. Oxidative stress induced by phenylketonuria in the rat: prevention by melatonin, vitamin E, and vitamin C. J Neurosci Res. 2002;69:550–558. doi: 10.1002/jnr.10307. [DOI] [PubMed] [Google Scholar]
- McDonald JD, Charlton CK. Characterization of mutations at the mouse phenylalanine hydroxylase locus. Genomics. 1997;39:402–405. doi: 10.1006/geno.1996.4508. [DOI] [PubMed] [Google Scholar]
- Paterna JC, Moccetti T, Mura A, Feldon J, Bueler H. Influence of promoter and WHV post-transcriptional regulatory element on AAV-mediated transgene expression in the rat brain. Gene Ther. 2000;7:1304–1311. doi: 10.1038/sj.gt.3301221. [DOI] [PubMed] [Google Scholar]
- Pietz J. Neurological aspects of adult phenylketonuria. Curr Opin Neurol. 1998;11:679–688. doi: 10.1097/00019052-199812000-00012. [DOI] [PubMed] [Google Scholar]
- Pietz J, Fatkenheuer B, Burgard P, Armbruster M, Esser G, Schmidt H. Psychiatric disorders in adult patients with early-treated phenylketonuria. Pediatrics. 1997;99:345–350. doi: 10.1542/peds.99.3.345. [DOI] [PubMed] [Google Scholar]
- Pietz J, Dunckelmann R, Rupp A, Rating D, Meinck HM, Schmidt H, Bremer HJ. Neurological outcome in adult patients with early-treated phenylketonuria. Eur J Pediatr. 1998;157:824–830. doi: 10.1007/s004310050945. [DOI] [PubMed] [Google Scholar]
- Puglisi-Allegra S, Cabib S, Pascucci T, Ventura R, Cali F, Romano V. Dramatic brain aminergic deficit in a genetic mouse model of phenylketonuria. NeuroReport. 2000;11:1361–1364. doi: 10.1097/00001756-200004270-00042. [DOI] [PubMed] [Google Scholar]
- Radi R, Cassina A, Hodara R. Nitric oxide and peroxynitrite interactions with mitochondria. Biol Chem. 2002;383:401–409. doi: 10.1515/BC.2002.044. [DOI] [PubMed] [Google Scholar]
- Saravanan KS, Sindhu KM, Mohanakumar KP. Acute intranigral infusion of rotenone in rats causes progressive biochemical lesions in the striatum similar to Parkinson's disease. 2005. pp. 147–155. [DOI] [PubMed] [Google Scholar]
- Schambach A, Wodrich H, Hildinger M, Bohne J, Krausslich HG, Baum C. Context dependence of different modules for posttranscriptional enhancement of gene expression from retroviral vectors. Mol Ther. 2000;2:435–445. doi: 10.1006/mthe.2000.0191. [DOI] [PubMed] [Google Scholar]
- Scriver CR. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill, Health Professions Division; New York: 1995. [Google Scholar]
- Shedlovsky A, McDonald JD, Symula D, Dove WF. Mouse models of human phenylketonuria. Genetics. 1993;134:1205–1210. doi: 10.1093/genetics/134.4.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra C, Vilaseca MA, Moyano D, Brandi N, Campistol J, Lambruschini N, Cambra FJ, Deulofeu R, Mira A. Antioxidant status in hyperphenylalaninemia. Clin Chim Acta. 1998;276:1–9. doi: 10.1016/s0009-8981(98)00091-6. [DOI] [PubMed] [Google Scholar]
- Sirtori LR, Dutra-Filho CS, Fitarelli D, Sitta A, Haeser A, Barschak AG, Wajner M, Coelho DM, Llesuy S, Bello-Klein A, Giugliani R, Deon M, Vargas CR. Oxidative stress in patients with phenylketonuria. Biochim Biophys Acta. 2005;1740:68–73. doi: 10.1016/j.bbadis.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Smith I, Wolff OH. Natural history of phenylketonuria and influence of early treatment. Lancet. 1974;2:540–544. doi: 10.1016/s0140-6736(74)91873-x. [DOI] [PubMed] [Google Scholar]
- Song S, Embury J, Laipis PJ, Berns KI, Crawford JM, Flotte TR. Stable therapeutic serum levels of human alpha-1 antitrypsin (AAT) after portal vein injection of recombinant adeno-associated virus (rAAV) vectors. Gene Ther. 2001;8:1299–1306. doi: 10.1038/sj.gt.3301422. [DOI] [PubMed] [Google Scholar]
- Swaiman KF, Wu SR. Phenylalanine and phenylacetate adversely affect developing mammalian brain neurons. Neurology. 1984;34:1246–1250. doi: 10.1212/wnl.34.9.1246. [DOI] [PubMed] [Google Scholar]
- Wajner M, Latini A, Wyse AT, Dutra-Filho CS. The role of oxidative damage in the neuropathology of organic acidurias: insights from animal studies. J Inherit Metab Dis. 2004;27:427–448. doi: 10.1023/B:BOLI.0000037353.13085.e2. [DOI] [PubMed] [Google Scholar]
- Wyse AT, Bavaresco CS, Bandinelli C, Streck EL, Franzon R, Dutra-Filho CS, Wajner M. Nitric oxide synthase inhibition by L-NAME prevents the decrease of Na+, K+-ATPase activity in midbrain of rats subjected to arginine administration. Neurochem Res. 2001;26:515–520. doi: 10.1023/a:1010912929042. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wang T, Qin L, Gao HM, Wilson B, Ali SF, Zhang W, Hong JS, Liu B. Neuroprotective effect of dextromethorphan in the MPTP Parkinson's disease model: role of NADPH oxidase. FASEB J. 2004;18:589–591. doi: 10.1096/fj.03-0983fje. [DOI] [PubMed] [Google Scholar]
- Zolotukhin S, Potter M, Zolotukhin I, Sakai Y, Loiler S, Fraites TJ, Jr, Chiodo VA, Phillipsberg T, Muzyczka N, Hauswirth WW, Flotte TR, Byrne BJ, Snyder RO. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28:158–167. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]