

Abstract
O6-alkylguanine-DNA alkyltransferase (AGT) is a crucial target both for the prevention of cancer and for chemotherapy, since it repairs mutagenic lesions in DNA, and it limits the effectiveness of alkylating chemotherapies. AGT catalyzes the unique, single-step, direct damage reversal repair of O6-alkylguanines by selectively transferring the O6-alkyl adduct to an internal cysteine residue. Recent crystal structures of human AGT alone and in complex with substrate DNA reveal a two-domain a/β fold and a bound zinc ion. AGT uses its helix-turn-helix motif to bind substrate DNA via the minor groove. The alkylated guanine is then flipped out from the base stack into the AGT active site for repair by covalent transfer of the alkyl adduct to Cys145. An asparagine hinge (Asn137) couples the helix-turn-helix DNA binding and active site motifs. An arginine finger (Arg128) stabilizes the extrahelical DNA conformation. With this newly improved structural understanding of AGT and its interactions with biologically relevant substrates, we can now begin to unravel the role it plays in preserving genetic integrity and discover how it promotes resistance to anticancer therapies.
Keywords: O6-alkylguanine-DNA alkyltransferase, AGT, O6-methylguanine-DNA methyltransferase, MGMT, DNA repair, protein crystal structure
1. Introduction
The genome is under constant attack from both exogenous and endogenous factors. The resulting DNA lesions must be continuously repaired to preserve genomic integrity. Many types of DNA lesions can result, requiring a variety of DNA repair systems, including nucleotide excision repair (NER), base excision repair (BER), and mismatch repair. DNA alkyl lesions arise from endogenous sources (such as S-adenosylmethionine) [1], environmental toxins [2], and anticancer chemotherapies [3]. Alkylation at the O6-position of guanine can result in G:C to A:T transition mutations [4], which are both mutagenic and cytotoxic. This is because guanine, which normally base pairs with cytosine, will often mispair during replication with thymine when alkylated at the O6-position (Fig. 1A). The DNA repair protein O6-alkylguanine-DNA alkyltransferase (AGT; E.C.2.1.1.63), also known as O6-methylguanine-DNA methyltransferase (MGMT), catalyzes the repair of alkylated guanines by stoichiometrically and irreversibly transferring O6-alkyl adducts to an active site cysteine (Cys145 in human AGT) in a suicide reaction, without inducing DNA strand breaks (Fig. 2). Aside from AGT, this type of DNA alkylation removal, sometimes referred to as direct damage reversal, is known in humans for only the ABH2 and ABH3 proteins, both of which are Fe(II)/2-oxoglutarate-dependent dioxygenases that repair the cytotoxic 1-methyladenine and 3-methylcytosine alkylation lesions by coupling oxidative demethylation of these substrates to conversion of 2-oxoglutarate into succinate and CO2 [5]. AGT-mediated DNA repair differs from other DNA repair pathways, in the following ways: AGT 1) can act alone 2) is both a transferase and an acceptor of the alkyl lesion, 3) is suicidal (inactivated after accepting the alkyl lesion), and 4) mediates stoichiometric repair [6]. These properties make AGT an important target in the design of anticancer strategies.
Fig. 1.

AGT substrates. (A) Normal guanine:cytosine base pair (left), O6-methylguanine:cytosine base pair (middle), and O6-methylguanine:thymine base pair (right). (B) O6-methylguanine, O6-benzylguanine, and O4-methylthymine.
Fig 2.

The AGT active site and proposed reaction mechanism. (A) Hydrogen bond network and proposed reaction mechanism for AGT. To facilitate attack at the O6-alkyl carbon, His146 acts as a water-mediated general base to deprotonate Cys145. The resultant imidazolium ion is stabilized by Glu172. Tyr114 donates a proton to N3 of O6-methylguanine. (B) Stereo view of the X-ray crystallographic structure of the AGT C145S-DNA complex (pdb 1t38) showing the active site, O6-methylguanine substrate, and hydrogen bond network.
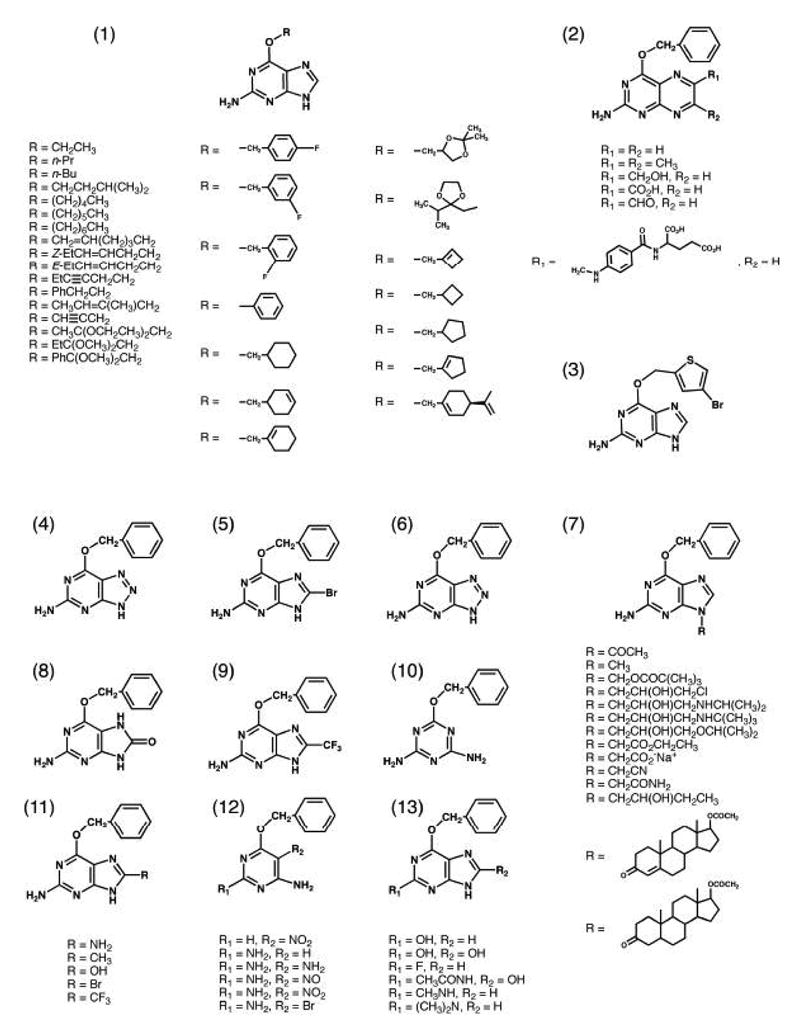
AGT is widely distributed amongst more than 100 different species, with known examples from Eubacteria, Archaea and Eukarya (Fig. 3) [7]. However, there is no AGT gene in plants, Schizosaccharomyces pombe, or Deinococcus radiodurans [7]. There are about 25 very highly conserved residues that provide a focus for structural and biochemical studies. Most of these residues have been implicated, through mutagenesis, in DNA binding, facilitation of alkyl transfer, and maintenance of AGT structure [8–18]. Despite significant variance of substrate selectivity across species, AGT is almost exclusively specific for repair of guanines with O6-alkyl adducts (Fig. 1B). The only other type of DNA lesion known to be repaired by AGT is O4-methylthymine (Fig. 1B). The repair of O4-methylthymine by human AGT is very slow and it actually impedes repair by NER, resulting in increased mutation [19,20]. Other AGTs, such as the E. coli Ogt, repair O4-methylthymine much more rapidly [21]. Human AGT is not limited to O6-methylguanine, but can also repair many other adducts at the O6-position of guanine, including ethyl-, 2-chloroethyl- and other aliphatic groups [22], and also benzyl- [23] and pyridyloxobutyl- [24] adducts. In fact, perhaps hundreds of AGT pseudosubstrates have been synthesized by various groups (Fig. 4) [25–35].
Fig. 3.

Sequence alignment of AGTs from select organisms. The active site motif and the DNA-binding region of the helix-turn-helix motif are outlined in blue. Human AGT numbering is used. The arginine finger (128), “Asn hinge” (137), and active site cysteine (145) are highlighted in yellow.
Fig. 4.

Selected AGT pseudosubstrates.
2. AGT structure
Several X-ray crystallographic and solution structures of unreacted AGT from humans [18,36], E. coli (Ada-C) [37], the archaebacterium Pyrococcus kodakaraensis (Pk) [38], and the thermophile Methanococcus jannaschii (MJ1529) [39] have revealed a two-domain a/β fold, with overall dimensions of approximately 20 × 35 × 40 Å for the human protein (Fig. 5A). Comparative analyses of these structures have allowed the identification of conserved structural motifs that are important for DNA recognition, substrate binding and selectivity, the alkyl-transfer mechanism, and the post-alkylation fate of AGT [40]. Although the human, E. coli, Pk, and MJ1529 AGTs display only 14% identity and 26% similarity [40], the overall domain structure and fold are almost identical among these four homologs [18,36–39].
Fig. 5.

Human AGT X-ray crystallographic structure, zinc site, and DNA binding. (A) Unreacted AGT structure (pdb 1eh6). The N-terminal domain is shown in green, the C-terminal domain is shown in yellow, and the HTH motif is shown in blue. The active site cysteine, arginine finger, “Asn hinge”, and zinc ligands are shown in ball-and-stick representation. (B) DNA-bound AGT structure (pdb 1t39). AGT uses the recognition helix of the HTH motif to bind the minor groove of DNA.
The N-terminal domains (approximately residues 1–85) for human, E. coli, Pk, and MJ1529 AGTs show no significant primary structural homology, yet they all exhibit a conserved a/β roll structure [39,40]. For all four structures, there is a three-stranded anti-parallel β-sheet followed by either two (human, E. coli, MJ1529) or three (Pk) helices. Remarkably, these secondary structures have relatively small variations in their lengths and orientations.
Human AGT contains a Zn(II) ion that is not present in any of the bacterial structures [18]. This Zn(II) ion binds the ligands Cys5, Cys24, His29, and His85 with tetrahedral coordination, and bridges the three strands of the N-terminal β-sheet with the coil immediately preceding the domain-spanning helix. The zinc ligands are completely conserved in the known mammalian sequences, suggesting that this zinc site may be general to higher eukaryotes. The zinc ion likely serves a structural role, and may stabilize the interface between the N- and C-terminal domains by securing the domain-spanning helix to the three strands of the N-terminal βsheet. Zinc binding causes increased stability of AGT and a small conformational change [41]. This is consistent with small distortions and slightly increased disorder of N-terminal residues in the apo-metal structures [36,42], as compared to the zinc-bound structures [18,43]. The removal of zinc from AGT does not inactivate this protein, but reduces the rate constant for repair by 60-fold; however, DNA binding is not affected [41].
The topology of the C-terminal domain is absolutely conserved in all known AGT structures. This domain (approximately residues 86–207) is composed of a short two-stranded, parallel β-sheet, four a -helices, and a 310 helix. Contained within this domain are the conserved active-site cysteine motif (PCHR), the O6-alkylguanine binding channel, and a helix-turn-helix (HTH) DNA-binding motif. Structures of methylated and benzylated human AGT have established that the active site is located near the recognition helix of the HTH motif [18]. The nucleophilic cysteine, part of the invariant active site PCHR motif of the 310 helix, is located near the bottom of a solvent accessible groove. This groove is approximately 8 Å wide, 9 Å deep and 14 Å long, and defines the O6-alkylguanine binding channel. Residues Tyr114-Ala121 and Ala127-Gly136 (human AGT numbering) form the first and second helices, respectively, of the HTH motif. The second HTH helix, called the recognition helix, interacts with DNA. The active site and HTH DNA binding motifs are linked by an “Asn-hinge” (Asn137) (Fig. 3), which stabilizes two overlapping tight turns. This Asn-hinge makes hydrogen bonds with the main chain atoms of Val139 and Ile143 and the thiol of Cys145, and makes contacts through the interdomain cleft with the N-terminal domain, accounting for more than 40% of the buried surface area between the two domains. The geometries of the HTH DNA-binding motif, the Asn-hinge, and the active site helix are essentially identical for all of these structures.
While the C-terminal domain of AGT contains the known necessary residues for DNA binding and alkyl transfer, the function of the N-terminal domain is still not known. In an attempt to better understand the role of the N-terminal domain, the two domains of human AGT have been separately expressed and purified [7]. Interestingly, the C-terminal domain alone retains no activity in the absence of the N-terminal domain and Zn2+. Surprisingly, both in vitro and in vivo experiments suggest that the N-terminal domain has weak AGT activity in the absence of the C-terminal domain. However, neither domain, either separately or together, retained activity in the absence of Zn2+. These data suggest an important structural role for the N-terminal domain in orienting the C-terminal domain for proper catalysis. Notably, two active AGT homologs, cAGT-2 from Caenorhabditis elegans [44] and AGTendoV from Ferroplasma acidarmanus [45], lack the N-terminal domain. Instead, cAGT-2 and AGTendoV each possess a long C-terminal extension protruding from the AGT domain that is similar to histone 1C and endonuclease V, respectively. These C-terminal extensions may be important for proper catalytic orientation of the active sites in these homologs.
3. AGT substrate binding and nucleotide flipping
Crystal structures of human AGT in the absence of DNA substrates provided a basis for addressing several questions about AGT function, but left some important unanswered questions. For example, by what mode does AGT bind DNA? How does AGT find and recognize an alkylated DNA lesion? Once AGT finds a site of damage, how does it gain access to the alkylated guanine? What is the basis for AGT’s substrate specificity? Since the AGT reaction is stoichiometric, how does it perform its repair in the most efficient manner? Recently, several human AGT-DNA complex crystal structures have become available (Fig. 6). One structure is of a catalytically inactive AGT mutant in a pre-transfer complex with DNA containing the biological substrate O6-methylguanine (Fig. 6A) [42]. A second structure is of a reaction intermediate consisting of native AGT covalently crosslinked to the N1,O6-ethanoxanthosine inhibitor (Fig. 6B) [42]. A third structure is of wild-type AGT in complex with a DNA substrate containing a chemically modified cytosine (N4-p-xylylenediaminecytosine) (Fig. 6C) [43]. These structures provide a molecular framework that can be used to answer some of the questions about AGT activity and aid in defining a plausible catalytic mechanism.
Fig. 6.

Human AGT substrate binding and nucleotide flipping. X-ray crystallographic structures of AGT in complex with DNA substrates containing (A) O6-methylguanine (pdb 1t38), (B) N1,O6-ethanoxanthosine (pdb 1t39), and (C) N4-p-xylylenediaminecytosine (pdb 1yfh). Tyr114 and Arg128 may promote 3′ phosphate rotation-induced nucleotide flipping.
Mutational data first implicated the HTH motif of AGT as the DNA-binding domain of the enzyme [13,15]. This was later supported by NMR structures of Ada-C, which provided the first direct experimental evidence of HTH-mediated DNA binding for this protein [46], followed by conclusive verification from crystal structures of AGT-DNA complexes [42,43]. HTH motifs are common elements present in roughly one-third of DNA-binding protein families, including DNA repair proteins [42]. Prior to the AGT-DNA complex structures, HTH motifs of DNA repair proteins were thought to interact with the DNA major groove for sequence-specific recognition. Surprisingly, structural data revealed that the HTH motif of human AGT promotes non-classical minor groove DNA binding (Fig. 5B). The well-conserved, small and hydrophobic nature of the recognition helix residues (Ala126, Ala127, Ala129, Gly131, and Gly132) allows this helix to pack closely with the DNA minor groove, minimizes sequence-specific interactions, and may also be advantageous for DNA repair and nucleotide flipping. Thus, AGT-DNA complex structures provided the first examples of HTH-mediated minor groove DNA binding. Other sequence independent DNA-binding proteins with HTH motifs may also exhibit minor groove-mediated DNA binding.
Originally, it was not known how AGT gained access to its alkylated guanine substrate. Initial models of AGT-mediated DNA repair proposed a significant protein conformational change through movement of the C-terminal α-helix to expose the recessed active site cysteine [37]. AGT crystal structures in the absence of DNA substrates, however, indicated that the alkylated guanine may be flipped out of the DNA helix [18,36]. Similarly, NMR structures of Ada-C in complex with DNA provided direct evidence that the conformational change for this protein is minimal upon binding methylated DNA, which suggested that base flipping was required for the repair function [46]. AGT-DNA complex structures confirmed that to gain access to alkylated guanines, AGT rotates the target base out from the DNA base stack and into its active site pocket (Fig. 6) [42,43]. This is not uncommon, since DNA repair proteins often flip target nucleotides to gain access to damaged bases that would otherwise be inaccessible within the DNA duplex structure. DNA binding does not cause much structural change to the human AGT protein itself. However, it does result in significant structural changes to the DNA [42,43]. Specifically, DNA binding causes the minor groove to widen more than 3 Å when compared to B-DNA without distortion, and causes the DNA to bend roughly 15° away from the protein [42]. These changes may help to flip out the damaged base from the DNA helix and to promote AGT cooperative binding to facilitate damage recognition.
Crystal structures of AGT in complex with DNA suggest a mechanism for 3′ phosphate rotation-induced nucleotide flipping that involves conserved residues Tyr114 and Arg128 (Fig. 6). The Tyr114 side chain is conformationally restricted, and therefore, by steric clash and charge repulsion, may promote nucleotide flipping via rotation of the 3′ phosphate [42]. Nucleotide base flipping with 3′ phosphate rotation has been directly observed in the protein-DNA co-crystal structures of other DNA repair proteins, such as the DNA glycosylases AlkA [47], mismatch-specific uracil DNA-glycosylase (MUG) [48], 3-methyladenine-DNA glycosylase [49], uracil-DNA glycosylase (UDG) [50], and human 8-oxoguanine glycosylase (hOGG1) [51], and the endonucleases apurinic/apyrimidinic endonuclease-1 (APE1) [52] and endonuclease IV [53]. AlkA, UDG, APE1, and endonuclease IV use a single aromatic side chain for phosphate rotation, similar to Tyr114 in human AGT, whereas MUG and hOGG1 use multiple residues for phosphate rotation. This sterically enforced 3′ phosphate rotation may be a mechanism general to nucleotide flipping for DNA repair proteins [42].
Arg128, located at the beginning of the recognition helix of the HTH motif, is an “arginine finger” that intercalates via the minor groove between the bases on either side of the substrate guanine (Fig. 6) [42]. This arginine interacts via a charged hydrogen bond with the orphaned cytosine (previously base paired to the flipped-out guanine) to stabilize the extrahelical DNA conformation. It may also be involved in scanning for damaged bases by playing an active role in pushing out bases. Mutations at Arg128 revealed that the length of this side chain is directly proportional to the alkyl transfer rate for methylated duplex DNA, but not for free O6-alkylguanine bases [40]. Replacement of arginine with lysine results in a modest loss in activity with methylated duplex DNA, since lysine is long enough to penetrate the base stack but has diminished stacking and hydrogen-bonding capability [40]. The R128L mutant has a five-fold reduction in dealkylation activity [40]. Leucine at this position may pack in the hydrophobic interior of the DNA base stack as seen for UDG [54] and AlkA [47], but it is not able to participate in any hydrogen bonds. Substitutions with even smaller amino acids result in a large loss of activity. The R128G mutation shows less than 0.02% of wild type activity [40]. These mutations have a minimal affect on DNA binding, and therefore do not slow repair through decreased DNA-binding affinity. These observations further support the importance of the role of Arg128 in stabilization of flipped-out bases.
The –PCHR– active site motif (residues 144–147 in human AGT), conserved throughout AGTs, (Fig. 3) [7], contains the active site cysteine (residue 145 for human AGT) (Fig. 2A). This reactive cysteine, located approximately 9 Å from the surface of the protein, [18], has a low pKa and high reactivity [55]. The cysteine thiolate and methyl group to be transferred are well-positioned for the dealkylation reaction, since they are almost directly opposite one another (Fig. 2) [42]. For the C145S AGT mutant-DNA complex structure, the C145S side chain is approximately 3.4 Å away from the methyl lesion, and rotates more than 90° to accommodate O6-methylguanine in the active site (Fig. 2B,6A) [42].
A Glu-His-water-Cys hydrogen bond network (Fig. 2), similar to the Asp-His-Ser catalytic triad of serine proteases, may increase reactivity of Cys145 in AGT [18,40]. This hydrogen bond network is conserved in AGTs, including Ada-C [46]. Like the serine protease catalytic triad, the AGT hydrogen bond network also places its histidine side chain in a hydrophobic environment. It is thought that the low pKa and high reactivity of Cys145 is due to generation of a thiolate anion via proton transfer through the network. According to this mechanism for the dealkylation reaction, His146 acts as a water-mediated base, while Cys145 acts as the nucleophile. Reactivity may also be promoted by reduction of the negative charge on the repaired guanine through the donation of a hydrogen bond by Tyr114 to N3 of O6-methylguanine (Fig. 2) [42].
Structures of AGT in complex with DNA substrates and inhibitors provide a basis with which to better understand recognition and selectivity by AGT. Selectivity for guanine is provided by hydrogen bonds and steric interactions [42]. The extrahelical base fits into a hydrophobic cleft made up of the Met134 side chain and Val155-Gly160 of the active site loop. Cys145 and Val148 carbonyls accept hydrogen bonds from guanine’s exocyclic amine. Tyr114 hydroxyl and Ser159 N atoms donate hydrogen bonds to guanine’s N3 and O6 atoms, respectively. These interactions provide AGT’s selectivity of guanine over adenine. The Gly131 Ca atom is only 3.5 Å away from C2′ atom of the ribose, leaving insufficient space for an O2′ ribose atom and accounting for the lack of repair of ribonucleotides by AGT. No direct contacts are made with the extrahelical ribose. Although the basis for recognition of O4-methylthymine by AGT has yet to be determined, the structure of AGT in complex with N4-p-xylylenediaminecytosine suggests that a Tyr114 hydrogen bond to the cytosine O2 atom, may also be present for thymine [43]. However, this structure does not yield insight into protonation of the N3 atom of the damaged base, as for guanine.
Selectivity of O6-methylguanine over guanine seems to be based upon hydrophobic interactions [42]. Tyr158 packs against small O6-alkyl adducts, and Pro140 can interact with larger O6-alkyl adducts [18,42]. There is only about a three-fold increase in affinity for O6-methylguanine over guanine [56], which may be the reason that O6-alkylguanines are repaired by a unique direct pathway, rather than a more typical base excision pathway. This direct damage reversal may act as a safe-guarding mechanism against the cytotoxic and mutagenic excision of undamaged guanines [42].
While AGT can react with a variety of O6-benzylguanine analogs, it prefers para-substituted derivatives to meta-, and it displays poor recognition for ortho-substituted derivatives [27,57]. The poor recognition for ortho-substituted O6-benzylguanine analogs can be explained by the relatively close packing (3.5 Å) of Tyr158 and the recognition helix along the edges of the benzyl group. The Gly160 Ca atom packs against one meta-position at a distance of 4.2 Å. This explains why AGT tolerates a single meta-substituent, but has a decreased rate for two meta-substituents. The para-carbon position is exposed near the inter-domain cleft, and this allows for a variety of bulky substituents at this position. The methyl adduct in the methylated AGT product complex overlays with the corresponding benzylic carbon of the benzyl adduct, but since it is smaller, does not exhibit the extensive hydrophobic packing interactions of the benzyl group [18]. This is consistent with the approximately 2000-fold lower activity of AGT toward O6-methylguanine [30].
It is known that for AGT to repair DNA, it must first bind DNA and detect the alkyl lesion. It is not known, however, how AGT efficiently finds sites of damage. Three mechanisms have been proposed for how AGT locates damaged DNA [58]: (1) AGT migrates along DNA and detects lesions by actively flipping out every base into its active site; (2) AGT selectively detects intrahelical lesions resulting from unstable or non-Watson-Crick base pairs; (3) AGT captures a hypothetical transient extrahelical base lesion that would result from the inability of the damaged base to form a stable Watson-Crick base pair with the opposing base of the complementary strand. Unfortunately, there are no direct methods available to distinguish between these scenarios. To better understand base flipping by AGT, Duguid and coworkers used a disulfide cross-linking strategy with a chemically modified cytosine to trap bases in the Ada-C and AGT active sites [58]. Their results suggest that Ada-C only detects and flips out unstable base pairs, whereas human AGT can flip out both stable and unstable base pairs. However, fluorescence and kinetic experiments of repair by human AGT indicate that this protein selectively flips out O6-alkylguanines from unstable base pairs, but does not affect normal G:C or A:T base pairs [59]. It has been recently proposed that AGT detects DNA lesions by searching for weakened and/or distorted base pairs rather than for the actual adduct [43].
AGT binds to single- and double-stranded DNA in a highly cooperative manner [56]. This could promote directional bias in repair and may aid the search for damaged DNA through localization of multiple AGT molecules. AGT repairs O6-methylguanine lesions at the 5′ end of DNA roughly 3.3 times faster than at the 3′ end in single stranded oligonucleotides with lesions near both ends [42]. This preference for repair at the 5′ end can be eliminated by putting a streptavidin block in the center of the oligonucleotide [42], further emphasizing the bias in the search mechanism. The 3′-to-5′ kinetic scanning bias, along with the cooperative DNA binding, suggest efficient AGT binding at the 5′ side of a DNA-binding nucleating AGT molecule [42]. This may be through some conformational change, such as further widening of the DNA minor groove observed in AGT-DNA complex structures [42]. Nucleation by AGT of template DNA soon to be transcribed or replicated slightly ahead of the transcription or replication machinery could result in the cooperative recruitment of other AGT molecules 5′ of the initial nucleation site [60]. This would allow for the 3′-to-5′ bias and for AGT to remain slightly ahead of transcription or replication machinery. This directional scanning and bias would allow for template DNA lesions to be removed before the polymerases reach them, thereby maximizing the efficiency of repair. By repeating this cycle of cooperative binding and repair, DNA could be constantly refreshed to provide nondamaged template DNA for transcription or replication. Also targeting AGT to regions of DNA where it is most needed would maximize efficient use of AGT and may help to compensate for the suicidal mechanism. However, experiments to determine if AGT can keep up with elongation rates by DNA and RNA polymerases have not yet been done. In support of the model proposed by Daniels et al. [42], genetic and biochemical data indicate that AGT selectively targets DNA undergoing transcription [61].
Once alkylated, mammalian AGT is unstable [62] and is rapidly degraded via the ubiquitin/proteasomal system [63,64]. The cause of ubiquitination is unknown, but may be promoted by alkylated AGT’s significant conformational change, which results from a steric clash between the S-alkylcysteine and Met134 [18]. The destabilized protein is rapidly degraded by the 26S proteosome [65]. This rapid degradation may be needed to prevent the inactive alkylated form of the protein from interfering with repair of additional O6-methylguanine lesions [19]. Inactivated AGT can also prevent transcriptional inactivation of genes that promote cell proliferation by binding the estrogen receptor [66].
The AGT gene contains a number of single nucleotide polymorphisms (SNP), several of which are located in the coding region of the gene: W65C, L84F, I143V, G160R and K178R [67,68]. Two of these SNPs, I143V and G160R, are near the active site Cys 145. The main chain of Ile 143, for example, makes hydrogen bonds with the Asn hinge (Asn 137), which connects the active site and DNA binding motifs. This could affect the substrate specificity but Ile 143 is replaced by Val in multiple known AGTs (Fig. 3). G160R confers AGT resistance to O6-benzylguanine [69]. Substitution of glycine for the longer arginine side chain at this position likely obstructs bulky substrate binding through steric or charge clashes. The W65C polymorphism leads to instability of the protein. Trp 65 is located at the interface between N- and C-terminal domains, and it is possible that replacement of the bulky tryptophan side chain with that of the smaller cysteine affects stability by disrupting p-interactions with Tyr 69 of the N-terminal domain and interrupting hydrophobic interactions with the Asn-hinge region in the C-terminal domain [67]. The L84F may affect Zn2+ binding, since it is exactly adjacent to the zinc binding ligand His 85. Similar to W65C, this residue is located at the interface between the two domains, but in this case, a large phenylalanine residue is replacing a small leucine residue. AGT structures, therefore, help to explain how these SNPs may have subtle effects on AGT-mediated repair of DNA alkyl adducts, influencing susceptibility to cancer and other diseases.
4. Implications for cancer chemotherapy
In addition to the direct repair of O6-alkylguanine lesions in DNA, AGT promotes tumor resistance to certain alkylating agents, commonly used in cancer treatments. These fall into two classes. The first are methylating agents such as procarbazine, dacarbazine (DTIC), streptozotocin and temozolomide. The second are chloroethylating agents such as 1,3-bis (2-chloroethyl)-1-nitrosurea (BCNU), ACNU, MeCCNU and CCNU. Both classes cause apoptosis in tumor cells [70–73] but via different mechanisms. Chloroethylating agents form lethal DNA crosslinks via a 1-(3-cytosinyl)-2-(1-guanyl)-ethane bridge. This results from chloroethylation at the O6-position of guanine, followed by spontaneous cyclization to form N1,O6-ethylguanine. This intermediate is unstable and readily reacts with N3 of the opposing cytosine to form the toxic crosslink between N1 of guanine and N3 of cytosine [74–78]. These crosslinks inhibit replication and transcription of DNA and lead to double strand breaks of DNA and apoptosis. Although there are other DNA and protein adducts formed by therapeutic chloroethylating agents, the G-C crosslink is a major factor in the killing of tumor cells. The ability of AGT to repair O6-chloroethylguanine rapidly and thus prevent the formation of this toxic damage renders the AGT content a critical factor in promoting resistance to these agents. Several studies have documented the importance of AGT in the outcome of therapy with BCNU by showing that a favorable response to treatment is inversely correlated with tumor AGT content [79–81]. Chloroethylating agents have been used for CNS tumors, multiple myeloma, melanoma, lymphoma, gastrointestinal tumors and other solid tumors. Although some remarkable responses have been obtained, their efficacy is limited with less than 20% of patients showing a major response. It is likely that the presence of AGT is a major factor in this limitation.
Therapeutic methylating agents form a wide variety of DNA monoadducts all of which may contribute to cell killing but O6-methylguanine appears to be of particular importance in leading to cell cycle arrest and apoptosis [73]. Its presence in DNA causes apoptotic cell death via a mechanism that depends on the activity of the mismatch repair (MMR) system [82,83]. One plausible explanation for this is that MMR recognizes O6-methylguanine:thymine as a mispair but removes the thymine. This causes a futile cycle of nucleotide removal and synthesis that generates single- and double-strand breaks in DNA and leads to apoptosis [84,85]. This explanation was supported by studies showing that O6-methylguanine was not toxic but highly mutagenic in the absence of MMR due to loss of protein components of MutSα or MutLα. However, studies using point mutations of MSH2 and MSH6 that disrupt the ATP processing ability of MutSα but do not prevent its binding to mismatched base pairs, did not block the apoptotic response to methylation damage [86,87]. Also, MutSα tightly binds O6-methylguanine:thymine mispairs and the resulting MutSαMutLα complex recruits ATR-ATRIP and activates ATR kinase [88]. These studies indicate that MMR proteins can act to cause cell killing by binding at sites of O6-methylguanine and signalling the presence of DNA damage. The two mechanisms are not mutually exclusive and both may contribute to the overall effect.
Therapeutic methylating agents are used to treat Hodgkin’s disease, brain tumors, melanoma and lymphomas. Temozolomide, with better pharmacokinetic properties than procarbazine or DTIC, was found to be effective in treating anaplastic astrocytomas and glioblastoma multiforme [89–91]. The absence or reduction in AGT activity due to the silencing of the MGMT gene by promoter methylation correlates with the efficacy of the treatment of glioblastoma by temozolomide [92,93].
In addition to the studies described above, many experiments using methylating and chloroethylating agents have showed greater cell killing of cultured cells lacking AGT and a better response of human tumor xenografts in nude mice occurs with tumors lacking AGT [22,94–96]. Therefore, a good case can be made for the development of AGT inhibitors that can be used to expand the range and efficacy of agents such as temozolomide and BCNU. A serious drawback with this approach is that alkylating agents are powerful mutagens and, in the absence of AGT, this property, which is known to be associated with the development of secondary tumors, will be enhanced. Thus, approaches that provide tumor specificity to AGT inactivation have particular merit and studies with more general AGT reduction are likely to be justifiable only when no alternatives are available.
Strategies to reduce AGT levels by regulatory approaches such as antisense or ribozymes have been suggested [97,98] and there is a great deal of evidence showing that AGT levels can be reduced by methylation-induced promoter silencing [92,93]. However, no viable clinical approaches have yet resulted from the development of methods to block AGT synthesis. In contrast, the direct inactivation of AGT has been a very active field, which has resulted in a number of clinical trials.
An important factor in developing anticancer therapies that target AGT is that AGT-mediated repair is stoichiometric. Therefore, since one AGT molecule is needed for each offending alkyl molecule, levels of AGT in cells can be completely depleted by an excess of O6-alkylated guanines. Additional factors that make AGT ideal for depletion are that covalent reaction with any O6-alkylguanine pseudosubstrate can inactivate it, and once removed, AGT is absent from cells until the synthesis of additional protein. Also, AGT reacts with O6-alkylguanine adducts in both free nucleobases and oligomeric deoxyribonucleotides, thus facilitating its inhibition. With this knowledge, several strategies have been used for countering the effects of AGT in cancer chemotherapies.
Attempts to deplete AGT by simply providing larger doses of alkylating agents or combining multiple agents so that the number of O6-guanine adducts in DNA exceeds the number of AGT molecules have been largely unsuccessful due to the toxicity [99,100]. Therefore, non-toxic AGT pseudosubstrates have been synthesized. The first of these to have adequate potency was O6-benzylguanine, which inactivates AGT through covalent transfer of its benzyl group to the active site cysteine [101,102]. O6-Benzylguanine inactivates AGT in vitro and in vivo and sensitizes tumor cells and xenografts to killing by BCNU or temozolomide [22,94–96]. It is rapidly converted to another equally potent AGT inhibitor, 8-oxo-O6-benzylguanine in humans due to its metabolism by CYP1A2 and CYP3A4 [103]. This metabolism renders O6-benzylguanine unsuitable for combination with DTIC which is activated by the same enzymes [104]. Adequate doses of O6-benzylguanine (c.120 mg/m2) for inactivation of tumor human AGT were readily achievable [105,106]. However, data from ongoing Phase II/III trials suggest that the dose of the alkylating agent that can be given without bone marrow damage is limited due to the lack of specificity of the AGT inhibitor towards the tumor. Despite some responses in these trials, particularly in brain tumors, and in multiple myeloma and subcutaneous T cell lymphomas the value of this therapy has been restricted [103,107–113]. This is most likely due to the lowered dose of BCNU that can be given in combination with O6-benzylguanine to avoid myelosuppression and success of these combinations will probably require regional administration approaches [114].
Preliminary results of trials of O6-benzylguanine and temozolomide seem more promising. Some responses in adult and pediatric patients with refractory CNS tumors including anaplastic astrocytoma and glioblastoma multiforme were observed [100,115,116]. However, it is clear that improved AGT inhibitors are needed. Since the rate of inactivation of AGT by O6-benzylguanine is >10,000 slower than the rate of the normal repair reaction, there is great potential to produce more potent inhibitors. Other O6-benzylguanine analogs have been synthesized as potential AGT inactivators, but unfortunately, the potencies of most of these inhibitors show only modest increases and none of these inhibitors exhibit greater potential tumor specificity than O6-benzylguanine. One such alternative is O6-(4-bromothenyl)guanine (PaTrin-2; Lomeguatrib) (See Fig. 4 (3)), which is in clinical trials in combination with temozolomide in England [95,117–121]. This molecule has a higher reactivity toward AGT and an oral formulation has been developed giving it two potential advantages over O6-benzylguanine. Numerous other non-selective AGT inhibitors also based on O6-benzylguanine have been described (Fig. 4) but have not been tested clinically. It is noteworthy that O6-benzyl-2′-deoxyguanosine was highly active in sensitizing tumor cells and xenografts to BCNU and temozolomide [122]. This is likely to be due to its improved pharmacokinetic properties since it is less potent than O6-benzylguanine in the inactivation of purified AGT. Oligonucleotides containing O6-benzylguanine [123] or other O6-alkylguanines [25] are much more potent than the corresponding free bases. Another advantage is that multiple AGT-inactivating adducts can be contained in a single oligonucleotide [123] but delivery of these inhibitors presents problems which have not yet been addressed.
The major drawback to the use of O6-(4-bromothenyl)guanine or O6-benzylguanine is their lack of tumor specificity. Another problem is their poor solubility in aqueous solution. Three approaches to deal with these problems are in progress. Several D-glucose-conjugated inhibitors have been synthesized [124,125]. These water soluble derivatives may be tumor targeted due to increased consumption of glucose by these cells [126] and the up-regulation of the glucose transporter [127]. The most efficient glucose-conjugated inhibitor so far tested is O6-4-bromothenylguanine-C8-βD-glucoside [128]. A second tactic has been to generate β-glucuronidase cleavable prodrugs of O6-benzyl-2′-deoxyguanosine and O6-benzylguanine [129]. These water-soluble derivatives are inactive until activated by β-glucuronidase and may be useful for prodrug monotherapy of necrotic tumors that liberate β-glucuronidase or for antibody-directed enzyme prodrug therapy with antibodies that can deliver β-glucuronidase to target tumor cells. Perhaps the most promising current approach is based on the synthesis of 2-amino-O4-benzylpteridine derivatives (See Fig. 4 (2)) which are very potent human AGT inactivators [26]. The most active of these was O4-benzylfolic acid. Preliminary studies in cultured cells and in xenografts indicate that O4-benzylfolic acid is taken up by the tumor-specific folate receptor system [26], which is an established route to deliver anti-tumor agents selectively [130,131].
Another potential problem in the use of AGT inhibitors such as O6-benzylguanine is the selection for AGT mutants that are resistant to inactivation. Alterations of the substrate binding pocket that either reduce the size available to exclude O6-benzylguanine or disrupt the hydrophobic binding between O6-benzylguanine and the protein are quite readily produced and at least 140 known single point mutations imparting resistance were found. Thirty distinct codons provide sites at which point mutations can cause resistance [94,132,133]. Some of these mutants, particularly those at positions Pro140, Gly156, Tyr158 and Lys165, render AGT almost totally refractory to O6-benzylguanine and O6-(4-bromothenyl)guanine. When expressed in mammalian cells, these mutant AGT proteins are highly effective in providing protection against BCNU or temozolomide that was not abolished by these drugs. Although they were obtained first in bacteria expressing human AGT, such resistant mutants have now been found in tumor cells treated in vitro with BCNU and O6-benzylguanine [134,135]. The extent to which newer more potent inactivators such as O4-benzylfolate can overcome this problem is not yet clear. O4-Benzylfolate is able to inactive the P140K mutant [26]. This mutant is totally resistant to O6-benzylguanine due to the combination of the loss of the interaction of the benzyl group with the Pro140 side chain and the presence of a charged group near to the active site. It is probable that O4-benzylfolate binds to additional residues in the AGT active site but these have not yet been determined.
Alkylating agents are toxic to both normal and cancer cells. In normal cells, they are especially toxic to highly proliferative tissues, such as hematopoietic cells and myelosuppression is frequently a dose-limiting toxicity of alkylation therapies. Gene therapy approaches to increase AGT levels in the sensitive bone marrow cells have been suggested [reviewed by [96,136–139]]. The O6-benzylguanine-resistant AGT mutants are a logical choice for such studies and may have the additional advantage that selection of stem cells expressing exogenous AGT would be favored in patients treated with O6-benzylguanine or O6-(4-bromothenyl)guanine. These gene transfers therefore have potential applications not only in the protection of patients treated with chemotherapeutic doses of alkylating agents but for selection of genetically altered stem cells for other nonmalignant diseases. Although some earlier laboratory studies were carried out with the G156A mutant and other AGT mutants with multiple alterations have been suggested [133], the P140K mutant seems to be the most promising and is the current focus of most of these studies [121,137,139–145].
5. Concluding Remarks
The detailed understanding of base repair processes that is being achieved by integration of structural, biochemical, and biological results is central to an informed basis for understanding and control of genome integrity relevant to more effective cancer interventions [146–148]. The search for inhibitors that are more potent, more readily bioavailable, and can be more specifically targeted to cancer cells continues. Structural analyses of protein interactions with inhibitors and DNA is now becoming possible in solution by small angle X-ray scattering combined with crystallography [149], so progress toward more effective inhibitors is likely to accelerate substantially. Currently, structures of AGT alone, in complex with small molecule inhibitors, and in complex with DNA substrates, provide a detailed knowledge of the active site structure and reaction mechanism. Building upon this knowledge will facilitate efforts to synthesize inhibitors with increased potency and tumor-targeting, making knowledge-based improvements to cancer therapies increasingly effective. More broadly, these DNA base repair structures are valuable as master keys to the understanding and control of human pathologies [150].
Acknowledgments
Many thanks to I.W. Khor for reading the manuscript and K. Hitomi and D. Shin for helpful discussion and suggestions. Research on O6-alkylguanine DNA-alkyltransferase in the Pegg and Tainer laboratories is supported by National Institutes of Health grant R01CA097209.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rydberg B, Lindahl T. Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-L-methionine is a potentially mutagenic reaction. EMBO J. 1982;1:211–216. doi: 10.1002/j.1460-2075.1982.tb01149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beranek DT. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat Res. 1990;231:11–30. doi: 10.1016/0027-5107(90)90173-2. [DOI] [PubMed] [Google Scholar]
- 3.Colvin DM. Alkylating agents and platinum antitumor compounds. In: Frei HJFE, Bast RC Jr, Kufe DW, Morton DL, Weichselbaum RR, editors. Cancer Medicine. Willams & Wilkins; Media, PA: 1997. pp. 949–975. [Google Scholar]
- 4.Kyrtopoulos SA, Anderson LM, Chhabra SK, Souliotis VL, Pletsa V, Valavanis C, Georgiadis P. DNA adducts and the mechanism of carcinogenesis and cytotoxicity of methylating agents of environmental and clinical significance. Cancer Detect Pev. 1997;21:391–405. [PubMed] [Google Scholar]
- 5.Sundheim O, Vagbo CB, Bjoras M, Sousa MM, Talstad V, Aas PA, Drablos F, Krokan HE, Tainer JA, Slupphaug G. Human ABH3 structure and key residues for oxidative demethylation to reverse DNA/RNA damage. Embo J. 2006;25:3389–3397. doi: 10.1038/sj.emboj.7601219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu L, Gerson SL. Targeted modulation of MGMT: clinical implications. Clin Cancer Res. 2006;12:328–331. doi: 10.1158/1078-0432.CCR-05-2543. [DOI] [PubMed] [Google Scholar]
- 7.Fang Q, Kanugula S, Pegg AE. Function of domains of human O6-alkylguanine-DNA alkyltransferase. Biochemistry. 2005;44:15396–15405. doi: 10.1021/bi051460d. [DOI] [PubMed] [Google Scholar]
- 8.Ling-Ling C, Nakamura T, Nakatsu Y, Sakumi K, Hayakawa H, Sekiguchi M. Specific amino acid sequences required for O6-methylguanine-DNA methyltransferase activity: analyses of three residues at or near the methyl acceptor site. Carcinogenesis. 1992;13:837–843. doi: 10.1093/carcin/13.5.837. [DOI] [PubMed] [Google Scholar]
- 9.Ihara K, Kawate H, Chueh LL, Hayakawa H, Sekiguchi M. Requirement of the Pro- Cys-His-Arg sequence for O6-methylguanine-DNA methyltransferase activity revealed by saturation mutagenesis with negative and positive screening. Mol Gen Genet. 1994;243:379–389. doi: 10.1007/BF00280468. [DOI] [PubMed] [Google Scholar]
- 10.Harris LC, Potter PM, Margison GP. Site directed mutagenesis of two cysteine residues in the E. coli ogt O6-alkylguanine DNA alkyltransferase protein. Biochem Biophys Res Commun. 1992;187:425–431. doi: 10.1016/s0006-291x(05)81510-4. [DOI] [PubMed] [Google Scholar]
- 11.Crone TM, Pegg AE. A single amino acid change in human O6-alkylguanine-DNA alkyltransferase decreasing sensitivity to inactivation by O6- benzylguanine. Cancer Res. 1993;53:4750–4753. [PubMed] [Google Scholar]
- 12.Crone TM, Goodtzova K, Pegg AE. Amino acid residues affecting the activity and stability of human O6- alkylguanine-DNA alkyltransferase. Mutat Res. 1996;363:15–25. doi: 10.1016/0921-8777(95)00058-5. [DOI] [PubMed] [Google Scholar]
- 13.Kanugula S, Goodtzova K, Edara S, Pegg AE. Alteration of arginine-128 to alanine abolishes the ability of human O6- alkylguanine-DNA alkyltransferase to repair methylated DNA but has no effect on its reaction with O6-benzylguanine. Biochemistry. 1995;34:7113–7119. doi: 10.1021/bi00021a024. [DOI] [PubMed] [Google Scholar]
- 14.Spratt TE, Wu JD, Levy DE, Kanugula S, Pegg A. Reaction and binding of oligos containing analogues of O6-methylguanine with wild-type and mutant human O6-alkylguanine-DNA alkyltransferase. Biochemistry. 1999;38:6801–6806. doi: 10.1021/bi982908w. [DOI] [PubMed] [Google Scholar]
- 15.Goodtzova K, Kanugula S, Edara S, Pegg AE. Investigation of the role of tyrosine-114 in the activity of human O6- alkylguanine-DNA alkyltranferase. Biochemistry. 1998;37:12489–12495. doi: 10.1021/bi9811718. [DOI] [PubMed] [Google Scholar]
- 16.Pieper RO, Morgan SE, Kelley MR. The role of two conserved amino acids, glutamine 90 and asparagine 137, in O6-methylguanine-DNA methyltransferase stability, activity and substrate specificity. Carcinogenesis. 1994;15:1895–1902. doi: 10.1093/carcin/15.9.1895. [DOI] [PubMed] [Google Scholar]
- 17.Rafferty JA, Tumelty J, Skorvaga M, Elder RH, Margison GP, Douglas KT. Sitedirected mutagenesis of glutamic acid 172 to glutamine completely inactivated human O6-alkylguanine- DNA-alkyltransferase. Biochem Biophys Res Commun. 1994;199:285–291. doi: 10.1006/bbrc.1994.1226. [DOI] [PubMed] [Google Scholar]
- 18.Daniels DS, Mol CD, Arvai AS, Kanugula S, Pegg AE, Tainer JA. Active and alkylated human AGT structures: a novel zinc site, inhibitor and extrahelical base binding. EMBO J. 2000;19:1719–1730. doi: 10.1093/emboj/19.7.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edara S, Kanugula S, Pegg AE. Expression of the inactive C145A mutant human O6-alkylguanine-DNA alkyltransferase in E.coli increases cell killing and mutations by N-methyl-N′-nitro-N-nitrosoguanidine. Carcinogenesis. 1999;20:103–108. doi: 10.1093/carcin/20.1.103. [DOI] [PubMed] [Google Scholar]
- 20.Samson L, Han S, Marquis JC, Rasmussen LJ. Mammalian DNA repair methyltransferases shield O4MeT from nucleotide excision repair. Carcinogenesis. 1997;18:919–924. doi: 10.1093/carcin/18.5.919. [DOI] [PubMed] [Google Scholar]
- 21.Sassanfar M, Dosanjh MK, Essigmann JM, Samson L. Relative efficiencies of the bacterial, yeast, and human DNA methyltransferases for the repair of O6-methylguanine and O4- methylthymine. Suggestive evidence for O4-methylthymine repair by eukaryotic methyltransferases. J Biol Chem. 1991;266:2767–2771. [PubMed] [Google Scholar]
- 22.Pegg AE, Dolan ME, Moschel RC. Structure, function and inhibition of O6-alkylguanine-DNA alkyltransferase. Progr Nucleic Acid Res Mol Biol. 1995;51:167–223. doi: 10.1016/s0079-6603(08)60879-x. [DOI] [PubMed] [Google Scholar]
- 23.Goodtzova K, Kanugula S, Edara S, Pauly GT, Moschel RC, Pegg AE. Repair of O6-benzylguanine by the Escherichia coli Ada and Ogt and the human O6-alkylguanine-DNA alkyltransferases. J Biol Chem. 1997;272:8332–8339. doi: 10.1074/jbc.272.13.8332. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Spratt TE, Pegg AE, Peterson LA. Synthesis of DNA oligonucleotides containing site-specifically incorporated O6-[4-oxo-4-(3-pyridyl)butyl]guanine and their reaction with O6-alkylguanine-DNA alkyltransferase. Chem Res Toxicol. 1999;12:127–131. doi: 10.1021/tx980251+. [DOI] [PubMed] [Google Scholar]
- 25.Shibata T, Glynn N, McMurry TB, McElhinney RS, Margison GP, Williams DM. Novel synthesis of O6-alkylguanine containing oligodeoxyribonucleotides as substrates for the human DNA repair protein, O6-methylguanine DNA methyltransferase (MGMT) Nucleic Acids Res. 2006;34:1884–1891. doi: 10.1093/nar/gkl117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nelson ME, Loktionova NA, Pegg AE, Moschel RC. 2-Amino-O4-benzylpteridine derivatives: potent inactivators of O6-alkylguanine-DNA alkyltransferase. J Med Chem. 2004;47:3887–3891. doi: 10.1021/jm049758+. [DOI] [PubMed] [Google Scholar]
- 27.Chae MY, McDougall MG, Dolan ME, Swenn K, Pegg AE, Moschel RC. Substituted O6-benzylguanine derivatives and their inactivation of human O6-alkylguanine-DNA alkyltransferase. Journal of Medicinal Chemistry. 1994;37:342–347. doi: 10.1021/jm00029a005. [DOI] [PubMed] [Google Scholar]
- 28.Chae M, Swenn K, Kanugula S, Dolan M, Pegg A, Moschel R. 8-Substituted O6-benzylguanine, substituted 6(4)-(benzyloxy)pyrimidine, and related derivatives as inactivators of human O6-alkylguanine-DNA alkyltransferase. J Med Chem. 1995;38:359–365. doi: 10.1021/jm00002a018. [DOI] [PubMed] [Google Scholar]
- 29.Griffin RJ, Arris CE, Bleasdale C, Boyle FT, Calvert AH, Curtin NJ, Dalby C, Kanugula S, Lembicz NK, Newell DR, Pegg AE, Golding BJ. Resistance-Modifying Agents. 7. Inhibition of O6-alkylguanine-DNA alkyltransferase by O6-alkenyl-, O6-cycloalkenyl- and O6-(2-oxoalkyl)-guanines, and potentiation of temozolomide cytotoxicity in vitro by O6-(1-cyclopentenylmethyl)guanine. J Med Chem. 2000;43:4071–4083. doi: 10.1021/jm000961o. [DOI] [PubMed] [Google Scholar]
- 30.Moschel RC, McDougall MG, Dolan ME, Stine L, Pegg AE. Structural features of substituted purine derivatives compatible with depletion of human O6-alkylguanine-DNA alkyltransferase. J Med Chem. 1992;35:4486–4491. doi: 10.1021/jm00101a028. [DOI] [PubMed] [Google Scholar]
- 31.Ewesuedo RB, Wilson LR, Friedman HS, Moschel RC, Dolan ME. Inactivation of O6-alkylguanine-DNA alkyltransferase by 8-substituted O6-benzylguanine analogs in mice. Cancer Chemother Pharmacol. 2001;47:63–69. doi: 10.1007/s002800000202. [DOI] [PubMed] [Google Scholar]
- 32.McElhinney RS, Donnelly DJ, McCormick JE, Kelly J, Watson AJ, Rafferty JA, Elder RH, Middleton MR, Willington MA, McMurry TBH, Margison GP. Inactivation of O6-alkylguanine-DNA alkyltransferase 1. Novel O6-(hetarylmethyl)guanines having basic rings in the side chain. J Med Chem. 1998;41:5265–5271. doi: 10.1021/jm9708644. [DOI] [PubMed] [Google Scholar]
- 33.Reinhard J, Hull WE, von der Lieth CW, Eichorn U, Kliem HC, Kaina B, Wiessler M. Monosaccharide-linked inhibitors of O6-methylguanine-DNA methyltransferase (MGMT): synthesis, molecular modeling, and structure-activity relationships. J Med Chem. 2001;44:4050–4061. doi: 10.1021/jm010006e. [DOI] [PubMed] [Google Scholar]
- 34.Reinhard J, Eichorn U, Wiessler M, Kaina B. Inactivation of O6-methylguanine-DNA methyltransferase by glucose-conjugated inhibitors. Int J Cancer. 2001;93:373–379. doi: 10.1002/ijc.1336. [DOI] [PubMed] [Google Scholar]
- 35.Mineura K, Fukuchi M, Kowada M, Hitomi K, Terashima I, Kohda K. Enhancement effect of O6-fluorobenzylguanines on chloroethylnitrosourea cytotoxicity in tumor cells. Life Sci. 1996;58:PL303–308. doi: 10.1016/0024-3205(96)00145-2. [DOI] [PubMed] [Google Scholar]
- 36.Wibley JEA, Pegg AE, Moody PCE. Crystal structure of the human O6-alkylguanine-DNA alkyltransferase. Nucleic Acids Res. 2000;28:393–401. doi: 10.1093/nar/28.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore MH, Gulbis JM, Dodson EJ, Demple B, Moody PC. Crystal structure of a suicidal DNA repair protein: the Ada O6- methylguanine-DNA methyltransferase from E. coli. EMBO J. 1994;13:1495–1501. doi: 10.1002/j.1460-2075.1994.tb06410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hashimoto H, Inoue T, Nishioka M, Fujiwara S, Takagi M, Imanaka T, Kai Y. Hyperthermostable protein structure maintained by intra and inter-helix ion-pairs in archaeal O6-methylguanine-DNA methyltransferase. J Mol Biol. 1999;292:707–716. doi: 10.1006/jmbi.1999.3100. [DOI] [PubMed] [Google Scholar]
- 39.Roberts A, Pelton JG, Wemmer DE. Structural studies of MJ1529, an O(6)-methylguanine-DNA methyltransferase. Magn Reson Chem. 2006;44:71–82. doi: 10.1002/mrc.1823. [DOI] [PubMed] [Google Scholar]
- 40.Daniels DS, Tainer JA. Conserved structural motifs governing the stoichiometric repair of alkylated DNA by O6-alkylguanine-DNA alkyltransferase. Mutat Res. 2000;460:151–163. doi: 10.1016/s0921-8777(00)00024-0. [DOI] [PubMed] [Google Scholar]
- 41.Rasimas JJ, Kanugula S, Dalessio PM, Ropson IJ, Fried MG, Pegg AE. Effects of zinc occupancy on human O6-alkylguanine-DNA alkyltransferase. Biochemistry. 2003a;42:980–990. doi: 10.1021/bi026970b. [DOI] [PubMed] [Google Scholar]
- 42.Daniels DS, Woo TT, Luu KX, Noll DM, Clarke ND, Pegg AE, Tainer JA. DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nat Struct Mol Biol. 2004;11:714–720. doi: 10.1038/nsmb791. [DOI] [PubMed] [Google Scholar]
- 43.Duguid EM, Rice PA, He C. The structure of the human AGT protein bound to DNA and its implications for damage detection. J Mol Biol. 2005;350:657–666. doi: 10.1016/j.jmb.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 44.Kanugula S, Pegg AE. Novel DNA repair alkyltransferase from Caenorhabditis elegans. Environ Mol Mutagen. 2001;38:235–243. doi: 10.1002/em.1077. [DOI] [PubMed] [Google Scholar]
- 45.Kanugula S, Pauly GT, Moschel RC, Pegg AE. A bifunctional DNA repair protein from Ferroplasma acidarmanus exhibits O6-alkylguanine-DNA alkyltransferase and endonuclease V activities. Proc Natl Acad Sci U S A. 2005;102:3617–3622. doi: 10.1073/pnas.0408719102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Verdemato PE, Brannigan JA, Damblon C, Zuccotto F, Moody PC, Lian LY. DNA-binding mechanism of the Escherichia coli Ada O(6)-alkylguanine-DNA alkyltransferase. Nucleic Acids Res. 2000;28:3710–3718. doi: 10.1093/nar/28.19.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hollis T, Ichikawa Y, Ellenberger T. DNA bending and a flip-out mechanism for base excision by the helix-hairpin-helix DNA glycosylase, Escherichia coli AlkA. EMBO J. 2000;19:758–766. doi: 10.1093/emboj/19.4.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barrett TE, Savva R, Panayotou G, Barlow T, Brown T, Jiricny J, Pearl LH. Crystal structure of a G:T/U mismatch-specific DNA glycosylase: mismatch recognition by complementary-strand interactions. Cell. 1998;92:117–129. doi: 10.1016/s0092-8674(00)80904-6. [DOI] [PubMed] [Google Scholar]
- 49.Lau AY, Scharer OD, Samson L, Verdine GL, Ellenberger T. Crystal structure of a human alkylbase-DNA repair enzyme complexed to DNA: mechanisms for nucleotide flipping and base excision. Cell. 1998;95:249–258. doi: 10.1016/s0092-8674(00)81755-9. [DOI] [PubMed] [Google Scholar]
- 50.Slupphaug G, Mol CD, Kavli B, Arvai AS, Krokan HE, Tainer JA. A nucleotideflipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996;384:87–92. doi: 10.1038/384087a0. [DOI] [PubMed] [Google Scholar]
- 51.Bruner SD, Norman DP, Verdine GL. Structural basis for recognition and repair of the endogenous mutagen 8-oxoguanine in DNA. Nature. 2000;403:859–866. doi: 10.1038/35002510. [DOI] [PubMed] [Google Scholar]
- 52.Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 53.Hosfield DJ, Guan Y, Haas BJ, Cunningham RP, Tainer JA. Structure of the DNA repair enzyme endonuclease IV and its DNA complex: double-nucleotide flipping at abasic sites and three-metal-ion catalysis. Cell. 1999;98:397–408. doi: 10.1016/s0092-8674(00)81968-6. [DOI] [PubMed] [Google Scholar]
- 54.Parikh SS, Mol CD, Slupphaug G, Bharati S, Krokan HE, Tainer JA. Base excision repair initiation revealed by crystal structures and binding kinetics of human uracil-DNA glycosylase with DNA. EMBO J. 1998;17:5214–5226. doi: 10.1093/emboj/17.17.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guengerich FP, Fang Q, Liu L, Hachey DL, Pegg AE. O6-alkylguanine-DNA alkyltransferase: low pKa and high reactivity of cysteine 145. Biochemistry. 2003 ;42:10965–10970. doi: 10.1021/bi034937z. [DOI] [PubMed] [Google Scholar]
- 56.Rasimas JJ, Pegg AE, Fried MG. DNA-binding mechanism of O6-alkylguanine-DNA alkyltransferase. Effects of protein and DNA alkylation on complex stability. J Biol Chem. 2003b;278:7973–7980. doi: 10.1074/jbc.M211854200. [DOI] [PubMed] [Google Scholar]
- 57.Mineura K, Fukuchi M, Kowada M, Terashima I, Kohda K. Differential inactivation of O6-methylguanine-DNA methyltransferase activity by O6-arylmethylguanines. Int J Cancer. 1995;63:148–151. doi: 10.1002/ijc.2910630126. [DOI] [PubMed] [Google Scholar]
- 58.Duguid EM, Mishina YHC. How do DNA repair proteins locate potential base lesions? A chemical crosslinking method to investigate O6-alkylguanine-DNA alkyltransferases. Chem Biol. 2003;10:827–835. doi: 10.1016/j.chembiol.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 59.Zang H, Fang Q, Pegg AE, Guengerich FP. Kinetic analysis of steps in the repair of damaged DNA by human O6-alkylguanine-DNA alkyltransferase. J Biol Chem. 2005;280:30873–30881. doi: 10.1074/jbc.M505283200. [DOI] [PubMed] [Google Scholar]
- 60.Begley TJ, Samson LD. Reversing DNA damage with a directional bias. Nat Struct Mol Biol. 2004;11:688–690. doi: 10.1038/nsmb0804-688. [DOI] [PubMed] [Google Scholar]
- 61.Ali RB, Teo AK, Oh HK, Chuang LS, Ayi TC, Li BF. Implication of localization of human DNA repair enzyme O6-methylguanine-DNA methyltransferase at active transcription sites in transcription-repair coupling of the mutagenic O6-methylguanine lesion. Mol Cell Biol. 1998;18:1660–1669. doi: 10.1128/mcb.18.3.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pegg AE, Wiest L, Mummert C, Stine L, Moschel RC, Dolan ME. Use of antibodies to human O6-alkylguanine-DNA alkyltransferase to study the content of this protein in cells treated with O6-benzylguanine or N-methyl-N′-nitro-N-nitrosoguanidine. Carcinogenesis. 1991;12:1679–1683. doi: 10.1093/carcin/12.9.1679. [DOI] [PubMed] [Google Scholar]
- 63.Srivenugopal KS, Yuan XH, Friedman HS, Ali-Osman F. Ubiquitination-dependent proteolysis of O6-methylguanine-DNA methyltransferase in human and murine tumor cells following inactivation with O6-benzylguanine or 1,3-bis(2-chloroethyl)-1- nitrosourea. Biochemistry. 1996;35:1328–1334. doi: 10.1021/bi9518205. [DOI] [PubMed] [Google Scholar]
- 64.Xu-Welliver M, Pegg AE. Degradation of the alkylated form of the DNA repair protein O6-alkylguanine-DNA alkyltransferase. Carcinogenesis. 2002;23:823–830. doi: 10.1093/carcin/23.5.823. [DOI] [PubMed] [Google Scholar]
- 65.Rasimas JJ, Dalessio PA, Ropson IJ, Pegg AE, Fried MG. Active-site alkylation destabilizes human O6-alkylguanine DNA alkyltransferase. Protein Sci. 2004;13:301–305. doi: 10.1110/ps.03319404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Teo AK, Oh HK, Ali RB, Li BF. The modified human DNA repair enzyme O(6)-methylguanine-DNA methyltransferase is a negative regulator of estrogen receptor-mediated transcription upon alkylation DNA damage. Mol Cell Biol. 2001;21:7105–7114. doi: 10.1128/MCB.21.20.7105-7114.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schwarzl SM, Smith JC, Kaina B, Efferth T. Molecular modeling of O6-methylguanine-DNA methyltransferase mutant proteins encoded by single nucleotide polymorphisms. Int J Mol Med. 2005;16:553–557. [PubMed] [Google Scholar]
- 68.Pegg AE, Fang Q, Loktionova NA. Human variants of O6-alkylguanine-DNA alkyltransferase. DNA Repair. 2007 doi: 10.1016/j.dnarep.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu-Welliver M, Leitao J, Kanugula S, Meehan WJ, Pegg AE. Role of codon 160 in the sensitivity of human O6-alkylguanine-DNA alkyltransferase to O6-benzylguanine. Biochem Pharmacol. 1999;58:1279–1285. doi: 10.1016/s0006-2952(99)00216-6. [DOI] [PubMed] [Google Scholar]
- 70.Meikrantz W, Bergom MA, Memisoglu A, Samson L. O6-alkylguanine DNA lesions trigger apoptosis. Carcinogenesis. 1998;19:369–372. doi: 10.1093/carcin/19.2.369. [DOI] [PubMed] [Google Scholar]
- 71.Hickman MJ, Samson LD. Role of DNA mismatch repair and p53 in signaling induction of apoptosis by alkylating agents. Proc Natl Acad Sci USA. 1999;96:10764–10769. doi: 10.1073/pnas.96.19.10764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.D’Atri S, Tentori L, Lacal PM, Graziani G, Pagani E, Benincasa E, Zambruno G, Bonmassar E, Jiricny J. Involvement of the mismatch repair system in temozolomide-induced apoptosis. Mol Pharmacol. 1998;54:334–341. doi: 10.1124/mol.54.2.334. [DOI] [PubMed] [Google Scholar]
- 73.Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12:440–450. doi: 10.1016/j.molmed.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 74.Erickson LC, Bradley MO, Ducore JM, Ewig RA, Kohn KW. DNA crosslinking and cytotoxicity in normal and transformed human cells treated with antitumor nitrosoureas. Proc Natl Acad Sci U S A. 1980;77:467–471. doi: 10.1073/pnas.77.1.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brent TP. Isolation and purification of O6-alkylguanine-DNA alkyltransferase from human leukemic cells. Prevention of chloroethylnitrosourea-induced cross-links by purified enzyme. Pharmacol Ther. 1985;31:121–140. doi: 10.1016/0163-7258(85)90040-3. [DOI] [PubMed] [Google Scholar]
- 76.Ludlum DB. The chloroethylnitrosoureas: Sensitivity and resistance to cancer chemotherapy at the molecular level. Cancer Invest. 1997;15:588–598. doi: 10.3109/07357909709047601. [DOI] [PubMed] [Google Scholar]
- 77.Fischbauer PL, Gall AS, Duncan JA, Hopkins PB. Direct demonstration in synthetic oligonucleotides that N,N1-bis(2-chloroethyl)-nitrosourea cross-links N1 of deoxyguanosine to N3 of deoxycytidine on opposite strands of duplex DNA. Cancer Res. 1999;59:4363–4368. [PubMed] [Google Scholar]
- 78.Bodell WJ, Tokuda K, Ludlum DB. Differences in DNA alkylation products formed in sensitive and resistant human glioma cells treated with N-(2-chloroethyl)-N-nitrosourea. Cancer Res. 1988;48:4489–4492. [PubMed] [Google Scholar]
- 79.Belanich M, Pastor M, Randall T, Guerra D, Kibitel J, Alas L, Li B, Citron M, Wasserman P, White A, Eyre H, Jaeckle K, Schulman S, Rector D, Prados M, Coons S, Shapiro W, Yarosh D. Retrospective study of the correlation between the DNA repair protein alkyltransferase and survival of brain tumor patients treated with carmustine. Cancer Res. 1996;56:783–788. [PubMed] [Google Scholar]
- 80.Jaeckle KA, Eyre HJ, Townsend JJ, Schulman S, Knudson HM, Belanich M, Yarosh DB, Bearman SI, Giroux DJ, Schold SC. Correlation of tumor O6 methylguanine-DNA methyltransferase levels with survival of malignant astrocytoma patients treated with bis-chloroethylnitrosourea: a Southwest Oncology Group study. J Clin Oncol. 1998;16:3310–3315. doi: 10.1200/JCO.1998.16.10.3310. [DOI] [PubMed] [Google Scholar]
- 81.Chen ZP, Yarosh D, Garcia Y, Tampieri D, Mohr G, Malapetsa A, Langleben A, Panasci LC. Relationship between O6-methylguanine-DNA methyltransferase levels and clinical response induced by chloroethylnitrosourea therapy in glioma patients. Can J Neurol Sci. 1999;26:104–109. [PubMed] [Google Scholar]
- 82.Kat A, Thilly WG, Fang WH, Longley MJ, Li GM, Modrich P. An alkylation-tolerant, mutator human cell line is deficient in strand-specific mismatch repair. Proc Natl Acad Sci USA. 1993;90:6424–6428. doi: 10.1073/pnas.90.14.6424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takagi Y, Takahashi M, Sanada M, Ito R, Yamaizumi M, Sekiguchi M. Roles of MGMT and MLH1 proteins in alkylation-induced apoptosis and mutagenesis. DNA Repair. 2003;7:1135–1146. doi: 10.1016/s1568-7864(03)00134-4. [DOI] [PubMed] [Google Scholar]
- 84.Karran P. Mechanism of tolerance to DNA damaging therapeutic drugs. Carcinogenesis. 2001;22:1931–1937. doi: 10.1093/carcin/22.12.1931. [DOI] [PubMed] [Google Scholar]
- 85.York SJ, Modrich P. Mismatch repair-dependent iterative excision at irreparable O6-methylguanine lesions in human nuclear extracts. J Biol Chem. 2006;281:22674–22683. doi: 10.1074/jbc.M603667200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lin DP, Wang Y, Scherer SJ, Clark AB, Yang K, Avdievich E, Jin B, Werling U, Parris T, Kurihara N, Umar A, Kucherlapati R, Lipkin M, Kunkel TA, Edelmann W. An Msh2 point mutation uncouples DNA mismatch repair and apoptosis. Cancer Res. 2004;64:517–522. doi: 10.1158/0008-5472.can-03-2957. [DOI] [PubMed] [Google Scholar]
- 87.Yang G, Scherer SJ, Shell SS, Yang K, Kim M, Lipkin M, Kucherlapati R, Kolodner RD, Edelmann W. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell. 2004;6:139–150. doi: 10.1016/j.ccr.2004.06.024. [DOI] [PubMed] [Google Scholar]
- 88.Yoshioka K, Yoshioka Y, Hsieh P. ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol Cell. 2006;22:501–510. doi: 10.1016/j.molcel.2006.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Newlands ES, Stevens MFG, Wedge SR, Wheelhouse RTBC. Temozolomide: a review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treatment Rev. 1997;23:35–61. doi: 10.1016/s0305-7372(97)90019-0. [DOI] [PubMed] [Google Scholar]
- 90.Friedman HS, McLendon RE, Dolan ME, Pegg AE, Moschel RC, Colvin OM, Cokgor I, Friedman AH, Schold SC, Bigner DD, Modrich PL. The biology of sensitivity and resistance to the molecule (temozolomide) Proc Am Assoc Cancer Res. 1999;40:754–755. [Google Scholar]
- 91.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 92.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JE, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 93.Esteller M, Herman JG. Generating mutations but providing chemosensitivity: the role of O6-methylguanine DNA methyltransferase in human cancer. Oncogene. 2004;23:1–8. doi: 10.1038/sj.onc.1207316. [DOI] [PubMed] [Google Scholar]
- 94.Pegg AE, Xu-Welliver M, Loktionova NA. The DNA repair protein O6-alkylguanine-DNA alkyltransferase as a target for cancer chemotherapy. In: Ehrlich EM, editor. DNA alterations in cancer: genetic and epigenetic changes. Eaton Publishing; Natick, MA: 2000. pp. 471–488. [Google Scholar]
- 95.Margison GP, Santibáñez-Koref MF. O6-Alkylguanine-DNA alkyltransferase: role in carcinogenesis and chemotherapy. BioEssays. 2002;24:255–266. doi: 10.1002/bies.10063. [DOI] [PubMed] [Google Scholar]
- 96.Gerson SL. MGMT: its role in cancer aetiology and cancer therapeutics. Nat Rev Cancer. 2004;4:296–307. doi: 10.1038/nrc1319. [DOI] [PubMed] [Google Scholar]
- 97.Potter PM, Harris LC, Remack JS, Edwards CC, Brent TP. Ribozyme-mediated modulation of human O6-methylguanine-DNA methyltransferase expression. Cancer Res. 1993;53:1731–1734. [PubMed] [Google Scholar]
- 98.Zhang Q, Ohannesian DW, Erickson LC. Hammerhead ribozyme-mediated sensitization of human tumor cells after treatment with 1,3-bis(2-chloroethyl)-1-nitrosourea. J Pharmacol Exp Ther. 2004;309:506–514. doi: 10.1124/jpet.103.061507. [DOI] [PubMed] [Google Scholar]
- 99.Micetich KC, Futscher B, Koch D, Fisher RI, Erickson LC. Phase I study of streptozotocin- and carmustine-sequenced administration in patients with advanced cancer. J Natl Cancer Inst. 1992;84:256–260. doi: 10.1093/jnci/84.4.256. [DOI] [PubMed] [Google Scholar]
- 100.Hammond LA, Eckardt JR, Kuhn JG, Gerson SL, Johnson T, Smith L, Drengler RL, Campbell E, Weiss GR, Von Hoff DD, Rowinsky EK. A randomized phase I and pharmacological trial of sequences of 1,3-bis(2-chloroethyl)-1-nitrosourea and temozolomide in patients with advanced solid neoplasms. Clin Cancer Res. 2004;10:1645–1656. doi: 10.1158/1078-0432.ccr-03-0174. [DOI] [PubMed] [Google Scholar]
- 101.Dolan ME, Moschel RC, Pegg AE. Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents. Proc Natl Acad Sci U S A. 1990;87:5368–5372. doi: 10.1073/pnas.87.14.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pegg AE, Boosalis M, Samson L, Moschel RC, Byers TL, Swenn K, Dolan ME. Mechanism of inactivation of human O6-alkylguanine-DNA alkyltransferase by O6-benzylguanine. Biochemistry. 1993;32:11998–12006. doi: 10.1021/bi00096a009. [DOI] [PubMed] [Google Scholar]
- 103.Dolan ME, Pegg AE. O6-benzylguanine and its role in chemotherapy. Clin Cancer Res. 1997;3:837–847. [PubMed] [Google Scholar]
- 104.Long L, Moschel RC, Dolan ME. Debenzylation of O6-benzyl-8-oxoguanine in human liver: implications for O6-benzylguanine metabolism. Biochem Pharmacol. 2001;61:721–726. doi: 10.1016/s0006-2952(01)00523-8. [DOI] [PubMed] [Google Scholar]
- 105.Dolan ME, Posner M, Karrison T, Radosta J, Steinberg G, Bertucci D, Vujasin L, Ratain MJ. Determination of the optimal modulatory dose of O6-benzylguanine in patients with surgically resectable tumors. Clin Cancer Res. 2002;8:2519–2523. [PubMed] [Google Scholar]
- 106.Tserng KY, Ingalls ST, Boczko EM, Spiro TP, Li X, Majka S, Gerson SL, Willson JK, Hoppel CL. Pharmacokinetics of O6-benzylguanine (NSC637037) and its metabolite, 8-oxo-O6-benzylguanine. J Clin Pharmacol. 2003;43:881–893. doi: 10.1177/0091270003256060. [DOI] [PubMed] [Google Scholar]
- 107.Quinn JA, Pluda J, Dolan ME, Delaney S, Kaplan R, Rich JN, Friedman AH, Reardon DA, Sampson JH, Colvin OM, Haglund MM, Pegg AE, Moschel RC, McLendon RE, Provenzale JM, Gururangan S, TourtUhlig S, Herndon JE, Bigner DD, Friedman HS. Phase II trial of carmustine plus O6 -benzylguanine for patients with nitrosourea-resistant recurrent or progressive malignant glioma. J Clin Oncol. 2002;2:22277–22283. doi: 10.1200/JCO.2002.09.084. [DOI] [PubMed] [Google Scholar]
- 108.Friedman HS, Keir S, Pegg AE, Houghton PJ, Colvin OM, Moschel RC, Bigner DD, Dolan ME. O6-Benzylguanine-mediated enhancement of chemotherapy. Mol Cancer Ther. 2002;1:943–948. [PubMed] [Google Scholar]
- 109.Dolan ME. Inhibition of DNA repair as a means of increasing the antitumor activity of DNA reactive agents. Adv Drug Delivery Reviews. 1997;26:105–118. doi: 10.1016/s0169-409x(97)00028-8. [DOI] [PubMed] [Google Scholar]
- 110.Dolan ME, Roy SK, Fasanmade A, Paras PR, Schilsky RL, Ratain MJ. O6-Benzylguanine in humans: metabolic, pharmacokinetic and pharmacodynamic findings. J Clin Oncol. 1998;16:1803–1810. doi: 10.1200/JCO.1998.16.5.1803. [DOI] [PubMed] [Google Scholar]
- 111.Spiro TP, Gerson SL, Liu L, Majka S, Haaga J, Hoppel CL, Ingalls ST, Pluda JM, Willson JK. O6-benzylguanine: a clinical trial establishing the biochemical modulatory dose in tumor tissue for alkyltransferase-directed DNA repair. Cancer Res. 1999;59:2402–2410. [PubMed] [Google Scholar]
- 112.Ryan CW, Dolan ME, Brockstein BB, McLendon R, Delaney SM, Samuels BL, Agamah ES, Vokes EE. A phase II trial of O6-benzylguanine and carmustine in patients with advanced soft tissue sarcoma. Cancer Chemother Pharmacol. 2006;58:634–639. doi: 10.1007/s00280-006-0210-0. [DOI] [PubMed] [Google Scholar]
- 113.Gajewski TF, Sosman J, Gerson SL, Liu L, Dolan E, Lin S, Vokes EE. Phase II trial of the O6-alkylguanine DNA alkyltransferase inhibitor O6-benzylguanine and 1,3-bis(2-chloroethyl)-1-nitrosourea in advanced melanoma. Clin Cancer Res. 2005;11:7861–7865. doi: 10.1158/1078-0432.CCR-05-0060. [DOI] [PubMed] [Google Scholar]
- 114.Rhines LD, Sampath P, Dolan ME, Tyler BM, Weingart BHJ. O6-Benzylguanine potentiates the antitumor effect of locally delivered carmustine against an intracranial rat glioma. Cancer Res. 2000;60:6307–6310. [PubMed] [Google Scholar]
- 115.Quinn JA, Desjardins A, Weingart J, Brem H, Dolan ME, Delaney SM, Vredenburgh J, Rich J, Friedman AH, Reardon DA, Sampson JH, Pegg AE, Moschel RC, Birch R, McLendon RE, Provenzale JM, Gururangan S, Dancey JE, Maxwell J, Tourt-Uhlig S, Herndon JE, 2nd, Bigner DD, Friedman HS. Phase I trial of temozolomide plus O6-benzylguanine for patients with recurrent or progressive malignant glioma. J Clin Oncol. 2005;23:7178–7187. doi: 10.1200/JCO.2005.06.502. [DOI] [PubMed] [Google Scholar]
- 116.Warren KE, Aikin AA, Libucha M, Widemann BC, Fox E, Packer RJ, Balis FM. Phase I study of O6-benzylguanine and temozolomide administered daily for 5 days to pediatric patients with solid tumors. J Clin Oncol. 2005;23:7646–7653. doi: 10.1200/JCO.2005.02.0024. [DOI] [PubMed] [Google Scholar]
- 117.Middleton MR, Margison GP. Improvement of chemotherapy efficacy by inactivation of a DNA-repair pathway. Lancet Oncol. 2003;4:37–44. doi: 10.1016/s1470-2045(03)00959-8. [DOI] [PubMed] [Google Scholar]
- 118.Ranson M, Middleton MR, Bridgewater J, Lee SM, Dawson M, Jowle D, Halbert G, Waller S, McGrath H, Gumbrell L, McElhinney RS, Donnelly D, McMurry TB, Margison GP. Lomeguatrib, a potent inhibitor of O6-alkylguanine-DNA-alkyltransferase: phase I safety, pharmacodynamic, and pharmacokinetic trial and evaluation in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2006 ;12:1577–1584. doi: 10.1158/1078-0432.CCR-05-2198. [DOI] [PubMed] [Google Scholar]
- 119.Middleton MR, Thatcher N, McMurry TBH, McElhinney RS, Donnelly DJ, Margison GP. Effect of O6-(4-bromothenyl)guanine on different temozolomide schedules in a human melanoma xenograft model. Int J Cancer. 2002;100:615–617. doi: 10.1002/ijc.10532. [DOI] [PubMed] [Google Scholar]
- 120.Clemons M, Kelly J, Watson AJ, Howell A, McElhinney RS, McMurry TB, Margison GP. O6-(4-bromothenyl)guanine reverses temozolomide resistance in human breast tumour MCF-7 cells and xenografts. Br J Cancer. 2005;93:1152–1156. doi: 10.1038/sj.bjc.6602833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Woolford LB, Southgate TD, Margison GP, Milsom MD, Fairbairn LJ. The P140K mutant of human O(6)-methylguanine-DNA-methyltransferase (MGMT) confers resistance in vitro and in vivo to temozolomide in combination with the novel MGMT inactivator O(6)-(4-bromothenyl)guanine. J Gene Med. 2006;8:29–34. doi: 10.1002/jgm.816. [DOI] [PubMed] [Google Scholar]
- 122.Kokkinakis DM, Bocangel DB, Schold SC, Moschel RC, Pegg AE. Thresholds of O6-alkylguanine-DNA alkyltransferase which confer significant resistance of human glial tumor xenografts to treatment with 1,3-bis(2-chloroethyl)-1-nitrosourea or temozolomide. Clin Cancer Res. 2001;7:421–428. [PubMed] [Google Scholar]
- 123.Luu KX, Kanugula S, Pegg AE, Pauly GT, Moschel RC. Repair of oligodeoxyribonucleotides by O6-alkylguanine-DNA alkyltransferase. Biochemistry. 2002;41:8689–8697. doi: 10.1021/bi025857i. [DOI] [PubMed] [Google Scholar]
- 124.Reinhard J, Hull WE, von der Lieth CW, Eichhorn U, Kliem HC, Kaina B, Wiessler M. Monosaccharide-linked inhibitors of O6-methylguanine-DNA methyltransferase (MGMT): synthesis, molecular modeling and structure-activity relationships. J Med Chem. 2001a;44:4050–4061. doi: 10.1021/jm010006e. [DOI] [PubMed] [Google Scholar]
- 125.Reinhard J, Eichhorn U, Wiessler M, Kaina B. Inactivation of O6-methylguanine-DNA methyltransferase by glucose-conjugated inhibitors. Int J Cancer. 2001b;93:373–379. doi: 10.1002/ijc.1336. [DOI] [PubMed] [Google Scholar]
- 126.Argiles JM, Lopez-Soriano FJ. Why do cancer cells have such a high glycolytic rate? Med Hypotheses. 1990;32:151–155. doi: 10.1016/0306-9877(90)90039-h. [DOI] [PubMed] [Google Scholar]
- 127.Yamamoto T, Seino Y, Fukumoto H, Koh G, Yano H, Inagaki N, Yamada Y, Inoue K, Manabe T, Imura H. Over-expression of facilitative glucose transporter genes in human cancer. Biochem Biophys Res Commun. 1990;170:223–230. doi: 10.1016/0006-291x(90)91263-r. [DOI] [PubMed] [Google Scholar]
- 128.Kaina B, Muhlhausen U, Piee-Staffa A, Christmann M, Garcia Boy R, Rosch F, Schirrmacher R. Inhibition of O6-methylguanine-DNA methyltransferase by glucose-conjugated inhibitors: comparison with nonconjugated inhibitors and effect on fotemustine and temozolomide-induced cell death. J Pharmacol Exp Ther. 2004;311:585–593. doi: 10.1124/jpet.104.071316. [DOI] [PubMed] [Google Scholar]
- 129.Wei G, Loktionova NA, Pegg AE, Moschel RC. Beta-glucuronidase-cleavable prodrugs of O6-benzylguanine and O6-benzyl-2′-deoxyguanosine. J Med Chem. 2005;48:256–261. doi: 10.1021/jm0493865. [DOI] [PubMed] [Google Scholar]
- 130.Leamon CP, Reddy JA. Folate-targeted chemotherapy. Adv Drug Deliv Rev. 2004;56:1127–1141. doi: 10.1016/j.addr.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 131.Low PS, Antony AC. Folate receptor-targeted drugs for cancer and inflammatory diseases. Adv Drug Deliv Rev. 2004;56:1055–1058. doi: 10.1016/j.addr.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 132.Loktionova NA, Pegg AE. Interaction of mammalian O6-alkylguanine-DNA alkyltransferases with O6-benzylguanine. Biochem Pharmacol. 2002;63:1431–1442. doi: 10.1016/s0006-2952(02)00906-1. [DOI] [PubMed] [Google Scholar]
- 133.Fontes AM, Davis B, Encell L, Lingas K, Comas DT, Zago MA, Loeb L, Pegg AE, Gerson SL. Differential competitive resistance to methylating versus chloroethylating agents among five O6-alkylguanine DNA alkyltransferases in human hematopoietic cells. Mol Cancer Therap. 2006;6:121–129. doi: 10.1158/1535-7163.MCT-05-0236. [DOI] [PubMed] [Google Scholar]
- 134.Liu L, Schwartz S, Davis BM, Gerson SL. Chemotherapy-induced O6-benzylguanine-resistant alkyltransferase mutations in mismatch-deficient colon cancer. Cancer Res. 2002;62:3070–3076. [PubMed] [Google Scholar]
- 135.Bacolod MD, Johnson SP, Pegg AE, Dolan ME, Moschel RC, Bullock NS, Fang Q, Colvin OM, Modrich P, Bigner DD, Friedman HS. Brain tumor cell lines resistant to O6-benzylguanine/1,3-bis(2-chloroethyl)-1-nitrosoure chemotherapy have O6-alkylguanine-DNA alkyltransferase mutations. Mol Cancer Ther. 2004;3:1127–1135. [PubMed] [Google Scholar]
- 136.Milsom MD, Fairbairn LJ. Protection and selection for gene therapy in the hematopoietic system. J Gene Med. 2004;6:133–146. doi: 10.1002/jgm.533. [DOI] [PubMed] [Google Scholar]
- 137.Southgate TD, Garside E, Margison GP, Fairbairn LJ. Dual agent chemoprotection by retroviral co-expression of either MDR1 or MRP1 with the P140K mutant of O6-methylguanine-DNA-methyl transferase. J Gene Med. 2006;8:972–979. doi: 10.1002/jgm.914. [DOI] [PubMed] [Google Scholar]
- 138.Cornetta K, Croop J, Dropcho E, Abonour R, Kieran MW, Kreissman S, Reeves L, Erickson LC, Williams DA. A pilot study of dose-intensified procarbazine, CCNU, vincristine for poor prognosis brain tumors utilizing fibronectin-assisted, retroviral-mediated modification of CD34+ peripheral blood cells with O6-methylguanine DNA methyltransferase. Cancer Gene Ther. 2006;13:886–895. doi: 10.1038/sj.cgt.7700963. [DOI] [PubMed] [Google Scholar]
- 139.Schambach A, Bohne J, Chandra S, Will E, Margison GP, Williams DA, Baum C. Equal potency of gammaretroviral and lentiviral SIN vectors for expression of O6-methylguanine-DNA methyltransferase in hematopoietic cells. Mol Ther. 2006;13:391–400. doi: 10.1016/j.ymthe.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 140.Zielske SP, Reese JS, Lingas KT, Donze JR, Gerson SL. In vivo selection of MGMT(P140K) lentivirus-transduced human NOD/SCID repopulating cells without pretransplant irradiation conditioning. J Clin Invest. 2003;112:1561–1570. doi: 10.1172/JCI17922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Persons DA, Allay ER, Sawai N, Hargrove PW, Brent TP, Hanawa H, Nienhuis AW, Sorrentino BP. Successful treatment of murine beta-thalassemia using in vivo selection of genetically modified, drug-resistant hematopoietic stem cells. Blood. 2003;102:506–513. doi: 10.1182/blood-2003-03-0677. [DOI] [PubMed] [Google Scholar]
- 142.Kreklau EL, Pollok KE, Bailey BJ, Liu N, Hartwell JR, Williams DA, Erickson LC. Hematopoietic expression of O6-methylguanine DNA methyltransferase-P140K allows intensive treatment of human glioma xenografts with combination O6-benzylguanine and 1,3-bis-(2-chloroethyl)-1-nitrosourea. Mol Cancer Ther. 2003;2:1321–1329. [PubMed] [Google Scholar]
- 143.Richard E, Robert E, Cario-Andre M, Ged C, Geronimi F, Gerson SL, de Verneuil H, Moreau-Gaudry F. Hematopoietic stem cell gene therapy of murine protoporphyria by methylguanine-DNA-methyltransferase-mediated in vivo drug selection. Gene Ther. 2004;11:1638–1647. doi: 10.1038/sj.gt.3302335. [DOI] [PubMed] [Google Scholar]
- 144.Chinnasamy D, Fairbairn LJ, Neuenfeldt J, Treisman JS, Hanson J, Margison JPGP, Chinnasamy N. Lentivirus-mediated expression of mutant MGMT P140K protects human CD34+ cells against the combined toxicity of O6-benzylguanine and 1,3-bis(2-chloroethyl)-nitrosourea or temozolomide. Hum Gene Ther. 2004;15:758–769. doi: 10.1089/1043034041648417. [DOI] [PubMed] [Google Scholar]
- 145.Milsom MD, Woolford LB, Margison GP, Humphries RK, Fairbairn LJ. Enhanced in vivo selection of bone marrow cells by retroviral-mediated coexpression of mutant O6-methylguanine-DNA-methyltransferase and HOXB4. Mol Ther. 2004;10:862–873. doi: 10.1016/j.ymthe.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 146.Hitomi K, Iwai S, Tainer JA. The intricate structural chemistry of base excision repair machinery: Implications for DNA damage recognition, removal, and repair. DNA Repair. 2007 doi: 10.1016/j.dnarep.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 147.Shin DS, Chahwan C, Huffman JL, Tainer JA. Structure and function of the double-strand break repair machinery. DNA Repair. 2004;3:863–873. doi: 10.1016/j.dnarep.2004.03.022. [DOI] [PubMed] [Google Scholar]
- 148.Huffman JL, Sundheim O, Tainer JA. DNA base damage recognition and removal: new twists and grooves. Mutat Res. 2005;577:55–76. doi: 10.1016/j.mrfmmm.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 149.Tsutakawa SE, Hura GL, Frankel KA, Cooper PK, Tainer JA. Structural analysis of flexible proteins in solution by small angle X-ray scattering combined with crystallography. J Struct Biol. 2006 doi: 10.1016/j.jsb.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 150.Perry JJ, Fan L, Tainer JA. Developing master keys to brain pathology, cancer and aging from the structural biology of proteins controlling reactive oxygen species and DNA repair. Neuroscience. 2006 doi: 10.1016/j.neuroscience.2006.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]