

Abstract
Cyclooxygenase-2 (COX-2) is highly expressed in osteoblasts, and COX-2 produced prostaglandins (PGs) can increase osteoblastic differentiation in vitro. The goal of this study was to examine effects of COX-2 expression on calvarial osteoblastic proliferation and apoptosis. Primary osteoblasts (POBs) were cultured from calvariae of COX-2 wild type (WT) and knockout (KO) mice. POB proliferation was evaluated by 3H-thymidine incorporation and analysis of cell replication and cell cycle distribution by flow cytometry. POB apoptosis was evaluated by annexin and PI staining on flow cytometry. As expected, PGE2 production and alkaline phosphatase (ALP) activity were increased in WT cultures compared to KO cultures. In contrast, cell numbers were decreased in WT compared to KO cells by day 4 of culture. Proliferation, measured on days 3–7 of culture, was 2-fold greater in KO than in WT POBs and associated with decreased Go/G1 and increased S cell cycle distribution. There was no significant effect of COX-2 genotype on apoptosis under basal culture conditions on day 3–5 of culture. Cell growth was decreased in KO POBs by addition of PGE2 or a protein kinase A agonist and increased in WT POBs by addition of NS398, a selective COX-2 inhibitor. In contrast, differentiation and cell growth in marrow stromal cell (MSC) cultures, evaluated by ALP and crystal violet staining respectively, was increased in MSCs from WT mice compared to MSCs from KO mice, and exogenous PGE2 increased cell growth in KO MSC cultures. We conclude that PGs secondary to COX-2 expression decrease osteoblastic proliferation in cultured calvarial cells but increase growth of osteoblastic precursors in MSC cultures.
Keywords: prostaglandins, apoptosis, nonsteroidal anti-inflammatory drugs, marrow stromal cells, mitogenesis
INTRODUCTION
Cyclooxygenase (COX) is a rate-limiting enzyme in conversion of arachidonic acid (AA) to prostanoids. It converts AA to PGG2 via a cyclooxygenase reaction and then reduces PGG2 to PGH2 in a peroxidase reaction [1]. Nonsteroidal anti-inflammatory drugs (NSAIDs) inhibit the cyclooxygenase activity of COX. There are two isoforms of COX: COX-1, which is constitutively expressed, and COX-2, which is inducible by multiple factors and involved in prostaglandin (PG) production during inflammation and other acute responses [2]. PGE2 is one of the most abundant PGs produced in bone, and most PGE2 produced by bone cells is associated with induction of COX-2 [3].
PGE2 acts via interactions with a subfamily of G-protein-coupled receptors (GPCRs), called EP1, EP2, EP3, and EP4 [4]. EP1 releases intracellular calcium and EP3 can inhibit cyclic 3,5-adenosine monophosphate (cAMP) formation. EP2 and EP4 can both stimulate cAMP formation, and EP4 may also act via a phosphatidylinositol 3-kinase-dependent pathway and activate extracellular signal-regulated kinases (ERKs) [5]. In addition to initiating different signaling pathways, recent data suggest EP receptors may undergo nuclear compartmentalization [6] or heterodimerize with other GCPRs [7], leading to more complex actions.
PGE2 is a potent stimulator of bone resorption and osteoclastogenesis [8], and both EP2 and EP4 have been implicated in these effects [9–11]. PGE2 is also anabolic for bone. In vitro, exogenous PGE2 stimulates osteoblastic differentiation in marrow stromal and calvarial osteoblast cultures [12–15]. In agreement with these studies, osteoblastic differentiation is decreased in marrow stromal cell cultures from COX-2 knockout (KO) mice compared to cultures from COX-2 wild type (WT) mice [16,17]. In vivo, administration of PGs in rats, dogs and humans increases cortical and cancellous bone mass [18–21]. PGE2 given to rats in vivo stimulates osteoblastic differentiation in ex vivo cultured bone marrow [22]. Both EP2 and EP4 have been implicated in the anabolic effects of PGE2. Local infusion of PGE2 adjacent to the femur was shown to produce callus formation in EP1, EP2 or EP3 receptor KO mice but not in EP4 KO mice, and marrow from EP4 KO mice did not mineralize in response to in vitro treatment with PGE2 [11]. EP2 and EP4 agonists have been shown to stimulate bone formation and/or enhance fracture healing in vivo [23,24], and bone mechanical properties may be reduced in EP2 and EP4 KO mice [25,26].
Some of the effects of PGs to increase new bone formation could be the result of PG-induced increases in growth of osteoblastic cells, secondary either to increased proliferation or decreased apoptosis. Studies of the effects of PGE2 on osteoblastic proliferation have been contradictory, showing both increased [27–29] and decreased proliferation [15, 30, 31], as well as biphasic effects [32, 33]. Although COX-2 expression and PGs have been consistently associated with increased resistance to apoptosis and tumor promotion in other tissues [34, 35], there have been few studies of the effects of PGE2 on apoptosis in osteoblasts and these have not produced consistent results. Inhibition of PG production with NSAIDs increased apoptosis in fetal rat calvarial osteoblast cultures [36], but treatment with PGE2 increased apoptosis in conditionally immortalized human osteoblasts [37]. We found that overexpression of COX-2 increased apoptosis in human osteosarcoma cells, although the effect was PGE2 independent [38].
NSAIDs have been used to study the role of PGs in many systems, but they can clearly have potent effects unrelated to their inhibition of COX activity [31, 36, 39]. We used primary calvarial cells from COX-2 WT and KO mice to study the role of endogenous COX-2 in the growth of osteoblasts in vitro. Osteoblastic growth was increased in KO cells relative to WT cells and the increased growth was due to increased proliferation, not decreased apoptosis. Differences between WT and KO cells were eliminated by addition of NS398, a selective inhibitor of COX-2 activity, to WT cells or by addition of PGE2 to KO cells and, hence, appear to be PG-mediated.
MATERIALS AND METHODS
Materials
PGE2 and NS398, a selective inhibitor of COX-2 activity, were purchased from Cayman Chemical (Ann Arbor, MI). Culture media were purchased from Gibco-BRL (Grand Island, NY) unless otherwise specified. Other reagents were purchased from Sigma (St. Louis, MO).
Animals
The COX-2 KO mice used in this study were developed at the University of North Carolina in a C57Bl/6, 129SV background [40, 41]. COX-2 WT and KO mice were bred for experiments by mating mice heterozygous for the disrupted COX-2 allele in a mixed C57Bl/6,129SV background. Mice were genotyped as described previously [8]. Mice were sacrificed at 5–8 weeks of age. All animal protocols were approved by the Animal Care and Use Committee of the University of Connecticut Health Center, Farmington, CT.
Primary Calvarial Osteoblast Cultures
Calvariae were dissected from 2–5 mice, washed with PBS and sequentially digested with 0.5 mg/ml of crude collagenase P (Roche Molecular Biochemical, Indianapolis, IN) in a solution of 1 ml trypsin/EDTA and 4 ml PBS at 37°C with gentle rocking. Five digests were performed, all for 10 min except the last one, which was for 90 min. After each digest, released cells were collected, the reaction stopped with 10% FCS, and the solution filtered through a Nitex membrane (Millipore Corp., Bedford, MA) to ensure a single cell suspension. Digests 2–5 were pooled and plated, grown to confluence, and replated at 5000 cells/cm2 before use. Cells were cultured at 37°C in a humidified atmosphere of 5% CO2, in phenol red-free DMEM with 10% heat-inactivated fetal calf serum (FCS), and 100 U/ml penicillin, 50 μg/ml streptomycin. PGE2, NS398 and vehicle (EtOH, 0.1%) were added at the beginning of the cultures. For differentiation studies, 50 μg/ml of L-ascorbic acid phosphate (Wako Pure Chemical Industries, Japan) and 10 μM beta-glycerophosphate (Sigma) were added at the first medium change.
For alkaline phosphatase (ALP) staining, cultures were fixed with citrate, acetone and formaldehyde for 1 min and stained for ALP using a kit from Sigma according to manufacturer’s instructions. For Von Kossa staining, cultures were fixed formaldehyde and washed with 0.1 M cacodylate buffer (pH 7.4). Cells are incubated sequentially with saturated LiCO3 (20 min), 5% AgNO3 (60 min under lamp), and 5% Na thiosulfate (5 min), with H2O washes between incubations.
To examine cell number, cells were rinsed with PBS, suspended with trypsin-EDTA (0.25%), centrifuged, and resuspended in 500 μl of culture medium. An aliquot of 100 μl of cell suspension was counted with Coulter Counter (Coulter Corporation, Miami, FL), lower limit set at 8.0 μm. Triplicate wells were counted for each experimental group.
Marrow Stromal Cell (MSC) Cultures
Marrow from both tibiae and femurs of a single mouse was flushed with a total of 1 ml of α-minimum essential medium (α-MEM). Marrow cells were plated at 1×106 cells/well in 6-well plates in α-MEM containing 10% FCS and 50 μg/ml L-ascorbic acid phosphate (Wako Pure Chemical Industries). Cells were cultured at 37°C in a humidified atmosphere of 5% CO2 in air with medium changes twice a week. PGE2 (1 μM) or vehicle (EtOH, 0.1%) was added at the beginning of the cultures and at each medium change. To stain for ALP, cultures were fixed with citrate, acetone and formaldehyde for 1 min and stained using a kit from Sigma according to manufacturer’s instructions. To estimate the area covered by all cells, cultures were counterstained with crystal violet according to the manufacturer’s protocol (Sigma). To calculate the area of crystal violet or alkaline phoshatase staining, dishes were scanned with a Hewlett Packard ScanJet and stained areas manually circumscribed and quantified using NIH Image 1.6.
ALP Activity
Cultures were lysed in 10 mM Tris solution (pH 7.5) supplemented with 0.1% Triton X-100. Supernatants were incubated with an alkaline buffer (pH 10.5) containing 5 mM of p-nitrophenol phosphate as substrate and 2 mM MgCl2. Absorbance was determined at 405 nM and compared with a p-nitrophenol standard titration curve (Sigma). ALP activity was normalized to total protein measured with BCA protein kit (Pierce Chemical Company, Rockford, IL).
PGE2 Assay
Medium was removed from cultures, frozen until use, and PGE2 accumulation measured by radioimmunoassay (RIA) as previously described [42]. PGE2 antibody for this assay was purchased from Dr. Lawrence Levine (Brandeis University, Waltham, MA). 100 μl of unmodified medium was assayed for each sample and 3 wells were assayed for each group. Unknowns and standards contained equal amounts of culture medium. 3[H]-PGE2 was used as tracer and the assays were run at antibody dilutions providing 20–40% binding. The assays were carried out at 4°C and free prostaglandins were removed with dextran-coated charcoal. Values for unknowns were calculated from a standard curve using a 4-parametric curve-fitting computer program (Softmax Pro, Molecular Devices Corporation, Sunnyvale, CA). The lower detection limit of the assay for PGE2 is 50 pg/ml or 0.14 nM.
3H-Thymidine Incorporation Assay
Cultures were pulsed with 5 mCi/ml 3H-thymidine (NEN Life Science Products, Boston, MA) and incubated for 2 hours. Cells were washed twice with PBS and then extracted twice with 10% TCA. The TCA insoluble fraction was dissolved in 0.5 M NaOH and counted by liquid scintillation. The amount of 3H-thymidine incorporated into each sample was normalized to cell number in parallel cultures.
5-(and-6)-Carboxyfluorescein Diacetate, Succinimidyl Ester (CFDA, SE) Staining
Cell replication can be examined by staining cells with CFDA,SE (CFSE). CFSE irreversibly couples to cellular proteins by reaction with lysine side chains and other available amines without damaging side effects, and when cells divide, CFSE labeling is distributed equally between the daughter cells, making them half as fluorescent as their parents. The fluorescence of cells labeled with CFSE is stable for several months. First passage primary osteoblasts were trypsinized, washed twice with sterile PBS at room temperature and then incubated with CFSE (Molecular Probes, Eugene, OR) at a concentration of 25 μM in PBS for 10 min at 37 °C. After 10 min, the reaction was stopped by adding FCS and incubated for another 10 min. Finally, the cells were washed twice with sterile PBS with 1% FCS and the labeled cells were plated at 5,000 cells/cm2 in 6-well dishes and grown for 3–7 days. Before analysis, cells were prepared by trypsinization and washing with PBS. 10,000 cells were subjected to the FACS analysis. Fluorescence intensity was measured on flow cytometry, using excitation at 488 nm at the FL1 detection channel (FACSCalibur, Becton Dickinson, San Jose, CA), and analyzed with CellQuest software (Becton Dickinson).
Annexin V-FITC Apoptosis Detection
Apoptosis was measured using the annexin V-FITC apoptosis detection kit from Calbiochem (San Diego, CA). This method detects externalization of phospholipids, which are normally located on the cytoplasmic surface of the cell membrane, via binding with the modified anticoagulant annexin-V. Cells were plated at 5,000/cm2 in 100-mm dishes and cultured for 5 days. Both adherent and floating cells were collected and resuspended in 1 X cold binding buffer (10 mM Hepes, pH 7.4, 150 mM NaCl, 2.5 mM CaCl2, 1 mM MgCl2, 4% BSA) for analysis. Cells were also stained with propidium iodide (PI) to detect dead cells. Analysis was on the FACSCalibur flow cytometer (Becton Dickinson) using CellQuest software (Becton Dickinson). 10,000 cells were subjected to the FACS analysis. Unstained cells were classified as “live”, cells stained for annexin-V only (early apoptotic) and cells stained for both annexin-V and PI (“late apoptotic” were combined and called “apoptotic, and cells stained for PI only were “dead.” The experiment was performed 3 times to obtain a mean ± SEM for n=3.
Cell Cycle Analysis
Cell cycle progression was analyzed by allophycocyanin (APC) BrdU/7-amino-actinomycin D (7-AAD) incorporation, using a kit from BD Pharmingen (San Diego, CA), according to the manufacturer’s instructions. BrdU is incorporated into replicating DNA, and 7-AAD binds total DNA. With this combination, two-color flow cytometric analysis can characterize cells that are actively synthesizing DNA in terms of their cell cycle position. Cells were plated at 5,000/cm2 in 100 mm cell culture dishes and cultured for 4–5 days with medium change at day 3. On the day of experiment, culture medium was changed and 10 μl/ml of 10 μM BrdU was added for 1–4 hours. Cells were resuspended and analyzed on a FACSCalibur flow cytometer (Becton Dickinson). 10,000 cells were subjected to the FACS analysis. APC was detected by FL4 channel, and 7-AAD was detected by FL3 channel. After gating out cellular aggregates and debris, cell cycle distribution was analyzed using CellQuest software (Becton Dickinson).
Statistical Analysis
Statistical significance of differences was determined by ANOVA with post-hoc comparison of more than two groups by Bonferroni (SigmaStat software, Jandel Scientific, San Rafael, CA)
RESULTS
PGE2 production and alkaline phosphatase activity
Calvarial osteoblasts were cultured for 14 days and medium PGE2 accumulation measured at intervals (Fig. 1A). COX-2 is generally expressed at low levels in osteoblasts but can be induced by addition of fresh serum when media are changed [43]. By day 2 of culture, 15 ng/ml PGE2 had accumulated in COX-2 WT POB cultures. The greatest PGE2 production occurred within the first week of culture, consistent with our observations that serum induction of COX-2 is greatest at early times in culture (unpublished data). In contrast, medium PGE2 levels were usually less than 50 pg/ml (undetectable by our RIA) in COX-2 KO cultures at all time points, indicating that COX-1 does not compensate for the absence of COX-2 under these conditions.
Figure 1. Medium PGE2 and alkaline phosphatase (ALP) activity in cultured calvarial osteoblasts from COX-2 WT and KO mice.
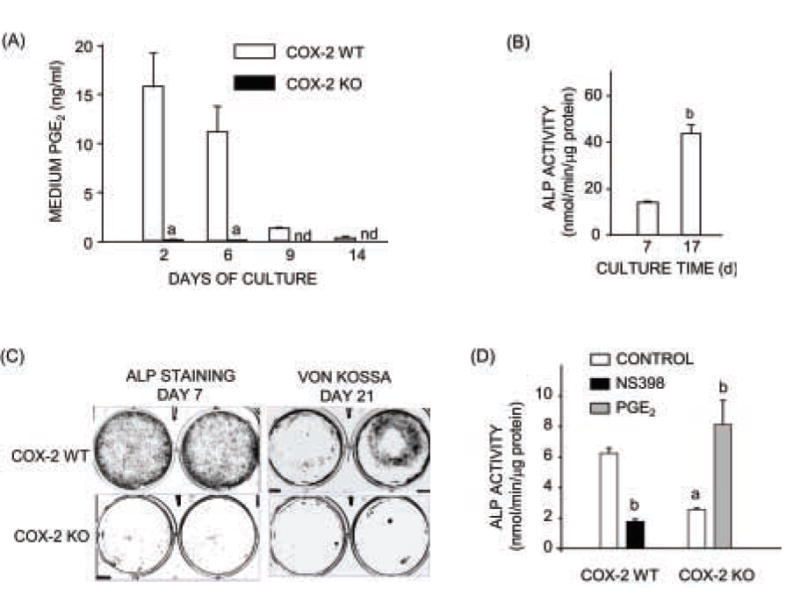
(A) Cumulative PGE2 measured at each medium change. (B) ALP activity calculated at 7 and 17 days of culture in WT cells. (C) ALP staining at 7 days of culture and Von Kossa staining at 21 days of culture. (D) ALP activity in cells cultured for 7 days. Cultures were treated with vehicle (Control), PGE2 (1 μM) or NS398 (0.1 μM). Bars are means ± SEM for n = 3 wells of cells. aSignificant effect of genotype, P<0.01. bSignificant effect of treatment, P<0.01. nd = not detectable.
POB cultures were made from calvariae of 6–8 wk old mice because COX-2 WT and KO mice were bred by crossing mice with only one allele of COX-2 disrupted and we wanted to wean mice before genotyping them. Similar to POBs from calvariae of neonatal mice, POBs from the older mice differentiated with time in culture, as reflected by increased ALP activity and staining and could mineralize (Fig. 1B,C). We previously showed that these POBs increases ALP activity and osteocalcin mRNA expression in response to bone morphogenetic protein (BMP)-2 [44]. As expected, ALP activity measured after a week of culture was decreased in COX-2 KO POBs compared to WT POBs (Fig. 1D). This difference was reversed by addition of PGE2 (1 μM) to KO cells and mimicked by adding NS-398 (0.1 μM) to WT cells.
Cell growth in calvarial osteoblast cultures
Cells were plated at 5000/cm2 and counted at multiple time points thereafter. Increased cell count in COX-2 KO cultures compared to WT cultures was apparent at day 4 of culture (Fig. 2A). The increase in cell growth reached a plateau by day 14 (Fig. 2B). The difference between WT and KO cells was maintained for up to 21 days of culture (Fig. 2B). Culture of COX-2 WT cells with NS-398 (0.1 μM), a selective inhibitor of COX-2 activity, increased cell numbers to the level of COX-2 KO cultures (Fig. 2B). Culture of COX-2 KO cells with PGE2 (1 μM) decreased cell numbers to the level of COX-2 WT cultures (Fig. 2B). Because PGE2 at 1 μM (352 ng/ml) is more than 20-fold higher than the highest cumulative endogenous PGE2 measured in COX-2 WT cultures, we treated COX-2 KO cultures with 10 and 100 nM (3.5 and 35 ng/ml) of PGE2 for comparison. Similar decreases in cell number were seen at all PGE2 concentrations on day 4 (Fig. 2C). These results, plus the ability of NS398 to mimic effects of COX-2 disruption, indicate that endogenous PG production is sufficient to explain the differences between WT and KO cultures.
Figure 2. Cell growth in calvarial osteoblast cultures from COX-2 WT and KO mice.
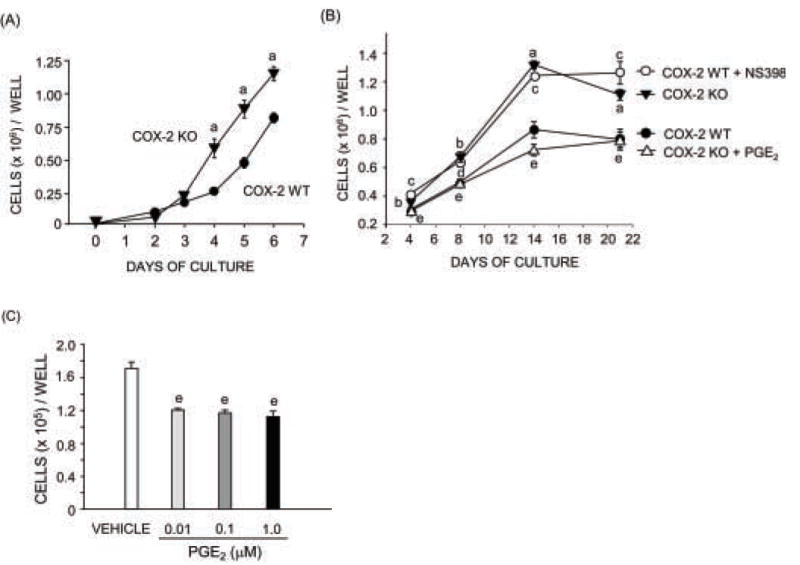
Cells were plated at 5000/cm2. (A) Cell growth during first week of culture. (B) Cell growth over 21 days of culture. Cultures were treated with vehicle (Control), PGE2 (1 μM) or NS398 (0.1 μM) for the duration of culture. (C) Cell counts in COX-2 KO cells at 4 days of culture after treatment with varying doses of PGE2. Symbols are means ± SEM for n=3 wells of cells. aSignificant effect of genotype, P<0.01; bP <0.05. cSignificant effect of NS398, P < 0.01; dP <0.05. eSignificant effect of PGE2, P<0.01.
Cell proliferation in calvarial osteoblast cultures
The incorporation of 3H-thymidine (TdR) into replicating DNA was measured on day 4 of culture following a 2 h pulse of 3H-thymidine. In order to take the difference in cell number between COX-2 WT and KO cultures into account, TdR was normalized to cell number in parallel cultures. Normalized TdR was significantly increased in COX-2 KO cultures relative to COX-2 WT cultures. A representative experiment is shown in Fig. 3A. The mean increase in TdR in COX-2 KO cells relative to COX-2 WT cells in 3 separate experiments was 1.8 ± 0.1 fold. Treatment with NS398 significantly increased TdR in COX-2 WT cultures, while addition of PGE2 decreased TdR in COX-2 KO cultures (Fig. 3A).
Figure 3. Cell proliferation and cell cycle progression in calvarial osteoblast cultures from COX-2 WT and KO mice.
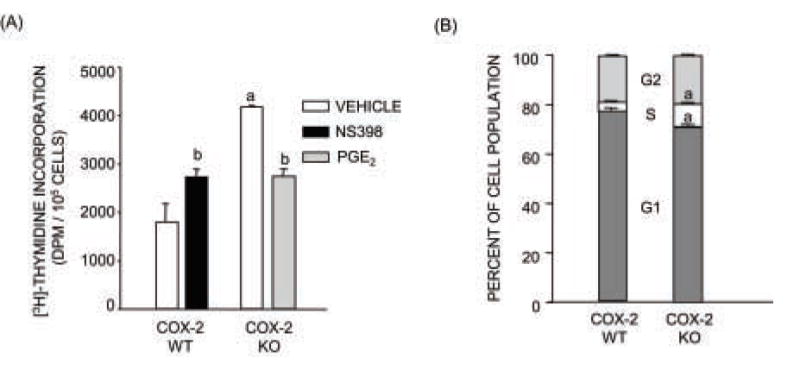
(A) 3H-thymidine incorporation (TdR). Cultures were treated with vehicle, PGE2 (1 μM) or NS398 (0.1 μM) throughout culture. TdR was measured on day 4 of culture after a 2-h pulse of 3H-thymidine. TdR was normalized to cell number in parallel cell cultures. Bars are means ± SEM for n=3. aSignificant effect of genotype, P<0.01. bSignificant effect of treatment, P<0.05. (B) Cell cycle analysis by flow cytometry after APC-BrdU/7-AAD staining. Results were calculated from three independent experiments. Cells were cultured for 4–5 days with BrdU incubation 2–4 hours after medium change. Bars are means ± SEM for n=3 experiments. aSignificant effect of genotype, P<0.01.
Cell cycle distribution was examined on days 4–5 of culture after staining cells with BrdU and 7-AAD staining. Even though cells were not synchronized, there was a significant shift from Go/G1 into S phase in the COX-2 KO cells compared to the WT cells, consistent with their increased proliferation (Fig. 3B). Cell replication was also evaluated by CFSE staining. The peak CFSE fluorescence intensity at day 7 of culture was shifted to the left in COX-2 KO cells relative to COX-2 WT cells, indicating faster cell division in COX-2 KO cultures (Fig. 4A). In another experiment, NS398 (1 μM) shifted the CFSE fluorescence intensity of part of the population to the left in COX-2 WT cultures and PGE2 (1 μM) shifted fluorescence of the population to the right in COX-2 KO cultures (Fig. 4B,C). PGE2 is thought to act predominantly via the cAMP pathway, and the protein kinase A agonist, forskolin (10 μM), had effects similar to PGE2. There was little or no shift in CFSE of COX-2 KO cells relative to COX-2 WT cells at day 3 of culture in 2 out of 3 experiments (data not shown), consistent with no difference in cell number at early time points (Fig. 2A).
Figure 4. Assessment of cell division by CFSE staining.
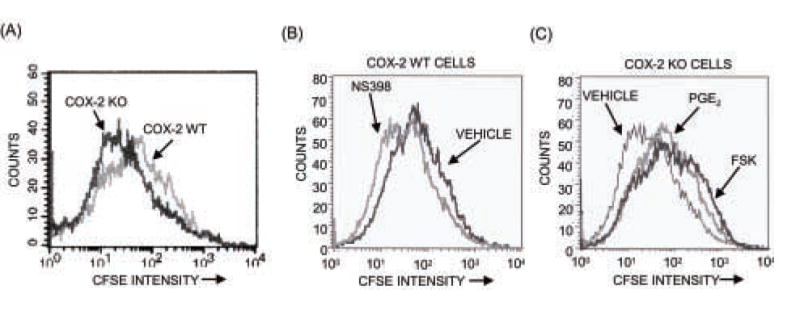
Cells were stained with CFSE before plating, cultured for 7 days and analyzed by flow cytometry, as described in Materials and Methods. (A) Comparison of cell division in COX-2 WT and KO cells. (B) Effects of NS398 (0.1 μM) on cell division in COX-2 WT cells. (C) Effects of PGE2 (1 μM) or forskolin (10 μM) on cell division in COX-2 KO cultures.
Apoptosis in calvarial osteoblast cultures
An increase in cell survival might also contribute to the increased cell number in COX-2 KO cultures. We measured cell viability on flow cytometry with staining of cells with annexin-V-FITC and propidium iodide. Our goal was to explain the increased cell count in COX-2 KO cultures compared to WT cultures. Because differences in cell number were detected under normal culture conditions, we did not add inducers of apoptosis. Data pooled from three independent flow cytometry experiments at 5 days of culture showed no statistically significant difference between COX-2 WT and KO cells in percent apoptotic cells or dead cells (Figure 5). However, there was a statistically non-significant trend toward decreased survival in KO cells. Hence, increased viability does not play a role in the increased numbers of KO cells relative to WT cells.
Figure 5. Cell apoptosis in calvarial osteoblasts from COX-2 WT and KO mice.
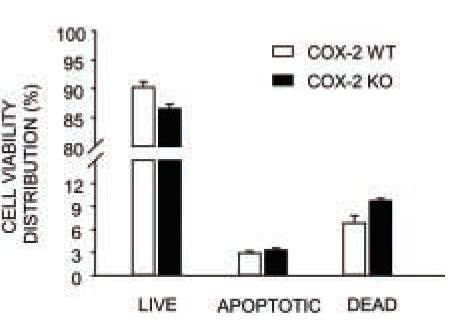
Cells were cultured for 5 days, double stained with annexin-V-FITC and PI and analyzed by flow cytometry. Bars are mean ± SEM of 3 independent experiments.
Comparison with marrow stromal cell (MSC) cultures
To compare the effects of COX-2 expression in POB cultures with the effects on osteoblastic progenitors in MSC cultures, we cultured marrow from COX-2 WT and KO mice for 14 days (Fig. 6A,B,C). Cumulative PGE2, measured in medium from day 7–10 of culture, was 12.5 ± 2 nM (n =3) in COX-2 WT wells and undetectable in COX-2 KO wells (data not shown). On day 14, cultures were stained for ALP, to mark the population of cells with potential to become osteoblasts, and counterstained with crystal violet, which stains all cells (Fig. 6A). The areas of ALP staining and crystal violet staining were decreased 75% and 50%, respectively, in KO cells compared to WT cells (Fig. 6B,C). Absence of COX-2 had more effect on ALP positive cells than on the total cell population, decreasing the ratio of ALP staining to crystal violet staining by 48%. Treatment of KO cultures with PGE2 (1 μM) reversed the decreases (Fig. 6). Similar results were seen in two other experiments. Hence, COX-2 expression or addition of PGE2 increased growth of osteoblastic progenitors in MSC cultures.
Figure 6. Alkaline phosphatase (ALP) and crystal violet staining of marrow stromal cells cultured from COX-2 WT and KO mice.
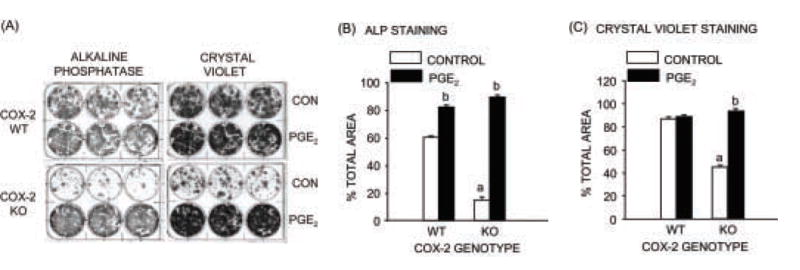
Cells were cultured for 14 days with vehicle (Control) or PGE2 (1 μM). (A) Cultures were stained for ALP and counterstained with crystal violet. Dishes were scanned and the area of ALP staining (B) and crystal violet staining (C) calculated. Bars are means ± SEM for 3 wells of cells. aSignificant effect of genotype, P<0.01. bSignificant effect of PGE2, P<0.01.
DISCUSSION
We used POB cells from the calvariae of COX-2 WT and KO mice to examine the role of endogenous COX-2 in osteoblastic growth in vitro. As shown previously [8,44–45], absence of COX-2 was associated with decreased PGE2 production, despite constitutive expression of COX-1. Absence of COX-2 decreased ALP activity and increased cell growth. The increase in cell number was associated with increased DNA synthesis and cell replication, with a decrease in G0/G1 and an increase in S in cell cycle distribution.
Most studies evaluating the role of endogenously produced PGs have used NSAIDs, which can have effects on cell growth in addition to their inhibition of COX activity, especially at high concentrations [31,36,39]. In our study, NS398 (0.1 μM), a selective inhibitor of COX-2 activity, added to COX-2 WT cells increased cell growth, similar to the disruption of COX-2 expression, but had no effect on COX-2 KO cells (data not shown). Although NS398 was able to mimic effects of COX-2 disruption, this does not necessarily mean that all effects of COX-2 KO on growth were due to decreased PG production. Both disruption of COX-2 expression and inhibition of COX-2 activity can increase the availability of substrate for COX-2, arachidonic acid, which might cause shunting of arachidonic acid into the lipoxygenase pathway to increase production of leukotrienes and HETEs. Free arachidonic acid has been reported to increase cell growth in osteoblastic cells via a noneicosanoid mechanism [46]. Leukotrienes and hydroxyeicosatetraenoic acids (HETEs) can also regulate cell growth [47]. However, the observation that exogenous PGE2 could reverse the effects of COX-2 gene disruption suggests that differences in growth between COX-2 WT and KO mice were PG-dependent.
Studies of exogenous PGE2 effects on osteoblast proliferation in vitro have shown both increased [27–29] and decreased proliferation [15, 30, 31], as well as biphasic dose-dependent effects [32, 33]. A number of factors can complicate such studies, including the autoamplification of PG effects by the PG-induction of COX-2 [48, 49] and the expression of different EP receptors on different osteoblastic cell types or at different differentiation stages of the cultures [50]. In two studies showing inhibition of proliferation by PGE2, the inhibition was associated with the cAMP pathway in MC3T3-E1 cells, which would suggest involvement of EP2 and EP4 receptors [50], while involvement of Ca2+ and phosphoinositide turnover, suggesting an EP1 receptor pathway [15], was reported in rat calvarial cells. In the current study, forskolin, a cAMP-PKA pathway agonist, had similar inhibitory effects as PGE2.
In contrast to the results in POB cultures, absence of COX-2 expression in MSC cultures decreased osteoblastogenesis, as previously reported [17], and general cell growth. POB cultures probably consist largely of osteoblastic precursors at the time of plating, while MSC cultures contain colony forming units with more potential to replicate and differentiate into several lineages. It is not yet possible to distinguish osteoblast progenitors with certainty in MSC cultures, but ALP staining should mark the population that includes osteoblastic progenitors, and this population was preferentially decreased by the absence of COX-2. Hence, it is possible that COX-2 derived PGs have stimulatory effects on the growth of early osteloblast progenitors and inhibitory effects on more mature osteoblastic cells. Similar conclusions were reached in a study that found PGE2 to stimulate DNA synthesis in the less differentiated population 1 of osteoblastic cells obtained by sequential digestion of fetal rat calvariae and to inhibit mitogenesis in the more mature populations 3–5 [51]. The more primitive osteoprogenitors are presumably the cells that contribute most significantly to replacement of osteoblasts in fracture healing [52], and decreased COX-2 expression or activity has been shown in multiple studies to inhibit or delay fracture healing [17,53–55].
COX-2 KO mice have been used to examine the role of COX-2 in the basal bone phenotype. Absence of COX-2 would be expected to decrease both bone formation and bone resorption [8]. One study reported that bulk material properties of bones from 3-mo old COX-2 KO mice in a C57Bl/6, DBA background were lower than those of WT mice, whereas stiffness and breaking force were similar [56]. Cortical bones of COX-2 KO mice were thinner and more porous than WT bones [56]. Another study of 4-mo old COX-2 KO mice in a 129 background found that males, but not females, had decreased cortical and trabecular femoral bone compared to WT mice [57]. However, COX-2 KO mice in a C57Bl/6, 129 background die 4 times faster than WT mice after weaning, likely due to progressive renal failure, and the renal failure can be associated with marked secondary hyperparathyroidism and decreased bone mass [58]. When only mice with intact renal function that had survived to 10 mo of age were studied, COX-2 KO mice still had elevated parathyroid levels [58]. Skeletal analysis showed no significant differences in cortical or trabecular bone density in KO mice compared to WT mice although rates of trabecular bone formation and mineral apposition were increased in KO mice relative to WT mice. A study of COX-2 KO mice in a DBA background, reported to have no renal abnormalities, found the KO mice to have lower bone mineral density and impaired bone strength relative to WT mice, but these KO mice also had hyperparathyroidism [59]. Hence, COX-2 KO mice may have systemic abnormalities that can make it difficult to determine the direct effects of COX-2 absence on bone in vivo.
PTH, which is also a potent anabolic agent for bone in vivo and works via similar G-protein signaling pathways as PGE2, has also been shown to inhibit osteoblastic cell proliferation in culture, in part via upregulation of cyclin kinase inhibitor protein p21(WAF1/Cip1) [60]. Our preliminary data indicate that p21 expression is decreased in the COX-2 KO calvarial osteoblast cells compared to WT cells (Pilbeam, unpublished data). Down regulation of the early osteoblastic proliferation stage is thought to be critical for proceeding to differentiation [61], and p21 may play an important role in this down regulation [62]. However, the relationship between cell proliferation and differentiation is probably not simply reciprocal [63].
COX-2 expression and PGs have been associated with increased resistance to apoptosis and tumor promotion in tissues other than bone [34,35]. The few studies on the effects of PGs on apoptosis of osteoblasts are contradictory, reporting that PGs can both decrease [36] and increase apoptosis [37]. We found no differences in survival between COX-2 WT and KO calvarial osteoblasts under basal culture conditions. Thus, the increased cell growth in COX-2 KO calvarial cells cannot be attributed to decreased apoptosis. Our results were obtained under basal culture conditions (no stressors except for changes of medium with fresh serum), and the effects of PGs in the presence of inducers of apoptosis, such as TNFα, may be different.
We conclude that COX-2 associated PGs have stimulatory effects on cell growth in early osteoblastic precursors and inhibitory effects on cell growth in later, more mature osteoblastic cells. We also conclude that the increased differentiation seen in COX-2 WT calvarial cultures compared to KO cultures cannot be attributed to increased proliferation or decreased apoptosis.
Acknowledgments
This work was funded by NIH RO1DK48361 and an award from the Donaghue Foundation of Connecticut to CP.
Grant funding: RO1DK48361 and the Donaghue Foundation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Smith WL, Song I. The enzymology of prostaglandin endoperoxide H synthases-1 and -2. Prostaglandins Other Lipid Mediat. 2002;68–69:115–28. doi: 10.1016/s0090-6980(02)00025-4. [DOI] [PubMed] [Google Scholar]
- 2.Herschman HR, Xie W, Reddy S. Inflammation, reproduction, cancer and all that... The regulation and role of the inducible prostaglandin synthase. Bioessays. 1995;17:1031–7. doi: 10.1002/bies.950171207. [DOI] [PubMed] [Google Scholar]
- 3.Pilbeam CC, Harrison JR, Raisz LG. Prostaglandins and bone metabolism. In: Bilezikian JP, Raisz LG, Rodan GA, editors. Principles of Bone Biology. New York: Academic Press; 2002. pp. 979–94. [Google Scholar]
- 4.Kobayashi T, Narumiya S. Function of prostanoid receptors: studies on knockout mice. Prostaglandins Other Lipid Mediat. 2002;68–69:557–73. 557–73. doi: 10.1016/s0090-6980(02)00055-2. [DOI] [PubMed] [Google Scholar]
- 5.Fujino H, Xu W, Regan JW. Prostaglandin E2 induced functional expression of early growth response factor-1by EP4, but not EP2, prostanoid receptors via the phosphatidylinositol 3-kinase and extracellular signal-regulated kinases. J Biol Chem. 2003;278:12151–6. doi: 10.1074/jbc.M212665200. [DOI] [PubMed] [Google Scholar]
- 6.Gobeil F, Jr, Vazquez-Tello A, Marrache AM, Bhattacharya M, Checchin D, Bkaily G, Lachapelle P, Ribeiro-da-Silva A, Chemtob S. Nuclear prostaglandin signaling system: biogenesis and actions via heptahelical receptors. Can J Physiol Pharmacol. 2003;81:196–204. doi: 10.1139/y02-163. [DOI] [PubMed] [Google Scholar]
- 7.Barnes PJ. Receptor heterodimerization: a new level of cross-talk. J Clin Invest. 2006;116:1210–2. doi: 10.1172/JCI28535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okada Y, Lorenzo JA, Freeman AM, Tomita M, Morham SG, Raisz LG, Pilbeam CC. Prostaglandin G/H synthase-2 is required for maximal formation of osteoclast-like cells in culture. J Clin Invest. 2000;105:823–32. doi: 10.1172/JCI8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Okada Y, Pilbeam CC, Lorenzo JA, Kennedy CR, Breyer RM, Raisz LG. Knockout of the murine prostaglandin EP2 receptor impairs osteoclastogenesis in vitro. Endocrinology. 2000;141:2054–61. doi: 10.1210/endo.141.6.7518. [DOI] [PubMed] [Google Scholar]
- 10.Miyaura C, Inada M, Suzawa T, Sugimoto Y, Ushikubi F, Ichikawa A, Narumiya S, Suda T. Impaired bone resorption to prostaglandin E2 in prostaglandin E receptor EP4-knockout mice. J Biol Chem. 2000;275:19819–23. doi: 10.1074/jbc.M002079200. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida K, Oida H, Kobayashi T, Maruyama T, Tanaka M, Katayama T, Yamaguchi K, Segi E, Tsuboyama T, Matsushita M, Ito K, Ito Y, Sugimoto Y, Ushikubi F, Ohuchida S, Kondo K, Nakamura T, Narumiya S. Stimulation of bone formation and prevention of bone loss by prostaglandin E EP4 receptor activation. Proc Natl Acad Sci U S A. 2002;99:4580–5. doi: 10.1073/pnas.062053399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flanagan AM, Chambers TJ. Stimulation of bone nodule formation in vitro by prostaglandins E1 and E2. Endocrinology. 1992;130:443–8. doi: 10.1210/endo.130.1.1309342. [DOI] [PubMed] [Google Scholar]
- 13.Nagata T, Kaho K, Nishikawa S, Shinohara H, Wakano Y, Ishida H. Effect of prostaglandin E2 on mineralization of bone nodules formed by fetal rat calvarial cells. Calcif Tissue Int. 1994;55:451–7. doi: 10.1007/BF00298559. [DOI] [PubMed] [Google Scholar]
- 14.Scutt A, Bertram P. Bone marrow cells are targets for the anabolic actions of prostaglandin E2 on bone: induction of a transition from nonadherent to adherent osteoblast precursors. J Bone Miner Res. 1995;10:474–87. doi: 10.1002/jbmr.5650100320. [DOI] [PubMed] [Google Scholar]
- 15.Kaneki H, Takasugi I, Fujieda M, Kiriu M, Mizuochi S, Ide H. Prostaglandin E2 stimulates the formation of mineralized bone nodules by a cAMP-independent mechanism in the culture of adult rat calvarial osteoblasts. J Cell Biochem. 1999;73:36–48. [PubMed] [Google Scholar]
- 16.Okada Y, Tomita M, Gronowicz G, Kawaguchi H, Sohn J, Tanaka Y, Morimoto I, Nakamura T, Raisz L, Pilbeam C. Effects of cyclooxygenase-2 gene disruption on osteoblastic function. J Bone Miner Res. 2000;15(S1):S217. [Google Scholar]
- 17.Zhang X, Schwarz EM, Young DA, Puzas JE, Rosier RN, O'Keefe RJ. Cyclooxygenase-2 regulates mesenchymal cell differentiation into the osteoblast lineage and is critically involved in bone repair. J Clin Invest. 2002;109:1405–15. doi: 10.1172/JCI15681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ueda K, Saito A, Nakano H, Aoshima M, Yokota M, Muraoka R, Iwaya T. Cortical hyperostosis following long-term administration of prostaglandin E1 in infants with cyanotic congenital heart disease. J Pediatr. 1980;97:834–6. doi: 10.1016/s0022-3476(80)80282-4. [DOI] [PubMed] [Google Scholar]
- 19.Norrdin RW, Shih MS. Systemic effects of prostaglandin E2 on vertebral trabecular remodeling in beagles used in a healing study. Calcif Tissue Int. 1988;42:363–8. doi: 10.1007/BF02556354. [DOI] [PubMed] [Google Scholar]
- 20.Faye-Petersen OM, Johnson WH, Jr, Carlo WA, Hedlund GL, Pacifico AD, Blair HC. Prostaglandin E1-induced hyperostosis: clinicopathologic correlations and possible pathogenetic mechanisms. Pediatr Pathol Lab Med. 1996;16:489–507. doi: 10.1080/15513819609168686. [DOI] [PubMed] [Google Scholar]
- 21.Jee WS, Ma YF. The in vivo anabolic actions of prostaglandins in bone. Bone. 1997;21:297–304. doi: 10.1016/s8756-3282(97)00147-6. [DOI] [PubMed] [Google Scholar]
- 22.Weinreb M, Suponitzky I, Keila S. Systemic administration of an anabolic dose of PGE2 in young rats increases the osteogenic capacity of bone marrow. Bone. 1997;20:521–6. doi: 10.1016/s8756-3282(97)00033-1. [DOI] [PubMed] [Google Scholar]
- 23.Ke HZ, Crawford DT, Qi H, Simmons HA, Owen TA, Paralkar VM, Li M, Lu B, Grasser WA, Cameron KO, Lefker BA, Silva-Jardine P, Scott DO, Zhang Q, Tian XY, Jee WS, Brown TA, Thompson DD. A nonprostanoid EP4 receptor selective prostaglandin E2 agonist restores bone mass and strength in aged, ovariectomized rats. J Bone Miner Res. 2006;21:565–75. doi: 10.1359/jbmr.051110. [DOI] [PubMed] [Google Scholar]
- 24.Li M, Ke HZ, Qi H, Healy DR, Li Y, Crawford DT, Paralkar VM, Owen TA, Cameron KO, Lefker BA, Brown TA, Thompson DD. A novel, non-prostanoid EP2 receptor-selective prostaglandin E2 agonist stimulates local bone formation and enhances fracture healing. J Bone Miner Res. 2003;18:2033–42. doi: 10.1359/jbmr.2003.18.11.2033. [DOI] [PubMed] [Google Scholar]
- 25.Akhter MP, Cullen DM, Gong G, Recker RR. Bone biomechanical properties in prostaglandin EP1 and EP2 knockout mice. Bone. 2001;29:121–5. doi: 10.1016/s8756-3282(01)00486-0. [DOI] [PubMed] [Google Scholar]
- 26.Akhter MP, Cullen DM, Pan LC. Bone biomechanical properties in EP4 knockout mice. Calcif Tissue Int. 2006;78:357–62. doi: 10.1007/s00223-005-0186-5. [DOI] [PubMed] [Google Scholar]
- 27.Chyun YS, Raisz LG. Stimulation of bone formation by prostaglandin E2. Prostaglandins. 1984;27:97–103. doi: 10.1016/0090-6980(84)90223-5. [DOI] [PubMed] [Google Scholar]
- 28.Feyen JH, Di BA, van dP, Lowik CW, Nijweide PJ. Effects of exogenous prostanoids on the proliferation of osteoblast- like cells in vitro. Prostaglandins. 1985;30:827–40. doi: 10.1016/0090-6980(85)90011-5. [DOI] [PubMed] [Google Scholar]
- 29.Hakeda Y, Yoshino T, Natakani Y, Kurihara N, Maeda N, Kumegawa M. Prostaglandin E2 stimulates DNA synthesis by a cyclic AMP-independent pathway in osteoblastic clone MC3T3-E1 cells. J Cell Physiol. 1986;128:155–61. doi: 10.1002/jcp.1041280204. [DOI] [PubMed] [Google Scholar]
- 30.Fang MA, Kujubu DA, Hahn TJ. The effects of prostaglandin E2, parathyroid hormone, and epidermal growth factor on mitogenesis, signaling, and primary response genes in UMR 106–01 osteoblast-like cells. Endocrinology. 1992;131:2113–9. doi: 10.1210/endo.131.5.1330491. [DOI] [PubMed] [Google Scholar]
- 31.Ho ML, Chang JK, Chuang LY, Hsu HK, Wang GJ. Effects of nonsteroidal anti-inflammatory drugs and prostaglandins on osteoblastic functions. Biochem Pharmacol. 1999;58:983–90. doi: 10.1016/s0006-2952(99)00186-0. [DOI] [PubMed] [Google Scholar]
- 32.Fujimori A, Tsutsumi M, Fukase M, Fujita T. Cyclooxygenase inhibitors enhance cell growth in an osteoblastic cell line, MC3T3-E1. J Bone Miner Res. 1989;4:697–704. doi: 10.1002/jbmr.5650040508. [DOI] [PubMed] [Google Scholar]
- 33.Baylink TM, Mohan S, Fitzsimmons RJ, Baylink DJ. Evaluation of signal transduction mechanisms for the mitogenic effects of prostaglandin E2 in normal human bone cells in vitro. J Bone Miner Res. 1996;11:1413–8. doi: 10.1002/jbmr.5650111007. [DOI] [PubMed] [Google Scholar]
- 34.Trifan OC, Hla T. Cyclooxygenase-2 modulates cellular growth and promotes tumorigenesis. J Cell Mol Med. 2003;7:207–22. doi: 10.1111/j.1582-4934.2003.tb00222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Backlund MG, Mann JR, DuBois RN. Mechanisms for the prevention of gastrointestinal cancer: the role of prostaglandin E2. Oncology. 2005;69 (Suppl 1):28–32. doi: 10.1159/000086629. [DOI] [PubMed] [Google Scholar]
- 36.Chang JK, Wang GJ, Tsai ST, Ho ML. Nonsteroidal anti-inflammatory drug effects on osteoblastic cell cycle, cytotoxicity, and cell death. Connect Tissue Res. 2005;46:200–10. doi: 10.1080/03008200500344025. [DOI] [PubMed] [Google Scholar]
- 37.Bodine PV, Billiard J, Moran RA, Ponce-de-Leon H, McLarney S, Mangine A, Scrimo MJ, Bhat RA, Stauffer B, Green J, Stein GS, Lian JB, Komm BS. The Wnt antagonist secreted frizzled-related protein-1 controls osteoblast and osteocyte apoptosis. J Cell Biochem. 2005;96:1212–30. doi: 10.1002/jcb.20599. [DOI] [PubMed] [Google Scholar]
- 38.Xu Z, Choudhary S, Voznesensky O, Mehrotra M, Woodard M, Hansen M, Herschman H, Pilbeam C. Overexpression of COX-2 in human osteosarcoma cells decreases proliferation and increases apoptosis. Cancer Res. 2006;66:6657–64. doi: 10.1158/0008-5472.CAN-05-3624. [DOI] [PubMed] [Google Scholar]
- 39.Jang TJ, Kang HJ, Kim JR, Yang CH. Nonsteroidal anti-inflammatory drug activated gene (NAG-1) expression is closely related to death receptor-4 and -5 induction, which may explain sulindac sulfide-induced gastric cancer cell apoptosis. Carcinogenesis. 2004;10:1853–8. doi: 10.1093/carcin/bgh199. [DOI] [PubMed] [Google Scholar]
- 40.Morham SG, Langenbach R, Loftin CD, Tiano HF, Vouloumanos N, Jennette JC, Mahler JF, Kluckman KD, Ledford A, Lee CA. Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell. 1995;83:473–82. doi: 10.1016/0092-8674(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 41.Langenbach R, Morham SG, Tiano HF, Loftin CD, Ghanayem BI, Chulada PC, Mahler JF, Lee CA, Goulding EH, Kluckman KD. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83:483–92. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 42.Raisz LG, Simmons HA. Effects of parathyroid hormone and cortisol on prostaglandin production by neonatal rat calvaria in vitro. Endocr Res. 1985;11:59–74. doi: 10.3109/07435808509035425. [DOI] [PubMed] [Google Scholar]
- 43.Pilbeam CC, Kawaguchi H, Hakeda Y, Voznesensky O, Alander CB, Raisz LG. Differential regulation of inducible and constitutive prostaglandin endoperoxide synthase in osteoblastic MC3T3-E1 cells. J Biol Chem. 1993;268:25643–9. [PubMed] [Google Scholar]
- 44.Chikazu D, Li X, Kawaguchi H, Sakuma Y, Voznesensky OS, Adams DJ, Xu M, Hoshio K, Katavic V, Herschman HR, Raisz LG, Pilbeam CC. Bone morphogenetic protein 2 induces cyclo-oxygenase 2 in osteoblasts via a Cbfal binding site: role in effects of bone morphogenetic protein 2 in vitro and in vivo. J Bone Miner Res. 2002;17:1430–40. doi: 10.1359/jbmr.2002.17.8.1430. [DOI] [PubMed] [Google Scholar]
- 45.Choudhary S, Wadhwa S, Raisz LG, Alander C, Pilbeam CC. Extracellular calcium is a potent inducer of cyclo-oxygenase-2 in murine osteoblasts through an ERK signaling pathway. J Bone Miner Res. 2003;18:1813–24. doi: 10.1359/jbmr.2003.18.10.1813. [DOI] [PubMed] [Google Scholar]
- 46.Fujimori A, Tsutsumi M, Yamada H, Fukase M, Fujita T. Arachidonic acid stimulates cell growth in an osteoblastic cell line, MC3T3-E1, by noneicosanoid mechanism. Calcif Tissue Int. 1989;44:186–91. doi: 10.1007/BF02556563. [DOI] [PubMed] [Google Scholar]
- 47.Ren W, Dziak R. Effects of leukotrienes on osteoblastic cell proliferation. Calcif Tissue Int. 1991;49:197–201. doi: 10.1007/BF02556118. [DOI] [PubMed] [Google Scholar]
- 48.Pilbeam CC, Raisz LG, Voznesensky O, Alander CB, Delman BN, Kawaguchi K. Autoregulation of inducible prostaglandin G/H synthase in osteoblastic cells by prostaglandins. J Bone Miner Res. 1994;10:406–14. doi: 10.1002/jbmr.5650100311. [DOI] [PubMed] [Google Scholar]
- 49.Suda M, Tanaka K, Yasoda A, Natsui K, Sakuma Y, Tanaka I, Ushikubi F, Narumiya S, Nakao K. Prostaglandin E2 (PGE2) autoamplifies its production through EP1 subtype of PGE receptor in mouse osteoblastic MC3T3-E1 cells. Calcif Tissue Int. 1998;62:327–31. doi: 10.1007/s002239900440. [DOI] [PubMed] [Google Scholar]
- 50.Suda M, Tanaka K, Natsui K, Usui T, Tanaka I, Fukushima M, Shigeno C, Konishi J, Narumiya S, Ichikawa A, Nakao N. Prostaglandin E receptor subtypes in mouse osteoblastic cell line. Endocrinology. 1996;137:1698–705. doi: 10.1210/endo.137.5.8612504. [DOI] [PubMed] [Google Scholar]
- 51.Centrella M, Casinghino S, McCarthy TL. Differential actions of prostaglandins in separate cell populations from fetal rat bone. Endocrinology. 1994;135:1611–20. doi: 10.1210/endo.135.4.7925124. [DOI] [PubMed] [Google Scholar]
- 52.Aubin JE. Advances in the osteoblast lineage. Biochem Cell Biol. 1998;76:899–910. [PubMed] [Google Scholar]
- 53.Simon AM, Manigrasso MB, O'Connor JP. Cyclo-oxygenase 2 function is essential for bone fracture healing. J Bone Miner Res. 2002;17:963–76. doi: 10.1359/jbmr.2002.17.6.963. [DOI] [PubMed] [Google Scholar]
- 54.Gerstenfeld LC, Thiede M, Seibert K, Mielke C, Phippard D, Svagr B, Cullinane D, Einhorn TA. Differential inhibition of fracture healing by non-selective and cyclooxygenase-2 selective non-steroidal anti-inflammatory drugs. J Orthop Res. 2003;21:670–5. doi: 10.1016/S0736-0266(03)00003-2. [DOI] [PubMed] [Google Scholar]
- 55.Brown KM, Saunders MM, Kirsch T, Donahue HJ, Reid JS. Effect of COX-2-specific inhibition on fracture-healing in the rat femur. J Bone Joint Surg Am. 2004;86-A:116–23. doi: 10.2106/00004623-200401000-00017. [DOI] [PubMed] [Google Scholar]
- 56.Chen Q, Rho JY, Fan Z, Laulederkind SJ, Raghow R. Congenital lack of COX-2 affects mechanical and geometric properties of bone in mice. Calcif Tissue Int. 2003;73:387–92. doi: 10.1007/s00223-002-0009-x. [DOI] [PubMed] [Google Scholar]
- 57.Robertson G, Xie C, Chen D, Awad H, Schwarz EM, O'Keefe RJ, Guldberg RE, Zhang X. Alteration of femoral bone morphology and density in COX-2-/- mice. Bone. 2006;39:767–772. doi: 10.1016/j.bone.2006.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu M, Choudhary S, Goltzman D, Ledgard F, Adams D, Gronowicz G, Koczon-Jaremko B, Raisz L, Pilbeam C. Do cyclooxygenase knockout mice have primary hyperparathyroidism? Endocrinology. 2005;146:1843–53. doi: 10.1210/en.2004-0734. [DOI] [PubMed] [Google Scholar]
- 59.Myers LK, Bhattacharya SD, Herring PA, Xing Z, Goorha S, Smith RA, Bhattacharya Sk, Carbone L, Faccio R, Kang AH, Ballou LR. The isozyme-specific effects of cyclooxygenase-deficiency on bone in mice. Bone. 2006;39:1048–1052. doi: 10.1016/j.bone.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 60.Qin L, Li X, Ko JK, Partridge NC. Parathyroid hormone uses multiple mechanisms to arrest the cell cycle progression of osteoblastic cells from g1 to s phase. J Biol Chem. 2005;280:3104–11. doi: 10.1074/jbc.M409846200. [DOI] [PubMed] [Google Scholar]
- 61.Stein GS, Lian JB. Molecular mechanisms mediating proliferation/differentiation interrelationships during progressive development of the osteoblast phenotype. Endocrine Rev. 1993;14:424–42. doi: 10.1210/edrv-14-4-424. [DOI] [PubMed] [Google Scholar]
- 62.Drissi H, Hushka D, Aslam F, Nguyen Q, Buffone E, Koff A, van WA, Lian JB, Stein JL, Stein GS. The cell cycle regulator p27kip1 contributes to growth and differentiation of osteoblasts. Cancer Res. 1999;59:3705–11. [PubMed] [Google Scholar]
- 63.Brown G, Hughes PJ, Michell H. Cell differentiation and proliferation--simultaneous but independent? Exp Cell Res. 2003;291:282–8. doi: 10.1016/s0014-4827(03)00393-8. [DOI] [PubMed] [Google Scholar]