

Abstract
Sulfite oxidizing enzymes are essential mononuclear molybdenum (Mo) proteins involved in sulfur metabolism of animals, plants and bacteria. There are three such enzymes presently known: (1) sulfite oxidase (SO) in animals, (2) SO in plants, and (3) sulfite dehydrogenase (SDH) in bacteria. X-ray crystal structures of enzymes from all three sources (chicken SO, Arabidopsis thaliana SO, and Starkeya novella SDH) show nearly identical square pyramidal coordination around the Mo atom, even though the overall structures of the proteins and the presence of additional cofactors vary. This structural information provides a molecular basis for studying the role of specific amino acids in catalysis. Animal SO catalyzes the final step in the degradation of sulfur-containing amino acids and is critical in detoxifying excess sulfite. Human SO deficiency is a fatal genetic disorder that leads to early death, and impaired SO activity is implicated in sulfite neurotoxicity. Animal SO and bacterial SDH contain both Mo and heme domains, whereas plant SO only has the Mo domain. Intraprotein electron transfer (IET) between the Mo and Fe centers in animal SO and bacterial SDH is a key step in the catalysis, which can be studied by laser flash photolysis in the presence of deazariboflavin. IET studies on animal SO and bacterial SDH clearly demonstrate the similarities and differences between these two types of sulfite oxidizing enzymes. Conformational change is involved in the IET of animal SO, in which electrostatic interactions may play a major role in guiding the docking of the heme domain to the Mo domain prior to electron transfer. In contrast, IET measurements for SDH demonstrate that IET occurs directly through the protein medium, which is distinctly different from that in animal SO. Point mutations in human SO can result in significantly impaired IET or no IET, thus rationalizing their fatal effects. The recent developments in our understanding of sulfite oxidizing enzyme mechanisms that are driven by a combination of molecular biology, rapid kinetics, pulsed electron paramagnetic resonance (EPR), and computational techniques are the subject of this review.
Keywords: sulfite oxidase, sulfite dehydrogenase, electron transfer, laser flash photolysis, sulfite oxidase deficiency, EPR
1. Introduction
Molybdenum (Mo)1 is the only second row transition metal that has known biological functions in all forms of life; over 50 molybdoenzymes are known that catalyze oxidation-reduction reactions that are essential in the metabolism of carbon, nitrogen and sulfur [1]. The mononuclear Mo enzymes fall into three distinct groups comprising the xanthine oxidase, DMSO reductase, and sulfite oxidase families. A number of them are of clinical significance for human health (e.g. xanthine and aldehyde oxidoreductases, sulfite oxidase).
The sulfite oxidase family comprises plant assimilatory nitrate reductases (NR) and sulfite-oxidizing enzymes from animals, plants and bacteria. The sulfite-oxidizing enzymes can be separated into two classes (Table 1), the sulfite oxidases (SO, found in animals and plants, EC 1.8.3.1), and the sulfite dehydrogenases (SDH, found in bacteria, EC 1.8.2.1), based on their ability to transfer electrons to molecular oxygen [2]. Arabidopsis thaliana SO is the smallest eukaryotic Mo enzyme consisting of a Mo cofactor-binding domain but lacking the heme domain that is present in the animal SO [3]. While animal SO is a mitochondrial enzyme with cytochrome (cyt) c as the physiological electron acceptor, plant SO is localized in peroxisomes and does not react with cyt c. It has been shown that oxygen acts as the terminal electron acceptor for plant SO [4].
Table 1.
Three types of sulfite oxidizing enzymes
In animals the SO enzyme catalyzes the oxidation of sulfite to sulfate, with ferricytochrome c ((cyt c)ox) as the physiological electron acceptor [1, 5–7]:
This is the final step in the oxidative degradation of the sulfur-containing amino acids cysteine and methionine. The enzyme also plays an important role in detoxifying exogenously supplied sulfite and sulfur dioxide.
Sulfite oxidase deficiency is an inherited sulfur metabolic disorder in humans that results in profound birth defects, severe neonatal neurological problems, and early death, with no effective therapies known [8]. The inborn error is characterized by dislocation of ocular lenses, mental retardation, and in severe cases, attenuated growth of the brain [9]. These severe neurological symptoms result from either point mutations in the SO protein itself (so-called isolated SO deficiency, in which only SO activity is affected), or the inability to properly produce the pyranopterindithiolate cofactor, which results in deficiencies in all Mo-containing enzymes (so-called Mo cofactor deficiency) [10–12]. The development of methods to clone and express human SO has revealed several different clinical point mutations that result in isolated SO deficiency [13–16]. The X-ray structure of the human SO is not yet available, although the structure of the human SO heme domain was reported [17]. However, the structure of the highly homologous chicken liver SO provides a molecular basis for interpreting the fatal point mutations of the human enzyme and a foundation for the biochemical and biophysical studies discussed in this review.
The biochemical basis of the pathology of sulfite oxidase deficiency is unclear, and merits further investigation. Fatal brain damage may be due to the accumulation of a toxic metabolite, possibly SO32−, which is a strong nucleophile that can react with a wide variety of cell components. It has been reported that sulfite reacts with protein disulfides to form sulfonated cysteine derivatives, and since the integrity of disulfide bonds is crucial to the tertiary structure and thus protein function, the disruption of protein structure by sulfitolysis may result in altered cellular activities leading to biochemical lesions [18, 19]. Alternatively, a deficiency in the reaction product (sulfate, SO42−) may disturb normal fetal and neonatal development of the brain [6]. In addition, the nature of the lesion in human sulfite oxidase deficiency (with the central nervous system (CNS) being disproportionately affected) suggests that the principal problem is likely to be lipid peroxidation rather than amino acid metabolism [20]. Specifically, the cell membranes of the CNS myelin sheath are unique in possessing high concentrations of sulfatides and related lipids, which is likely the root cause of the sensitivity of the CNS to SO deficiency.
In the past decade or so, significant advances in SO enzymology have taken place in several important aspects. These include, but are not limited to, the following:
Successful cloning and expression of human [16, 21–23], chicken [24] and rat [25] SO proteins and mutants by Rajagopalan and coworkers;
The newly identified sulfite oxidizing enzymes in plants by Mendel and coworkers [3], and bacteria by Kappler [26];
X-ray crystal structures of all three sulfite-oxidizing enzymes have been obtained: wild type proteins of chicken SO [27], Arabidopsis thaliana SO [28], and S. novella SDH [29]); chicken SO R138Q mutant [24] and S. novella SDH Y236F mutant [30];
Rapid kinetics studies on physiologically relevant human SO mutants by stopped flow [31, 32] and laser flash photolysis [33–35] techniques;
Structural determination of enzyme intermediates by advanced spectroscopy techniques: pulsed electron paramagnetic resonance (EPR) [36–39] (for a recent review, see [40]), X-ray absorption [41–43], magnetic circular dichroism [44], and resonance Raman spectroscopies [45].
Direct measurement by protein film voltammetry of midpoint potentials of catalytically significant redox couples in SO [46] and SDH [2].
Exciting developments in the mechanism of Mo cofactor biosynthesis [10, 11, 47–54] have been summarized in recent excellent reviews [6, 55, 56]. These elegant studies were carried out by Rajagopalan, Mendel, Schwarz and their coworkers.
All sulfite oxidizing enzymes, except plant SO, possess two redox centers located in the Mo and heme domains. Intraprotein interdomain electron transfer (IET) processes between these two centers are critical in enzymatic turnover. The scope of this review is primarily about structural and functional relationships in the IET between the Mo and Fe centers in animal SO and S. novella SDH. We will focus on eukaryotes because our understanding of electron transfer is much more advanced in these systems. Most of this knowledge derives from studies with chicken and human SO proteins.
2. Structural characterization of sulfite oxidizing enzymes
X-ray crystal structures of wild type enzymes from all three sources (chicken SO [27], Arabidopsis thaliana SO [28], and S. novella SDH [29]) show nearly identical square pyramidal coordination around the Mo atom (Fig. 1), even though the overall structures of the proteins and the presence of additional cofactors vary (Table 1).
Fig. 1.
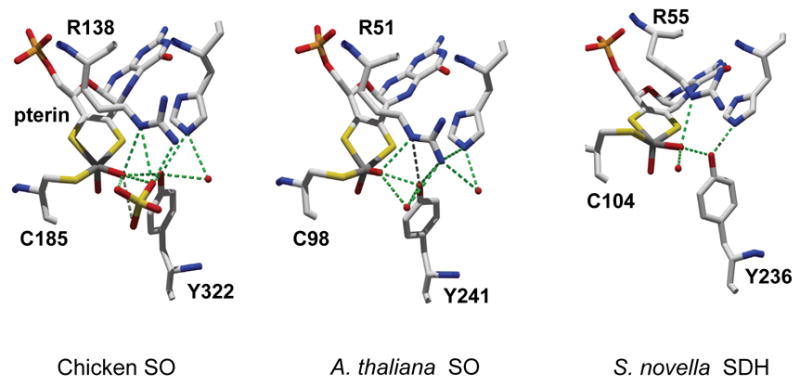
Selected amino acids and ligands near Mo centers of wild type proteins of chicken SO (left), A. thaliana SO (middle), and S. novella SDH (right). Water molecules are shown in red spheres. Hydrogen bonds are shown in dashed green lines. Coordinates for this figure are from the Brookhaven Protein Data Bank; PDB entries for chicken SO, A. thaliana SO and S. novella SDH are 1SOX, 1OGP, and 2BLF, respectively. The structures of chicken SO R138Q [24] and S. novella SDH Y236F [30] mutants have been recently reported.
Very recently, structures of resting and sulfate-bound recombinant chicken SO proteins (wild type and R138Q) were reported [24]. The assignment of residue 6 in the heme domain to glutamine or glutamic acid in the first crystal structure of native chicken SO was incorrect, and has been re-assigned to arginine in the recently reported structure of recombinant chicken SO, based upon sequencing analysis of native chicken SO [24]. The authors could not grow crystals of the recombinant chicken SO encoding the incorrect residue. No heme domain was observed in the crystal structures of the recombinant chicken SO proteins [24], and the native chicken SO is thus still the only animal SO whose full length structure has been solved (see below) [27].
The X-ray structure of native chicken SO has established the relationship of the heme and Mo domains [27]. Each subunit of the homodimeric chicken SO contains a small N-terminal b5-type cytochrome domain, a large central Mo-binding domain, and a large C-terminal interface domain. In each subunit the Mo domain and the b5-type heme domain are linked by a flexible peptide loop of 10 amino acids. It is noteworthy that the dispositions of the heme domains within the dimeric protein of the unit cell are not in an equivalent position relative to their respective molybdenum domains. This observed variation in heme orientation has previously been interpreted as evidence of domain-domain flexibility [27], and supports the hypothesis that conformational change is involved in the electron transfer between the Mo and heme centers (see below).
In the crystal structure of chicken SO, the Mo atom is coordinated by five ligands with pseudo square pyramidal coordination geometry (Fig. 1). The terminal oxo group occupies the axial position, and the equatorial positions are occupied by three sulfur atoms (one from C185, two from pterin) and one water/hydroxo ligand. The exchangeable equatorial MoV–OH group can be directly detected by continuous wave (CW) and pulsed electron paramagnetic resonance (EPR) spectroscopy [40]. Recent pulsed EPR studies on 17O enriched chicken SO protein [37] and model compounds [57] suggest that the axial oxo ligand is also exchangeable. As expected for binding an anionic substrate, the sulfite-binding site is highly positively charged, and consists of three arginines (R138, R190 and R450), W204 and Y322.
The bacterial SDH is a heterodimer consisting of a large Mo-binding subunit (SorA) and a small heme c-containing subunit (SorB). The heme and Mo centers of the bacterial SDH are in close proximity (Mo···Fe distance: ~ 16 Å), and thus the crystal structure of this enzyme has for the first time allowed direct insights into potential electron transfer pathways between the two redox centers [29]. Intriguingly, the SorB subunit and the heme b domain of the chicken liver enzyme have a similar overall shape, and in chicken SO the latter can be modeled in the position occupied by SorB in the SorAB structure with only minor steric hindrances (Fig. 2).
Fig. 2.
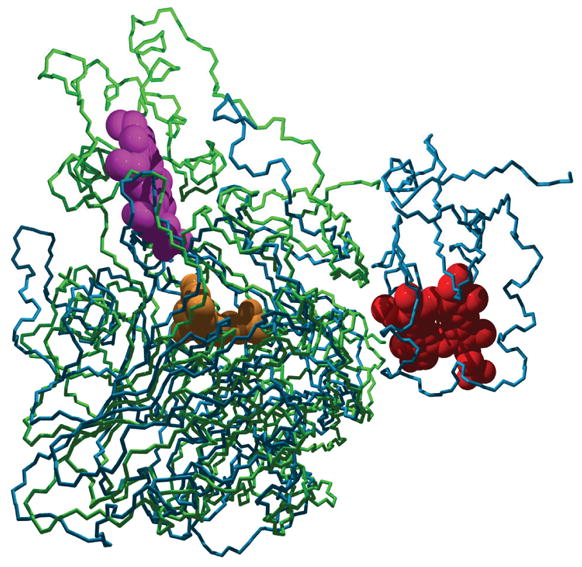
Cα traces of superimposed S. novella SDH (green) and chicken SO (blue) crystal structures with the heme moieties shown in a space filling mode (red and magenta in SO and SDH, respectively), and the Mo cofactor in yellow. The superposition is in an orientation to demonstrate the very different cytochrome interaction sites of the two models. Note that the loop connecting the Mo and heme domains in chicken SO is disordered in the crystal structure, indicating flexibility of this loop as a tether for the heme motion during SO catalysis.
Very recently, the structures of the recombinant chicken SO R138Q mutant [24] and the S. novella SDH Y236F mutant [41] have provided new structural insights into the impact of point mutations on sulfite-oxidizing activities. Significant alterations in the substrate-binding pocket were detected in the structure of the chicken SO R138Q mutant, and a comparison between the wild type and mutant proteins revealed that the active site Arg-450 residue adopts different conformations in the presence and absence of bound sulfate [24]. The size of the binding pocket is thereby considerably reduced, and its position relative to the cofactor is shifted, causing an increase in the distance of the sulfur atom of the bound sulfate to the Mo.
3. Catalytic mechanism of animal SO
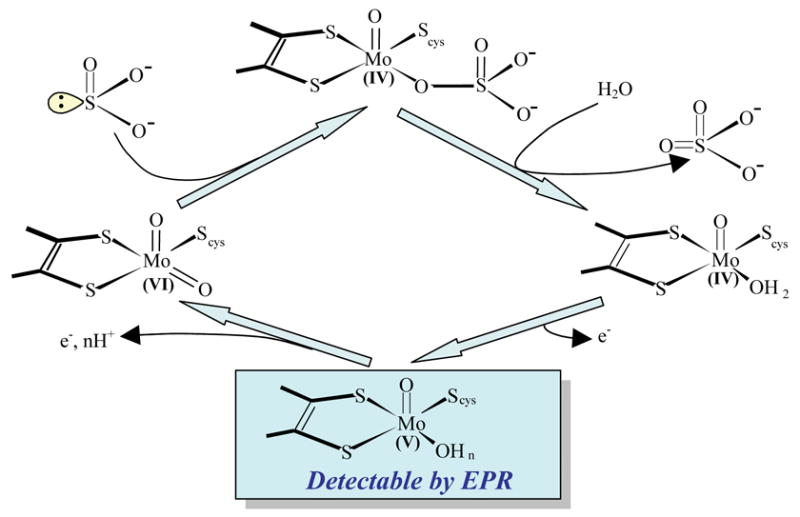
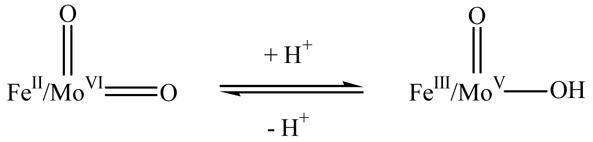
The overall mechanism of animal SO, originally proposed by Hille, has now become generally accepted: sulfite is oxidized to sulfate at the Mo center, and the reducing equivalents are passed on to the b5 heme, where, in turn, the terminal electron carrier (cyt c)ox is reduced (Fig. 3) [58–62]. We refer here to that portion of the catalytic sequence involving the reaction of sulfite with oxidized enzyme to yield the reduced enzyme (and sulfate) as the reductive half reaction, and that involving the reaction of the reduced enzyme thus obtained with cyt c to yield oxidized enzyme (and reduced cyt c) as the oxidative half reaction. The reductive half reaction starts with the reaction of the Mo(VI) center in the fully oxidized SO with sulfite to produce sulfate. The transient two-electron reduced form of Mo(IV)/Fe(III) undergoes IET to generate the Mo(V)/Fe(II) form that is detectable by EPR spectroscopy (Fig. 3) [36, 37, 63–65]. In the oxidative half reaction, a one-electron transfer to exogenous (cyt c)ox, accomplishes re-oxidation of the Fe(II) center, and leaves the enzyme in a one-electron reduced form of Mo(V)/Fe(III). A second Mo→Fe IET step (giving Mo(VI)/Fe(II)), followed by reduction of a second equivalent of (cyt c)ox, regenerates the enzyme to the fully oxidized state of Mo(VI)/Fe(III). Recent attempts to mimic the physiological electron acceptor cyt c by a modified electrode are gaining more interest in order to develop novel sulfite biosensors [66].
Fig. 3.

Proposed oxidation state changes occurring at the Mo and Fe centers of native animal SO during the catalytic oxidation of sulfite and the concomitant reduction of (cyt c)ox. Only the equatorial oxygen atom among the ligands of Mo is shown for clarity. Note that the intermediate FeII/MoV–OH in the reductive half reaction is EPR detectable. The one-electron reduction of FeIII, indicated by a dotted arrow connecting MoVIFeIII and MoVIFeII, can be initiated with a laser pulse in a solution containing dRF and the sacrificial electron donor semicarbazide. The subsequent IET between MoVIFeII and MoVFeIII, which is of particular interest in this review, is highlighted in red. The rate constants of forward and reverse IET (kf and kr, respectively) are defined in the text.
A stopped flow kinetic study of oxidized chicken SO with sulfite [62] indicates that, on a much slower time scale than the initial reduction of SO, an intermolecular dismutation reaction takes place in which two equivalents of two-electron reduced enzyme (i.e. Mo(V)/Fe(II)) are converted to one equivalent each of three- and one-electron reduced enzyme (i.e. Mo(IV)Fe(II) and Mo(VI)/Fe(II), respectively). The one-electron reduced form Mo(VI)/Fe(II) is then able to react with a second equivalent of sulfite, ultimately yielding fully reduced enzyme. This dismutation process is an extremely slow further reduction of enzyme after the fast phase of the reaction (i.e. oxidized SO reduction by sulfite), taking place on a time scale of minutes. In the steady-state, the fate of the one-electron reduced enzyme will be determined by the concentrations of sulfite and cyt c along with their rates of reaction with enzyme. It is an intriguing possibility that the enzyme can cycle between one- and three-electron reduced forms (as suggested in the stopped flow kinetic studies [62]), as well as between oxidized and one-electron reduced enzyme (Fig. 3).
By analogy to mechanisms proposed for the reaction of model Mo(VI)O2 compounds with phosphines [67, 68], Hille proposed a reaction mechanism for animal SO in which catalysis is initiated by attack of the sulfite lone pair on one of the Mo=O units of the Mo center [60], as shown in Fig. 4. A transient Mo(IV)-sulfate complex is explicitly proposed in this earlier work. The indirect role played by the substrate’s oxyanion groups in facilitating reaction has been demonstrated by the fact that methylation of these groups to give dimethylsulfite results in a substantial (300-fold) increase in Kd for the reductive half-reaction at pH 8.0, but has only a negligible effect on kred for the reaction [61]. The pH independence of kred seen in subsequent work is entirely consistent with this chemistry and further indicates that acid-base chemistry is not involved in the breakdown of the oxidized enzyme/sulfite complex [62].
Fig. 4.
Proposed chemical mechanism for the reductive half reaction of SO [60].
The kinetics of reductive half reaction can be determined by stopped flow methods in which oxidized SO is mixed with sodium sulfite by monitoring the reduction of the heme at 426 and 430 nm [31, 62]. The oxidative half reaction kinetics can be determined by stopped flow approaches in which fully reduced SO (i.e. Mo(IV)/Fe(II)) is mixed with cyt c by following the reaction at either 547 (an isosbestic point for chicken SO) or 562 nm (an isosbestic point for cyt c), which permits cyt c reduction and SO reoxidation to be followed independently in the course of the reaction [62]. All experiments need to be performed anaerobically. Rajagopalan et al. recently reported an elegant study on the reductive half reaction by stopped flow in a comparative study on the truncated Mo domain and the full length human SO [31]. This study gives rates for the discrete step of SO reduction by sulfite and the overall reactions (which include the sulfite-reduction step followed by IET between Mo(IV) and Fe(III), see Fig. 3), and thus provides convincing evidence that reduction of the Mo center is quite fast, indicating that this is not the rate-limiting step in the catalytic cycle.
There are two EPR-active intermediates in the catalytic cycle: Mo(V)(Fe(II) and Mo(V)Fe(III) (Fig. 3) [63]. The first form can be readily generated by reduction of animal SO and S. novella SDH with excess sulfite [36, 39]. The latter form needs a more complicated procedure in which titanium(III) citrate is used to reduce animal SO to Mo(V)Fe(III) [69]. This form is well suited for Mo···Fe distance measurement by pulsed electron-electron double resonance (ELDOR) spectroscopy [69]. For plant SO, in order to generate the Mo(V) form, one-half equivalent of ferricyanide needs to be added after sulfite-reduction in order to reoxidize the Mo(IV) form to Mo(V) because this protein lacks a heme domain [38, 45]. We have proposed a sulfate-bound model for the structure of this form of plant SO based upon comprehensive pulsed EPR studies [38]. Very recently this proposal has been verified by pulsed EPR of the Mo(V) form generated from 33S-labeled sulfite [70]. It is interesting that reduction of plant SO with titanium(III) citrate yields a distinct form [38].
The CW EPR signal of Mo(V) depends on pH [71, 72], anion species [73], reductants [38], and protein mutations [35]. Pulsed EPR spectroscopies have been used to elucidate the structure of the Mo(V) forms, in order to provide unique insights into the catalytic mechanism at the Mo active site [36–38, 64, 74, 75]. Comparative EPR studies on deuterated [63] and 17O-enriched samples [76] can detect nearby protons and oxygen atoms in the intermediates, which are involved in SO catalysis. Pulsed EPR studies on the SO intermediates have been discussed in a recent review [40].
4. IET in sulfite oxidizing enzymes
As a multi-redox-center enzyme, IET between the Mo and Fe centers in animal SO and S. novella SDH is fundamental to the function of the enzymes. We have previously shown that exogenous deazariboflavin radicals generated in situ with a laser pulse will rapidly reduce the Fe(III) center of animal SO or S. novella SDH by one electron (see dotted arrow in Fig. 3), followed by intramolecular equilibration between the redox centers. Note that this latter process corresponds to the second physiologically essential IET step highlighted in red in Fig. 3, and the laser flash photolysis technique follows the IET process in the reverse direction of the enzymatic turnover. Note that this method is a perturbation one, with the observed rate constant reflecting the sum of the forward and reverse rate constants for electron transfer regardless of the direction in which the redox equilibrium is shifted experimentally (i.e., whether it is in the physiological direction or the reverse). Since this reaction is reversible, microscopic rate constants for forward and reverse reactions can be obtained from knowing their sum (i.e. the observable rate constant from the flash photolysis experiment) and their ratio (i.e. the equilibrium constant, as calculated from the transient trace, see below). Extensive flash photolysis studies on native chicken SO and wild type recombinant human SO have shown that the first-order IET rate constants between the reduced Fe(II) and oxidized Mo(VI) centers depend on the following experimental conditions: solution viscosity [34, 77], sulfate concentrations [78–80], and pH [34, 78, 79]; this information is important for understanding the IET mechanism in the enzymatic catalysis (see below).
In this section, we will first briefly review the laser flash photolysis technique that has allowed the attainment of new levels of insight into the IET between the Mo and Fe centers in sulfite oxidizing enzymes. We will then review studies on the role of conformational change in the IET. Finally, the effects of point mutations in human SO on the IET process will be discussed.
4.1. Determination of the IET kinetics by laser flash photolysis
Laser flash photolysis of animal SO or S. novella SDH in the presence of 5-deazariboflavin (dRF) and a sacrificial donor (EDTA or freshly made semicarbazide) results in the rapid one-electron reduction of the Fe(III) heme center, followed by IET between the Mo and Fe centers. Fig. 5 shows a typical transient kinetic trace obtained at 555 nm upon laser flash photoexcitation of a solution containing oxidized wild type human SO, dRF and semicarbazide (AH2). The kinetic behavior can be fully described in terms of Eqs. 1–4 shown below.
Fig. 5.
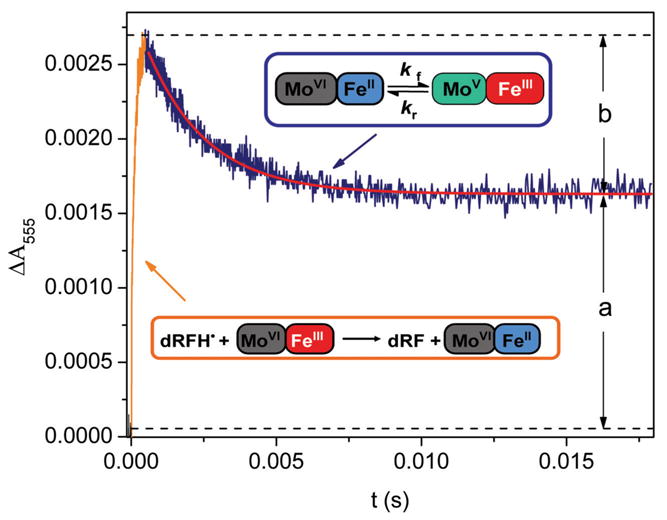
Transient obtained at 555 nm upon photoexcitation of a solution containing wild type human SO, dRF, and 0.5 mM fresh semicarbazide hydrochloride (pH 7.4). The portion of the figure outlined by the orange box points to heme reduction by dRFH•; this process is pseudo first order, and its rate depends on protein concentration. The dark blue box points to heme reoxidation due to the subsequent IET between the Mo and Fe centers; this process is independent of protein concentration, consistent with its intraprotein nature. The red solid line indicates a single-exponential fit to the IET phase. Keq = b/a.
| Eq. 1 |
| Eq. 2 |
| Eq. 3 |
| Eq. 4 |
The laser pulse produces 3dRF by absorption and intersystem crossing (Eq. 1), which abstracts a hydrogen atom from semicarbazide or EDTA to form the highly reducing dRFH• (Eq. 2). The initial positive deflection of absorbance from zero in Fig. 5 is due to net reduction of the Fe(III) center to the Fe(II) form (Eq. 3), which has an absorbance maximum at 555 nm. The subsequent slow absorption decay is due to the net IET from Fe(II) to Mo(VI), i.e., the intramolecular re-oxidation of Fe(II) (Eq. 4). The transient does not fall back to the baseline, which clearly indicates the existence of an equilibrium between the Mo(VI)/Fe(II) and Mo(V)/Fe(III) forms of SO (Eq. 4). The kinetics of this process are independent of SO concentration, indicating that the heme re-oxidation is an intramolecular process. The observed rate constant is thus assigned to the sum of the rate constants for the forward and reverse processes.
For the case shown in Fig. 5, in which the flash-induced reduction of SO (Eq. 3) occurs much faster than the subsequent IET (Eq. 4), accurate values for the overall IET rate constant ket (= kf + kr) and parameters a and b (see Fig. 5) can be obtained by fitting the heme re-oxidation phase with the exponential function given in Eq. 5.
| Eq. 5 |
Based on Eq. 4, the parameters a and b in Eq. 5 have the meanings given in Eqs. 6 and 7 (where A0 is the absorbance extrapolated to t = 0, assuming that the photochemically-induced reduction of SO is instantaneous).
| Eq. 6 |
| Eq. 7 |
| Eq. 8 |
Thus the individual IET rate constants kf and kr can be calculated from ket and the value of Keq (= b/a, Eq. 8, Fig. 5). The IET rate constants and Keq values for the wild type human SO and the native chicken SO under the same conditions are shown in Table 2.
Table 2.
Flash photolysis kinetic parameters for human and chicken SO a
| ket (s−1) | Keq | kf (s−1) | kr (s−1) | |
|---|---|---|---|---|
| Wild-type human SO b | 491 ± 11 | 0.73 ± 0.08 | 207 ± 5 | 284 ± 6 |
| Chicken SO | 1318 ± 28 | 1.63 ± 0.04 | 817 ± 18 | 501 ± 10 |
Solutions for flash photolysis experiments contained 0.5 mM semicarbazide, 10 mM Tris; pH was adjusted to 7.4 with HCl. Data taken from ref. [34].
Some additional features of the methodology are also worthy of note.
The intensity of the pulsed laser that is used to initiate the photochemical reduction is purposely kept low, so that substoichiometric quantities of dRFH• are generated relative to the protein. This assures that pseudo-first-order conditions apply (Eq. 3), and that only a single electron can be added to each protein molecule.
The irradiated volume in the sample cuvette is kept small (< 1% of the total volume) so that only minor net conversion to product occurs. This allows samples to be subjected to multiple flashes for signal averaging, and for determination of protein concentration dependence by adding aliquots of concentrated protein to the sample.
One important control experiment is that traces with different signal amplitudes (i.e. different reduced protein concentrations) are always collected. If the traces give similar rates, the electron transfer process is thus of an intra-protein nature. This is important because other inter-protein processes may confound the absorption changes, and if so, the observed rate would depend upon protein concentration. Therefore, the kinetics must be determined at various protein concentrations to be sure that the intra-protein electron transfer process between the Mo and Fe centers is being measured.
4.2. Role of conformational change in the IET in animal SO
The distance between the heme and Mo centers in the chicken SO structure is large (~ 32 Å), and the redox potential difference between the cofactors is small (~ 10 mV at pH 6 [81]). Despite this, IET between the two centers can be quite rapid; using laser flash photolysis methods (see above), ket values as large as 2400 s−1 have been observed [78]. Arguments based on simple Marcus theory predict that ket should be less than 100 s−1 for this process (given the observed large distance and small driving forces) [82, 83]. This discrepancy has led to the suggestion that chicken SO may adopt a different conformation in solution from that observed in the crystal structure, that brings the heme and Mo centers much closer [78]. Such a rearrangement to a more “productive” orientation must occur before electron transfer, which suggests that fast electron transfer between Mo (VI) and Fe (II) centers requires subtle and precise positioning, involving reorientation and docking of the two redox partners with respect to each other. It is possible that this is the rate-limiting step in the IET process, which would correspond to a conformationally gated electron transfer.
Fig. 6 shows a schematic drawing of the relative positions of the Mo and Fe domains in the crystal structure, and depicts how the backbone rearrangement of the protein might bring the two redox-active centers sufficiently close together to permit rapid electron transfer between them. These two centers are linked by a flexible polypeptide loop, suggesting that conformational changes, which alter the Mo···Fe distance, may play an important role in the observed IET rates. Indeed, in the crystal structure, the two heme domains do not occupy identical positions in the dimer, suggesting that they may be quite mobile. SO is only one of many biological electron transfer systems for which specific protein conformations are required for optimal function [84, 85].
Fig. 6.
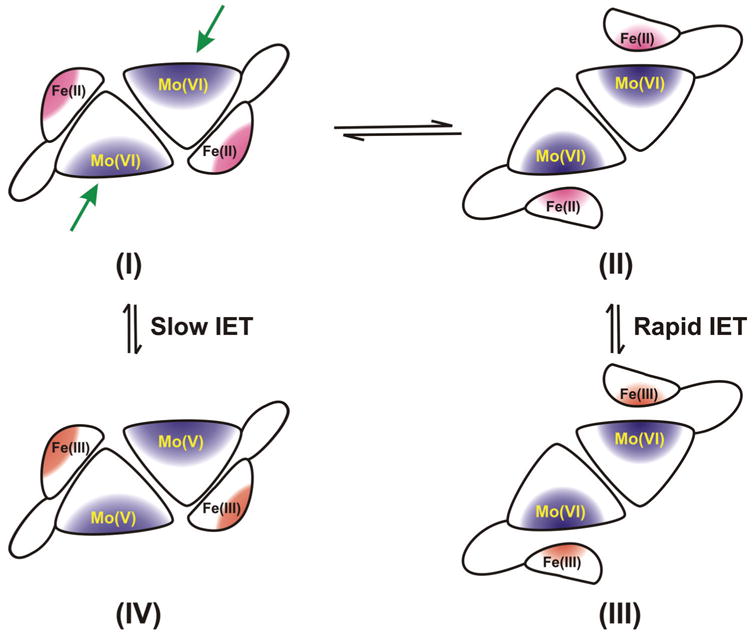
Proposed docking of the heme domain to the Mo domain in vertebrate SO that could move the Mo and Fe centers into closer proximity (structure II) than that observed in the crystal structure of dimeric SO (structure I). Subsequently, the transient internal complex (structure II) may facilitate rapid IET to generate structure III. A possible docking site near the Mo domain in the dimeric form is marked by a green arrow.
4.2.1. Viscosity dependence of the IET in animal SO
The effect of solution viscosity on the IET in chicken SO has been investigated using laser flash photolysis [77], in order to test the possibility that a large protein conformational change is involved in the reaction. The solution viscosity was varied over the range 1.0–2.0 cP by addition of either polyethylene glycol-400 (PEG-400) or sucrose. In the presence of either viscosogen, an appreciable decrease in the IET rate constant value was observed upon an increase in the solvent viscosity (1232 s−1 and 886 s−1 in the presence of 0 % and 10 % (v/v) PEG-400, respectively). All normalized values, determined in buffered mixtures of water with the two different solutes, PEG 400 or sucrose, fall on the same straight line when plotted versus the viscosity. The IET rate constant was shown to exhibit a linear dependence on the negative 0.7th power of the viscosity. Steady state kinetics data indicate that PEG-400 has at best a minor effect on the activity (i.e., kcat) of SO in the range of viscosity studied. In addition, the CW-EPR spectrum of sulfite-reduced SO in 20 % sucrose is the same as that without sucrose, which indicates that the active site structure of the molybdenum domain remains unchanged upon addition of sucrose. Based on these results, we conclude that it is solution viscosity, and not other properties of the added viscosogens and/or their effect on SO structure, that is responsible for the dependence of IET rates on the solvent composition. The results are consistent with the role of conformational changes on IET in animal SO (Fig. 6).
Very recently, the smaller turnover numbers obtained in protein film voltammetric studies (kcat 2–4 s−1, as compared to 70–100 s−1 in solution) suggest that only a small fraction of chicken SO is bound at the electrode in a manner that permits the conformational change necessary for fast interdomain electron transfer, again indicating that the motion of the heme domain is a controlling factor for chicken SO activity [46]. The conformational flexibility of chicken SO has been further supported by a thorough analysis of the ELDOR decay curves and simulations suggesting a distribution of intramolecular Mo…Fe distances [69]. Taken together, these results indicate that efficient IET between the Mo(VI) and Fe (II) centers requires precise orientation of the two redox partners with respect to each other, and that the consequent IET-productive conformations of animal SO are vital to facilitate rapid IET between the two metal centers. Interestingly, a comparison of the crystal structures of chicken SO and Arabidopsis thaliana SO reveals possible surface regions in chicken SO for docking of its heme domain to the Mo domain in the course of IET and catalysis [28]. This is further strengthened by the conservation of five out of seven animal SO-specific surface residues in NRs, which belong to the sulfite oxidase family. Like animal SOs, NRs also contain a cyt b5 domain that is essential for internal electron transfer, which is predicted to interact in a similar fashion with the Mo domain [28].
4.2.2. Viscosity independence of the IET in bacterial SDH
Heterodimeric bacterial SDH contains heme and Mo centers in separate tightly associated subunits, and thus this enzyme represents a distinctly different type of sulfite-oxidizing enzyme [26], in which a fairly rigid positioning of the redox centers relative to one another is essential to maintain the enzyme’s integrity. In contrast to animal SO, the IET rate constants in SDH (120 s−1 at pH 6.0) are not affected by either solution viscosity or the presence of the sulfate anion [86], indicating that IET in SDH proceeds directly within the protein medium and does not involve substantial motion of the two redox centers relative to each other, consistent with the presence of a tightly bound cytochrome subunit [29]. It has been proposed that sulfate binding (as well as that of other anions) near the Mo center in chicken SO decreases the positive charge on the surface of the Mo domain, thereby retarding the docking of the heme domain to the Mo domain, thus inhibiting IET [78]. The IET in SDH from S. novella does not involve significant protein motion/docking, and therefore sulfate cannot inhibit IET in this protein by masking surface charges necessary for the repositioning of the protein domains [86]. Sulfate binding does, however, inhibit the enzyme activity which shows a mixed-type non-competitive pattern with respect to sulfite. Kic and Kiu values were determined to be 24 and 16 mM for sulfite [26].
These results provide direct evidence that the effects of sulfate anion and solution viscosity on IET in animal SO are due to interference with inter-domain docking, and clearly demonstrate the differences between animal SO and bacterial SDH in terms of conformational gating effects. It is reasonable to surmise that SDH has a favorable Mo···Fe distance and/or intervening amino acid arrangement that allows IET to proceed directly through the protein medium without involving conformational changes. Indeed, in the crystal structure of SDH, the Mo···Fe distance is about 16 Å, and IET pathways through either aromatic residues or hydrogen bonds were proposed (Fig. 7) [29].
Fig. 7.
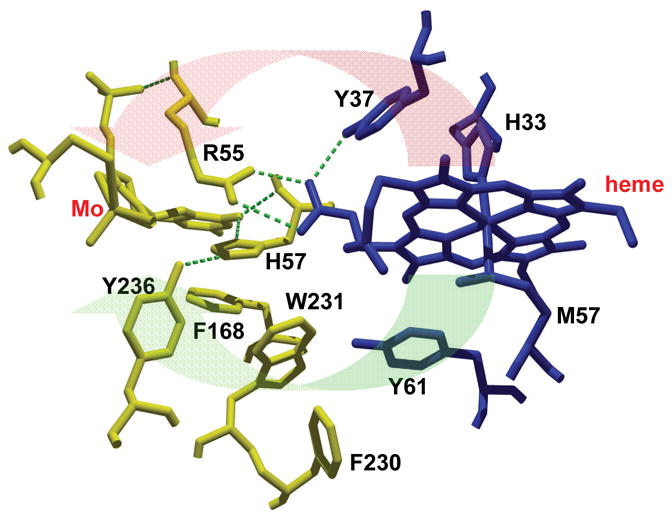
Potential pathways for electron transfer from the Fe to Mo centers in S. novella SDH. The red block arrow shows pathways through hydrogen bonds, whereas the green arrow shows pathways through aromatic residues. SorA residues are yellow, SorB residues are blue.
4.3. IET kinetics in mutants of human SO
4.3.1. Studies of point mutations identified in sulfite oxidase deficiency patients
As the IET between the Mo and Fe centers is fundamental to SO catalysis, it is reasonable to propose that fatal point mutations of human SO, identified in SO deficiency patients, result in impaired SO activity through retarding the IET process. It is also important to note that not all fatal point mutations in human SO are due to impaired internal electron transfer; thus a large subset of these mutations map to the so-called interface domain of SO (for example G473D), and presumably the effect is primarily on protein stability, rather than electron transfer.
Human SO has been successfully cloned and expressed in highly active forms in E. coli [16, 21–23], and thus this hypothesis can be tested. Due to the lack of an X-ray structure for human SO, the crystal structure of the highly homologous chicken SO provides a starting point for a molecular understanding of kinetic and spectroscopic studies of the effects of point mutations on recombinant human SO. As will be described below, flash photolysis studies on IET in human SO mutants R160Q, G473D and A208D have provided direct evidence of the vital roles of specific amino acids in efficient IET and SO catalysis.
Arginine 160 in human SO is conserved in all SO species sequenced to date. Previous steady-state kinetic studies of the R160Q human SO mutant showed a remarkable decrease in kcat/Kmsulfite of nearly 1000-fold [16]. In the crystal structure of chicken SO, Arg 138, the equivalent of Arg 160 in human SO, is involved in the formation of a positively charged sulfite binding site [27]. In order to further assess the role of Arg 160 in human SO, IET rates between the Fe and Mo centers in the wild type, R160Q and R160K human SO forms were investigated by laser flash photolysis [33]. In the R160Q mutant, the IET rate constant at pH 6.0 (0.6 s−1) was decreased by nearly three orders of magnitude relative to wild type (411 s−1), which indicates that the positive charge of Arg 160 is essential for efficient IET in human SO. Furthermore, the IET rate constant for the R160K mutant is about one-fourth that of the wild type enzyme, which strongly indicates that it is the loss of charge of Arg 160, and not its precise location, that is responsible for the much larger decrease in IET rates in the R160Q mutant. At pH 6.0, the kcat value of R160Q is 0.33 s−1, comparable to its ket value (0.64 s−1), suggesting that in this mutant IET may become the rate-limiting step during catalytic turnover. Thus, the large decrease in the IET rate constant rationalizes the fatal impact of this mutation in patients with this genetic disorder. The presence of a positive charge at Arg 160 of human SO may allow the most optimal orientations for IET between the heme and Mo domains through electrostatic interactions between these regions of the protein (i.e., a kinetic rather than a thermodynamic effect). The R160Q mutation is likely to impair the IET through decreasing the likehood of forming the IET-competent conformation rather than decreasing the intrinsic IET rate constant within this conformation.
Mutations G473D and A208D were identified in patients with isolated SO deficiency, and the equivalent amino acids (G451 and A186, respectively) have been localized to the vicinity of the molybdopterin active site in the X-ray structure of chicken SO [27]. Steady-state kinetic studies of enzyme turnover and laser flash photolysis measurements of the IET rate constants between the Fe and Mo centers were carried out in the recombinant G473D, G473A, G473W, G473D/R212A, and A208D human SO mutants [35]. In the G473D and A208D mutants, the IET rate constants at pH 6.0 (0.17 and 0.10 s−1, respectively) were decreased by three orders of magnitude relative to wild type. The ket values of G473D and A208D human SO at pH 6.0 are close to their kcat values at this pH (0.14 s−1 and 0.15 s−1, respectively). Thus, IET in these mutants becomes the rate-limiting step during catalytic turnover, similar to the results obtained with R160Q human SO [33]. Therefore, the large decreases in the IET rate constants and the kcat values again rationalize a kinetic basis for the fatal impact of these mutations. Additionally, far-UV CD spectra of G473D indicate that the protein backbone conformation is remarkably changed, and sedimentation equilibrium indicate that the protein is monomeric [32]. Furthermore, EPR studies also suggest that the active site structure of the Mo(V) form of A208D is different from that of the wild type: specifically the EPR spectrum of A208D at pH 9.5 has the same g values as the low pH form of wild type and shows splitting due to the strong coupling of the nearby exchangeable proton [35]. In contrast, similar studies on G473A show that it is dimeric, that its Mo(V) active site structure is similar to the wild type, and that its IET rate constant (188 s−1) is only 2.6-fold smaller than that of wild type. IET in G473W is severely impaired, and no IET at all is observed for G473D/R212A. In chicken SO the equivalent residues (G451 and A186) are both buried inside the protein. Thus, for human SO, the mutations to charged residues at the equivalent sites most likely cause crucial global or localized structural changes, thereby retarding IET and efficient catalytic turnover of the sulfite oxidation reaction. Recent steady state kinetics and CD spectroscopy results demonstrate that the interaction between the heme and Mo domains is important for the cyt c interaction [32], although the mutations are not in the heme domain, which is consistent with the laser flash photolysis work. As EPR data suggest that the Mo center is perturbed in the A208D and G473D mutants, a secondary reason for the impaired IET might be due to changes in the electronic structure of the Mo center. This factor might be important in the A208D mutant as its global conformation is not changed as much compared to the G473D mutant.
4.3.2. Y343F human SO mutant
Combining pH dependence of IET [78] and stopped flow [62] kinetics information with the crystal structure [27] and the pulsed EPR results [75] for the Mo(V) form of the two-electron reduced state of SO has led to a self-consistent proposal for a coupled electron proton transfer (CEPT) reaction at the Mo center, as follows:
In the crystal structure of chicken SO, Tyr 322 (the equivalent site of human SO Tyr 343) is within H-bonding distance of the equatorial Mo–O group and is also accessible to water molecules (Fig. 1), and was proposed to serve as a proton shuttle between Mo–O and OH− or H2O [78]. The mutation of this conserved tyrosine residue provides direct evidence for its important role in the IET [34]. Steady-state kinetic analysis of the Y343F mutant showed an increase in Km sulfite and a decrease in kcat resulting in a 23-fold attenuation in the specificity constant kcat/Km sulfite at the optimum pH of 8.25. This indicates that Tyr 343 is involved in the binding of the substrate and catalysis within the Mo active site. Furthermore, the IET rate constant in the mutant at pH 6.0 (46 s−1 at pH 6.0) is only about one-tenth that of the wild-type enzyme, suggesting that the OH group of Tyr 343 is important for efficient IET in SO. Note that only a small shift in Keq (i.e., a thermodynamic factor) was observed for the Y343F mutant at pH 6, which cannot account for such a large change in the IET rate constants (~10-fold). The most plausible explanation is that the hydrophobic phenylalanine side chain in the Y343F mutant may hinder direct access of water or H+ to the equatorial Mo=O group, thus retarding efficient CEPT (i. e., a kinetic factor). This is clearly shown by the shift in the dependence of Keq for IET to lower pH values in the Y343F mutant. The loss of the putative hydrogen bond to the apical Mo=O is likely to be the basis for the effect, particularly in view of the long-established association of proton uptake by ligands to molybdenum upon reduction of the metal, as originally discussed by Stiefel [87] and developed further by us in the context of CEPT within SO [75, 78]. Very recent stopped-flow analyses of the reductive half reaction of Y343F and wild type human SO along with the proteins lacking the N-terminal heme domain [31] demonstrated that Y343F SO is impaired in overall catalytic activity across the range of pH values measured, with lower activity at neutral and low pH. In addition, the Kmsulfite of Y343F was significantly higher than that of the wild-type protein, particularly at high pH. However, at high pH, sulfite binding and molybdenum center reduction probably contribute to the low rate observed in Y343F SO. Taken together, their results suggest that the Tyr343 residue has dual roles in both attraction and binding of the substrate (especially at high pH) and in product release and subsequent IET (especially at low pH).
5. Future perspectives
The aim of the present review is to highlight current mechanistic information about sulfite oxidizing enzymes. It has become clear from these studies that our understanding of sulfite oxidizing enzymes will be moved forward by increasing applications of a combined approach of molecular biology, rapid kinetics (laser flash photolysis and stopped flow), advanced spectroscopy, protein crystallography, and computational modeling. This is clearly a fertile area for future study. Although considerable progress has been obtained, some outstanding questions remain unanswered, and certainly deserve further investigation at the molecular level. These can be summarized as follows.
The results of the IET kinetic studies of human SO mutants suggest the importance of the conserved surface near the Mo site for efficient IET mediated by appropriate docking of the heme domain, and efforts to carry out extensive mutagenesis studies on conserved surface residues in SO enzymes, especially of the strictly conserved charged residues near the Mo center in human SO, should prove fruitful. Further flash photolysis studies of such mutants should provide additional insights into the role of surface charge in the docking of the heme domain and IET in SO. A potential additional strategy would be to label the domain(s) with fluorescence probe(s), and then examine the interdomain interaction with the fluorescence resonance energy transfer technique.
The bacterial SDH is a promising model for studying electron transfer in sulfite oxidizing enzymes without the complicating factor of domain movement (Figs. 2 and 7). Now that the structure of both chicken SO and S. novella SDH are available, it is critical to further identify amino acids involved in the direct IET through the protein medium in SDH. It will be also interesting to determine important residues for efficient IET in the productive conformation of animal SO (i.e. after the heme is docked to the Mo domain), to determine if there are significant differences between the two types of enzymes.
Given the importance of heme domain motion in animal SO catalysis, the flexible loop between the Mo and heme domains may act as a tether to give the heme domain both an appropriate freedom (to move closer to the Mo domain) and yet a necessary restriction to dock precisely. Indeed, isolated Mo and heme domains generated by tryptic cleavage of rat liver SO do not possess SO activity [88]. It will thus be interesting to examine the role of the loop in the IET and SO function by either shortening its length or by altering its flexibility.
Several new point mutations in human SO have been identified by genetic analysis of SO deficiency patients [89, 90]; all of them locate near the Mo active site. It is clearly important to clone and express these novel mutants and to carry out integrated structural and functional studies in order to understand the underlying mechanism of the fatal effects. It will be interesting to distinguish between the effects of modified protein structure and retarded IET on the enzyme activity. It is important to note that EPR spectra of human SO G473D and A208D mutants are distinct from those of wild type, indicating notable changes in the Mo environment upon the mutations [35]. This demonstrates the importance of quantifying the structural changes using advanced spectroscopy techniques including pulsed EPR and X-ray absorption.
Recent pulsed EPR studies in 17O-enriched water provided evidence for exchange of the axial oxo ligand in the high pH form of chicken SO [37]. Given that the reaction SO catalyzes is an oxygen atom transfer reaction, it is important to further investigate the mechanism of oxygen ligand exchange under conditions relevant to SO function (e.g. pH, anion species and conditions). In such studies, design of procedures to enrich the protein or model compounds with isotopes and development of appropriate pulsed EPR methodologies (in particular at high magnetic field) are critical.
Besides low pH, high pH and phosphate-bound forms of SO, a new EPR form at low pH (sulfate-bound form) was proposed for sulfite-reduced plant SO [38]. This form does not have a nearby exchangeable proton, and the presence of coordinated sulfate has been definitively confirmed very recently by pulsed EPR studies of A. thaliana SO generated by reduction with 33S-labeled sulfite [70]. These unusual EPR properties of A. thaliana SO are proposed to be directly related to the “open” and “closed” forms of the active site modulated by conformations of the active site arginine (Arg 374), in combination with a more restricted substrate and water access to the active site of the plant enzyme than that found in vertebrate SOs [34]. It will thus be interesting to conduct pulsed EPR studies on the plant SO mutants of R374.
Computational modeling and simulation are certainly required to provide potential explanations and rationales for the role of specific amino acids in the IET and SO catalysis process. A recent interesting simulation study suggests that the tether length may control the transition between the electron tunneling and diffusion-limited regimes in the IET reaction [91].
Sulfite toxicity has been implicated in an increasing number of pathological conditions [92]. It has been shown that a combined exposure of tungsten and arsenic causes leukemia, which may be related to impaired SO activity and overproduction of reactive oxygen species [93]. At present, we know very little about the mechanism underlying sulfite toxicity, and thus it is important to expand studies of sulfite oxidizing enzymes to elucidate the pathological mechanism in vivo.
Acknowledgments
We thank many colleagues who have worked with us over the years on sulfite oxidizing enzymes. In particular we are grateful to Prof. K.V. Rajagopalan and Dr. Heather L. Wilson at Duke University for providing SO mutants and Prof. Ulrike Kappler at Queensland for providing SDH, as well as for critical discussions during our collaborations. We thank the reviewers for the helpful comments. The research was supported by grant GM 37773 to JHE.
Abbreviations
- Mo
molybdenum
- SO
sulfite oxidase
- SDH
sulfite dehydrogenases
- NR
nitrate reductase
- cyt
cytochrome
- (cyt c)ox and (cyt c)red
ferricytochrome c and ferrocytochrome c, respectively
- IET
intramolecular electron transfer
- ket
rate constant for IET
- EPR
electron paramagnetic resonance
- CW
continuous wave
- CD
circular dichroism
- dRF and dRFH•
5-deazariboflavin and 5-deazariboflavin semiquinone, respectively
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hille R. The mononuclear molybdenum enzymes. Chem Rev. 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]
- 2.Aguey-Zinsou KF, Bernhardt PV, Kappler U, McEwan AG. Direct electrochemistry of a bacterial sulfite dehydrogenase. J Am Chem Soc. 2003;125:530–535. doi: 10.1021/ja028293e. [DOI] [PubMed] [Google Scholar]
- 3.Eilers T, Schwarz G, Brinkmann H, Witt C, Richter T, Nieder J, Koch B, Hille R, Hansch R, Mendel RR. Identification and biochemical characterization of Arabidopsis thaliana sulfite oxidase - A new player in plant sulfur metabolism. J Biol Chem. 2001;276:46989–46994. doi: 10.1074/jbc.M108078200. [DOI] [PubMed] [Google Scholar]
- 4.Hansch R, Lang C, Riebeseel E, Lindigkeit R, Gessler A, Rennenberg H, Mendel RR. Plant sulfite oxidase as novel producer of H2O2 - Combination of enzyme catalysis with a subsequent non-enzymatic reaction step. J Biol Chem. 2006;281:6884–6888. doi: 10.1074/jbc.M513054200. [DOI] [PubMed] [Google Scholar]
- 5.Kisker C. Sulfite oxidase. In: Messerschmidt A, Huber R, Poulos T, Wieghardt K, editors. Handbook of Metalloproteins. John Wiley and Sons, Ltd; New York: 2001. pp. 1121–1135. [Google Scholar]
- 6.Schindelin H, Kisker C, Rajagopalan KV. Advances in Protein Chemistry. Vol. 58. Academic Press Inc; San Diego: 2001. pp. 47–94. [DOI] [PubMed] [Google Scholar]
- 7.Rajagopalan KV, Johnson JL. Sulfite oxidase. In: Creighton TE, editor. Wiley Encyclopedia of Molecular Medicine. Wiley; New York: 2002. pp. 3048–3051. [Google Scholar]
- 8.Johnson JL. Prenatal diagnosis of molybdenum cofactor deficiency and isolated sulfite oxidase deficiency. Prenat Diagn. 2003;23:6–8. doi: 10.1002/pd.505. [DOI] [PubMed] [Google Scholar]
- 9.Dublin AB, Hald JK, Wootton-Gorges SL. Isolated sulfite oxidase deficiency: MR imaging features. Am J Neuroradiol. 2002;23:484–485. [PMC free article] [PubMed] [Google Scholar]
- 10.Matthies A, Rajagopalan KV, Mendel RR, Leimkuhler S. Evidence for the physiological role of a rhodanese-like protein for the biosynthesis of the molybdenum cofactor in humans. Proc Natl Acad Sci U S A. 2004;101:5946–5951. doi: 10.1073/pnas.0308191101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwarz G, Santamaria-Araujo JA, Wolf S, Lee HJ, Adham IM, Grone HJ, Schwegler H, Sass JO, Otte T, Hanzelmann P, Mendel RR, Engel W, Reiss J. Rescue of lethal molybdenum cofactor deficiency by a biosynthetic precursor from Escherichia coli. Hum Mol Genet. 2004;13:1249–1255. doi: 10.1093/hmg/ddh136. [DOI] [PubMed] [Google Scholar]
- 12.Johnson JL, Duran M. Molybdenum cofactor deficiency and isolated sulfite oxidase deficiency. In: Scriver C, Beaudet A, Sly W, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill; New York: 2001. pp. 3163–3177. [Google Scholar]
- 13.Tan WH, Eichler FS, Hoda S, Lee MS, Baris H, Hanley CA, Grant E, Krishnamoorthy KS, Shih VE. Isolated sulfite oxidase deficiency: A case report with a novel mutation and review of the literature. Pediatrics. 2005;116:757–766. doi: 10.1542/peds.2004-1897. [DOI] [PubMed] [Google Scholar]
- 14.Lee HF, Mak BSC, Chi CS, Tsai CR, Chen CH, Shu SG. A novel mutation in neonatal isolated sulphite oxidase deficiency. Neuropediatrics. 2002;33:174–179. doi: 10.1055/s-2002-34491. [DOI] [PubMed] [Google Scholar]
- 15.Johnson JL, Rajagopalan KV, Renier WO, Van der Burgt I, Ruitenbeek W. Isolated sulfite oxidase deficiency: mutation analysis and DNA-based prenatal diagnosis. Prenat Diagn. 2002;22:433–436. doi: 10.1002/pd.335. [DOI] [PubMed] [Google Scholar]
- 16.Garrett RM, Johnson JL, Graf TN, Feigenbaum A, Rajagopalan KV. Human sulfite oxidase R160Q: Identification of the mutation in a sulfite oxidase-deficient patient and expression and characterization of the mutant enzyme. Proc Natl Acad Sci U S A. 1998;95:6394–6398. doi: 10.1073/pnas.95.11.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rudolph MJ, Johnson JL, Rajagopalan KV, Kisker C. The 1.2 angstrom structure of the human sulfite oxidase cytochrome b(5) domain. Acta Crystallogr Sect D-Biol Crystallogr. 2003;59:1183–1191. doi: 10.1107/s0907444903009934. [DOI] [PubMed] [Google Scholar]
- 18.Bailey JL, Cole RD. Studies on the Reaction of Sulfite with Proteins. J Biol Chem. 1959;234:1733–1739. [PubMed] [Google Scholar]
- 19.KD Menzel DB, Leung KH. Covalent reactions in the toxicity of SO2 and sulfite. Adv Exp Med Biol. 1986;197:477–492. doi: 10.1007/978-1-4684-5134-4_46. [DOI] [PubMed] [Google Scholar]
- 20.Kucukatay V, Hacioglu G, Savcioglu F, Yargicoglu P, Agar A. Visual evoked potentials in normal and sulfite oxidase deficient rats exposed to ingested sulfite. NeuroToxicology. 2006;27:93–100. doi: 10.1016/j.neuro.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 21.Garrett RM, Bellissimo DB, Rajagopalan KV. Molecular cloning of human liver sulfite oxidase. Biochim Biophys Acta-Gene Struct Expression. 1995;1262:147–149. doi: 10.1016/0167-4781(95)00068-r. [DOI] [PubMed] [Google Scholar]
- 22.Temple CA, Graf TN, Rajagopalan KV. Optimization of expression of human sulfite oxidase and its molybdenum domain. Arch Biochem Biophys. 2000;383:281–287. doi: 10.1006/abbi.2000.2089. [DOI] [PubMed] [Google Scholar]
- 23.Leimkuhler S, Rajagopalan KV. In vitro incorporation of nascent molybdenum cofactor into human sulfite oxidase. J Biol Chem. 2001;276:1837–1844. doi: 10.1074/jbc.M007304200. [DOI] [PubMed] [Google Scholar]
- 24.Karakas E, Wilson HL, Graf TN, Xiang S, Jaramillo-Buswuets S, Rajagopalan KV, Kisker C. Structural insights into sulfite oxidase deficiency. J Biol Chem. 2005;280:33506–33515. doi: 10.1074/jbc.M505035200. [DOI] [PubMed] [Google Scholar]
- 25.Garrett RM, Rajagopalan KV. Molecular cloning of rat liver sulfite oxidase - Expression of a eukaryotic Mo-pterin-containing enzyme in Escherichia coli. J Biol Chem. 1994;269:272–276. [PubMed] [Google Scholar]
- 26.Kappler U, Bennett B, Rethmeier J, Schwarz G, Deutzmann R, McEwan AG, Dahl C. Sulfite : Cytochrome c oxidoreductase from Thiobacillus novellus - Purification, characterization, and molecular biology of a heterodimeric member of the sulfite oxidase family. J Biol Chem. 2000;275:13202–13212. doi: 10.1074/jbc.275.18.13202. [DOI] [PubMed] [Google Scholar]
- 27.Kisker C, Schindelin H, Pacheco A, Wehbi WA, Garrett RM, Rajagopalan KV, Enemark JH, Rees DC. Molecular basis of sulfite oxidase deficiency from the structure of sulfite oxidase. Cell. 1997;91:973–983. doi: 10.1016/s0092-8674(00)80488-2. [DOI] [PubMed] [Google Scholar]
- 28.Schrader N, Fischer K, Theis K, Mendel RR, Schwarz G, Kisker C. The crystal structure of plant sulfite oxidase provides insights into sulfite oxidation in plants and animals. Structure. 2003;11:1251–1263. doi: 10.1016/j.str.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 29.Kappler U, Bailey S. Molecular basis of intramolecular electron transfer in sulfite-oxidizing enzymes is revealed by high resolution structure of a heterodimeric complex of the catalytic molybdopterin subunit and a c-type cytochrome subunit. J Biol Chem. 2005;280:24999–25007. doi: 10.1074/jbc.M503237200. [DOI] [PubMed] [Google Scholar]
- 30.Kappler U, Bailey S, Feng CJ, Honeychurch MJ, Hanson GR, Bernhardt PV, Tollin G, Enemark JH. Kinetic and structural evidence for the importance of Tyr236 for the integrity of the Mo active site in a bacterial sulfite dehydrogenase. Biochemistry. 2006;45:9696–9705. doi: 10.1021/bi060058b. [DOI] [PubMed] [Google Scholar]
- 31.Wilson HL, Rajagopalan KV. The role of tyrosine 343 in substrate binding and catalysis by human sulfite oxidase. J Biol Chem. 2004;279:15105–15113. doi: 10.1074/jbc.M314288200. [DOI] [PubMed] [Google Scholar]
- 32.Wilson HL, Wilkinson SR, Rajagopalan KV. The G473D mutation impairs dimerization and catalysis in human sulfite oxidase. Biochemistry. 2006;45:2149–2160. doi: 10.1021/bi051609l. [DOI] [PubMed] [Google Scholar]
- 33.Feng CJ, Wilson HL, Hurley JK, Hazzard JT, Tollin G, Rajagopalan KV, Enemark JH. Essential role of conserved arginine 160 in intramolecular electron transfer in human sulfite oxidase. Biochemistry. 2003;42:12235–12242. doi: 10.1021/bi0350194. [DOI] [PubMed] [Google Scholar]
- 34.Feng CJ, Wilson HL, Hurley JK, Hazzard JT, Tollin G, Rajagopalan KV, Enemark JH. Role of conserved tyrosine 343 in intramolecular electron transfer in human sulfite oxidase. J Biol Chem. 2003;278:2913–2920. doi: 10.1074/jbc.M210374200. [DOI] [PubMed] [Google Scholar]
- 35.Feng CJ, Wilson HL, Tollin G, Astashkin AV, Hazzard JT, Rajagopalan KV, Enemark JH. The pathogenic human sulfite oxidase mutants G473D and A208D are defective in intramolecular electron transfer. Biochemistry. 2005;44:13734–13743. doi: 10.1021/bi050907f. [DOI] [PubMed] [Google Scholar]
- 36.Astashkin AV, Raitsimring AM, Feng CJ, Johnson JL, Rajagopalan KV, Enemark JH. Pulsed EPR studies of nonexchangeable protons near the Mo(V) center of sulfite oxidase: Direct detection of the alpha-proton of the coordinated cysteinyl residue and structural implications for the active site. J Am Chem Soc. 2002;124:6109–6118. doi: 10.1021/ja0115417. [DOI] [PubMed] [Google Scholar]
- 37.Astashkin AV, Feng CJ, Raitsimring AM, Enemark JH. O-17 ESEEM evidence for exchange of the axial oxo ligand in the molybdenum center of the high pH form of sulfite oxidase. J Am Chem Soc. 2005;127:502–503. doi: 10.1021/ja0461466. [DOI] [PubMed] [Google Scholar]
- 38.Astashkin AV, Hood BL, Feng CJ, Hille R, Mendel RR, Raitsimring AM, Enemark JH. Structures of the Mo(V) forms of sulfite oxidase from Arabidopsis thaliana by pulsed EPR spectroscopy. Biochemistry. 2005;44:13274–13281. doi: 10.1021/bi051220y. [DOI] [PubMed] [Google Scholar]
- 39.Raitsimring AM, Kappler U, Feng CJ, Astashkin AV, Enemark JH. Pulsed EPR studies of a bacterial sulfite-oxidizing enzyme with pH-invariant hyperfine interactions from exchangeable protons. Inorg Chem. 2005;44:7283–7285. doi: 10.1021/ic0509534. [DOI] [PubMed] [Google Scholar]
- 40.Enemark JH, Astashkin AV, Raitsimring AM. Investigation of the coordination structures of the molybdenum(v) sites of sulfite oxidizing enzymes by pulsed EPR spectroscopy. Dalton Trans. 2006:3501–3514. doi: 10.1039/b602919a. [DOI] [PubMed] [Google Scholar]
- 41.Doonan CJ, Kappler U, George GN. Structure of the active site of sulfite dehydrogenase from Starkeya novella. Inorg Chem. 2006;45:7488–7492. doi: 10.1021/ic0607944. [DOI] [PubMed] [Google Scholar]
- 42.Harris HH, George GN, Rajagopalan KV. High-resolution EXAFS of the active site of human sulfite oxidase: Comparison with density functional theory and X-ray crystallographic results. Inorg Chem. 2006;45:493–495. doi: 10.1021/ic0512274. [DOI] [PubMed] [Google Scholar]
- 43.George GN, Garrett RM, Prince RC, Rajagopalan KV. Coordination chemistry at the molybdenum site of sulfite oxidase: Redox-induced structural changes in the cysteine 207 to serine mutant. Inorg Chem. 2004;43:8456–8460. doi: 10.1021/ic0489847. [DOI] [PubMed] [Google Scholar]
- 44.Helton ME, Pacheco A, McMaster J, Enemark JH, Kirk ML. An MCD spectroscopic study of the molybdenum active site in sulfite oxidase: insight into the role of coordinated cysteine. J Inorg Biochem. 2000;80:227–233. doi: 10.1016/s0162-0134(00)00082-9. [DOI] [PubMed] [Google Scholar]
- 45.Hemann C, Hood BL, Fulton M, Hansch R, Schwarz G, Mendel RR, Kirk ML, Hille R. Spectroscopic and kinetic studies of Arabidopsis thaliana sulfite oxidase: Nature of the redox-active orbital and electronic structure contributions to catalysis. J Am Chem Soc. 2005;127:16567–16577. doi: 10.1021/ja0530873. [DOI] [PubMed] [Google Scholar]
- 46.Elliott SJ, McElhaney AE, Feng CJ, Enemark JH, Armstrong FA. A voltammetric study of interdomain electron transfer within sulfite oxidase. J Am Chem Soc. 2002;124:11612–11613. doi: 10.1021/ja027776f. [DOI] [PubMed] [Google Scholar]
- 47.Nichols JD, Xiang S, Schindelin H, Rajagopalan KV. Mutational Analysis of Escherichia coli MoeA: Two Functional Activities Map to the Active Site Cleft. Biochemistry. 2007;46:78–86. doi: 10.1021/bi061551q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Llamas A, Otte T, Multhaup G, Mendel RR, Schwarz G. The mechanism of nucleotide-assisted molybdenum insertion into molybdopterin - A novel route toward metal cofactor assembly. J Biol Chem. 2006;281:18343–18350. doi: 10.1074/jbc.M601415200. [DOI] [PubMed] [Google Scholar]
- 49.Fischer K, Llamas A, Tejada-Jimenez M, Schrader N, Kuper J, Ataya FS, Galvan A, Mendel RR, Fernandez E, Schwarz G. Function and structure of the molybdenum cofactor carrier protein from Chlamydomonas reinhardtii. J Biol Chem. 2006;281:30186–30194. doi: 10.1074/jbc.M603919200. [DOI] [PubMed] [Google Scholar]
- 50.Kuper J, Llamas A, Hecht HJ, Mendel RR, Schwarz G. Structure of the molybdopterin-bound Cnx1G domain links molybdenum and copper metabolism. Nature. 2004;430:803–806. doi: 10.1038/nature02681. [DOI] [PubMed] [Google Scholar]
- 51.Gutzke G, Fischer B, Mendel RR, Schwarz G. Thiocarboxylation of molybdopterin synthase provides evidence for the mechanism of dithiolene formation in metal-binding pterins. J Biol Chem. 2001;276:36268–36274. doi: 10.1074/jbc.M105321200. [DOI] [PubMed] [Google Scholar]
- 52.Leimkuhler S, Rajagopalan KV. A sulfurtransferase is required in the transfer of cysteine sulfur in the in vitro synthesis of molybdopterin from precursor Z in Escherichia coli. J Biol Chem. 2001;276:22024–22031. doi: 10.1074/jbc.M102072200. [DOI] [PubMed] [Google Scholar]
- 53.Johnson JL, Coyne KE, Rajagopalan KV, Van Hove JLK, Mackay M, Pitt J, Boneh A. Molybdopterin synthase mutations in a mild case of molybdenum cofactor deficiency. Am J Med Genet. 2001;104:169–173. doi: 10.1002/1096-8628(20011122)104:2<169::aid-ajmg1603>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 54.Kuper J, Palmer T, Mendel RR, Schwarz G. Mutations in the molybdenum cofactor biosynthetic protein Cnx1G from Arabidopsis thaliana define functions for molybdopterin binding, molybdenum insertion, and molybdenum cofactor stabilization. Proc Natl Acad Sci U S A. 2000;97:6475–6480. doi: 10.1073/pnas.110568497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schwarz G, Mendel RR. Molybdenum cofactor biosynthesis and molybdenum enzymes. Annu Rev Plant Biol. 2006;57:623–647. doi: 10.1146/annurev.arplant.57.032905.105437. [DOI] [PubMed] [Google Scholar]
- 56.Mendel RR, Bittner F. Cell biology of molybdenum. Biochim Biophys Acta-Mol Cell Res. 2006;1763:621–635. doi: 10.1016/j.bbamcr.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 57.Astashkin AV, Neese F, Raitsimring AM, Cooney JJA, Bultman E, Enemark JH. Pulsed EPR investigations of systems modeling molybdenum enzymes: Hyperfine and quadrupole parameters of OXO-O-17 in [(MOO)-O-17(SPh)4](-) J Am Chem Soc. 2005;127:16713–16722. doi: 10.1021/ja055472y. [DOI] [PubMed] [Google Scholar]
- 58.Rajagopalan KV. Sulphite oxidase. In: Coughlan MP, editor. Molybdenum and Molybdenum-Containing Enzymes. Pergamon Press; Oxford, U.K.: 1980. pp. 241–272. [Google Scholar]
- 59.Hille R. Essays in Biochemistry. Vol. 34. Portland Press Ltd; London: 1999. pp. 125–137. [DOI] [PubMed] [Google Scholar]
- 60.Hille R. The reaction mechanism of oxomolybdenum enzymes. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1994;1184:143–169. doi: 10.1016/0005-2728(94)90220-8. [DOI] [PubMed] [Google Scholar]
- 61.Brody MS, Hille R. The Reaction of Chicken Liver Sulfite Oxidase with Dimethylsulfite. Biochim Biophys Acta-Protein Struct Molec Enzym. 1995;1253:133–135. doi: 10.1016/0167-4838(95)00194-4. [DOI] [PubMed] [Google Scholar]
- 62.Brody MS, Hille R. The kinetic behavior of chicken liver sulfite oxidase. Biochemistry. 1999;38:6668–6677. doi: 10.1021/bi9902539. [DOI] [PubMed] [Google Scholar]
- 63.Enemark JH, Astashkin AV, Raitsimring AM. Variable frequency pulsed EPR studies of molybdenum enzymes: Structure of molybdenum enzymes. In: Telser JA, editor. Paramagnetic Resonance of Metallobiomolecules. ACS Symposium Series; 858, Washington: 2003. pp. 179–192. [Google Scholar]
- 64.Astashkin AV, Mader ML, Pacheco A, Enemark JH, Raitsimring AM. Direct detection of the proton-containing group coordinated to Mo(V) in the high pH form of chicken liver sulfite oxidase by refocused primary ESEEM spectroscopy: Structural and mechanistic implications. J Am Chem Soc. 2000;122:5294–5302. [Google Scholar]
- 65.Pacheco A, Basu P, Borbat P, Raitsimring AM, Enemark JH. Multifrequency ESEEM spectroscopy of sulfite oxidase in phosphate buffer: Direct evidence for coordinated phosphate. Inorg Chem. 1996;35:7001–7008. doi: 10.1021/ic9607806. [DOI] [PubMed] [Google Scholar]
- 66.Ferapontova EE, Ruzgas T, Gorton L. Direct electron transfer of heme- and molybdopterin cofactor-containing chicken liver sulfite oxidase on alkanethiol-modified gold electrodes. Anal Chem. 2003;75:4841–4850. doi: 10.1021/ac0341923. [DOI] [PubMed] [Google Scholar]
- 67.Caradonna JP, Reddy PR, Holm RH. Kinetics, Mechanisms, and Catalysis of Oxygen Atom Transfer-Reactions of S-Oxide and Pyridine N-Oxide Substrates with Molybdenum(Iv,Vi) Complexes - Relevance to Molybdoenzymes. J Am Chem Soc. 1988;110:2139–2144. [Google Scholar]
- 68.Pietsch MA, Hall MB. Theoretical studies on models for the oxo-transfer reaction of dioxomolybdenum enzymes. Inorg Chem. 1996;35:1273–1278. doi: 10.1021/ic951044p. [DOI] [PubMed] [Google Scholar]
- 69.Codd R, Astashkin AV, Pacheco A, Raitsimring AM, Enemark JH. Pulsed ELDOR spectroscopy of the Mo(V)/Fe(III) state of sulfite oxidase prepared by one-electron reduction with Ti(III) citrate. J Biol Inorg Chem. 2002;7:338–350. doi: 10.1007/s00775-001-0303-5. [DOI] [PubMed] [Google Scholar]
- 70.Astaskhin AV, Johnson-Winters K, Klein EL, Byrne RS, Hille R, Raitsimring AM, Enemark JH. Direct demonstration of the presence of coordinated sulfate in the reaction pathway of Arabidopsis thaliana sulfite oxidase using 33S labeling and ESEEM spectroscopy. 2007 doi: 10.1021/ja0704885. to be submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kessler DL, V RK. Purification and properties of sulfite oxidase from chicken liver - Presence of molybdenum in sulfite oxidase from diverse sources. J Biol Chem. 1972;247:6566–6573. [PubMed] [Google Scholar]
- 72.Bray RC, Gutteridge S, Lamy MT, Wilkinson T. Equilibria amongst different molybdenum (V)-containing species from sulfite oxidase - Evidence for a halide ligand of molybdenum in the low pH species. Biochem J. 1983;211:227–236. doi: 10.1042/bj2110227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gutteridge S, Lamy MT, Bray RC. The nature of the phosphate inhibitor complex of sulfite oxidase from electron paramagnetic resonance studies using O-17. Biochem J. 1980;191:285–288. doi: 10.1042/bj1910285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Astashkin AV, Raitsimring AM, Feng C, Johnson JL, Rajagopalan KV, Enemark JH. The Mo-OH proton of the low-pH form of sulfite oxidase: Comparison of the hyperfine interactions obtained from pulsed ENDOR, CW-EPR and ESEEM measurements. Appl Magn Reson. 2002;22:421–430. [Google Scholar]
- 75.Raitsimring AM, Pacheco A, Enemark JH. ESEEM investigations of the high pH and low pH forms of chicken liver sulfite oxidase. J Am Chem Soc. 1998;120:11263–11278. [Google Scholar]
- 76.Cramer SP, Johnson JL, Rajagopalan KV, Sorrell TN. Observation of O-17 effects on Mo-V EPR spectra in sulfite oxidase, xanthine dehydrogenase, and MoO(SC6H5)4. Biochem Biophys Res Commun. 1979;91:434–439. doi: 10.1016/0006-291x(79)91540-7. [DOI] [PubMed] [Google Scholar]
- 77.Feng CJ, Kedia RV, Hazzard JT, Hurley JK, Tollin G, Enemark JH. Effect of solution viscosity on intramolecular electron transfer in sulfite oxidase. Biochemistry. 2002;41:5816–5821. doi: 10.1021/bi016059f. [DOI] [PubMed] [Google Scholar]
- 78.Pacheco A, Hazzard JT, Tollin G, Enemark JH. The pH dependence of intramolecular electron transfer rates in sulfite oxidase at high and low anion concentrations. J Biol Inorg Chem. 1999;4:390–401. doi: 10.1007/s007750050325. [DOI] [PubMed] [Google Scholar]
- 79.Sullivan EP, Hazzard JT, Tollin G, Enemark JH. Electron transfer in sulfite oxidase - Effects of pH and anions on transient kinetics. Biochemistry. 1993;32:12465–12470. doi: 10.1021/bi00097a026. [DOI] [PubMed] [Google Scholar]
- 80.Sullivan EP, Hazzard JT, Tollin G, Enemark JH. Inhibition of intramolecular electron transfer in sulfite oxidase by anion binding. J Am Chem Soc. 1992;114:9662–9663. [Google Scholar]
- 81.Spence JT, Kipke CA, Enemark JH, Sunde RA. Stoichiometry of electron uptake and the effect of anions and pH on the molybdenum and heme reduction potentials of sulfite oxidase. Inorg Chem. 1991;30:3011–3015. [Google Scholar]
- 82.Gray HB, Winkler JR. Electron transfer in proteins. Annu Rev Biochem. 1996;65:537–561. doi: 10.1146/annurev.bi.65.070196.002541. [DOI] [PubMed] [Google Scholar]
- 83.Page CC, Moser CC, Chen XX, Dutton PL. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 84.Leys D, Scrutton NS. Electrical circuitry in biology: Emerging principles from protein structure. Curr Opin Struct Biol. 2004;14:642–647. doi: 10.1016/j.sbi.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 85.Hoffman BM, Celis LM, Cull DA, Patel AD, Seifert JL, Wheeler KE, Wang JY, Yao J, Kurnikov IV, Nocek JM. Differential influence of dynamic processes on forward and reverse electron transfer across a protein-protein interface. Proc Natl Acad Sci U S A. 2005;102:3564–3569. doi: 10.1073/pnas.0408767102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Feng CJ, Kappler U, Tollin G, Enemark JH. Intramolecular electron transfer in a bacterial sulfite dehydrogenase. J Am Chem Soc. 2003;125:14696–14697. doi: 10.1021/ja038197t. [DOI] [PubMed] [Google Scholar]
- 87.Stiefel EI. Proposed Molecular Mechanism for Action of Molybdenum in Enzymes - Coupled Proton and Electron-Transfer. Proc Natl Acad Sci U S A. 1973;70:988–992. doi: 10.1073/pnas.70.4.988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Johnson JL, Rajagopalan KV. Tryptic cleavage of rat liver sulfite oxidase - Isolation and characterization of molybdenum and heme domains. J Biol Chem. 1977;252:2017–2025. [PubMed] [Google Scholar]
- 89.Johnson JL, Coyne KE, Garrett RM, Zabot MT, Dorche C, Kisker C, Rajagopalan KV. Isolated sulfite oxidase deficiency: Identification of 12 novel SUOX mutations in 10 patients. Hum Mutat. 2002;20:74. doi: 10.1002/humu.9038. [DOI] [PubMed] [Google Scholar]
- 90.Karakas E, Kisker C. Structural analysis of missense mutations causing isolated sulfite oxidase deficiency. Dalton Trans. 2005:3459–3463. doi: 10.1039/b505789m. [DOI] [PubMed] [Google Scholar]
- 91.Kawatsu T, Beratan DN. Electron transfer between cofactors in protein domains linked by a flexible tether. Chem Phys. 2006;326:259–269. [Google Scholar]
- 92.Zhang X, Vincent AS, Halliwell B, Wong KP. A mechanism of sulfite neurotoxicity - Direct inhibition of glutamate dehydrogenase. J Biol Chem. 2004;279:43035–43045. doi: 10.1074/jbc.M402759200. [DOI] [PubMed] [Google Scholar]
- 93.Steinberg KK, Relling MV, Gallagher ML, Greene CN, Rubin CS, French D, Holmes AK, Carroll WL, Koontz DA, Sampson EJ, Satten GA. Genetic studies of a cluster of acute lymphoblastic leukemia cases in Churchill county, Nevada. Environmental Health Perspectives. 2007;115:158–164. doi: 10.1289/ehp.9025. [DOI] [PMC free article] [PubMed] [Google Scholar]