

Abstract
Candida albicans contains ten putative cytochrome P450 (CYP) genes coding for enzymes that appear to play important roles in fungal survival and virulence. Here, we report the characterization of CYP52A21, a putative alkane/fatty acid hydroxylase. The recombinant CYP52A21 protein containing a 6 × (His)-tag was expressed in Escherichia coli and was purified. The purified protein, reconstituted with rat NADPH-cytochrome P450 reductase, ω-hydroxylated dodecanoic acid to give 12-hydroxydodecanoic acid, but to a lesser extent also catalyzed (ω-1)-hydroxylation to give 11-hydroxydodecanoic acid. When 12,12,12-d3-dodecanoic acid was used as the substrate, there was a major shift in the oxidation from the ω- to the (ω-1)-hydroxylated product. The regioselectivity of fatty acid hydroxylation was examined with the 12-iodo-, 12-bromo-, and 12-chlorododecanoic acids. Although all three 12-halododecanoic acids bound to CYP52A21 with similar affinities, the production of 12-oxododecanoic acid decreased as the size of the terminal halide increased. The regioselectivity of CYP52A21 fatty acid oxidation is thus consistent with presentation of the terminal end of the fatty acid chain for oxidation via a narrow channel that limits access to other atoms of the fatty acid chain. This constricted access, in contrast to that proposed for the CYP4A family of enzymes, does not involve covalent binding of the heme to the protein.
Keywords: cytochrome P450, Candida albicans, fatty acid hydroxylase, covalent heme binding, halogen oxidation
Introduction
The cytochrome P450 monooxygenases (CYPs or P450s) are members of a superfamily of heme-containing enzymes involved in the oxidative metabolism of endo- and xenobiotic chemicals [1]. These P450 enzymes are found throughout nature, from archaebacteria to humans [2]. Despite differences in the phylogenetic origin, cellular localization, electron donor partners, and substrate specificities of P450 enzymes, their gross active sites and catalytic mechanisms appear to be remarkably conserved [3].
Candida albicans is a major pathogenic fungus that causes opportunistic oral and vaginal infections in humans [4,5]. Systemic fungal infections have emerged as important causes of morbidity and mortality in immunocompromised patients, such as those infected with HIV or undergoing cancer chemotherapy and/or organ/bone marrow transplantation [6,7]. The complete genome of C. albicans, was recently determined [8]. Previously, we reported the characterization of a fungal heme oxygenase, CaHmx1, from C. albicans and proposed that it might be a suitable target for agents directed at the treatment of Candida infections [9]. Furthermore, the C. albicans genome contains genes coding for approximately ten putative P450 enzymes, including CYP51 and CYP61, both of which are also found in S. cerevisiae (http://www.candidagenome.org/). It also possesses one member of the CYP52 family that is normally characterized by alkane-inducibility. CYP52 enzymes have been found in soil yeasts such as Candida tropicalis and Candida maltosa. These P450 enzymes are commonly responsible for the first and rate-limiting step in the ω-oxidation of n-alkanes and/or fatty acids [10-13]. The CYP52 from C. albicans was previously identified and was shown in vivo to possibly play a role in multidrug resistance, although it was not involved in azole drug resistance [14]. The oxidation of hydrocarbons or fatty acids by CYP52 thus appears to be important for lipid metabolism and for the pathogenesis and survival of C. albicans.
Fatty acids are commonly hydroxylated by the mammalian CYP4 enzymes, a family of proteins that are characterized by their unique ability to hydroxylate the thermodynamically disfavored terminal methyl group of fatty acids rather than the default, thermodynamically favored internal chain carbons that are hydroxylated by other P450 enzymes [1]. Clearly, the CYP4 enzymes have evolved a strategy that allows them to specifically direct the reaction to the terminal (ω-) carbon of the fatty acid chain. We have postulated that this strategy involves a constricted access channel through which the fatty acid terminus must be threaded for oxidation, have proposed that covalent binding of the heme to the protein facilitates the assembly of a sufficiently rigid access channel, and have used terminally halogenated fatty acids to obtain information on the effective diameter of the CYP4A channel [15-17].
In this study, we have overexpressed and purified CYP52A21, the first recombinant P450 from C. albicans, and have characterized the protein in terms of its structural, spectroscopic, and catalytic properties. The results indicate that covalent binding of the heme to the protein is not required for a narrow channel access to be used to control the fatty acid hydroxylation regiospecificity.
Materials and methods
Chemicals and enzymes
Dodecanoic acid, 12,12,12-d3-dodecanoic acid, sodium dithionite, glucose-6-phosphate, glucose-6-phosphate dehydrogenase, and NADP+ were purchased from Sigma (St. Louis, MO) or Aldrich Chemical Co. (Milwaukee, WI). The syntheses of 12-chlorododecanoic acid, 12-bromododecanoic acid, and 12-iodododecanoic acid have been reported elsewhere [17]. Other chemicals were of the highest grade commercially available. Recombinant rat NADPH-P450 reductase and CYP4A1, both expressed in E. coli, were kindly provided by Dr. Fengyun Xu (UCSF).
Construction of expression plasmids
The general approach has been described [9,18]. The genomic DNA from C. albicans was kindly provided by the laboratory of Alexander Johnson at UCSF. The open reading frame for CYP52A21, and an added 6 × His-C-terminal tag, were amplified using PCR and the amplified PCR fragment was cloned into the pCW(Ori+) vector using the NdeI and XbaI restriction sites. The cloned vectors were verified by nucleotide sequencing and restriction digestion. The open reading frame of CYP52A21 contains a CTG encoding a Ser that was corrected by site-directed mutagenesis to enable expression of the correct recombinant protein in E. coli as previously described for CaHmx1 [9,19]. Site-directed mutagenesis to change Leu40 to a Ser was carried out using Quick-Change mutagenesis (Stratagene, La Jolla, CA). The change in nucleotides was confirmed by nucleotide sequencing.
Enzyme expression and purification
Expression and purification of CYP52A21 were carried out, with minor modifications, by previously described procedures [20]. Briefly, the E. coli strains transformed with the pCW(Ori+) vectors were inoculated into TB medium containing 100 μg/ml ampicillin and 1.0 mM IPTG. The expression cultures were grown in 1 liter Fernbach flasks at 37 °C for 3 h and then at 28 °C with shaking at 200 rpm for 24 h. Bacterial inner membrane fractions containing CYP52A21 were isolated and prepared from 1 liter TB (with ampicillin, 100 μg/ml) expression cultures of E. coli DH5α. Purification of the CYP52A21 protein using a Ni2+-nitrilotriacetate column was carried out as previously described for a different protein [20,21]. Briefly, the membrane was solubilized at 4 °C overnight in 100 mM potassium phosphate buffer (pH 7.4) containing 20% glycerol, 0.5 M NaCl, 10 mM β-mercaptoethanol, and 1.5% CHAPS (w/v). The solubilized fraction was then loaded onto a Ni2+-nitrilotriacetate column (Qiagen, Valencia, CA) and the purified protein was obtained with elution buffer that contained 400 mM imidazole. The eluted fraction containing the P450 was dialyzed at 4 °C against 100 mM potassium phosphate buffer (pH 7.4) containing 20% glycerol and 0.1 mM EDTA.
Spectroscopic characterization
Sodium dithionite was added to reduce the purified ferric P450. The CO-ferrous P450 complex was then generated by passing CO gas through a solution of the ferrous P450. UV-visible spectra were collected on a CARY Varian spectrophotometer in 100 mM potassium phosphate buffer (pH 7.4) at room temperature.
Measurement of CYP52A21 oxidation activity by GC-MS
The fatty acid ω-/(ω-1)-hydroxylation activities of CYP52A21 were determined using a P450/NADPH-P450 reductase/phospholipid reconstituted systems [22]. The reaction mixture included 100 pmol of purified P450 protein, 250 pmol of rat NADPH-P450 reductase, and L-α-dilauroyl-sn-glycero-3-phosphocholine (45 μM) in 0.50 ml of 100 mM potassium phosphate buffer (pH 7.4), along with a specified amount of the substrate. An NADPH-generating system [22] was used to start the reactions. The incubations were generally done for 10-30 min at 37 °C and were terminated by addition of 0.5 ml of CH2Cl2. The CH2Cl2 extract was then dried under N2 before the residue was trimethylsilylated by incubation with N,O-bis(trimethylsilyl)trifluoroacetamide (50 μl) at 70 °C for 10-15 min. The derivatized samples were allowed to cool, vortexed, and transferred to sealed Teflon-capped glass vials for either manual or autoinjection into the GC-MS. Analyses were performed on an Agilent 6850 gas chromatograph coupled to an Agilent 5973 Network Mass Selective Detector in the electron impact mode as previously described [17].
Heme staining on SDS-gel electrophoresis
Heme detection by SDS-PAGE was previously reported [23]. Briefly, SDS-gel electrophoresis was carried out for purified CYP52A21 and CYP4A1 (as a positive control). The gel was then immersed in the dark in a mixture of 3 parts of 3,3’,5,5’-tetramethylbenzidine (1.5 mg/mL MeOH) and 7 parts of 250 mM sodium acetate buffer (pH 5.0) for 1-2 h before H2O2 was added to a final concentration of 30 mM. Over the next 30 min, light blue bands emerged, indicating the presence of heme.
Spectral Binding Titrations
Purified CYP52A21 was diluted to 2 μM in 100 mM potassium phosphate buffer (pH 7.4) and divided between two 0.5 ml glass cuvettes. Spectra (350 – 500 nm) were then recorded with subsequent additions of substrates (from a methanol stock [24]) using a CARY Varian spectrophotometer. The difference in absorbance between the absorption maximum (390 nm) and minimum (420 nm) was then plotted versus the substrate concentration [25].
Results
Sequence alignment with other P450s
The gene for CYP52A21 from C. albicans was named ALK8 because it encodes a putative protein highly homologous to the alkane-inducible P450s from C. maltosa and C. tropicalis [14]. Previous in vivo experiments suggested that the protein encoded by ALK8 could hydroxylate fatty acids [14]. The well-known P450 enzymes that catalyze fatty acid hydroxylation are the members of the mammalian P450 4A family and P450BM3 (CYP102) [1]. Amino acid sequence alignment of CYP52A21 with P450 4A1 and 4A3 revealed a high sequence similarity with a score of more than 70%, while the alignment with P450BM3 was only 30% (Fig. 1) [26]. The good sequence alignment of CYP52A21 with CYP4A3 suggests that they may have similar enzymatic specificities. However, it is significant that the conserved Glu residue responsible for covalent attachment of the heme to the protein in the P450 4A family is not found in CYP52A21 (Fig. 1) [27]. Indeed, heme staining by adding 3,3’,5,5’-tetramethylbenzidine and H2O2 to an SDS-Page gel showed that CYP52A21 does not have a covalently bound heme, in contrast to CYP4A1 which, as expected, does contain the protein-bound heme (supplementary Fig. S1).
Fig. 1.
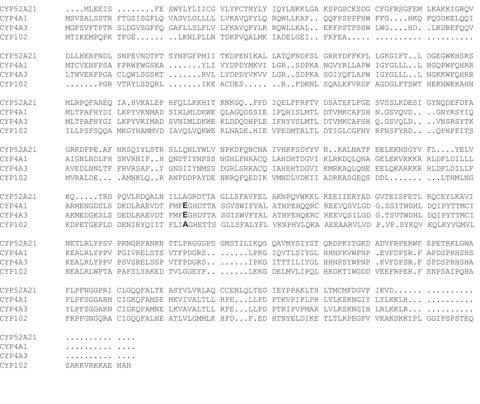
Sequence alignment of CYP52A21 with CYP4A1, CYP4A3, and CYP101 (P450BM3). The amino acid sequences were aligned using the software T-Coffee from the Swiss Institute of Bioinformatics (http://www.ch.embnet.org/software/TCoffee.html). The residues corresponding to the glutamate involved in heme-covalent binding in the CYP4 family are shown in bold.
Expression and purification of CYP52A21
The typical P450 expression level spectrally determined in the whole cell culture was around 60 nmol P450 holoenzyme per liter of culture medium. After separation by ultracentrifugation, there was no detectable P450 in the soluble fraction and all the detectable P450 was located in the membrane fraction. CHAPS, an anionic surfactant, successfully solubilized the P450 protein from the membrane fraction. Purified CYP52A21, obtained after chromatography on a Ni-column, gave a single band on SDS-PAGE at 52 kDa, as expected from the open reading frame of the gene (Fig. 2).
Fig. 2.
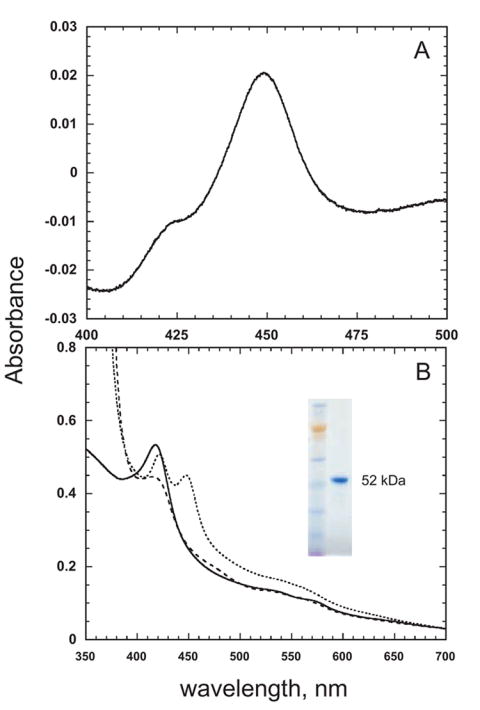
Spectra of purified CYP52A21. A, The Fe2+ · CO vs Fe2+ difference spectrum. B, The absolute spectra of the ferric (—), ferrous (– –), and ferrous carbon monoxide protein (----). The inset shows an SDS-PAGE gel of the purified CYP52A21 protein.
Spectral properties of CYP52A21
The reduced CO difference spectrum of CYP52A21 exhibited an absorption maximum at 449 nm (Fig. 2A). Examination of the absolute spectra showed that the ferric form of the protein was almost completely in the low spin state (Fig. 2B).
Binding of dodecanoic acid to CYP52A21
Titration of purified CYP52A21 with dodecanoic acid (lauric acid) showed a typical type I substrate binding spectral change (increase at 390 nm, and decrease at 420 nm). This is consistent with displacement of the distal iron water ligand upon binding of the dodecanoic acid to the protein (Fig. 3). The calculated Kd value is 5.1 ± 0.4 μM (Fig. 3). In contrast, no spectral change was observed when the enzyme was titrated with either dodecane or hexadecane, which suggests that simple hydrocarbons are not bound by this enzyme (result not shown).
Fig. 3.
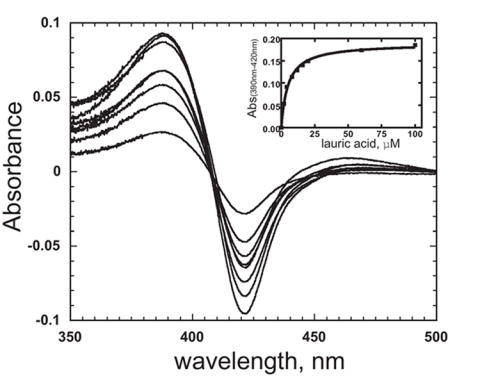
Titration of CYP52A21 with dodecanoic acid. Increasing concentrations of dodecanoic acid were added to both the sample and reference cuvettes. The inset shows the plot of ΔA390-420nm vs concentration of dodecanoic acid.
Catalytic activities of CYP52A21
A reconstituted system consisting of CYP52A21 and rat NADPH-P450 reductase catalyzed the oxidation of dodecanoic acid (Fig. 4). An NADPH generating system supported this reaction more efficiently than NADPH itself (data not shown). The major product of the reaction was the ω-hydroxylated 12-hydroxydodecanoic acid (12.26 min), although the (ω-1)-hydroxylated 11-hydroxydodecanoic acid (11.49 min) was also obtained as a minor product (Fig. 4). In general terms, this product pattern is similar to that obtained with the mammalian CYP4A enzymes. The concentration-dependence of dodecanoic acid oxidation is shown Fig. 5. The Hill equation instead of the Michaelis-Menten equation was used to obtain the best fit for the data, from which a kcat of approximately 33 min-1 and a Km of 57 μM were calculated (Fig. 5). Analysis of the data with the Hill equation indicated apparent cooperativity with n = 2.3. In accord with the finding that neither dodecane nor hexadecane caused a spectral shift, no detectable products were formed in incubations of these hydrocarbons with the reconstituted CYP52A21 system (data not shown).
Fig. 4.
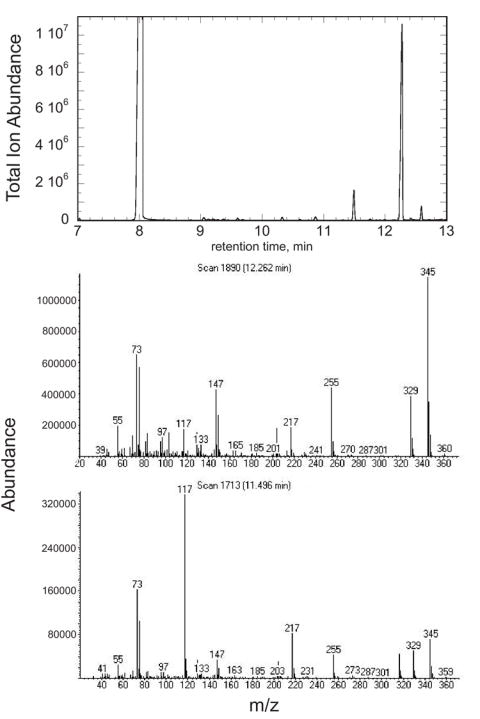
GC-MS of products formed in the reaction of CYP52A21 with dodecanoic acid. A. GC-MS total ion trace of the reaction products. B. Mass-spectrum of the product at 12.26 min, C. Mass-spectrum of the product at 11.49 min.
Fig. 5.
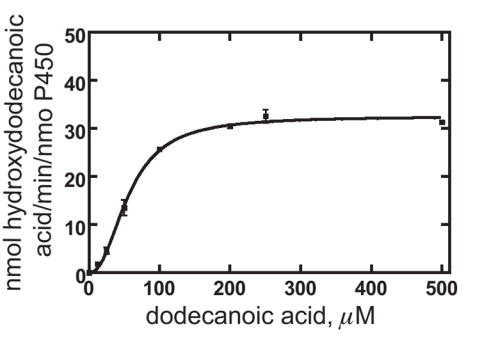
Steady-state kinetics of dodecanoic acid hydroxylation by CYP52A21. Each point is a mean ± SD of triplicate assays. The estimated parameters were kcat 33 ± 1 nmol of total hydroxylated dodecanoic acid formed/min/nmol of P450, Km 57 ± 2 μM, and kcat/Km 0.58 ± 0.03.
ω-Hydroxylation by CYP52A21 was greatly attenuated while formation of the (ω-1)-hydroxylated product was increased when 12,12,12-d3-dodecanoic acid was used as the substrate instead of unlabeled dodecanoic acid (Table 1). This large shift in the hydroxylation regiospecificity caused by the replacement of the terminal methyl hydrogens by deuteriums indicates that both the ω- and (ω-1)-positions are accessible to the oxidizing species, although the intrinsically disfavored ω-hydroxylation is favored over the intrinsically easier (ω-1)-hydroxylation. However, as oxidation of a C-D bond has a higher energy barrier than that of a C-H bond, the consequent decrease in the rate of ω-hydroxylation with the deuterated substrate allows the (ω-1)-hydroxylation to compete more effectively, resulting in increased formation of 11-hydroxydodecanoic acid at the expense of 12-hydroxydodecanoic acid.
Table 1.
Isotope-dependent Shift of Dodecanoic Acid Hydroxylation Catalyzed by CYP52A21
| Substrates | 12-hydroxydodecanoic acid | 11-hydroxydodecanoic acid conversion, % | Total | ω/(ω-1) |
|---|---|---|---|---|
| d0-dodecanoic acid | 49.0 ± 7.6 | 4.4 ± 0.9 | 53.4 ± 8.5 | 11.1 ± 2.9 |
| d3-dodecanoic acid | 19.5 ± 1.5 | 22.3 ± 1.9 | 41.8 ± 3.5 | 0.87 ± 0.10 |
Oxidation of halododecanoic acids by CYP52A21
12-Halododecanoic acids have been used to investigate the dimensions of the putative channel through which substrates are presented for oxidation in the mammalian enzyme CYP4A1 [17]. The P450-catalyzed oxidation of halogen-substituted carbons normally proceeds via hydroxylation of the carbon followed by elimination of the halide ion to give the carbonyl product. However, in the CYP4A enzymes, which preferentially oxidize the terminal atom of the hydrocarbon chain, the oxidation results to a substantial extent in direct oxidation of the terminal halide atom. In the CYP4A1 study, it was found that the 12-chloro and 12-bromododecanoic acids underwent halide atom oxidation, but the 12-iodo substrate did not. As the inherent reactivity of the halogen atoms predicts the reverse reactivity order (i.e., I > Br > I), product formation was not governed by intrinsic halogen oxidizability. The oxidation order correlates, however, with the size of the halogen atom, as the chlorine is the smallest and the iodine the largest. From these results it was concluded that a “channel” limits substrate access to the oxidizing species, and that the channel was wide enough for presentation of a bromo but not iodo terminus.
As shown here, CYP52A21 also preferentially oxidizes the terminal carbon atom of fatty acid substrates. We have therefore investigated the ability of this enzyme to oxidize the 12-chloro-, 12-bromo-, and 12-iododedecanoic acids. Difference binding studies, which monitor the spin state change caused by displacement of the distal water ligand from the iron on binding of the fatty acid, first established that all three substrates are readily bound by the enzyme. The three 12-halododecanoic acids had comparable Kd values and were bound 1.2-2-fold tighter than the parent dodecanoic acid (Table 2). The catalytic studies established that the total hydroxylation (conversion) of the 12-halododecanoic acids by CYP52A21 decreased as the size of the terminal atom increased (Table 3). All three 12-halododecanoic acids underwent comparable oxidation of the halide atom, as indicated by formation of 12-hydroxydodecanoic acid (Table 3). Previous work has shown that the hydroxyl oxygen in this product derives from water, not molecular oxygen, and is introduced by hydrolysis of an initially formed oxohalonium (R-X+-O-) metabolite (17). However, the extent of “;ω-1” hydroxylation, which leads to the aldehyde 12-oxododecanoic acid, decreased in proportion to the size of the terminal halogen atom (i.e., Cl > Br ≫ I). This suggests that the ability of the oxidizing species to reach around the terminal halogen atom to react with the last carbon in the chain is increasingly impaired as the terminal halogen atom gets larger. The results differ from those obtained with CYP4A1 in that the 12-iodosubstrate was oxidized t osome extent by CYP52A21. The access channel in this enzyme thus appears to be modestly larger than in CYP4A1. It is not that large, however, as access to the last carbon of the chain (i.e., that adjacent to the halogen atom) was severely impaired as the halogen atom increased in size. As indicated in Table 4, the turnover numbers for the 12-halododecanoic acids decreased as the size of the halogen atom increased, and were much lower than the turnover numbers for the unhalogenated lauric acid, as might be expected from the greater difficulty in oxidizing substrates in which the available oxidation sites are a poorly oxidizable halogen atom or a carbon atom that is increasingly sterically masked as the size of the halogen atom increases.
Table 2.
Estimated Substrate Binding Affinities to CYP52A21
| Substrates | Kd (μM) |
|---|---|
| CYP52A21 | |
| dodecanoic acid | 5.1 ± 0.4 |
| 12-chlorododecanoic acid | 2.7 ± 0.2 |
| 12-bromododecanoic acid | 4.1 ± 0.2 |
| 12-iodododecanoic acid | 2.5 ± 0.2 |
Table 3.
Conversion of 12-Halododecanoic Acids by CYP52A21
| Substrates | 12-hydroxydodecanoic
acid |
12-oxododecanoic
acid |
11-hydroxydodecanoic
acid |
Total conversion | ω/(ω-1) |
|---|---|---|---|---|---|
| % | |||||
| 12-chlorododecanoic
acid |
1.5 ± 0.4 | 4.2 ± 0.2 | 5.7 ± 0.2 | 0.36 ± 0.27 | |
| 12-bromododecanoic
acid |
1.5 ± 0.1 | 1.1 ± 0.1 | 2.6 ± 0.1 | 1.36 ± 0.11 | |
| 12-iodododecanoic
acid |
1.1 ± 0.2 | < 0.1 | 1.1 ± 0.2 | ||
Table 4.
Turnover Numbers for Halododecanoic Acid Oxidation by CYP52A21
| Substrates | 12-hydroxydodecanoic acid | 12-oxododecanoic
acid |
11- hydroxydodecanoic
acid |
Total |
|---|---|---|---|---|
| min-1 | ||||
| dodecanoic acid | 28 | 2 | 30 | |
| 12-chlorododecanoic
acid |
0.25 | 0.70 | 0.95 | |
| 12-bromododecanoic
acid |
0.25 | 0.18 | 0.43 | |
| 12-iodo-dodecanoic
acid |
0.18 | 0.18 | ||
Inhibition of CYP52A21 by azole compounds
C. albicans infections are treated with azole antifungal agents that are generally considered to act by inhibiting P450 enzymes involved in steroid and possibly lipid metabolism. We did not find any inhibition of CYP52A21 dodecanoic acid hydroxylation by fluconazole or econazole, and ketoconazole only showed a weak inhibition with IC50 = 104 μM (Fig. 6). CYP52A21 thus does not appear to be one of the targets of azole antifungal agents in C. albicans.
Fig. 6.
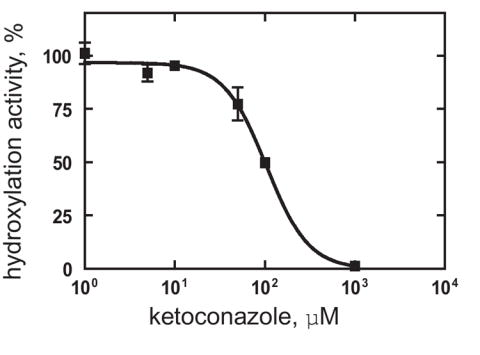
Inhibition of CYP52A21 dodecanoic acid hydroxylation by ketoconazole.
Discussion
Baker’s yeast S. cerevisiae has only three P450 genes: CYP51, a lanosterol demethylase; CYP57, which is responsible for synthesis of the dityrosine required in cell wall assembly; and CYP61, which has been identified by proteomic studies as a sterol 22-desaturase [28]. Fungal P450s, unlike the bacterial enzymes but like the mammalian proteins, are generally membrane-associated and their turnover is supported by a single NADPH-dependent flavoprotein reductase. Analysis of the genomic database (http://www.candidagenome.org/) showed that the pathogenic fungus C. albicans possesses approximately ten putative P450 enzymes, including a CYP52 protein. The CYP52 family encompasses the alkane-oxidizing proteins found in soil/environmental yeasts like C. tropicalis and C. maltosa, but homologues of these enzymes are not found in S. cerevisiae or S. pombe. An analysis by David Nelson indicates that the CYP52 sequences most closely resemble a cluster of bacterial P450 enzymes called the eukaryote-like bacterial P450s (http://drnelson.utmem.edu/CytochromeP450.html). These include CYP102 (a fatty acid hydroxylase) and CYP110. This cluster next joins with the CYP4 clan that includes the mammalian fatty acid hydroxylases.
The metabolites of arachidonic acid produced by the mammalian CYP4 family have important physiological roles. 20-Hydroxyeicosatetraenoic acid (20-HETE), the ω-hydroxylated product of arachidonic acid, is a vasoconstrictor [29,30], whereas 19-HETE, the (ω-1)-hydroxylated product, as well as some of the arachidonic acid epoxides are vasodilators [30-32]. Therefore, the ω-hydroxylation regiospecificity of CYP4A enzymes is of critical physiological importance. We previously (a) postulated that a constricted access channel controls ω-hydroxylation, (b) discovered that the heme is covalently bound to the protein in most CYP4 enzymes, (c) identified the residue involved in the covalent linkage, and (d) proposed that covalent heme binding is important for the architecture of the narrow channel [17,33,34]. It is thus somewhat unexpected that CYP52A21 from C. albicans predominantly hydroxylates fatty acids at the ω-position (Figure 4) despite the fact that the Glu residue involved in heme-covalent binding in CYP4A enzymes is replaced in CYP52A21 by an Ala and its heme is not covalently bound (Fig. 1 and supplementary Fig. S1).
Nevertheless, the basic strategy used to enforce ω-hydroxylation in CYP52A21 appears to be similar to that used by the CYP4A enzymes: sterically restricting access to the oxidizing species so that oxidation of carbons other than the relatively unreactive terminal carbon is difficult. The observation that replacement of the terminal methyl in the fatty acid by a trideuteriomethyl group shifts the oxidation to the ω-1 position demonstrates that ω- and (ω-1)-hydroxylation occur in competition, and as ω-hydroxylation becomes more difficult due to the deuterium isotope effect, (ω-1)-hydroxylation increases in importance (Table 1). The evidence provided by the oxidation of 12-halododecanoic acids supports the existence of a sterically restricted channel for presentation of the terminal atom of the substrate to the oxidizing species. The channel may be slightly larger than in CYP4 enzymes, as CYP52A21 oxidizes not only the 12-chloro- and 12-bromo-, but also the 12-iodododecanoic acids to 12-hydroxydodecanoic acid. CYP4A1 only oxidizes the chloro and bromo analogues, presumably because the 12-iodo is too large to enter the channel. The fact that the (ω-1)-hydroxylation that leads to the terminal aldehyde decreases as the halide becomes larger supports the inference of a sterically restricted channel. Thus, if heme covalent binding contributes to the assembly of a rigid access channel in the CYP4 enzymes, its contribution is not a prerequisite to the formation of such a channel.
CYP52A21 oxidizes myristic (C14) and palmitic (C16) acids preferentially at the ω-position (Table 5). The conversion of myristic acid to the hydroxylated product is comparable to that of dodecanoic acid, but the hydroxylation of palmitic acid was decreased by 70%. In contrast, CYP52A21 did not detectably oxidize dodecane or hexadecane. This observation, the fact that the rate of hydroxylation of dodecanoic acid was the same in the presence or absence of hexadecane or dodecane, and the absence of a binding spectral shift when the hydrocarbons were added to ferric CYP52A21, clearly establish that the hydrocarbons do not bind and are not substrates for this enzyme. Two conclusions follows from these results: (a) the enzyme can process fatty acids of different lengths, and (b) the fatty acid carboxyl group is absolutely required for binding to CYP52A21. A differential specificity for alkanes versus fatty acids has been reported for the CYP52 enzymes of C. maltosa [12]. Alk5A was found to preferentially hydroxylate fatty acids whereas Alk1A preferentially hydroxylated alkanes [12]. CYP52A21 thus resembles Alk5A more than it does Alk1A.
Table 5.
Oxidation of Longer Fatty Acids by CYP52A21
| Substrates | ω-hydroxylation | (ω-1)-hydroxylation | Total | ω/(ω-1) |
|---|---|---|---|---|
| conversion, % | ||||
| lauric acid (C12) | 49.0 ± 7.6 | 4.4 ± 0.9 | 53.4 ± 8.5 | 11.1 ± 2.9 |
| myristic acid (C14) | 42.3 ± 0.4 | 4.4 ± 0.4 | 46.7 ± 4.6 | 9.6 ± 0.9 |
| palmitic acid (C16) | 14.2 ± 1.1 | 1.0 ± 0.1 | 15.3 ± 1.1 | 14.2 ± 1.8 |
Supplementary Material
Staining of covalently attached heme with 3,3’,5,5’-tetramethylbenzidine.
Acknowledgments
We thank Suzanne Noble of the Alexander Johnson group (UCSF) for providing the cDNA of C. albicans. This study was supported by United States Public Health Service grant RO1 GM25515.
The abbreviations used are
- CYP
cytochrome P450
- PCR
polymerase chain reaction
- TB
Terrific Broth
- Kd
A dissociation constant estimated by measurement of the UV-visible changes in the P450 heme spectrum
Footnotes
The on-line version of this article (available at http://eselect.sciencedirect.com/) contains Supplementary Data.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. 3. Plenum Press; New York: 2005. [Google Scholar]
- 2.Guengerich FP. Nature Rev Drug Discov. 2002;1:359–366. doi: 10.1038/nrd792. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Z, Sibbesen O, Johnson RA, Ortiz de Montellano PR. Bioorg Med Chem. 1998;6:1501–1508. doi: 10.1016/s0968-0896(98)00091-1. [DOI] [PubMed] [Google Scholar]
- 4.Odds FC. Crit Rev Microbiol. 1987;15:1–5. doi: 10.3109/10408418709104444. [DOI] [PubMed] [Google Scholar]
- 5.Pendrak ML, Chao MP, Yan SS, Roberts DD. J Biol Chem. 2004;279:3426–3433. doi: 10.1074/jbc.M311550200. [DOI] [PubMed] [Google Scholar]
- 6.Fidel PL., Jr Oral Dis. 2002;8(Suppl 2):69–75. doi: 10.1034/j.1601-0825.2002.00015.x. [DOI] [PubMed] [Google Scholar]
- 7.Kullberg BJ, Oude Lashof AM. Eur J Med Res. 2002;7:183–191. [PubMed] [Google Scholar]
- 8.d’Enfert C, Goyard S, Rodriguez-Arnaveilhe S, Frangeul L, Jones L, Tekaia F, Bader O, Albrecht A, Castillo L, Dominguez A, Ernst JF, Fradin C, Gaillardin C, Garcia-Sanchez S, de Groot P, Hube B, Klis FM, Krishnamurthy S, Kunze D, Lopez MC, Mavor A, Martin N, Moszer I, Onesime D, Perez Martin J, Sentandreu R, Valentin E, Brown AJ. Nucleic Acids Res. 2005;33:D353–D357. doi: 10.1093/nar/gki124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim D, Yukl ET, Moenne-Loccoz P, Ortiz de Montellano PR. Biochemistry. 2006;45:14772–14780. doi: 10.1021/bi061429r. [DOI] [PubMed] [Google Scholar]
- 10.Craft DL, Madduri KM, Eshoo M, Wilson CR. Appl Environ Microbiol. 2003;69:5983–5991. doi: 10.1128/AEM.69.10.5983-5991.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sutter TR, Sanglard D, Loper JC. J Biol Chem. 1990;265:16428–16436. [PubMed] [Google Scholar]
- 12.Zimmer T, Ohkuma M, Ohta A, Takagi M, Schunck WH. Biochem Biophys Res Commun. 1996;224:784–789. doi: 10.1006/bbrc.1996.1100. [DOI] [PubMed] [Google Scholar]
- 13.Seghezzi W, Sanglard D, Fiechter A. Gene. 1991;106:51–60. doi: 10.1016/0378-1119(91)90565-s. [DOI] [PubMed] [Google Scholar]
- 14.Panwar SL, Krishnamurthy S, Gupta V, Alarco AM, Raymond M, Sanglard D, Prasad R. Yeast. 2001;18:1117–1129. doi: 10.1002/yea.762. [DOI] [PubMed] [Google Scholar]
- 15.CaJacob CA, Chan WK, Shephard E, Ortiz de Montellano PR. J Biol Chem. 1988;263:18640–18649. [PubMed] [Google Scholar]
- 16.Hoch U, Ortiz De Montellano PR. J Biol Chem. 2001;276:11339–11346. doi: 10.1074/jbc.M009969200. [DOI] [PubMed] [Google Scholar]
- 17.He X, Cryle MJ, De Voss JJ, de Montellano PR. J Biol Chem. 2005;280:22697–22705. doi: 10.1074/jbc.M502632200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim D, Guengerich FP. Biochemistry. 2004;43:981–988. doi: 10.1021/bi035593f. [DOI] [PubMed] [Google Scholar]
- 19.Ohama T, Suzuki T, Mori M, Osawa S, Ueda T, Watanabe K, Nakase T. Nucleic Acids Res. 1993;21:4039–4045. doi: 10.1093/nar/21.17.4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim D, Wu ZL, Guengerich FP. J Biol Chem. 2005;280:40319–40327. doi: 10.1074/jbc.M508171200. [DOI] [PubMed] [Google Scholar]
- 21.Yun C-H, Miller GP, Guengerich FP. Biochemistry. 2000;39:11319–11329. doi: 10.1021/bi000869u. [DOI] [PubMed] [Google Scholar]
- 22.Guengerich FP. In: Principles and Methods of Toxicology. Hayes AW, editor. Taylor & Francis; Philadelphia: 2001. pp. 1625–1687. [Google Scholar]
- 23.Henne KR, Kunze KL, Zheng YM, Christmas P, Soberman RJ, Rettie AE. Biochemistry. 2001;40:12925–12931. doi: 10.1021/bi011171z. [DOI] [PubMed] [Google Scholar]
- 24.Yun CH, Kim KH, Calcutt MW, Guengerich FP. J Biol Chem. 2005;280:12279–12291. doi: 10.1074/jbc.M411019200. [DOI] [PubMed] [Google Scholar]
- 25.Schenkman JB, Remmer H, Estabrook RW. Mol Pharmacol. 1967;3:113–123. [PubMed] [Google Scholar]
- 26.Notredame C, Higgins DG, Heringa J. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- 27.Girvan HM, Marshall KR, Lawson RJ, Leys D, Joyce MG, Clarkson J, Smith WE, Cheesman MR, Munro AW. J Biol Chem. 2004;279:23274–23286. doi: 10.1074/jbc.M401716200. [DOI] [PubMed] [Google Scholar]
- 28.Kelly SL, Kelly DE, Jackson CJ, Warrilow AGS, Lamb DC. In: Cytochrome P450: Structure, Mechanism, and Biochemistry. 3. Ortiz de Montellano PR, editor. Plenum Press; New York: 2005. pp. 585–617. [Google Scholar]
- 29.Imig JD, Navar LG. Am J Physiol. 1996;271:F87–F93. doi: 10.1152/ajprenal.1996.271.1.F87. [DOI] [PubMed] [Google Scholar]
- 30.Ma YH, Gebremedhin D, Schwartzman ML, Falck JR, Clark JE, Masters BS, Harder DR, Roman RJ. Circ Res. 1993;72:126–136. doi: 10.1161/01.res.72.1.126. [DOI] [PubMed] [Google Scholar]
- 31.Zeldin DC. J Biol Chem. 2001;276:36059–36062. doi: 10.1074/jbc.R100030200. [DOI] [PubMed] [Google Scholar]
- 32.Escalante B, Falck JR, Yadagiri P, Sun LM, Laniado-Schwartzman M. Biochem Biophys Res Commun. 1988;152:1269–1274. doi: 10.1016/s0006-291x(88)80422-4. [DOI] [PubMed] [Google Scholar]
- 33.Dierks EA, Zhang Z, Johnson EF, Ortiz de Montellano PR. J Biol Chem. 1998;273:23055–23061. doi: 10.1074/jbc.273.36.23055. [DOI] [PubMed] [Google Scholar]
- 34.Hoch U, Falck JR, Ortiz de Montellano PR. J Biol Chem. 2000;275:26952–26958. doi: 10.1074/jbc.M004841200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Staining of covalently attached heme with 3,3’,5,5’-tetramethylbenzidine.