

Abstract
The prevalence of resistance to known anti-malarial drugs has resulted in the expansion of anti-malarial drug discovery efforts. Academic and non-profit institutions are partnering with the pharmaceutical industry to develop new anti-malarial drugs. Several new anti-malarial agents are undergoing clinical trials, mainly those resurrected from previous anti-malarial drug discovery programs. Novel anti-malarials are being advanced through the drug development process, of course with the anticipated high failure rate typical of drug discovery. Many of these are summarized in this review. Mechanisms for funding anti-malarial drug discovery and genomic information to aid drug target selection have never been better. It remains to be seen whether ongoing efforts will be sufficient for reducing malaria burden in the developing world.
It is common knowledge that malaria is a serious worldwide health problem due to the emergence of parasites that are resistant to well-established anti-malaria drugs. While continued attempts to develop a vaccine for malaria are ongoing, drugs continue to be the only treatment option [1]. In this review, we will highlight some of the anti-malaria drug discovery efforts that are currently being developed at universities, research institutions and pharmaceutical companies worldwide. Over the past decade, hundreds of reports of anti-malarial substances have appeared in the primary and patent literature; however, many of these reports disclose compounds that block the growth of malaria parasites in human erythrocyte cultures at concentrations in the micromolar range. Based on the fact that values of ED50 (the concentration of compound that causes 50% growth inhibition of cultured malaria parasites) of well established anti-malarial drugs, such as chloroquine and artemisinin, are in the low nanomolar range and emergence of clinically significant resistant parasites occurs when the ED50 for the drug increases by ~10-fold, it is probably the case that only compounds that have values of ED50 in the low nanomolar range can be considered as significant anti-malarial leads. Thus, this review will focus on reports where low nanomolar anti-malarial potency has been demonstrated. A second point is that compounds with values of ED50 in the micromolar range probably stunt the growth of parasites by binding to multiple targets. In these cases, it will be difficult to take a rational and iterative approach for improving the potency of lead compounds. An example of studies from the author’s laboratory is revealing. It has been shown that compounds that are sub-nanomolar inhibitors of the protein farnesyltransferase enzyme from the malaria parasite are cytotoxic to parasites with values of ED50 in the low nanomolar range [2]. Five structurally-distinct low nanomolar inhibitors of human protein farnesyltransferase were found to have no effect on malarial protein farnesyltransferase when tested at concentrations up to 10 μM, yet all of these compounds blocked the growth of parasites in culture with values of ED50 in the 2–10 μM range [2], very likely due to off-target effects.
Drug discovery in general is a very challenging process but it is espeically challenging for anti-malarials for several reasons [1]: 1) It is generally agreed among physicians in malaria endemic countries that drugs for malaria treatment need to well tolerated and safe in humans, with side effects no worse than some of the best tolerated drugs. This is because of the large number of people that will take anti-malarials and the fact that follow-up health care is under-developed in places where malaria is prevalent; 2) Anti-malarials need to be orally bioavailable for ease of administration in a non-hospital setting; 3) Because of concern about compliance and the development of resistance, a 3 day maximum therapy for cure with once or twice a day dosing is desirable; 4) Drugs need to be used in combination to reduce the development of resistance, which increases the number of new drugs that need to be developed; 5) Anti-malarial drugs need to have a low cost of goods. This last factor is nontrivial since most drugs in use in developed countries are beyond the cost of goods requirement for anti-malarials; 6) A good part of anti-malarial drug development occurs at research centers that are not ideally structured for drug discovery (i.e. academic and non-profit research institutions).
Next generation chloroquine analogs
Current thinking is that the 4-aminoquinoline chloroquine (Fig. 1) kills malaria by accumulating in the food vacuole of parasites where it interferes with the polymerization of heme into hemazoin. Parasites resistant to chloroquine have acquired a mutant transporter PfCRT that is capable of reducing intracellular exposure to the drug. Remaining controversies with these ideas are not discussed here. Since resistance is controlled by the binding of chloroquine to one or more parasite macromolecules that are distinct from the parasite-killing target, it seems reasonable that molecules can be found that maintain potency for disrupting heme polymerization while having reduced affinity for the resistance factors. Krogstad and co-workers have been pursuing AQ-13 (Fig. 1), which is a close structural analog of chloroquine. AQ-13, like chloroquine, shows low nanomolar potency against cultures of P. falciparum-infected erythrocytes, even those that are have acquired resistance to chloroquine. Recent phase I clinical trials have shown that AQ-13 displays pharmacokinetics and toxicities that are only minimally different than those for chloroquine [3]. Continued searching for chloroquine analogs that work against chloroquine resistant parasites seems like a very logical plan. Inspired by this line or reasoning, Guy and co-workers have developed robust parallel synthetic methods for the preparation of libraries of 4-aminoquinolines and related molecules [4,5]. Several compounds were found that display ED50 values similar to that of chloroquine. These hits need to now pass through pharmacokinetic and toxicology filters to find the most promising candidates.
Figure 1.
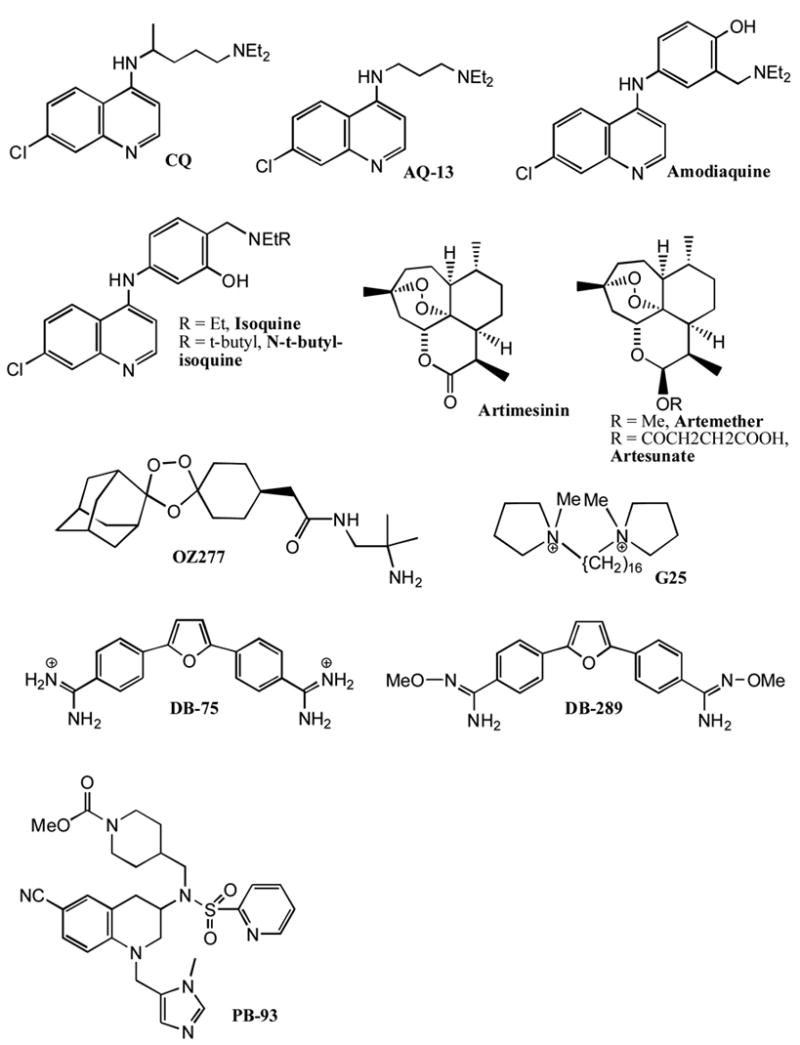
Structure of anti-malarial agents. See text for further discussion.
Amodiaquine (Fig. 1) is a 4-aminoquinoline structurally related to chloroquine but effective at killing chloroquine resistant parasites. Although amodiaquine is an effective anti-malarial in patients, it use is compromised by its hepatotoxicity and its ability to cause agranulocytosis [6]. Current thinking is that amodiaquine is oxidized by one or more cytochrome P450 enzymes to the quinone imine and that the later is responsible for the toxicity owing to its ability to react with a variety of nucleophiles. Ward and colleagues studied the regioisomer of amodiaquine in which the positions of the groups on the hydroxy aniline ring have been swapped. Isoquine (Fig. 1) emerged from this study as a promising anti-malarial agent that cannot be oxidized to the quinone imine [6]. Studies with radioactive compounds showed that, compared to amodiaquine, much less isoquine accumulated in the liver of rats. This suggests that isoquine does not form covalent adducts with liver components. Further optimization led to the discovery of N-t-butyl-isoquine (Fig. 1) [6], which is now being transitioned toward anti-malarial clinical trials.
Artemisinin and related organic peroxides
The plant derived, natural product artemisinin (Fig. 1) is now in wide use for treatment of malaria. Artemisinin-based combination therapies are now recommended by the World Health Organization for treatment of uncomplicated malaria in regions where resistance to other anti-malarials is prevalent. Further optimization of artemisinin based therapy for malaria is ongoing. A number of semi-synthetic routes to prepare artemisinin analogs, such as artemether and artesunate (Fig. 1) with changes to the δ-lactone portion have been developed with the goal of improving the pharmacokinetic prosperities [7,8]. New combination therapies where one of the components is an artemisinin-derived antimalarial are either in clinical development or are recently approved therapies. These include: Pyramax, the artemisinin analog artesunate in combination with the 4-aminoquinoline pyronaridine; Coartem, the artemisinin analog artemether in combination with lumefantrine, an anti-malarial drug developed several years ago in China and with an unknown mode of action; DHA-piperaquine, the artemisinin analog dihydroartemisin in combination with the quinoline-based drug piperaquine; the triple drug combination DCA composed of the dihydrofolate reductase inhibitor pro-drug chloroproguanil, the sulfa-drug dapsone and the artemisinin analog artesunate (www.mmv.org); ASAQ the artemisinin analog artesunate in combination with the 4-aminoquinoline amodiaquine (www.dndi.org).
The demand for plant-derived artemisinin may soon exceed the supply. In a remarkable series of bioengineering experiments, Keasling’s lab has transplanted plant biosynthetic genes into yeast to allow production of the artemisinin precursor artemisinic acid in yields that appear suitable for large scale fermentation [9]. The oxygenation of artemisinic acid to artemisinin will probably have to be carried out by non-fermentation methods since large amounts of the latter are probably toxic to cells. It remains to be seen whether a short and practical conversion of artemisinic acid to artemisinin can be achieved by chemical synthesis.
A second approach to meet the demands for artemisinin-based therapies is to develop a totally synthetic artemisinin analog that can be manufactured at a price competitive with that of the agricultural process. In a remarkable series of studies and inspired by the presence of the peroxide function of artemisinin, Vennerstrom, Charman and their co-workers developed the 1,2,4-trioxolane OZ277 (Fig. 1) as a potent antimalarial that has recently transitioned in to phase II clinical trials [10] (www.mmv.org). OZ277 lacks chiral centers and is synthesized in a short and economical fashion by Griesbaum co-ozonolysis involving the joining of O-methyl adamantanone oxime with a substituted cyclohexanone in the presence of ozone followed by post-ozonolysis side chain elaboration [10].
The mode of parasite killing by artemisinin and related peroxides is heavily debated. At one extreme is the idea that carbon-centered radicals derived from metal-induced scission of the peroxide bond lead to alkylation of a large number of intracellular targets (see Fig. 2 for a proposal for OZ277 reactivity). At the other extreme is the proposal that artemisinin acts at a single target, a Ca2+-ATPase known as PfATP6 [11]. Evidence for the former comes from the “loose” structure-activity relationships observed for anti-malarial activity of artemisinin analogs, i.e. activity is maintained even when a large number of diverse structural changes are made to OZ277. Evidence in support of PfATP6 as the major target comes from recent studies showing that parasites harboring a single point mutant in PfATP6 are resistant to artemisinin [12]. Both sets of data seem compelling, so the controversy continues. It remains possible that artemisinin and OZ277 kill parasites by different mechanisms.
Figure 2.
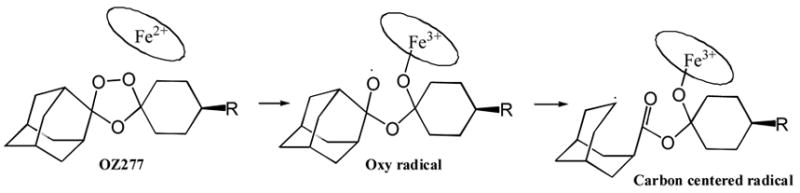
Proposed mechanism for heme iron-catalyzed generation of a carbon-centered radical from the anti-malarial agent OZ277. The carbon-centered radical is proposed to alkylate a number of parasite proteins. See text for further discussion.
Novel cationic molecules
It is well established that during rapid parasite growth in infected erythrocytes, phosphatidylcholine biosynthesis is ramped up, presumably as a result of increased membrane biosynthesis by dividing parasites. It appears that choline uptake is required for this phospholipid biosynthesis since choline uptake in infected erythrocytes is much higher than in non-infected cells. Vial and colleagues have been exploring a number of cationic molecules as choline analogs with the ability to block choline uptake. In early studies, the compound G25 (Fig. 1) emerged as a bis-cationic molecule with exceptional in vitro anti-malarial activity (ED50 = 0.6 nM) [13]. G25 is ~1000-fold less toxic to mammalian cells lines. Later studies showed that G25 selectively accumulates in malaria-infected erythrocytes [14]. Treatment of malaria-infected monkeys with low doses of G25 (i.m. 0.15 mg/kg twice daily for 8 days) was sufficient to achieve a cure in all 5 treated animals [14]. Compounds such as G25 display poor oral bioavailability presumably because they contain permanent positive charges. In a remarkable series of studies, Vial and co-workers reported that the bis-cationic compound T3 (Fig. 3) displays potent in vitro anti-malarial activity with ED50s in the 2–9 nM range [15]. In the same report they found that the pro-drug TE3 (Fig. 3) is stable in intestinal fluid, slowly breaks down to T3 in gastric fluid (half-time ~ 8 hr), and rapidly breaks down in human plasma (half-time ~ 5 min). The oral bioavailability of TE3 in rats is 16%, and this pro-drug is able to cure malaria in monkeys when dosed orally in the 3–27 mg/kg range once a day for 4 days [15]. These compounds are exciting leads for anti-malarial drug discovery. Whether a malarial choline transporter is the target of these compounds remains to be conclusively established.
Figure 3.
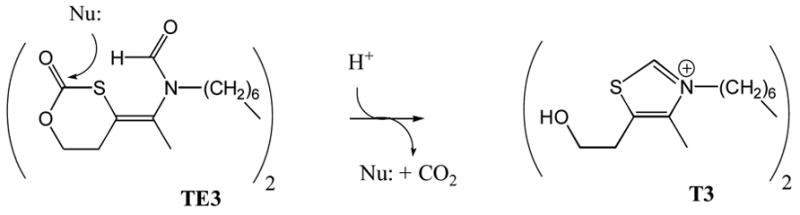
Generation of the active anti-malarial agent T3 from the pro-drug TE3. See text for further discussion.
Tidwell and co-workers have been studying a family of bis-cationic molecules as anti-microbial agents for several years. One compound, DB-75 (Fig. 1) is a broad spectrum anti-microbial that displays low nanomolar potency as an inhibitor of the growth of Trypanosoma brucei, the parasite that causes African sleeping sickness. DB-75 is structurally related to pentamidine, a compound known to posses anti-protozoal activity since the 1930s. A major breakthrough came when it was discovered that the diamidoxime pro-drug, DB-289 (Fig. 1) is orally absorbed and then converted to the active DB-75 in the bloodstream [16]. DB-289 is currently undergoing clinical trials for the treatment of African sleeping sickness. In the meantime, DB-289 was found to display good potency against malaria [17]. Given the fact that DB-289 had already gone through phase I clinical trials for the treatment of African sleeping sickness, a clinical trial to test this compound for efficacy in malaria was initiated in Thailand [17]. The study showed that DB-289 exhibited good efficacy in clearing patients of P. falciparum and P. vivax infections. However, because efficacy required ~7 days, it was decided to halt clinical testing of DB-289 for malaria with the hope that a more active analog can be found that can achieve a cure of malaria in ~3 days (in order to meet the anti-malarial drug target parameters listed above).
The mode of microbial killing by bis-amidines like pentamidine and DB-75 is unknown. It is well established that these compounds accumulate to high concentrations in Trypanosoma brucei [18] and likely in P. falciparum-infected human red blood cells. Given the high intracellular concentration, multiple targets seem likely.
Piggyback medicinal chemistry
The classical approach for the development of anti-malarials is to focus on parasite targets for which no human host homolog exists, thereby minimizing the chance of drug toxicity. While this concept certainly is rational, the downside is that the process of drug development starting only from a parasite-specific target is a very long and expensive process. The piggyback approach is to explore classes of drug molecules as anti-malarials that have already been thoroughly evaluated as drug leads for diseases that have been addressed by major pharma. One example is protein farnesyltransferase inhibitors that have been extensively developed over the past 15 years as anti-cancer agents. It was shown several years ago shown that P. falciparum contains protein farnesyltransferase, and that inhibitors of this enzyme are significantly more toxic to parasites than to human cells. Extensive clinical trials of protein farnesyltransferase inhibitors have shown that they are well tolerated in man during 2–3 weeks of continuous dosing, so it seems that an inhibitor that is selective for the malaria enzyme is not needed, especially since malaria treatment is typically a few day course of drug administration. After testing all of the major classes of protein farnesyltransferase inhibitors that have been developed at major drug companies, Gelb, Van Voorhis and their colleagues found that the tetrahydroquinoline class of compounds, developed at Bristol Myers Squibb, display low nanomolar potency for killing cultured P. falciparum [2] (PB-93 in Fig. 1 is one example). The mode of parasite killing is very likely due to inhibition of protein farnesylation since parasites that have become resistant to tetrahydroquinolines contain a mutant protein farnesyltransfearse that displays a ~10-fold reduction in affinity for these inhibitors [19]. The current status of protein farnesyltransferase inhibitors as anti-malarials is that the can cure malaria-infected rodents [2], but further work is needed to reduce the metabolic instability of this class of drug leads.
Miscellaneous drug discovery efforts
A number of promising anti-malarial drug discovery projects are being funded by the Medicines for Malaria Venture (MMV), a non-profit organization focused on anti-malarial drug discovery. These projects are only listed here since results have not been published. These include: 1) Inhibitors of cysteine proteases involved in the degradation of hemoglobin (falcipain inhibitors); Inhibitors of dihydrofolate reductase; 3) Inhibitors of the enoyl-acyl carrier protein component of type II fatty acid synthase. Additional information about these projects can be found at http://mmv.org.
Closing Remarks
There is considerable progress in the development of new anti-malarials owing in large part to the availability of new funding mechanisms for drug discovery research (i.e. The Bill and Melinda Gates Foundation, The Wellcome Trust, Irish Aid, USAID, and others). What seems to be working more than anything else in the past decade is the development of Public-Private Partnerships (PPPs) in which Pharma contributes drug discovery skills, whereas academic and non-profit institutions contribute basic biology leading to drug target selection and field expertise [20]. Many of the most successful anti-malaria drug development programs are proceeding through the Medicines for Malaria Venture [21], which started in 1999. It is fair to say that most of the promising new anti-malarial agents in clinical trials are either resurrected agents from past anti-malarial programs that were halted years ago, often in Pharma because of budget restrictions, or are structural analogs of known anti-malarials (i.e. chloroquine and artemisinin analogs). The failure rate for novel anti-malarials is high and will probably remain high for the foreseeable future. There is valid concern that the number of programs focusing on novel anti-malarials is insufficient to sustain the anti-malarial drug pipeline. Having said all of that, the anti-malarial drug discovery funding mechanisms and availability of information for drug target selection have never been better.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (AI54384) and by the Medicines for Malaria Venture (Geneva, Switzerland).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nwaka S, Hudson A. Innovative lead discovery strategies for tropical diseases. Nat Rev Drug Discov. 2006;5:941–955. doi: 10.1038/nrd2144. • This is a recent review article that talks about various aspects of drug development for neglected tropical diseases including the special requirement for a low cost of goods and the unusual mechanisms for funding projects. [DOI] [PubMed] [Google Scholar]
- 2.Nallan L, Bauer KD, Bendale P, Rivas K, Yokoyama K, Horney CP, Pendyala PR, Floyd D, Lombardo LJ, Williams DK, et al. Protein Farnesyltransferase Inhibitors Exhibit Potent Antimalarial Activity. J Med Chem. 2005;48:3704–3713. doi: 10.1021/jm0491039. • This article is a nice example of the “piggy back” drug discovery method. [DOI] [PubMed] [Google Scholar]
- 3.Mzayek F, Deng H, Mather FJ, Wasilevich EC, Liu H, Hadi CM, Chansolme DH, Murphy HA, Melek BH, Tenaglia AN, et al. Randomized Dose-Ranging Controlled Trial of AQ-13, a Candidate Antimalarial, and Chloroquine in Healthy Volunteers. PLoS Clin Trials. 2007;2:e6. doi: 10.1371/journal.pctr.0020006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Madrid PB, Wilson NT, DeRisi JL, Guy RK. Parellel Synthesis and Antimalarial Screening of a 4-Aminoquinoline Library. J Comb Chem. 2004;6:437–442. doi: 10.1021/cc0340473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson MO, Sherrill J, Madrid PB, Liou AP, Weisman JL, DeRisi JL, Guy RK. Parallel synthesis of 9-aminoacridines and their evaluation against chloroquine-resistant Plasmodium falciparum. Bioorg Med Chem. 2006;14:334–343. doi: 10.1016/j.bmc.2005.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Neill PM, Mukhtar A, Stocks PA, Randle LE, Hindley S, Ward SA, Storr RC, Bickley JF, O’Neil IA, Maggs JL, et al. Isoquine and Related Amodiaquine Analogues: A New Generation of Improved 4-Aminoquinoline Antimalarials. J Med CHem. 2003;46:4933–4945. doi: 10.1021/jm030796n. •• This article describes a highly successful anti-malarial drug discovery program that is approach clinical trials. It also nicely illustrates how knowledge of the mechanism of drug toxicity can lead to a rational plan for drug improvement. [DOI] [PubMed] [Google Scholar]
- 7.Posner GH, O’Neill PM. Knowledge of the proposed chemical mechanism of action and cytochrome p450 metabolism of antimalarial trioxanes like artemisinin allows rational design of new antimalarial peroxides. Acc Chem Res. 2004;37:397–404. doi: 10.1021/ar020227u. [DOI] [PubMed] [Google Scholar]
- 8.Paik IH, Shapiro TA, Labonte T, Narducci SAA, Baege AC, Posner GH. Second generation, orally active, antimalarial, artemisinin-derived trioxane dimers with high stability, efficacy, and anticancer activity. J Med Chem. 2006;49:2731–2734. doi: 10.1021/jm058288w. [DOI] [PubMed] [Google Scholar]
- 9.Ro DK, Paradise EM, Quellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature. 2006;440:940–943. doi: 10.1038/nature04640. •• This is a nice article that illustrates the possibility of production of natural products in a readily fermentable microorganism using modern bioengineering methods. It describes a very important area of synthetic methodology that has to be considered along with conventional, non-fermentatation organic synthesis. [DOI] [PubMed] [Google Scholar]
- 10.Vennerstrom JL, Arbe-Barnes S, Brun R, Charman SA, Chiu FC, Chollet J, Dong Y, Dorn A, Hunziker D, Matile H, et al. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature. 2004;430:900–904. doi: 10.1038/nature02779. •• This article describes a culmination of a tour de force effort to make analogs of the well established artemisinin-based anti-malarials using a relatively simple synthetic route. [DOI] [PubMed] [Google Scholar]
- 11.Eckstein-Ludwig U, Webb RJ, Van Goethem ID, East JM, Lee AG, Kimura M, O’Neill PM, Bray PG, Ward SA, Krishna S. Artemisinins target the SERCA of Plasmodium falciparum. Nature. 2003;424:957–961. doi: 10.1038/nature01813. [DOI] [PubMed] [Google Scholar]
- 12.Uhlemann AC, Cameron A, Eckstein-Ludwig U, Fischbarg J, Iserovich P, Zuniga FA, East M, Lee A, Brady L, Haynes RK, et al. A single amino acid residue can determine the sensitivity of SERCAs to artemisinins. Nat Struct Mol Biol. 2005;12:628–629. doi: 10.1038/nsmb947. [DOI] [PubMed] [Google Scholar]
- 13.Calas M, Ancelin ML, Cordina G, Portefaix P, Piquet G, Vidal-Sailhan V, Vial HJ. Antimalarial activity of compounds interfering with Plasmodium falciparum phospholipid metabolism: comparison between mono- and bisquaternary ammonium salts. J Med Chem. 2000;43:505–516. doi: 10.1021/jm9911027. [DOI] [PubMed] [Google Scholar]
- 14.Wengelnik KVV, Ancelin ML, Cathiard AM, Morgat JL, Kocken CH, Calas M, Herrera S, Thomas AW, Vial HJ, Related Articles L, Wengelnik K, Vidal V, Ancelin ML, Cathiard AM, Morgat JL, Kocken CH, Calas M, Herrera S, et al. A class of potent antimalarials and their specific accumulation in infected erythrocytes. Science. 2002;295:1311–1314. doi: 10.1126/science.1067236. •This article describes some of the most highy potent anti-malarials every reported. [DOI] [PubMed] [Google Scholar]
- 15.Vial HJ, Wein S, Farenc C, Kocken C, Nicolas O, Ancelin ML, Bressolle F, Thomas A, Calas M. Prodrugs of bisthiazolium salts are orally potent antimalarials. PNAS. 2004;101:15458–15463. doi: 10.1073/pnas.0404037101. ••This article shows a clever prodrug approach for rendering charged compounds into orally bioavailable drug precursors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang MZ, Saulter JY, Usuki E, Cheung YL, Hall M, Bridges AS, Loewen G, Parkinson OT, Stephens CE, Allen JL, et al. CYP4F enzymes are the major enzymes in human liver microsomes that catalyze the O-demethylation of the antiparasitic prodrug DB289 [2,5-bis(4-amidinophenyl)furan-bis-O-methylamidoxime] Drug Metab Dispos. 2006;34:1985–1994. doi: 10.1124/dmd.106.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yeramian P, Meshnick SR, Krudsood S, Chalermrut K, Silachamroon U, Tangpukdee N, Allen J, Brun R, Kwiek JJ, Tidwell R, et al. Efficacy of DB289 in Thai patients with Plasmodium vivax or acute, uncomplicated Plasmodium falciparum infections. J Infect Dis. 2005;192:319–322. doi: 10.1086/430928. ••This article describes a class of structrually simple and highly potent anti-malarials that are promising candidates for new drugs. [DOI] [PubMed] [Google Scholar]
- 18.Mathias AM, Holman JL, Sturk LM, Ismail MA, Boykin DW, Tidwell RR, Hall JE. Accumulation and intracellular distribution of antitrypanosomal diamidine compounds DB75 and DB820 in African trypanosomes. Antimicrob Agents Chemother. 2006;50:2185–2191. doi: 10.1128/AAC.00192-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eastman RT, White J, Hucke O, Bauer K, Yokoyama K, Nallan L, Chakrabarti D, Verlinde CL, Gelb MH, Rathod PK, et al. Resistance to a protein farnesyltransferase inhibitor in Plasmodium falciparum. J Biol Chem. 2005;280:13554–13559. doi: 10.1074/jbc.M413556200. [DOI] [PubMed] [Google Scholar]
- 20.Croft SL. Public-private partnership: from there to here. Trans R Soc Trop Med Hyg. 2005;99:S9–S14. doi: 10.1016/j.trstmh.2005.06.008. •This article describes the most successful current funding mechanism for the development of drugs for neglected tropical diseases. [DOI] [PubMed] [Google Scholar]
- 21.Bathurst I, Hentschel C. Medicines for Malaria Venture: sustaining antimalarial drug development. Trends Parasitol. 2006;22:301–307. doi: 10.1016/j.pt.2006.05.011. •This article also describes the most successful current funding mechanism for the development of drugs for malaria. [DOI] [PubMed] [Google Scholar]