

Abstract
Heterotetrameric sarcosine oxidase (TSOX) is a complex bifunctional enzyme that catalyzes the oxidation of the methyl group in sarcosine (N-methylglycine) and transfer of the oxidized methyl group into the 1-carbon metabolic pool. In addition to four different subunits, TSOX contains three coenzymes (FAD, FMN, NAD) and a binding site for tetrahydrofolate, the coenzyme acceptor of the oxidized methyl group from sarcosine. Based on preliminary success in crystallization of the natural enzyme, the genes encoding the subunits for TSOX from Pseudomonas maltophila (pTSOX) were cloned by functional screening of a genomic library. Recombinant enzyme exhibiting the same specific activity as natural pTSOX could not be isolated using a similar or identical purification procedure. This difficulty was overcome by affinity purification of recombinant pTSOX containing a C-terminal (His)6 tag on the subunit (γ) encoded by soxG, the gene located at the 3′ end of the pTSOX operon. Affinity purified pTSOX could not be crystallized, a problem traced to microheterogeneity in the recombinant enzyme where about half of the FMN is present in a modified form that is not found in the natural enzyme and may be a biosynthetic intermediate. The modified flavin was eliminated by expression of the recombinant enzyme in the presence of sarcosine, the same reagent used to induce expression of the natural enzyme. Homogenous recombinant pTSOX was isolated from cells grown in the presence of sarcosine by chromatography on affinity and hydrophobic interaction matrices. High quality crystals that diffract to 1.85 Å resolution have been obtained.
Heterotetrameric sarcosine oxidase (TSOX1) is an inducible bacterial enzyme that is important in the catabolism of sarcosine (N-methylglycine), a common soil metabolite that can act as sole source of carbon and energy for many microorganisms [1]. TSOX contains four different subunits (α, β, γ, δ) that range in size from about 10 to 100 kDa, three coenzymes (NAD, FMN, FAD) and a binding site for a fourth coenzyme (tetrahydrofolate, H4folate) that acts as a substrate [2–7]. NAD is noncovalently bound to the α subunit [8]. FMN is covalently attached to a histidine residue in the β subunit but the flavin ring appears to be located at a subunit interface [5,8]. FAD is noncovalently bound, probably to the β subunit [8].
TSOX is a bifunctional enzyme that catalyzes both sarcosine oxidation and the synthesis of 5,10-methylenetetrahydrofolate (5,10-CH2-H4 folate). Sarcosine binds near the noncovalently bound FAD and is oxidized to the corresponding imine, accompanied by the reduction of FAD to 1,5-dihydroFAD (FADH2). Electrons from FADH2 are transferred, one at a time, to the covalently bound FMN which then reduces oxygen to hydrogen peroxide [9,10]. In a second reaction, TSOX catalyzes the transfer of the oxidized 1-carbon fragment from the sarcosine imine to H4folate, forming 5,10-CH2-H4folate plus glycine [6,7] (Scheme 1). The enzyme is also active in the absence of H4folate. Under these conditions, the sarcosine imine is hydrolyzed to yield glycine and formaldehyde. NAD does not participate in sarcosine oxidation [3]. It has been suggested that this coenzyme plays an important structural role in TSOX [8].
Scheme 1.
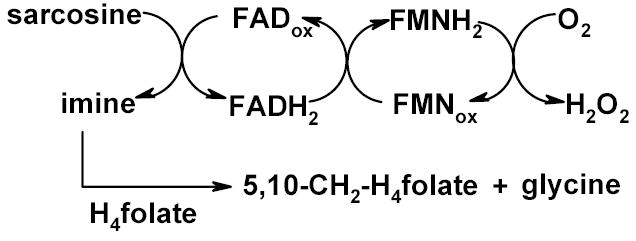
Reactions catalyzed by TSOX
TSOX is the most complex member of a family of amine oxidizing flavoenzymes. This family is mainly composed of relatively small (~44 kDa), monomeric proteins with covalently bound FAD as the only prosthetic group, such as monomeric sarcosine oxidase (MSOX), N-methyltryptophan oxidase, pipecolate oxidase and nikD [11–18]. Interestingly, MSOX also catalyzes sarcosine oxidation but is a monofunctional enzyme and does not use H4folate as a substrate [6], unlike TSOX. Crystal structures of free MSOX and complexes with substrate analogs are available [11,14].
TSOX has been purified from at least a half dozen different bacteria [19–24] but no crystal structure has been reported despite its clear importance in unraveling the structural and functional organization of this complex enzyme. Arguably the most highly characterized TSOX is the enzyme from Corynebacterium sp. P-1 (cTSOX) [1–10,19]. Despite considerable and prolonged effort, our attempts to crystallize this enzyme were not successful. We therefore initiated a survey of different bacteria in an attempt to identify a source of crystallizable TSOX. Promising results were obtained with the enzyme (pTSOX) isolated from Pseudomonas maltophilia. Purification of the natural form of this enzyme was first described in a 1991 US patent [23]. In 2001 we briefly noted the presence of modified flavin in recombinant pTSOX [8]. In this paper, we report the cloning of the pTSOX genes and expression and purification of recombinant pTSOX, including techniques for removal of microhetereogeneity that result in the formation of well-ordered crystals that diffract to 1.85 Å resolution.
MATERIALS AND METHODS
Plasmids and bacteria.
Pseudomonas maltophilia (DSM 2701, renamed as Stenotrophomomas maltophilia) was obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH. pBluescript II SK(+) and E. coli strain XL-1 Blue were purchased from Stratagene. Plasmids pET17b and pET28b were purchased from Novagen. TA cloning vector pCR2.1 and E. coli INVαF’ were obtained from Invitrogen. E. coli XL-1 Blue/pLJC400 cells were previously developed in this laboratory [25].
General procedures.
PCR was conducted using Pfu DNA polymerase (Stratagene) and a Hybaid Touchdown Thermocycler. PCR products were purified using one of the following methods: 1) QIAquick PCR Purification Kit (Qiagen) or 2) agarose gel (0.8 or 1.5%) electrophoresis, followed by recovery using the QIAquick Gel Extraction Kit (Qiagen). The second method was also used to purify DNA fragments from restriction digests. Ligations were performed using T4 DNA ligase (Promega). Oligonucleotides for PCR amplification were prepared by MWG-Biotech. Sequencing of P. maltophilia DNA and PCR products was conducted by the Nucleic Acid/Protein Research Core Facility at the Children’s Hospital of Philadelphia. All restriction enzymes were purchased from New England Biolabs.
Activity and Protein Assays.
Sarcosine oxidase activity in the absence of H4folate was determined on the basis of the amount of formaldehyde formed using the Nash procedure [26], as described previously [27]. Protein was determined using the Bio-Rad micro protein assay [13].
Purification of pTSOX from P. maltophilia and NH2-terminal sequence analysis.
P. maltophilia was grown at 29 °C in the same medium used for growth of Corynebacterium sp. P-1 [19], except the amount of yeast extract was increased to 2.5 g/L. The natural P. maltophilia enzyme was isolated by a procedure similar to that described for the isolation of cTSOX from C. sp. P-1 [25] except: 1. The concentration of MgSO4 in the cell lysis buffer was increased to 2.5 mM; 2. pTSOX was precipitated from the cell lysate with 85% ammonium sulfate; 3. the Ultrogel ACA 34 chromatography step was omitted; 4. DEAE-cellulose chromatography was performed before, rather than after, Phenyl-Sepharose CL-4B chromatography. For NH2-terminal sequence analysis, the purified enzyme was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The separated subunits then were transferred to PVDF membranes. Sequence analysis was conducted at the Harvard Microchemistry Facility.
Construction and screening a P. maltophilia genomic library.
P. maltophilia was grown as described above. Genomic DNA was isolated using a Qiagen DNeasy tissue kit and then partially digested with Sau3AI. The digest was fractionated on a 0.6% agarose gel to yield a pool of DNA fragments of approximately 4.4 to 9.0 kb. pBluescript II SK(+) was digested with BamHI, dephosphorylated with calf intestinal alkaline phosphatase (Promega) and then mixed with the 4.4 to 9.0 kb pool of P. maltophilia DNA fragments. After treatment with T4 DNA ligase, the sample was used to transform E. coli XL1-Blue cells to ampicillin resistance. The library was screened for cells that expressed sarcosine oxidase activity using indicator plates that contained sarcosine, o-dianasidine, horseradish peroxidase and isopropyl β-D-thiogalactopyranoside (IPTG), as previously described [25]. The plates were incubated at 34 °C for 19 h and then kept at room temperature (~22°C). Under these conditions, cells expressing sarcosine oxidase activity generate hydrogen peroxide and form brown colonies. Plasmid pME4 (see Figure 1) was isolated from a clone that formed a brown colony on indicator plates.
Figure 1.
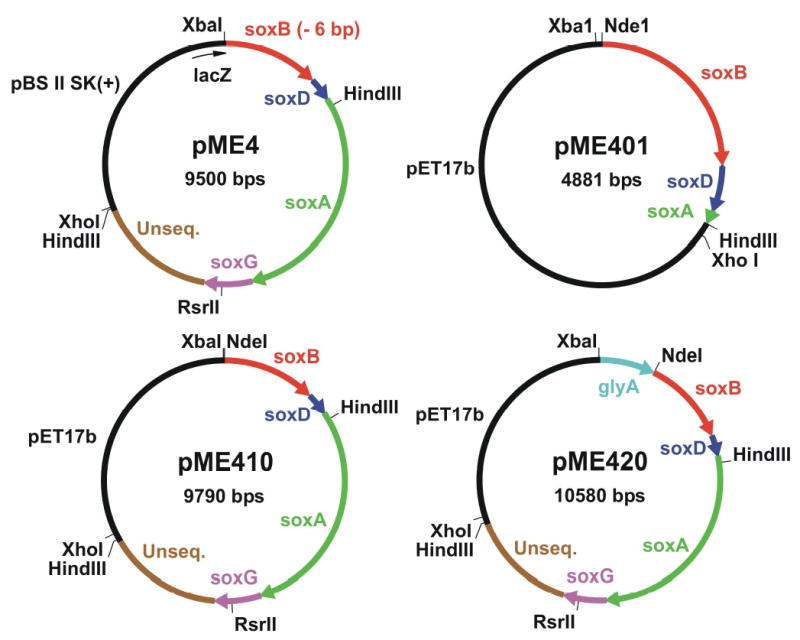
Plasmid pME4 was isolated by functional screening of a P. maltophilia DNA library for colonies expressing sarcosine oxidase activity and is missing 6 bp at the 5′ end of soxB. Plasmid pME401 is an intermediate construct that contains the complete soxB and soxD genes and a small portion from the 5′ end of soxA. Plasmid pME410 contains complete genes for all four pTSOX subunits plus an unsequenced region (unseq.) of P. maltophia DNA. Plasmid pME420 is a derivative of pME410, obtained after insertion of a fragment from the 3′ end of the corynebacterial glyA gene. For clarity, the vector-encoded lacZ gene fragment in pME4 is shown at 5 times its actual size. Arrowheads indicate the direction of transcription.
Construction of plasmid pME410.
As will be described, the P. maltophilia DNA insert in pME4 contained all four pTSOX genes, arranged in the order: soxB, soxD, soxA, soxG (coding for the β, δ, α, and γ subunits, respectively). The sequence was complete except for 6 bases coding for the first two amino acids at the N-terminus of the β subunit. PCR was conducted to add the missing 6 bases and introduce an NdeI restriction site at the 5’ end of the soxB gene. The P. maltophilia insert in pME4 contains a single HindIII site, located near the 5′ end of soxA (Figure 1). Owing to the large size of the pTSOX coding region, primers were designed to generate a PCR product extending from the newly introduced NdeI site at the beginning of soxB to the HindIII site in soxA. Reactions were performed using plasmid pME4 as template, 5′- CGCTCTAcAtaTgGctGATCTGCTGCCTGA-3′ as forward primer (NdeI site is underlined, codons for the two missing amino acids are shown in bold, lower case letters indicate mutations introduced into vector DNA immediately upstream of the P. maltophilia DNA insert) and 5′-AGCAGTGCGCTGGCTACGGTGTCG-3′ as backward primer. The backward primer was designed to hybridize to a region immediately 3′ to the HindIII site in soxA. The PCR product was cut with NdeI and HindIII to yield a desired 1,669 bp fragment that was subcloned between the NdeI and HindIII sites of plasmid pET17b. The resulting construct (pME401, see Figure 1) contained soxB, soxD and a small piece of the 5′ end of soxA.
The following approach was used to restore the missing pTSOX genes in plasmid pME401. Plasmid pME4 contains two HindIII sites: 1) a site within soxA, as described above; 2) a site within the vector-derived multicloning region, immediately downstream of the P. maltophilia insert. Plasmid pME4 was digested with HindIII to yield a 4.9 kb HindIII fragment that contained the missing part of soxA, soxG, a ~1.5 kb unsequenced region at the 3′ end of the P. maltophilia insert and a small portion of the vector multicloning site. This fragment was subcloned into the unique HindIII site of plasmid pME401. Transformed BL21(DE3) cells were plated on indicator plates to screen for clones where the 4.9 kb fragment had inserted in the proper orientation. Plasmid pME410 (see Figure 1) was isolated from a clone that formed a brown color and was sequenced to confirm the orientation of the 4.9 kb insert.
Construction of plasmid pME430.
As will be described, E. coli BL21(DE3)/pME410 proved unsatisfactory as a source of high qualtity recombinant pTSOX. The following strategy was designed to generate an expression system for pTSOX similar to that used successfully in the production of recombinant cTSOX (E. coli XL-1 Blue/pLJC400) [25]. The plasmid used for cTSOX expression (pLJC400) is a derivative of pBluescript II SK(+). The multicloning site in this vector is located within the lacZ gene, a feature that promotes expression of lacZ fusions with the products of genes inserted at this site. The cTSOX operon contains a gene coding for serine hydroxymethyltransferase (glyA), located immediately upstream of the soxB gene. A portion of glyA is found at the 5′ end of the corynebacterial DNA insert in pLJC400 where it is expressed as a lacZ-fusion protein. PCR was conducted to generate a product containing the glyA gene fragment from pLJC400. The primers were designed to include a short segment of vector DNA (harboring a unique XbaI site) at the 5′ end and to introduce a NdeI site at the 3′ end of glyA. The reaction was conducted with pLJC400 as template, 5′-GCGCAATTAACCCTCACTA-3′ as forward primer and 5′-ATCAGCCATatgCTATTCTCCGATC-3′ as backward primer (NdeI site is underlined and mutations are shown in lower case). The PCR product was cut with XbaI and NdeI. The desired 868 bp fragment was subcloned between the XbaI and NdeI sites of plasmid pME410 to yield a new construct (pME420, see Figure 1) containing the glyA gene fragment just upstream of soxB, with 3 bp separating the end of glyA from the start of soxB. Plasmid pME420 was used to transform XL-1 Blue cells to ampicillin resistance. In a final step, the entire insert in pME420 (glyA plus the P. maltophilia DNA) was transferred into pBluescript II SK(+). Plasmid pME420 was digested with XbaI and XhoI to yield a desired 7419 bp product that was subcloned between the XbaI and XhoI sites of pBluescript II SK(+). The resulting plasmid (pME430, see Figure 2) was used to transform XL-1 Blue cells to ampicillin resistance.
Figure 2.
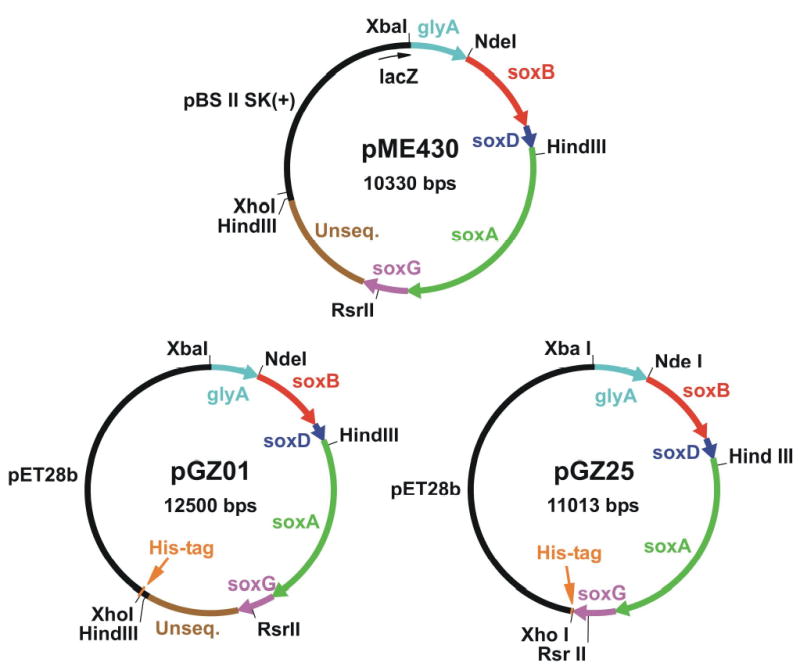
Plasmid pME430 was obtained by transferring the entire insert from pME420 (see Figure 1) into pBluescript II SK(+). Plasmid pGZ01 is an intermediate construct obtained after transfer of the insert from pME430 into a vector encoding a (His)6 affinity tag. Plasmid pGZ25 is a derivative of pGZ01 where the vector-encoded (His)6 tag is positioned immediately downstream of soxG. For clarity, the vector-encoded lacZ gene fragment in pME430 is shown at 5 times its actual size. Arrowheads indicate the direction of transcription.
Construction of plasmid pGZ25.
The following approach was designed to introduce an affinity tag at the C-terminus of the γ subunit. Firstly, the entire insert in pME430 was transferred into pET28b, a vector designed to introduce either a N‐ or C-terminal His tag. Plasmid pME430 was digested with XbaI and XhoI to yield a desired 7419 bp fragment that was subcloned between the XbaI and XhoI sites of plasmid pET28b. The resulting plasmid (pGZ01, see Figure 2) was used to transform E. coli BL21 (DE3) to kanamycin resistence. The next step was designed to remove an unsequenced region of P. maltophilia DNA (~1.5 kB, located between the 3′ end of soxG and the vector-encoded His tag), eliminate the soxG stop codon and introduce a XhoI restriction site at the 3’ end of the soxG coding sequence. PCR was used to synthesize a DNA fragment starting from a unique RsrII site near the 3′ end of soxG, continuing through the coding region of soxG and ending with a XhoI site introduced by the backward primer. Reactions were performed using plasmid pGZ01 as template, 5′-CGGACCGGATGCGCCGCTGGTGCTGCGCAAG-3′ as forward primer (RsrII site is underlined) and 5′-CTCGAGGATCACTGCTTCCGAGGCGAAC-3′ as backward primer (XhoI site is underlined). The PCR product (235 pb) was purified, incubated at 37 °C for 3 h with Taq polymerase (Qiagen) and dATP, re-purified and then ligated with TA cloning vector pCR2.1 in an overnight incubation at 14 °C. The resulting plasmid was transferred into E. coli INVαF’. Transformants were subjected to blue-white screening. Plasmid DNA was isolated from a solid white colony using Plasmid Midi Kit (Qiagen) and digested with XhoI and RsrII to yield a desired 232 bp fragment (DNA-Y). Plasmid pGZ01 was digested with XhoI and RsrII to yield a 10784 kB fragment that contained all of the DNA from pGZ01 except for a region extending from the RsrII site within the soxG gene to the 3′ end of the P. maltophilia DNA insert. The 10784 kb fragment was ligated with the 232 bp DNA-Y fragment. The resulting construct was used to transform E. coli BL21(DE3) to kanamycin resistance. A clone containing the desired plasmid (pGZ25, see Figure 2) was selected based on restriction analysis and sequenced across the DNA-Y insert.
Purification of recombinant pTSOX using pME410 or pME430 as expression vector.
E. coli BL21 (DE3)/pME410 or XL-I Blue/pME430 cells were grown at 29 °C in Luria-Bertani medium (LB) (6 L) containing ampicillin (100 μg/mL) until A600 ~0.6. pTSOX expression was then induced with 0.4 [BL21 (DE3)/pME410] or 0.2 (XL-I Blue/pME430) mM IPTG. The cells were harvested after an overnight incubation and lysed by sonication, as previously described [25]. pTSOX was purified by a procedure involving precipitation with 80% ammonium sulfate, followed by chromatography on Ultrogel ACA 34, Phenyl-Sepharose and DEAE-Sephacel, similar to that described for the isolation of recombinant cTSOX [25]. Recombinant pTSOX from E. coli XL-I Blue/pME430 was also purified using the procedure described above for the isolation of the natural enzyme from P. maltophilia.
Purification of His-tagged pTSOX using pGZ25 as expression vector.
E. coli BL21(DE3)/pGZ25 cells were grown at 25 °C in LB (5 L) containing kanamycin (100 μg/mL) and 0, 10 or 50 g/L of sarcosine until A595 ~0.6. pTSOX expression was then induced with 0.5 mM IPTG. The cells were harvested after an overnight incubation and lysed by sonication, as previously described [25], except the MgSO4 concentration was increased to 2.5 mM and the lysis buffer was changed to 50 mM sodium phosphate, pH 8.0, containing 500 mM NaCl and 20% glycerol. The soluble extract, obtained after centrifuging the cell lysate, was dialyzed against lysis buffer and then mixed with 25 ml of Co2++ affinity matrix (BD Bioscience). (Dialysis removed components in the crude extract that interfered with affinity chromatography, as judged by the poor yields obtained when the dialysis step was omitted.) After gentle rocking for 60 min, the mixture was poured into a column and washed with additional lysis buffer until the eluate exhibited no absorbance at 280 nm. pTSOX was eluted using a 1-L linear gradient from 0 to 150 mM imidazole in lysis buffer. For enzyme expressed in the presence of 0 or 10 g/L of sarcosine, the eluate was dialyzed against 20 mM sodium phosphate buffer, pH 8.0, containing 1 mM ethylenediaminetetraacetic acid (EDTA), (storage buffer), concentrated using a Centricon YM-30 ultrafiltration cell and then stored in aliquots at −80 °C. An additional step was added for enzyme expressed in the presence of 50 g/L of sarcosine since these preparations were intended for use in crystallization trials. In this case, the eluate from the affinity matrix was mixed (4:1) with a saturated solution of ammonium sulfate in 20 mM sodium phosphate buffer, pH 8.0, containing 1 mM EDTA and applied to a Phenyl-Sepharose column (1.8 x 25 cm) equilibrated with 20% saturated ammonium sulfate in 20 mM sodium phosphate buffer, pH 8.0, containing 1 mM EDTA. After washing the column with the equilibration buffer, pTSOX was eluted with a 240-ml gradient from 20% to 0% saturated ammonium sulfate in 20 mM sodium phosphate buffer, pH 8.0, containing 1 mM EDTA. The enzyme was dialyzed against storage buffer, concentrated and then stored at −80 °C as described above.
Crystallization of His-tagged pTSOX.
Crystallization trials were conducted with recombinant enzyme expressed in the presence of sarcosine (50 g/L) and then purified by affinity and hydrophobic interaction chromatography, as detailed above. The trials were carried out by using the sparse-matrix sampling technique [28], a variety of screening kits (Hampton Research, Laguna Niguel CA; Emerald BioSystems, Bainbridge Island, WA) and the hanging drop vapor diffusion method. Diffraction quality crystals were obtained by using a protein stock solution (19.4 mg/mL in 20 mM Na2PO4 buffer, pH 8.0, 1 mM EDTA) that was diluted to 15 mg/mL with 10 mM Na2PO4 buffer, pH 7.2. Each drop (2 L) was mixed with an equal volume of a reservoir solution consisting of 17–20% PEG 20 K, 100 mM Tris buffer, pH 8.5, 40 mM sodium furoate and 10 mM Na2SO3 and allowed to equilibrate with the reservoir solution (1.0 mL) at room temperature. Prior to data collection, the crystal was incubated with a cryoprotection solution consisting of the reservoir solution plus 15% glycerol and then exposed to a nitrogen gas stream maintained at ~100 K. X-ray diffraction data were collected at the Biocars beamline 14BMC of the Advanced Photon Source, Argonne, IL.
RESULTS AND DISCUSSION
Cloning the genes encoding the four subunits of pTSOX.
Since we anticipated that the pTSOX genes might be organized into a single operon of about 5 kb, a P. maltophilia genomic library was constructed using DNA fragments that were mostly large enough to contain the complete operon and thereby enable functional screening of the library. The library was constructed using pBluescript II SK(+), a vector where open reading frames (ORFs) inserted into the multicloning site are expressed as lacZ fusions. The library was screened using indicator plates where clones expressing sarcosine oxidase activity form brown colonies. Assuming a typical bacterial genome size, we estimated that it would be necessary to screen about 4600 colonies in order to find a clone containing the desired 5 kb fragment. This value was calculated using the relationship, N = ln (1−P)/ln[1−(I/G) with P(probability) = 0.99, I (fragment size) = 5 kb, G (genome size) = 5000 kb [29]. In fact, a single brown colony was found after screening 3900 colonies. The sarcosine-dependence of the brown color was verified by re-plating the positive clone on indicator plates lacking sarcosine.
The plasmid isolated from the brown colony (pME4, see Figure 1) contained a 6.5 kb insert of P. maltophilia DNA, as judged by restriction analysis. We sequenced 5225 bp, starting from the 5′ end of the insert. This region contained 4 ORFs arranged in the order soxB, soxD, soxA, soxG and coding for the β, δ, α, and γ subunits of pTSOX, respectively (Gene Bank Accession No. AY953276). The gene arrangement and DNA sequence at the 5′ end of the genes and in the intergenic regions are shown in Figure 3. The ORFs were identified based on the expected size of the corresponding pTSOX subunits (Table 1) and the agreement between the deduced amino acid sequence and observed N-terminal sequence data (Figure 3). The soxB and soxD genes are separated by 11 bp. The stop codon of soxD overlaps with the start codon of soxA. The start codon of soxG is found 8 bp before the end of soxA. The organization of the pTSOX genes is strikingly similar to that observed for the corresponding cTSOX genes [2]. Amino acid sequence comparisons show a variable extent of sequence conservation between the two enzymes, depending on the subunit. The highest degree of homology is observed for the β subunit (96% identity). Less, albeit appreciable, homology is observed for the other three subunits which exhibit 72% to 84% sequence identity (Table 1).
Figure 3.
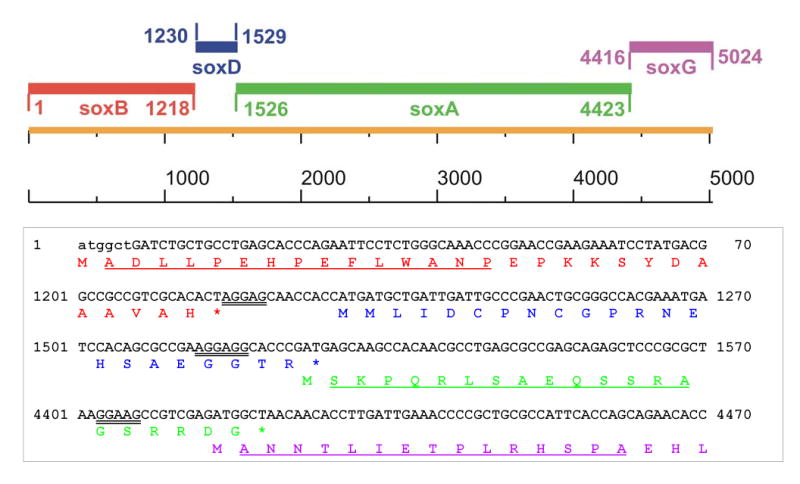
The top panel shows the organization of the pTSOX genes. The bottom panel shows the nucleotide sequence at the 5′ end of each of the pTSOX genes and the intergenic regions along with the corresponding deduced amino acid sequence. The first 6 bp of soxB were missing in the original clone (pME4) and are shown in lower case. The deduced amino acid sequences of the β, δ, α and γ subunits are shown in colors (red, blue, green and magenta, respectively) that match those used for the corresponding genes in the top panel. Putative ribosomal-binding sites are indicated by double underlining. N-terminal peptide sequence data were obtained for all subunits except δ. The observed peptide sequences (indicated by single underlining) matched the deduced amino acid sequence.
Table 1.
Comparison of the subunits in pTSOX and cTSOX
| subunit | number of amino acids
|
||
|---|---|---|---|
| P. maltophila | C. sp. P-1 | Identitya (%) | |
| alpha | 965 | 967 | 84 |
| beta | 405 | 405 | 96 |
| gamma | 202 | 203 | 72 |
| delta | 99 | 98 | 80 |
Similar results were obtained using the GAP or BESTFIT programs in the Genetics Computer Group package.
Purification of recombinant pTSOX using pME410 or pME430 as expression vector.
Comparison of the observed N-terminal sequence with the deduced sequence of the β subunit showed that 6 bp were missing from the 5′ end of soxB in pME4 (Figure 3). Sarcosine oxidizing activity was observed using pME4 as expression vector despite the fact that the β subunit contains essential redox cofactors [4,5,8]. The observed activity is explained by an in-frame insertion of soxB into the vector-encoded lacZ gene, resulting in the expression of the β subunit as a lacZ fusion. PCR was used to replace the missing 6 bp. The full-length copy of soxB, along with the other pTSOX genes, were incorporated into a new vector (pME410, Figure 1) where gene expression is controlled by the T7 promoter. The specific activity of recombinant pTSOX, isolated from cells transformed with pME410, was 35% lower than observed for the natural enzyme (Table 2). The specific activity could not be improved by adding steps to the purification procedure.
Table 2.
Purification of natural and recombinant pTSOX
| Preparationa | Purification Methodb | Yieldc (mg) | Specific Activity (U/mg) |
|---|---|---|---|
| Natural enzyme | AS/AX/HIC | 22 | 9.8 |
| pME410 | AS/Gel/HIC/AX | 140 | 6.4 |
| pME430 | AS/Gel/HIC/AX | 382 | 4.7 |
| pME430 | AS/AX/HIC | 198 | 6.9 |
| pGZ25 | |||
| no sarcosine | Co2+ | 498 | 9.8 |
| plus sarcosine (50 g/L) | Co2+/HIC | 49 (352)d | 10.4 |
Recombinant pTSOX was expressed under the control of the T7 (pME410, pGZ25) or the lac (pME430) promoter.
AS, ammonium sulfate precipitation; AX, anion exchange chromatography; HIC, hydrophobic interaction chromatography (Phenyl-Sepharose); Gel, gel filtration chromatography; Co2+, affinity chromatography.
The volume of cell culture varied between 5–10 L. Yields are normalized to 10 L of cell culture.
The value in parentheses is the yield obtained after the Co2+ affinity step.
We had previously observed similar specific activities for the natural form of cTSOX and recombinant cTSOX isolated from E. coli XL-1 Blue/pLJC400 [25]. We therefore decided to construct an analogous expression system for pTSOX. Plasmid pLJC400 is a pBluescript II SK(+) derivative containing the genes for cTSOX plus a 3′ fragment of glyA, a corynebacterial gene coding for serine hydroxymethyltransferase. Gene expression is controlled by the lac promoter. The glyA gene fragment in pLJC400 is positioned 2 bp upstream of the corynebacterial soxB gene where it is expressed as a lacZ-glyA-fusion. A similar feature was incorporated into plasmid pME430 (Figure 2), a pBluescript II SK(+) derivative where the pTSOX genes are positioned 3 bp downstream of the glyA gene fragment from pLJC400. A substantial amount of pTSOX was isolated using pME430 as expression vector but the specific activity of the preparation was even lower than observed with pME410 (Table 2). Again, the specific activity was not improved by adding an additional step to the purification procedure. Since somewhat different methods had been used for purification of recombinant and natural pTSOX, we repeated the isolation of the recombinant enzyme from XL-1 Blue/pME430 cells using the same procedure as for the natural enzyme. Although somewhat improved, the specific activity of recombinant pTSOX was still significantly lower (30%) than the natural enzyme (Table 2).
Expression and isolation of His-tagged pTSOX.
We sought to facilitate purification of recombinant pTSOX by introducing a (His)6 affinity tag. The organization of the pTSOX operon, with overlapping or closely spaced genes, placed restrictions on likely locations for an affinity tag. The observed gene arrangement is typically associated with translational coupling, a process where the same ribosome mediates translation of contiguous genes without dissociating from the mRNA [30]. Indeed, potential ribosome-binding sites are identifiable 6 to 8 bp before the start of soxD, soxA and soxG (Figure 3). To avoid potential adverse effects on translation, insertion of the extra bp for a (His)6 tag would be restricted to the 5′ end of soxB or the 3′ end of soxG. Since a N-terminal (His)6 tag on the β subunit might interfere with FAD binding at the nearby ADP-binding motif [8], the C-terminus of the γ subunit was selected for introduction of the affinity tag. The entire insert in pME430 was transferred to an appropriate vector, generating plasmid pGZ01 (see Figure 2). PCR was then conducted to position the vector-encoded (His)6 tag immediately downstream of soxG. The resulting construct (pGZ25, see Figure 2) adds 8 amino acids [LeuGlu(His)6] to the C-terminus the γ subunit. Recombinant pTSOX could be isolated from cells transformed with pGZ25 by using a single Co2+ affinity chromatography step. The purified enzyme exhibited the same specific activity as the natural enzyme (Table 2) but failed to crystallize.
Elimination of microheterogeneity.
Our previous studies showed that about half of the covalently bound FMN in recombinant cTSOX is in a modified form, tentatively identified as a 4a-adduct with a cysteine thiolate [25]. The adduct is not present in the natural enzyme. The adduct exhibits a single absorption maximum at 383 nm (ɛ383 = 7.3 mM−1 cm−1) and negligible absorption at 450 nm. Both natural and recombinant cTSOX exhibit visible absorption maxima at 368 and 450 nm but the adduct-containing recombinant enzyme is characterized by elevated values for the spectral ratios, A280/A450 and A368/A450. The adduct is converted to unmodified FMN by turnover of the recombinant enzyme with sarcosine, treatment with hydrogen peroxide (a product formed during turnover) or reaction with a reagent (methyl methanethiosulfonate, MMTS) that alkylates thiols. In each case, disruption of the adduct is accompanied by an increase in absorption at 450 nm and a downward shift in the spectral ratios to values similar to those observed for natural enzyme.
As observed with cTSOX, the adduct is not present in the natural form of pTSOX but is found in the recombinant His-tagged enzyme, as judged by the elevated spectral ratios (Table 3). Since microheterogeneity due to the presence of partially modified FMN might interfere with enzyme crystallization, we sought to eliminate the adduct in recombinant pTSOX by expression of the His-tagged enzyme in the presence of sarcosine. The natural enzyme is induced by growth of P. maltophilia in the presence of 10 g of sarcosine per liter of culture medium. However, the adduct in recombinant pTSOX is not eliminated under these conditions, as judged by the elevated spectral ratios and the increase in absorbance at 450 nm observed upon reaction of the enzyme with MMTS (Table 3, Figure 4). The adduct was, however, essentially eliminated by expression of the His-tagged enzyme in the presence of a 5-fold higher concentration of sarcosine (Table 3, Figure 4, inset), a difference attributed to the higher expression level of recombinant enzyme. SDS-PAGE analysis of recombinant pTSOX, expressed under these conditions and purified by a single Co2+ affinity column, revealed the presence of a smear of faint bands in between those observed for the α and β subunits. These bands might reflect proteolytic degradation products of the α subunit and could potentially interfere with crystallization. This source of microheterogeneity was eliminated by chromatography on Phenyl-Sepharose (Figure 5). Although the recovery was less than 15%, the final preparation exhibited a good specific activity (Table 3).
Table 3.
Spectral Properties of Natural pTSOX and His-tagged Recombinant Enzyme
| untreated
|
MMTS-treated
|
||||
|---|---|---|---|---|---|
| Preparation | A280/A450 | A368/A450 | A280/A450 | A368/A450 | 4a- adduct (%) |
| Natural pTSOXa | 13.7 | 0.82 | - | - | 0 |
| Recombinantb | |||||
| no sarcosine | 16.7 | 1.09 | ndc | ndc | ndc |
| 10 g sarcosine/L | 16.9 | 0.97 | 12.8 | 0.84 | 49d |
| 50 g sarcosine/L | 12.9 | 0.82 | 12.8 | 0.81 | 1.6d |
The natural enzyme is induced by growth of P. maltophilia in the presence of 10 g/L of sarcosine and does not contain adduct, as judged the fact that its spectral properties are not affected by H2O2 [8].
The recombinant enzyme was produced using pGZ25 as the expression vector.
nd, not determined
Estimated by comparing A280/A450 of the untreated enzyme with the value observed after reaction with MMTS.
Figure 4.

Reaction of His-tagged recombinant pTSOX with MMTS. Reactions were conducted in 10 mM potassium phosphate buffer, pH 8.0 at 20 °C. The main panel and inset show the reactions observed with enzyme isolated from cells grown in the presence of 10 and 50 g of sarcosine, respectively, per liter of culture medium. In each case, the solid curve is the spectrum of untreated enzyme; the dashed curve is the spectrum recorded after reaction with 1 mM MMTS for 30 min. The dotted curves in the main panel are spectra recorded at intermediate times (1, 2 and 10 min).
Figure 5.
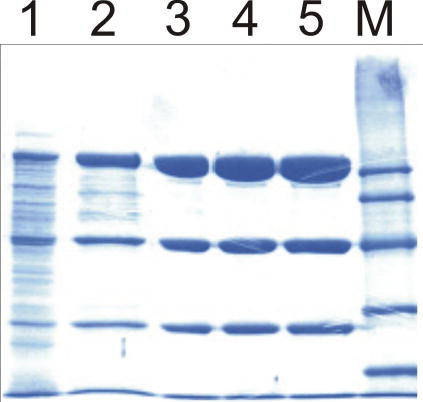
Coomassie blue stained SDS-PAGE gel of the purification of His-tagged recombinant pTSOX. Lane 1 is the soluble cell lysate (10 μg) obtained after growth of E. coli BL21(DE3)/pGZ25 in media containing 50 g sarcosine/L. Lane 2 (10 μg) is after purification on a Co2+ affinity column. Lane 3 (10 μg) and lanes 4 and 5 (15 μg) were obtained after further purification on a Phenyl-Sepharose column. Molecular markers are shown in lane M.
Crystallization of His-tagged pTSOX.
The highly purified, adduct-free His-tagged enzyme described above was subjected to crystallization trials using the sparse-matrix sampling technique [28], a variety of screening kits and the hanging drop vapor diffusion method. These studies showed that very thin plates could be obtained only with PEG 20 K as the precipitant and MES or Tris buffer, pH 6.5-8.5, and were somewhat difficult to reproduce. Diffraction quality crystals of average dimension 0.5x0.1x0.02 mm3 appeared after about five days under the conditions described in the methods section. X-ray diffraction data were collected to 1.85 A resolution from a thin crystal about 0.4x0.2x0.02 mm3 at the Advanced Photon Source. The crystal was orthorhombic, space group P212121 with unit cell dimensions a=73.2 Å, b=132.4 Å, c=197.4 Å. Assuming a single 185 kDa pTSOX molecule in the asymmetric unit, the solvent content is about 52%. In the outer resolution shell (1.92–1.85 Å) the data were about 91% complete with I/σ~2.7, and the overall Rmerge=8.9% (Table 4). The crystal structure of pTSOX will be published elsewhere.
Table 4.
Data Collection Statistics for pTSOXa
| Shell
|
|||||||
|---|---|---|---|---|---|---|---|
| Upper Limit (Å) | Lower Limit (Å) | No. of Reflec. | Redundancy | % Cover-age | <I> | <I/σ(I)> | Rmerge |
| 50.00 | 3.99 | 15186 | 3.9 | 88.9 | 95438 | 14.2 | 0.062 |
| 3.99 | 3.16 | 16346 | 4.3 | 98.6 | 79136 | 16.4 | 0.069 |
| 3.16 | 2.76 | 16393 | 4.2 | 99.6 | 32432 | 15.4 | 0.079 |
| 2.76 | 2.51 | 16298 | 4.2 | 99.6 | 17720 | 12.6 | 0.097 |
| 2.51 | 2.33 | 16215 | 4.1 | 99.1 | 12316 | 10.3 | 0.120 |
| 2.33 | 2.19 | 15978 | 4.0 | 97.9 | 9410 | 8.2 | 0.150 |
| 2.19 | 2.08 | 15699 | 4.0 | 96.5 | 7290 | 6.7 | 0.180 |
| 2.08 | 1.99 | 15445 | 3.9 | 94.8 | 5447 | 5.3 | 0.219 |
| 1.99 | 1.92 | 15204 | 3.8 | 93.8 | 3643 | 3.9 | 0.271 |
| 1.92 | 1.85 | 14815 | 3.4 | 91.1 | 2454 | 2.7 | 0.325 |
| All Data | 157579 | 3.9 | 95.0 | 26677 | 12.5 | 0.089 | |
Rmerge= ∑h∑i I(h)−Ii(h) /∑h∑iIi(h), where Ii(h) and I(h) are the ith and mean measurements of reflection h. I/σ(I) is the average signal to noise ratio for merged reflection intensities.
Conclusions.
Studies to clone and express the recombinant form of pTSOX were initiated on the basis of preliminary studies with the natural enzyme which formed crystals that diffracted to 2.8 Å. Initial difficulties in isolating high quality recombinant pTSOX appeared to be resolved by the introduction of a (His)6 affinity tag at a location that did not interfere with the presumed coupled translation of the pTSOX operon. This approach yielded recombinant pTSOX that exhibited the same specific activity as the natural enzyme but the affinity-purified recombinant enzyme failed to crystallize. The problem was traced to microhetereogeneity in recombinant enzyme where about half of the covalently bound FMN is present in a modified form, tentatively identified as a 4a-thiolate adduct. Indeed, small crystals of recombinant enzyme were obtained after disrupting the adduct with hydrogen peroxide. This microheterogeneity is not present in the natural enzyme where no adduct is detected. Expression of the natural enzyme is induced by sarcosine whereas IPTG is used to induce expression of the recombinant enzyme. Since the adduct in recombinant enzyme can be converted to unmodified FMN by in vitro turnover with sarcosine, we reasoned that the absence of the adduct in the natural enzyme is due to its disruption in vivo by reaction of the newly synthesized enzyme with the sarcosine used to induce enzyme expression. Consistent with this hypothesis, adduct-free preparations of recombinant enzyme could be obtained by including sarcosine in the growth medium. The results show that the presence of modified flavin is not an artifact of recombinant enzyme expression. Instead, expression of recombinant TSOX in the absence of sarcosine has let to the serendipitous discovery of a modified flavin that appears to be an intermediate in TSOX biosynthesis. In summary, high quality crystals of pTSOX were obtained by switching to a new bacterial source and elimination of microheterogeneity in affinity-purified recombinant enzyme associated with the presence of modified flavin and putative proteolytic degradation products.
Acknowledgments
This work was supported in part by Grant GM 31704 (M. S. J.) from the National Institutes of Health. We acknowledge initial attempts to crystallize TSOX by Maria Louisa Veisaga, Peter Trickey, Saumen Datta and Scott White.
Footnotes
Abbreviations: TSOX, heterotetrameric sarcosine oxidase; cTSOX, TSOX from Corynebacterium sp. P-1; pTSOX, TSOX from Pseudomonas maltophilia; FAD, flavin adenine dinucleotide; FMN, flavin mononucleotide; MSOX, monomeric sarcosine oxidase; MMTS, methyl methanethiosulfonate
References
- 1.K. Kvalnes-Krick, M. S. Jorns, Role of the covalent and noncovalent flavins in sarcosine oxidase, in F. Muller (Ed.) Chemistry and Biochemistry of Flavoenzymes, CRC Press Inc., Boca Raton, 1991, pp. 425–435.
- 2.Chlumsky LJ, Zhang L, Jorns MS. Sequence analysis of sarcosine oxidase and nearby genes reveals homologies with key enzymes of folate one-carbon metabolism. J. Biol. Chem. 1995;270:18252–18259. doi: 10.1074/jbc.270.31.18252. [DOI] [PubMed] [Google Scholar]
- 3.Willie A, Jorns MS. Discovery of a third coenzyme in sarcosine oxidase. Biochemistry. 1995;34:16703–16707. doi: 10.1021/bi00051a019. [DOI] [PubMed] [Google Scholar]
- 4.Willie A, Edmondson DE, Jorns MS. Sarcosine oxidase contains a novel covalently bound FMN. Biochemistry. 1996;35:5292–5299. doi: 10.1021/bi952995h. [DOI] [PubMed] [Google Scholar]
- 5.Chlumsky LJ, Sturgess AW, Nieves E, Jorns MS. Identification of the covalent flavin attachment site in sarcosine oxidase. Biochemistry. 1998;37:2089–2095. doi: 10.1021/bi972705s. [DOI] [PubMed] [Google Scholar]
- 6.Wagner MA, Jorns MS. Folate utilization by monomeric versus heterotetrameric sarcosine oxidases. Arch. Biochem. Biophys. 1997;342:176–181. doi: 10.1006/abbi.1997.0106. [DOI] [PubMed] [Google Scholar]
- 7.Kvalnes-Krick K, Jorns MS. Interaction of tetrahydrofolate and other folate derivatives with bacterial sarcosine oxidase. Biochemistry. 1987;26:7391–7395. doi: 10.1021/bi00397a029. [DOI] [PubMed] [Google Scholar]
- 8.Eschenbrenner M, Chlumsky LJ, Khanna P, Strasser F, Jorns MS. Organization of the multiple coenzymes and subunits and role of the covalent flavin link in the complex heterotetrameric sarcosine oxidase. Biochemistry. 2001;40:5352–5367. doi: 10.1021/bi010101p. [DOI] [PubMed] [Google Scholar]
- 9.Zeller H-D, Hille R, Jorns MS. Bacterial sarcosine oxidase: Identification of novel substrates and a biradical reaction intermediate. Biochemistry. 1989;28:5145–5154. doi: 10.1021/bi00438a035. [DOI] [PubMed] [Google Scholar]
- 10.Ali SN, Zeller HD, Calisto MK, Jorns MS. Kinetics of electron entry, exit, and interflavin electron transfer during catalysis by sarcosine oxidase. Biochemistry. 1991;30:10980–10986. doi: 10.1021/bi00109a024. [DOI] [PubMed] [Google Scholar]
- 11.Wagner MA, Trickey P, Chen Z, Mathews MS, Jorns MS. Monomeric sarcosine oxidase: 1. Flavin reactivity and active site binding determinants. Biochemistry. 2000;39:8813–8824. doi: 10.1021/bi000349z. [DOI] [PubMed] [Google Scholar]
- 12.Khanna P, Jorns MS. Characterization of the FAD-containing N-methyltryptophan oxidase from Escherichia coli. Biochemistry. 2001;40:1441–1450. doi: 10.1021/bi0024411. [DOI] [PubMed] [Google Scholar]
- 13.Wagner MA, Khanna P, Jorns MS. Structure of the flavocoenzyme of two homologous amine oxidases: Monomeric sarcosine oxidase and N-methyltryptophan oxidase. Biochemistry. 1999;38:5588–5595. doi: 10.1021/bi982955o. [DOI] [PubMed] [Google Scholar]
- 14.Trickey P, Wagner MA, Jorns MS, Mathews FS. Monomeric sarcosine oxidase: Structure of a covalently-flavinylated secondary amine oxidizing enzyme. Structure. 1999;7:331–345. doi: 10.1016/s0969-2126(99)80043-4. [DOI] [PubMed] [Google Scholar]
- 15.Reuber BE, Karl C, Reimann SA, Mihalik SJ, Dodt G. Cloning and functional expression of a mammalian gene for a peroxisomal sarcosine oxidase. J. Biol. Chem. 1997;272:6766–6776. doi: 10.1074/jbc.272.10.6766. [DOI] [PubMed] [Google Scholar]
- 16.Koyama Y, Ohmori H. Nucleotide sequence of the Escherichia coli solA gene encoding a sarcosine oxidase-like protein and characterization of its product. Gene. 1996;181:179–183. doi: 10.1016/s0378-1119(96)00500-8. [DOI] [PubMed] [Google Scholar]
- 17.Venci D, Zhao G, Jorns MS. Molecular characterization of nikD, a new flavoenzyme important in the biosynthesis of nikkomycin antibiotics. Biochemistry. 2002;41:15795–15802. doi: 10.1021/bi020515y. [DOI] [PubMed] [Google Scholar]
- 18.Bruckner RC, Zhao GH, Venci D, Jorns MS. Nikkomycin biosynthesis: Formation of a 4-electron oxidation product during turnover of nikD with its physiological substrate. Biochemistry. 2004;43:9160–9167. doi: 10.1021/bi0493618. [DOI] [PubMed] [Google Scholar]
- 19.Kvalnes-Krick K, Jorns MS. Bacterial sarcosine oxidase: Comparison of two multisubunit enzymes containing both covalent and noncovalent flavin. Biochemistry. 1986;25:6061–6069. doi: 10.1021/bi00368a034. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki M. Purification and some properties of sarcosine oxidase from Corynebacterium sp. U-96. J. Biochem. 1981;89:599–607. doi: 10.1093/oxfordjournals.jbchem.a133236. [DOI] [PubMed] [Google Scholar]
- 21.Ogushi S, Nagoao K, Emi S, Ando M, Tsuru D. Sarcosine oxidase from Arthrobacter ureafaciens: Purification and some properties. Chem. Pharm. Bull. 1988;36:1445–1450. [Google Scholar]
- 22.Harris RJ, Meskys R, Sutcliffe MJ, Scrutton NS. Kinetic studies of the mechanism of carbon-hydrogen bond breakage by the heterotetrameric sarcosine oxidase of Arthrobacter sp 1-IN. Biochemistry. 2000;39:1189–1198. doi: 10.1021/bi991941v. [DOI] [PubMed] [Google Scholar]
- 23.U. Mayr, H. Gauhl, H. Seidel, Process for obtaining sarcosine oxidase from microorganisms, United States Patent, Patent Number 5,024,945 (1991) 1–4.
- 24.U. Mayr, H. Mollering, J. Siedel, H. Seidel, Hydrogen peroxide-forming sarcosine oxidase, United States Patent, Patent Number 4,743,549 (1988) 1–4.
- 25.Chlumsky LJ, Zhang LN, Ramsey AJ, Jorns MS. Preparation and properties of recombinant corynebacterial sarcosine oxidase: Evidence for posttranslational modification during turnover with sarcosine. Biochemistry. 1993;32:11132–11142. doi: 10.1021/bi00092a024. [DOI] [PubMed] [Google Scholar]
- 26.Nash T. The colorimetric estimation of formaldehyde by means of the Hantzsch reaction. Biochem. J. 1953;55:416–421. doi: 10.1042/bj0550416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jorns MS. Properties and catalytic function of the two nonequivalent flavins in sarcosine oxidase. Biochemistry. 1985;24:3189–3194. doi: 10.1021/bi00334a017. [DOI] [PubMed] [Google Scholar]
- 28.Jancarik J, Kim S-H. Sparse matrix sampling: a screening method for crystallization of proteins. J. Appl. Cryst. 1991;24:409–411. [Google Scholar]
- 29.Clark L, Carbon J. A colony bank containing synthetic ColE1 hydrids representative of the entire E. coli genome. Cell. 1976;9:91–99. doi: 10.1016/0092-8674(76)90055-6. [DOI] [PubMed] [Google Scholar]
- 30.Normark S, Bergstrom S, Edlund T, Grundstrom T, Jaurin B, Lindberg FP, Olof O. Overlapping genes. Ann. Rev. Genet. 1983;17:499–525. doi: 10.1146/annurev.ge.17.120183.002435. [DOI] [PubMed] [Google Scholar]