

Abstract
Background
The mechanisms by which mesenchymal stem cells (MSCs) may protect native tissue are incompletely understood. Understanding the mechanisms by which these cells release factors such as vascular endothelial growth factor (VEGF), may lead to enhanced protection. We hypothesized that stress, in the form of hypoxia or TNF, activates MSCs to release VEGF by STAT3 and p38 MAPK dependent mechanisms.
Methods and Results
Mouse MSCs from wild type (WT) and STAT3 knockout mice (STAT3KO) were harvested and purified by a single-step method using adhesion. The release of VEGF was analyzed by using MSC conditioned media under hypoxia or TNF stimulation with or without p38 MAPK inhibition. Activation of STAT3 and p38 MAPK were determined by analysis of cell lysates. MSCs released VEGF under normoxia, which was associated with constitutive STAT3 activity. STAT3 deficiency resulted in decreased MSC production of VEGF. In response to hypoxia or TNF, MSCs produced more VEGF, which was correlated with hypoxia or TNF activated p38 MAPK and STAT3. The p38 MAPK inhibitor significantly decreased hypoxia-induced or TNF-stimulated VEGF production in WT. Additionally, STAT3 ablation neutralized hypoxia-induced MSC release of VEGF. No effect of p38 MAPK inhibitor alone was observed on MSC release of VEGF in WT. However, inhibition of p38 MAPK blocked release of VEGF in STAT3KO MSCs.
CONCLUSIONS
MSCs are a potent source of VEGF, the production of which is mediated by STAT3 under normoxia partly; however, following hypoxia or TNF exposure, MSC release of VEGF is mediated by both STAT3 and p38 MAPK.
Keywords: hypoxia, mesenchymal stromal cell, growth substances, STAT3, signal transduction
INTRODUCTION
The delivery of progenitor cells into ischemic myocardium is emerging as a potentially promising therapy. Although bone marrow derived mesechymal stem cells (MSCs) may differentiate into one or more types of cells including cardiac myocytes [1, 2], cardiac protection provided by MSCs may not only be mediated by transdifferentiation [3]. Indeed, accumulated studies demonstrate that transdifferentiation of stem cells into cardiomyocytes occurrs at very low levels (less than 0.05% cardiomyocytes derived from stem cell sources)[4, 5]. In addition, our previous study has shown that adult progenitor cell differentiation is not required for myocardial functional protection [6]. This has led to an important appreciation that other mechanisms may contribute to the protective effect provided by stem cells on ischemic tissue.
We recently reported that human progenitor cells are a potent source of vascular endothelial growth factor (VEGF) [7], which may be a critical growth factor in the protection of ischemic tissue. Bone marrow stem cells have been shown to protect myocardial function from ischemic injury through paracrine actions of an increase in local VEGF [8]. Additionally, it has been reported that MSCs mediate acute protective effects on ischemic myocardium through elevated VEGF [9]. Therefore, it can be proposed that stem/progenitor cells may exert protection by the secretion of protective factors such as VEGF. However, it is unclear by what mechanisms stem cells produce VEGF under stress, such as hypoxia or TNF stimulation. It has been shown that VEGF expression is associated with activation of signal transducer and activator of transcription 3 (STAT3) [10, 11] and p38 MAPK is required for VEGF production in fibroblast cell lines [12]. Recently, the STAT3 signaling pathway has been implicated in a variety of cellular functions, including the TNFR2-mediated survival pathway, apoptosis, proliferation and the immune response [13]. However, little information exists regarding the effect of STAT3 and p38 MAPK on MSC production of VEGF under hypoxia. Given these interesting unknowns, the purposes of this study were to determine the effects of STAT3 and p38 MAPK on MSC production of VEGF under hypoxia or TNF stimulation by using mice with a conditional deletion of STAT3 from bone marrow MSCs, as well as a p38 MAPK inhibitor.
MATERIALS AND METHODS
Animals
The STAT3 deficiency mouse line was in C57B6/129 mixed background, and has been described previously [14]. Four- to six- week-old wild type (WT) and conditioned STAT3 knockout mice were maintained in a quiet quarantine room for one month before the experiments. The animal protocol was reviewed and approved by the Indiana Animal Care and Use Committee of Indiana University. All animals received humane care in compliance with the “Guide for the Care and Use of Laboratory Animals” (NIH publication No. 85-23, revised 1985).
Preparation of Mouse Bone Marrow Mesenchymal Stromal Cells
In addition to wild type animals with an identical background, we used a mouse line in which STAT3 was deleted from bone marrow cells by crossing STAT3 allele floxed mouse with a Tie-2 promoter driving cre expression mouse [14]. A single-step purification method using adhesion to cell culture plastic is employed as previously described [15] with the following modifications: Mouse bone marrow mesenchymal stromal cells were harvested from bilateral femurs and tibias by removing the epiphyses and flushing the shaft with complete media [Iscove’s Modified Dulbecco’s Medium with 10% fetal bovine serum and 1% pen-strep (GIBCO Invitrogen, Carlsbad, CA)]. Cells were disaggregated by vigorous pipetting several times and were passed through 30-μm nylon mesh to remove remaining clumps of tissue. Cells were washed by adding complete media, centrifuging for 5 min at 300 rpm @ 24°C and removing supernatant. The cell pellet was then resuspended and cultured in 75 cm2 culture flasks with complete media at 37°C, 5% CO2 and 90% humidity. MSCs preferentially attached to the polystyrene surface; after 48 h, nonadherent cells in suspension were discarded. Fresh complete medium was added and replaced every three days thereafter. When the cultures reached 90% of confluence, MSCs were detached by the addition of a solution 0.25% trypsin-EDTA (GIBCO Invitrogen, Carlsbad, CA) and cells were passaged.
Experimental Groups
After five passages, MSCs were plated in 12 well plates at a concentration of 1 × 105 cells/well/ml. WT and STAT3KO MSCs were divided into 3 experimental groups (triplicate wells per group: 1) control; 2) 24-hour or 48-hour hypoxia with or without p38 MAPK inhibitor (p38 MKI) (10 μM of SB 203580); 3) 50 ng/ml of TNF with or without p38 MKI. After 24-hour (TNF and hypoxia) and 48-hour (hypoxia) incubation, supernatants were collected for VEGF assay (ELISA). Cells were harvested for western blot assay. The experiment was repeated on three separate occasions (n=6–11 wells/group).
VEGF ELISA
VEGF in the MSC supernatant was determined by enzyme-linked immunosorbent assay (ELISA) using a commercially available ELISA set (R&D Systems Inc., Minneapolis, MN). ELISA was performed according to the manufacturer’s instructions. All samples and standards were measured in duplicate. All reported VEGF values in cell supernatant were compared with cell number at the time of assay.
Western blotting
Western blot analysis was performed to measure STAT3 and p38 MAP kinase proteins. Cells were lysed in cold buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-Glycerophosphate, 1 mM Na3VO4, 1 μg/ml Leupeptin, 1 mM PMSF, and centrifuged at 12000 rpm for 10 minutes. The protein extracts (10 μg/lane) were subjected to electrophoresis on a 8–16% precise protein gel (Pierce, Rockford, IL) and transferred to a nitrocellulose membrane, which was stained by Naphthol Blue-Black to confirm equal protein loading. The membranes were incubated in 5% dry milk for 1 hour and then incubated with the following primary antibodies: STAT3, phosphor-STAT3 (Tyr705), p38 MAP kinase antibody and phosphor-p38 MAP kinase (Thr180/Tyr182) (Cell Signaling Technology, Beverly, MA). Membranes were then incubated with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse IgG secondary antibody and detection using supersignal west pico stable peroxide solution (Pierce, Rockford, IL). Films were scanned using an Epson Perfection 3200 Scanner (Epson America, Long Beach, CA) and band density was analyzed using ImageJ software (NIH).
Presentation of data and statistical analysis
All reported values are mean ± SEM (n=6–11 well/group). Data was compared using Student’s t-test. A two-tailed probability value of less than 0.05 was considered statistically significant.
RESULTS
Hypoxia or TNF stimulates MSC production of VEGF dependent on p38 MAPK
Under hypoxia or TNF stimulation, bone marrow stromal cells secreted significantly larger amounts of VEGF compared to controls (figure 1). MSC production of VEGF was increased nearly 50% in the 24-hour hypoxia group, 80% in the 48-hour hypoxia group and 65% in the 24-hour TNF stimulated group.
Figure 1.
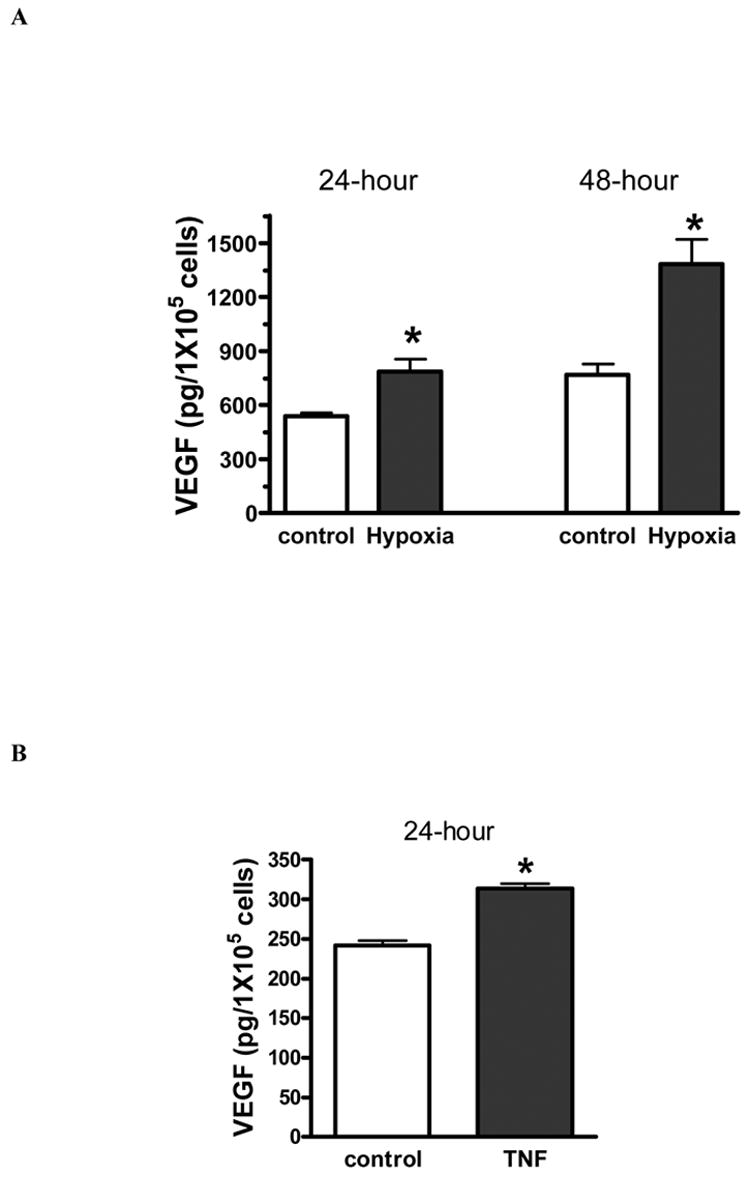
Hypoxia (A) or TNF stimulation (B) significantly increased bone marrow stromal cell (MSC) secretion of VEGF. Results are mean ± SEM, n=3/group (the experiment was repeated on three separate occasions) *p<0.05 vs. control.
In addition, hypoxia or TNF stimulation resulted in increased activation of p38 MAPK in MSC culture (figure 2). Activation of p38 MAPK was increased three fold with hypoxia and two fold with TNF stimulation. Administration of p38 MAPK inhibitor (10 μM of SB 203580) decreased production of VEGF in response to hypoxia or TNF in MSC (figure 3 A, B). There was no effect of p38 MAPK inhibitor alone on production of VEGF.
Figure 2.
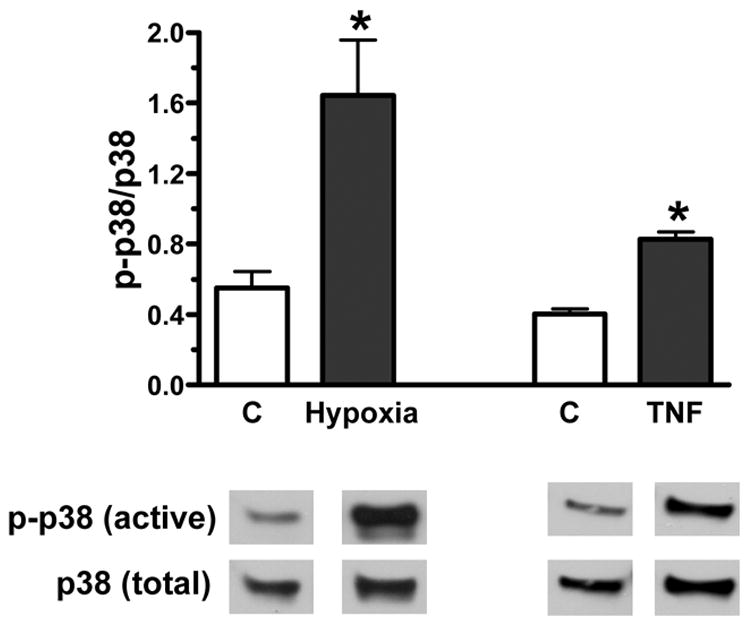
Activation of p38 MAPK in bone marrow stromal cells (MSCs) under hypoxia or TNF exposure. Both 24-hour hypoxia and TNF stimulation markedly elevated p38 MAPK activation in MSCs. Top shows densitometry data of p-p38 MAPK vs. total p38 MAPK (%) and bottom is representative immunoblots. Results are mean ± SEM, n=3–6/group, *p<0.05 vs. control.
Figure 3.
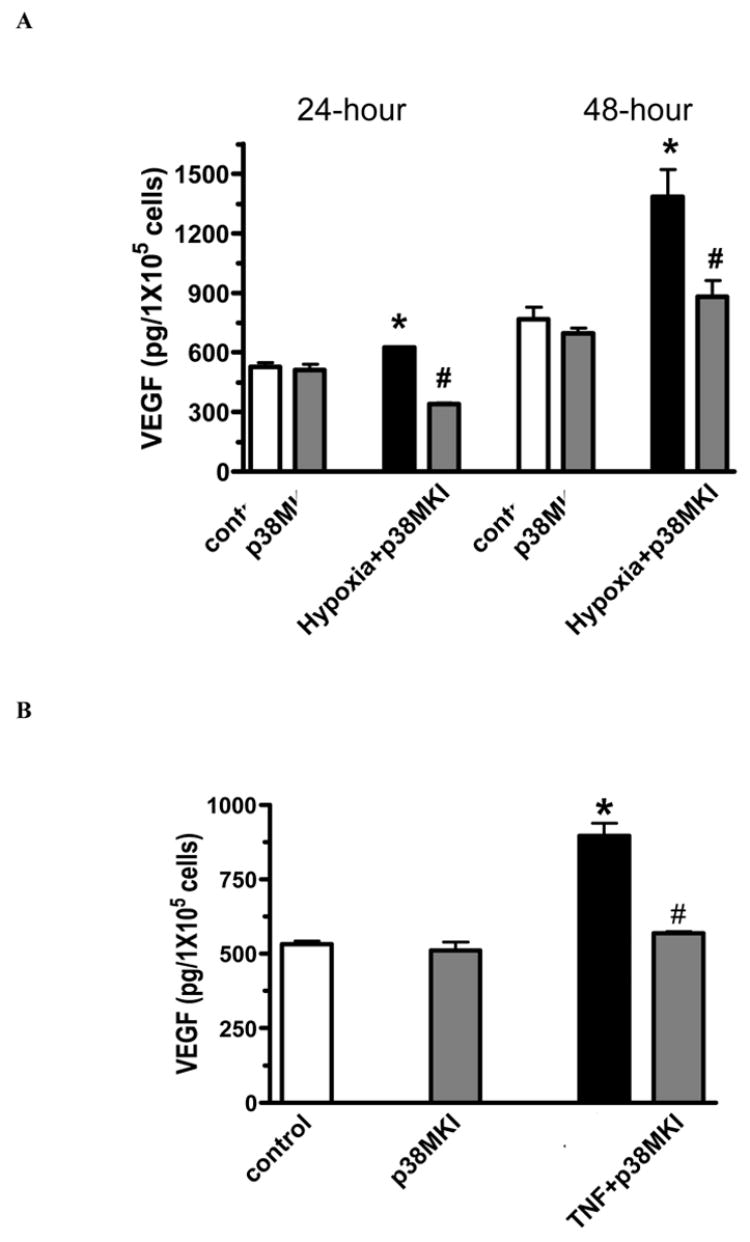
Production of VEGF in wild type MSCs in response to hypoxia (A) or TNF (B) with or without p38 MAPK inhibitor (10 μM of SB 203580) (p38MKI). p38 MAPK inhibitor significantly decreased hypoxia-induced or TNF-stimulated VEGF production in MSCs. Results are mean ± SEM, n=3/group (the experiment was repeated on three separate occasions) *p<0.05 vs. control and p38 MKI alone, #p<0.05 vs. hypoxia or TNF.
STAT3 mediates MSC release of VEGF under normoxia
After 24-hour or 48-hour incubation at 37°C, 5% CO2 and 90% humidity, STAT3 deficiency resulted in lower levels of VEGF secretion by MSC (figure 4A). VEGF production was decreased in STAT3KO MSC by nearly 40%. The western blot of STAT3 further indicated that STAT3 activity was not observed in STAT3KO MSC, and that lower levels of STAT3 protein existed in MSCs derived from STAT3KO mice (figure 4B).
Figure 4.
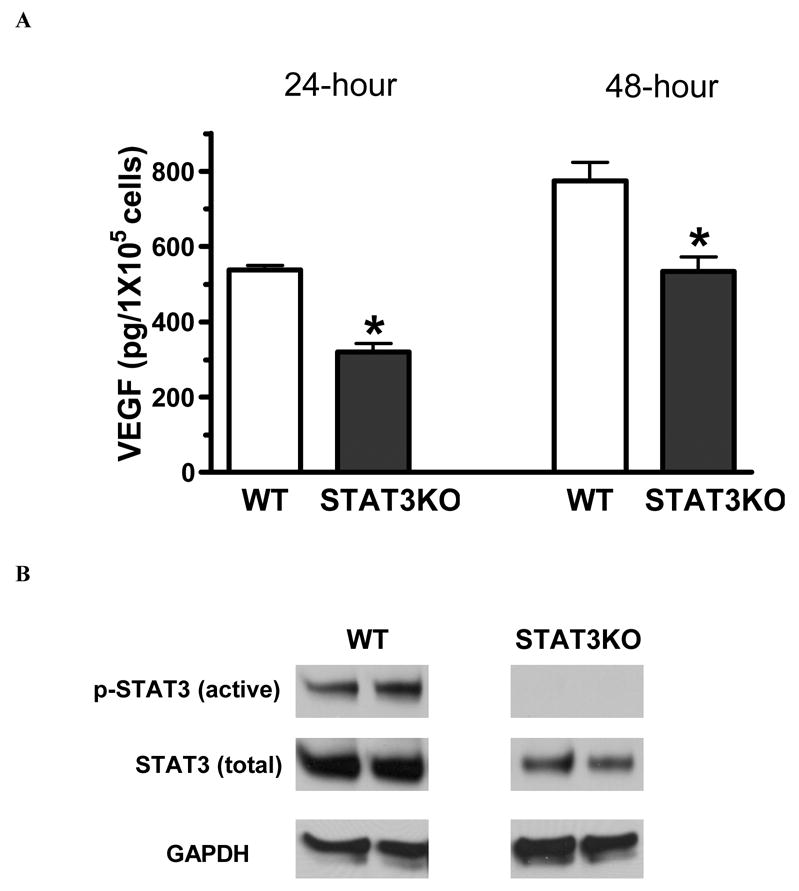
Effect of STAT3 on MSC production of VEGF under normoxia. A. MSCs with STAT3 ablation produced less VEGF under normal culture condition. B. Representative immunoblots (2 lanes/group) show that no activation of STAT3 and less total STAT3 were observed in STAT3KO MSCs compared with wild type under 24 hour normal culture condition. GAPDH is representative of loading control. Results are mean ± SEM, n=3/group (the experiment was repeated on three separate occasions) *p<0.05 vs. WT.
Effect of STAT3 on MSC in response to hypoxia or TNF stimulation
Hypoxia or TNF stimulation increased STAT3 activation in MSCs (figure 5A). STAT3 activation was increased by two fold with hypoxia and by three fold with TNF stimulation. However, STAT3 ablation neutralized hypoxia-induced VEGF production in MSCs (figure 5B). STAT3 deficient MSCs produced 30% more VEGF with TNF stimulation (figure 5C), however this was still lower than VEGF levels produced by WT MSCs.
Figure 5.
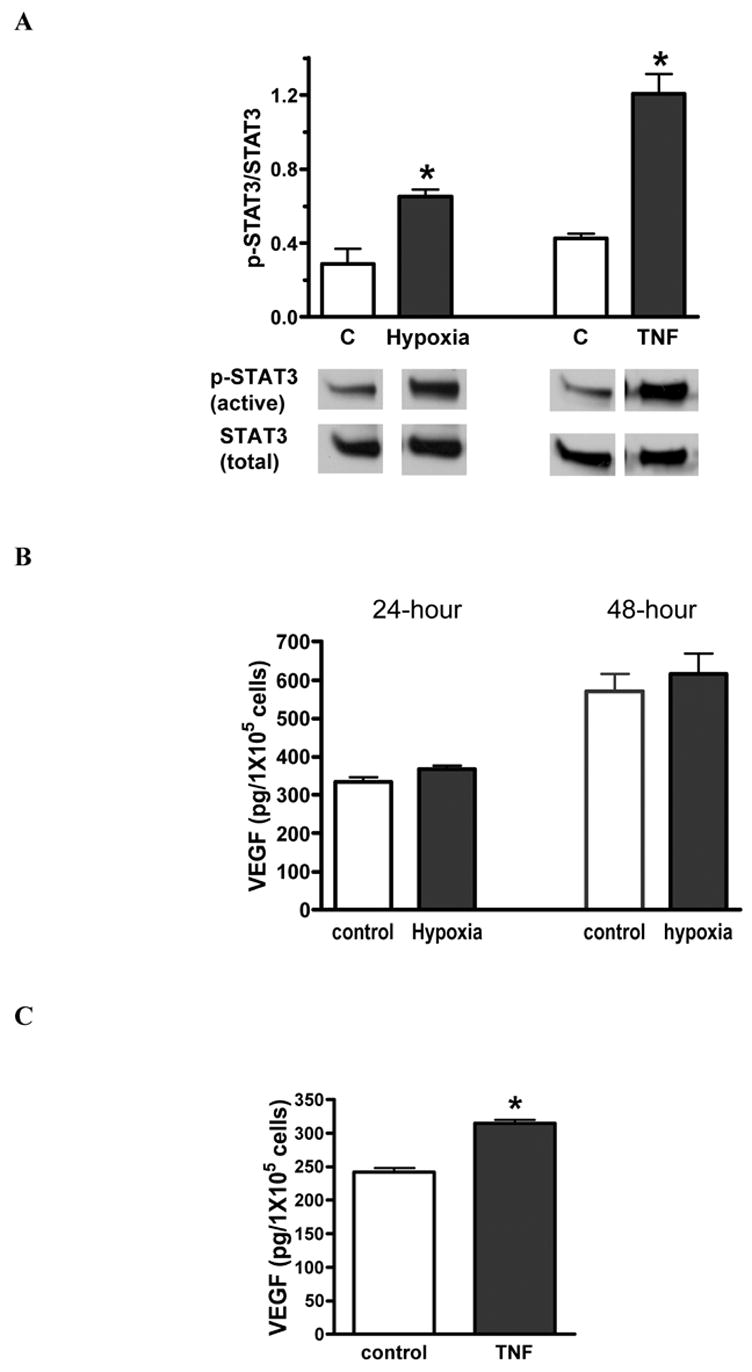
Effect of STAT3 on MSC release of VEGF under hypoxia or TNF. A. Both hypoxia and TNF stimulation significantly increased STAT3 activation in wild type MSCs. Top shows densitometry data of p-STAT3 (active) vs. total STAT3 (%) and bottom is representative immunoblots. B. STAT3 deficiency in MSCs neutralized hypoxia-induced VEGF production. C. Ablation of the STAT3 gene in MSCs partly blocked TNF-stimulated VEGF secretion. Results are mean ± SEM, n=3/group (the experiment was repeated on three separate occasions) *p<0.05 vs. control.
Effect of p38 MAPK on STAT3KO MSC production of VEGF under hypoxia or TNF
Although hypoxia or TNF stimulation resulted in less of an increase of p38 MAPK activation in STAT3KO MSCs compared to WT, p38 MAPK activation was elevated by 30% under hypoxia or TNF exposure (figure 6A). Interestingly, administration of p38 MAPK inhibitor resulted in lower levels of VEGF production not only under hypoxia or TNF stimulation, but also under normoxia in STAT3KO MSCs (figure 6B, C).
Figure 6.
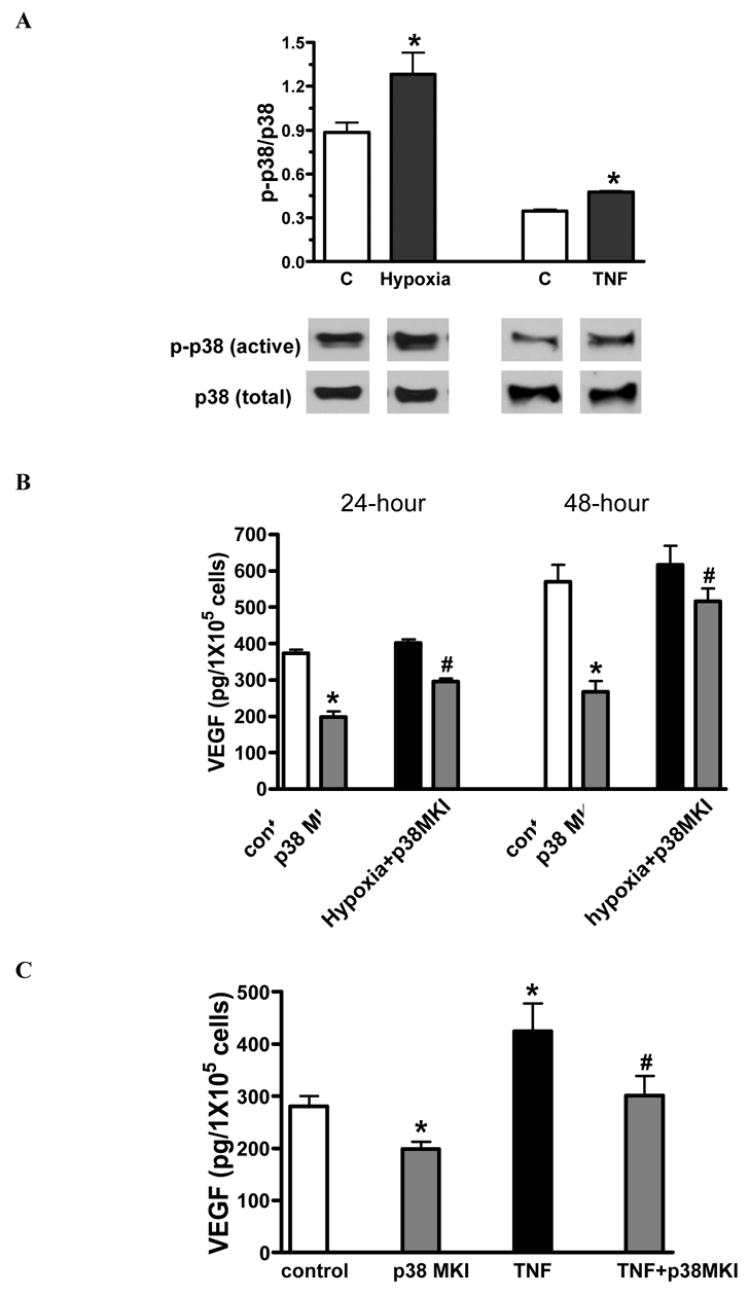
Effect of p38 MAPK on release of VEGF in STAT3KO MSC response to hypoxia or TNF. A. p38 MAPK activation was observed in STAT3KO MSC exposed to hypoxia or TNF. Top shows densitometry data of p-p38 (active) vs. total p38 (%) and bottom is representative immunoblots. B. It shows STAT3KO MSC secretion of VEGF under hypoxia with or without p38 MAPK inhibitor. C. TNF-stimulated VEGF production was analyzed in STAT3KO MSCs with or without p38 MAPK inhibitor. Results are mean ± SEM, n=3/group (the experiment was repeated on three separate occasions) *p<0.05 vs. control, #p<0.05 vs. hypoxia or TNF.
DISCUSSION
In the present study, we analyzed MSC function following hypoxia or TNF stimulation by using mouse bone marrow STAT3 knockouts (since the systemic STAT3 knockout is lethal), as well as p38 MAPK inhibition. Our results indicated that: 1) MSCs are a potent source of VEGF; 2) STAT3 mediates VEGF production under normoxia partly; 3) MSC release of VEGF is mediated by STAT3 and p38 MAPK following hypoxia or TNF stimulation.
Progenitor cell therapy has received much attention for its potential benefit in the treatment of diseases that result in the loss of viable tissue. However, some studies have questioned the differentiation of progenitor cells into cardiomyocytes in ischemic myocardium [3]. In addition, the magnitude of progenitor cells’ contribution to target tissue cells is very low, which rarely provides protection in most organs. Thus, there has been a growing supposition that progenitor cells may protect ischemic organs via paracrine actions [16, 17]. Previously, our group not only indicated that pretreatment of human progenitor cells into the isolated heart immediately before myocardial ischemia improved postischemic myocardial function through stem cell differentiation-independent mechanisms [6], but also demonstrated that human progenitor cells were shown to be a potent source of VEGF in response to TNF [7]. Local increases in VEGF production have been noted after bone marrow cells were delivered into ischemic tissue [3]. In addition, it has been reported that MSCs mediate acute protective effects on ischemic myocardium through elevated VEGF [9]. Altogether, it can be proposed that progenitor cells may exert protection by the secretion of protective factors such as VEGF. VEGF was selected to represent MSC function in this study for the following reasons: 1) VEGF is an angiogenic factor that benefits neovascularization and tissue remodeling [18, 19]; impaired myocardial angiogenesis and ischemic induced heart failure have been observed in mice with disruption of VEGF activity [20]; 2) VEGF exerts an anti-apoptotic effect as conditioned media containing VEGF has been shown to increase cell growth and to inhibit cell apoptosis [21]; 3) VEGF is able to recruit circulating bone marrow derived progenitor cells to necrotic tissue [22].
Hypoxia stimulated VEGF expression has been noted in tumor cells, vascular endothelial cells and fibroblasts [10]. Additionally, accumulated evidence has shown that TNF, as a local proinflammatory cytokine induced by various injuries, stimulates VEGF production in human monocytes, macrophages and fibroblasts [23]. However, it is unknown whether hypoxia or TNF will induce mouse MSC release of VEGF, and if so, by what mechanisms. In fact, it may be very important to understand the signaling mechanisms that MSCs utilize in order to prime progenitor cells for maximal growth factor production in vitro during therapeutic use.
p38 MAPK appears to be an important protein kinase that is involved in the production of several proinflammatory cytokines [24–27]. Activation of p38 MAPK is required for VEGF production in fibroblast cell lines [12]. In addition, a role for MAPK in the regulation of VEGF expression has been reported in mouse embryonic stem cells [28] and mesenchymal stem cells [29]. Our previous studies have indicated that p38 MAPK is involved in TNF-induced production of growth factors in human progenitor cells [7]. In the present study, our data demonstrate that TNF-stimulated VEGF production is associated with increased p38 MAPK activation, and this increase in VEGF production is blocked by administration of a p38 MAPK inhibitor in mouse MSCs. These findings not only corroborate our previous results, but they also demonstrate that hypoxia stimulates more release of VEGF in MSCs. Hypoxia-induced VEGF production also appears to require p38 MAPK.
Recent evidence has indicated that transcription of the VEGF promoter is mediated by active STAT3 in human renal carcinoma cells [10]. Several different types of tumor cells constitutively express active STAT3, which is associated with up-regulated angiogenesis and increased VEGF production [11, 30]. In addition, glycoprotein 130-mediated VEGF production requires STAT3 activation in cardiomyocytes [31]. Over-expression of active STAT3 in cardiomyocytes has been shown to increase myocardial VEGF production [32]. Therefore, it can be proposed that STAT3 signaling may mediate MSC production of VEGF. A systemic KO of STAT3 has been reported to result in early embryonic death [33]. Here, we used a mouse line in which STAT3 was deleted from bone marrow cells by crossing STAT3 allele floxed mouse with a Tie-2 promoter driving cre expression mouse [14]. Our data indicated that total STAT3 protein levels were much lower in STAT3KO MSCs compared with wild type, and that STAT3KO MSCs had no STAT3 activity. In parallel, VEGF production was significantly lower in KO MSCs compared with wild type. These results suggest that STAT3 is required to mediate MSC release of VEGF under normal culture conditions.
The question now becomes whether hypoxia-induced or TNF-stimulated release of VEGF is mediated by STAT3 signaling in MSCs. It has been reported that hypoxia-activated STAT3 plays an essential role in VEGF expression, and that hypoxia-stimulated VEGF production was inhibited by blockade of STAT3 in renal carcinoma cells [10]. Our results that hypoxia induced more activation of STAT3 correlated with higher VEGF production in hypoxic MSCs and that STAT3 deficiency neutralized hypoxia-induced MSC release of VEGF were in line with the previous study. On the other hand, TNF has been shown to mediate cytokine production via various types of signaling pathways, including the STAT3 and MAPK pathways [34]. Here, our data indicated that TNF-activated STAT3 was involved in regulating MSC production of VEGF, and that ablation of the STAT3 gene partly blocked TNF-stimulated release of VEGF. These results suggest that MSC production of VEGF may be mediated by different mechanisms with hypoxia and TNF stimulation. In response to hypoxia, STAT3 exerts essential effects on MSC release of VEGF, and the p38 MAPK may play a complementary role in STAT3-mediated VEGF production as the consequence of active STAT3. However, when exposed to TNF, both STAT3 and p38 MAPK are activated to regulate MSC production of VEGF in a parallel fashion. A subsequent observation is that p38 MAPK inhibitor alone had no effect on WT MSC release of VEGF, whereas VEGF production was decreased by inhibition of p38 MAPK in STAT3KO MSCs under normoxia. It can be postulated that p38 MAPK activity may exist in STAT3KO MSCs and may be responsible for VEGF production under normoxia. It may also potentially be an adaptation of MSCs to STAT3 ablation.
In summary, our study provides new evidence for interactions between STAT3 and p38 MAPK during hypoxia-induced or TNF-stimulated MSC production of VEGF (figure 7). Our global hypothesis is that ischemia/reperfusion or hypoxia induces cardiac myocytes to release proinflammatory cytokines, such as TNF. TNF then stimulates stem cell production of VEGF via STAT3 and p38 MAPK pathways. Locally produced VEGF by stem cells then protects cardiac myocytes against ischemia/hypoxia. Additionally, ischemia/reperfusion or hypoxia itself induces stem cells delivered into ischemic myocardium to release VEGF through STAT3 signaling. In the present study, our data suggests that hypoxia-induced or TNF-stimulated MSC release of VEGF is mediated by STAT3 and p38 MAPK signaling. However, the detailed mechanisms of interactions between STAT3 and p38 MAPK in MSC production of VEGF warrant further investigation.
Figure 7.
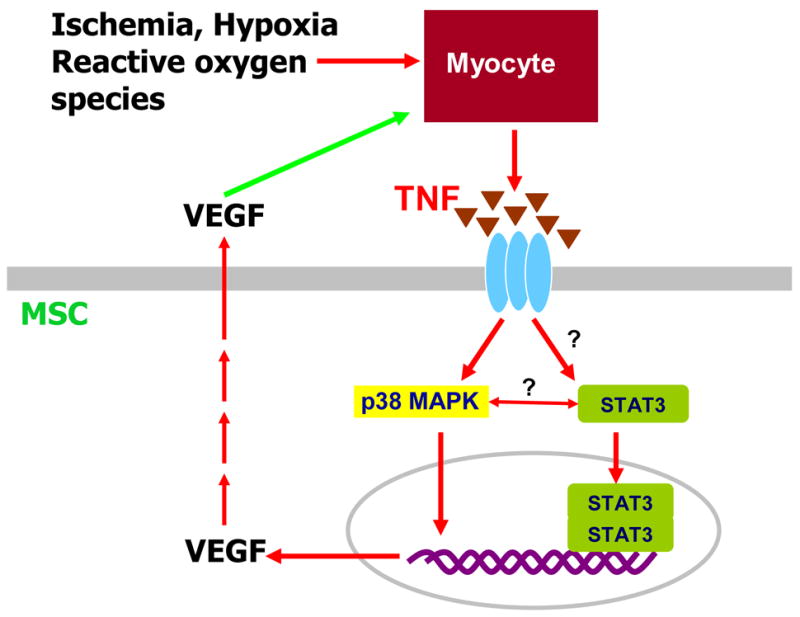
Hypothetical model of ischemic/hypoxic effects on cardiac myocytes and stem cells. Simplified pathways illustrating the relationship between the STAT3 signaling and p38 MAPK pathway under hypoxia or TNF stimulation in MSC production of VEGF.
Acknowledgments
This work was supported by NIH R01GM070628 (DRM), AHA Post-doctoral Fellowship 0526008Z (MW) and NIH K99 HL087607-01 (MW).
This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number C06 RR015481-01 from the National Center for Research Resources, National Institutes of Health.
Footnotes
DISCLOSURES
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Silva GV, Litovsky S, Assad JA, Sousa AL, Martin BJ, Vela D, et al. Mesenchymal stem cells differentiate into an endothelial phenotype, enhance vascular density, and improve heart function in a canine chronic ischemia model. Circulation. 2005;111:150–6. doi: 10.1161/01.CIR.0000151812.86142.45. [DOI] [PubMed] [Google Scholar]
- 2.Kawada H, Fujita J, Kinjo K, Matsuzaki Y, Tsuma M, Miyatake H, et al. Nonhematopoietic mesenchymal stem cells can be mobilized and differentiate into cardiomyocytes after myocardial infarction. Blood. 2004;104:3581–7. doi: 10.1182/blood-2004-04-1488. [DOI] [PubMed] [Google Scholar]
- 3.Nagaya N, Kangawa K, Itoh T, Iwase T, Murakami S, Miyahara Y, et al. Transplantation of mesenchymal stem cells improves cardiac function in a rat model of dilated cardiomyopathy. Circulation. 2005;112:1128–35. doi: 10.1161/CIRCULATIONAHA.104.500447. [DOI] [PubMed] [Google Scholar]
- 4.Laflamme MA, Myerson D, Saffitz JE, Murry CE. Evidence for cardiomyocyte repopulation by extracardiac progenitors in transplanted human hearts. Circ Res. 2002;90:634–40. doi: 10.1161/01.res.0000014822.62629.eb. [DOI] [PubMed] [Google Scholar]
- 5.Jackson KA, Majka SM, Wang H, Pocius J, Hartley CJ, Majesky MW, et al. Regeneration of ischemic cardiac muscle and vascular endothelium by adult stem cells. J Clin Invest. 2001;107:1395–402. doi: 10.1172/JCI12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang M, Tsai BM, Crisostomo PR, Meldrum DR. Pretreatment with Adult Progenitor Cells Improves Recovery and Decreases Native Myocardial Proinflammatory Signaling after Ischemia. Shock. 2006;25:454–9. doi: 10.1097/01.shk.0000209536.68682.90. [DOI] [PubMed] [Google Scholar]
- 7.Wang M, Crisostomo PR, Herring C, Meldrum KK, Meldrum DR. Human progenitor cells from bone marrow or adipose tissue produce VEGF, HGF, and IGF-I in response to TNF by a p38 MAPK-dependent mechanism. Am J Physiol Regul Integr Comp Physiol. 2006;291:R880–4. doi: 10.1152/ajpregu.00280.2006. [DOI] [PubMed] [Google Scholar]
- 8.Uemura R, Xu M, Ahmad N, Ashraf M. Bone marrow stem cells prevent left ventricular remodeling of ischemic heart through paracrine signaling. Circulation research. 2006;98:1414–21. doi: 10.1161/01.RES.0000225952.61196.39. [DOI] [PubMed] [Google Scholar]
- 9.Tang YL, Zhao Q, Zhang YC, Cheng L, Liu M, Shi J, et al. Autologous mesenchymal stem cell transplantation induce VEGF and neovascularization in ischemic myocardium. Regul Pept. 2004;117:3. doi: 10.1016/j.regpep.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 10.Jung JE, Lee HG, Cho IH, Chung DH, Yoon SH, Yang YM, et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. Faseb J. 2005;19:1296–8. doi: 10.1096/fj.04-3099fje. [DOI] [PubMed] [Google Scholar]
- 11.Xu Q, Briggs J, Park S, Niu G, Kortylewski M, Zhang S, et al. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene. 2005;24:5552–60. doi: 10.1038/sj.onc.1208719. [DOI] [PubMed] [Google Scholar]
- 12.Duyndam MC, Hulscher ST, van der Wall E, Pinedo HM, Boven E. Evidence for a role of p38 kinase in hypoxia-inducible factor 1-independent induction of vascular endothelial growth factor expression by sodium arsenite. J Biol Chem. 2003;278:6885–95. doi: 10.1074/jbc.M206320200. [DOI] [PubMed] [Google Scholar]
- 13.Fu XY. STAT3 in immune responses and inflammatory bowel diseases. Cell Res. 2006;16:214–9. doi: 10.1038/sj.cr.7310029. [DOI] [PubMed] [Google Scholar]
- 14.Welte T, Zhang SS, Wang T, Zhang Z, Hesslein DG, Yin Z, et al. STAT3 deletion during hematopoiesis causes Crohn’s disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci U S A. 2003;100:1879–84. doi: 10.1073/pnas.0237137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peister A, Mellad JA, Larson BL, Hall BM, Gibson LF, Prockop DJ. Adult stem cells from bone marrow (MSCs) isolated from different strains of inbred mice vary in surface epitopes, rates of proliferation, and differentiation potential. Blood. 2004;103:1662–8. doi: 10.1182/blood-2003-09-3070. [DOI] [PubMed] [Google Scholar]
- 16.Wagers AJ, Weissman IL. Plasticity of adult stem cells. Cell. 2004;116:639–48. doi: 10.1016/s0092-8674(04)00208-9. [DOI] [PubMed] [Google Scholar]
- 17.Jiang S, Haider H, Idris NM, Salim A, Ashraf M. Supportive interaction between cell survival signaling and angiocompetent factors enhances donor cell survival and promotes angiomyogenesis for cardiac repair. Circulation research. 2006;99:776–84. doi: 10.1161/01.RES.0000244687.97719.4f. [DOI] [PubMed] [Google Scholar]
- 18.Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147–57. doi: 10.1016/s0092-8674(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Haider HK, Ahmad N, Xu M, Ge R, Ashraf M. Combining pharmacological mobilization with intramyocardial delivery of bone marrow cells over-expressing VEGF is more effective for cardiac repair. Journal of molecular and cellular cardiology. 2006;40:736–45. doi: 10.1016/j.yjmcc.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 20.Carmeliet P, Ng YS, Nuyens D, Theilmeier G, Brusselmans K, Cornelissen I, et al. Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Nat Med. 1999;5:495–502. doi: 10.1038/8379. [DOI] [PubMed] [Google Scholar]
- 21.Rehman J, Traktuev D, Li J, Merfeld-Clauss S, Temm-Grove CJ, Bovenkerk JE, et al. Secretion of angiogenic and antiapoptotic factors by human adipose stromal cells. Circulation. 2004;109:1292–8. doi: 10.1161/01.CIR.0000121425.42966.F1. [DOI] [PubMed] [Google Scholar]
- 22.Grunewald M, Avraham I, Dor Y, Bachar-Lustig E, Itin A, Yung S, et al. VEGF-induced adult neovascularization: recruitment, retention, and role of accessory cells. Cell. 2006;124:175–89. doi: 10.1016/j.cell.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 23.Malaguarnera L, Imbesi R, Di Rosa M, Scuto A, Castrogiovanni P, Messina A, et al. Action of prolactin, IFN-gamma, TNF-alpha and LPS on heme oxygenase-1 expression and VEGF release in human monocytes/macrophages. Int Immunopharmacol. 2005;5:1458–69. doi: 10.1016/j.intimp.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 24.Thobe BM, Frink M, Choudhry MA, Schwacha MG, Bland KI, Chaudry IH. Src family kinases regulate p38 MAPK-mediated IL-6 production in Kupffer cells following hypoxia. American journal of physiology. 2006;291:C476–82. doi: 10.1152/ajpcell.00076.2006. [DOI] [PubMed] [Google Scholar]
- 25.Jarrar D, Wang P, Song GY, Cioffi WG, Bland KI, Chaudry IH. Inhibition of tyrosine kinase signaling after trauma-hemorrhage: a novel approach for improving organ function and decreasing susceptibility to subsequent sepsis. Ann Surg. 2000;231:399–407. doi: 10.1097/00000658-200003000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwacha MG, Chaudry IH, Alexander M. Regulation of macrophage IL-10 production postinjury via beta2 integrin signaling and the P38 MAP kinase pathway. Shock. 2003;20:529–35. doi: 10.1097/01.shk.0000095059.62263.56. [DOI] [PubMed] [Google Scholar]
- 27.Song GY, Chung CS, Chaudry IH, Ayala A. MAPK p38 antagonism as a novel method of inhibiting lymphoid immune suppression in polymicrobial sepsis. Am J Physiol Cell Physiol. 2001;281:C662–9. doi: 10.1152/ajpcell.2001.281.2.C662. [DOI] [PubMed] [Google Scholar]
- 28.Sauer H, Bekhite MM, Hescheler J, Wartenberg M. Redox control of angiogenic factors and CD31-positive vessel-like structures in mouse embryonic stem cells after direct current electrical field stimulation. Exp Cell Res. 2005;304:380–90. doi: 10.1016/j.yexcr.2004.11.026. [DOI] [PubMed] [Google Scholar]
- 29.Wang FS, Wang CJ, Chen YJ, Chang PR, Huang YT, Sun YC, et al. Ras induction of superoxide activates ERK-dependent angiogenic transcription factor HIF-1 alpha and VEGF-A expression in shock wave-stimulated osteoblasts. J Biol Chem. 2004;279:10331–7. doi: 10.1074/jbc.M308013200. [DOI] [PubMed] [Google Scholar]
- 30.Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000–8. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 31.Funamoto M, Fujio Y, Kunisada K, Negoro S, Tone E, Osugi T, et al. Signal transducer and activator of transcription 3 is required for glycoprotein 130-mediated induction of vascular endothelial growth factor in cardiac myocytes. J Biol Chem. 2000;275:10561–6. doi: 10.1074/jbc.275.14.10561. [DOI] [PubMed] [Google Scholar]
- 32.Osugi T, Oshima Y, Fujio Y, Funamoto M, Yamashita A, Negoro S, et al. Cardiac-specific activation of signal transducer and activator of transcription 3 promotes vascular formation in the heart. J Biol Chem. 2002;277:6676–81. doi: 10.1074/jbc.M108246200. [DOI] [PubMed] [Google Scholar]
- 33.Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A. 1997;94:3801–4. doi: 10.1073/pnas.94.8.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Theiss AL, Simmons JG, Jobin C, Lund PK. Tumor necrosis factor (TNF) alpha increases collagen accumulation and proliferation in intestinal myofibroblasts via TNF receptor 2. J Biol Chem. 2005;280:36099–109. doi: 10.1074/jbc.M505291200. [DOI] [PubMed] [Google Scholar]