

Summary
Expression of the scavenger receptor class B, type I (SR-BI) receptor facilitates high density lipoprotein cholesterol transport and correlates with protection against atherosclerosis. Studies have shown that SR-BI self-associates, but many of the techniques used to characterize SR-BI homo-oligomerization were wrought with the prospect of producing artifacts. Therefore, we employed fluorescence resonance energy transfer (FRET) to visualize SR-BI homo-oligomerization with the benefit of gaining information about its quaternary structure in the absence of typical membrane receptor artifacts. To this end, SR-BI was tagged at the N- or C-termini with either cyan (CFP) or yellow (YFP) fluorescent protein. To test whether SR-BI subunits oligomerize through N-N, N-C or C-C terminal interactions, we co-expressed the appropriate SR-BI fusion protein combinations in COS-7 cells and measured live-cell FRET following acceptor photobleaching. We did not observe FRET with co-transfection of SR-BI with CFP and YFP at the N-termini nor at the N- and C-termini, suggesting that the N-termini are not proximal to each other or to the C-termini. However, FRET was observed with co-transfection of SR-BI with CFP and YFP at the C-termini, suggesting that the C-terminal ends are within 10 nm of each other, consistent with SR-BI dimerization via its C-terminal region.
Keywords: scavenger receptor class B, type I, SR-BI, fluorescence resonance energy transfer, FRET, selective uptake, oligomerization, acceptor photobleaching
INTRODUCTION
Epidemiological studies show that the risk for developing coronary heart disease, including atherosclerosis, is inversely related to plasma concentrations of high density lipoproteins (HDL) [1, 2]. It is believed that HDL is athero-protective, primarily by virtue of its role in reverse cholesterol transport (RCT). RCT is defined as the process whereby cholesterol is transferred from peripheral tissues to HDL and then transported to the liver for bile acid synthesis and/or secretion into bile, or to steroidogenic tissues for steroid hormone synthesis. Scavenger receptor class B, type I (SR-BI), an 82 kDa cell surface glycoprotein, was identified as a physiologically relevant HDL receptor due to its ability to facilitate the selective uptake of HDL cholesteryl esters (CE) [3]. SR-BI-mediated selective uptake of HDL-CE is a two-step process: (i) HDL must bind to the extracellular domain of SR-BI and (ii) lipid alone is transferred from HDL to the plasma membrane without holoparticle uptake (reviewed in [4]). SR-BI is highly expressed in the liver, adrenals, ovaries and testes: tissues which exhibit high rates of HDL-CE selective uptake [3, 5–8] and represent the last step in RCT.
The flux of free cholesterol (FC) between cells and serum lipoproteins is important for the maintenance of whole body cholesterol homeostasis and RCT. The flux of FC between cells and extracellular acceptors is important for two steps in this process: (i) the removal of FC from peripheral cells, including macrophages, and (ii) the delivery of HDL-FC to the liver. Several studies have demonstrated that rates of FC efflux to HDL are correlated to SR-BI levels [9, 10]. In addition, SR-BI stimulates bidirectional FC flux with the direction of net FC movement determined by the cholesterol concentration gradient between cells and acceptor lipoproteins [11, 12]. The fact that SR-BI facilitates the bi-directional flux of cholesterol suggests the possibility that this receptor participates in both early and late steps in the RCT pathway.
The detailed mechanisms by which SR-BI mediates cholesterol movement between HDL and cells are poorly understood. Reaven et al. found that SR-BI existed in an elaborate cell surface compartment of HDL-filled microvillar channels that were formed by the stacking of microvilli or by juxtapositioning of microvilli against the cell surface [13, 14]. Recently, our lab demonstrated that the vast majority of SR-BI was present in patches or clusters that were predominantly on microvillar extensions of the plasma membrane [15]. However, to date, there is very little information detailing how the structural organization of SR-BI within the plasma membrane may affect its role as a conduit for the mass movement of CE and FC between cells and HDL.
SR-BI (509 amino acids) is a membrane protein with a large extracellular loop (408 residues), two transmembrane domains (22 and 23 residues) and two small cytoplasmic tails (C-term, 47 residues; N-term, 9 residues) [16]. Studies in rodent adrenal glands demonstrated the existence of SR-BI oligomers [7, 17, 18] and recently our laboratory provided further biochemical proof that SR-BI receptors exist as homo-dimers, tetramers and possibly higher order oligomers [19]. This is intriguing since the low aqueous solubility of CE and the rates of CE selective uptake (compared to those of various phospholipids) led to the proposal that HDL-CE uptake may occur via a non-aqueous pathway, possibly involving SR-BI and the formation of a “hydrophobic channel” [20]. It is possible that this “channel” may occur either by multimerization of SR-BI with itself or with other membrane proteins.
In the present study, we used fluorescence resonance energy transfer (FRET) spectroscopy between fluorescent protein-tagged SR-BI monomers to examine oligomerization in living cells. FRET is a distance-dependent physical process by which energy from an excited donor fluorophore is transferred to an energy acceptor fluorophore, provided the distance between the two is 1–10 nm ([21]; for reviews see [22–25]). Since the efficiency of FRET is dependent on the inverse sixth power of intermolecular separation [26, 27], a doubling of the distance between donor and acceptor fluorophores can reduce the FRET efficiency from 50% to 1.5%. Thus, the detection of FRET indicates the likelihood of protein-protein interaction between subunits that contain fluorophore pairs [28–32].
We chose to use monomeric enhanced cyan fluorescent protein (CFP) and monomeric enhanced yellow fluorescent protein (YFP) as the donor/acceptor fluorophore pair for studying SR-BI intermolecular interactions. The CFP/YFP pair: 1) has substantial overlap in the emission/excitation spectra, 2) is fluorescently stable, and can be repeatedly excited to take sequential images without substantial loss of fluorescence intensity, and 3) has a relatively large calculated Ro (the distance at which 50% energy transfer occurs) between randomly oriented fluorophores of 5.2 nm, thus allowing FRET to be detected at larger distances [25, 33, 34]. After coupling SR-BI with either CFP (donor) or YFP (acceptor), we exploited FRET technology to determine (i) whether SR-BI oligomerizes in intact cells, in the absence of detergents and (ii) which region of SR-BI might be responsible for oligomerization. Our findings revealed that the C-termini of SR-BI monomers, and not the N- termini, were within 10 nm of each other and were therefore close enough in proximity to elicit protein-protein interactions. These observations hold implications for the homo-oligomeric structure of SR-BI within the cell membrane.
MATERIALS AND METHODS
2.1 Chemicals
The following antibodies were used: polyclonal anti-SR-BI C-terminal and extracellular domain antibodies (Novus Biologicals, Inc., Littleton, CO); peroxidase-conjugated goat anti-rabbit secondary IgG (Jackson ImmunoResearch Laboratories, West Grove, PA); A.v. peptide antibody to Aequorea victoria green fluorescent protein (GFP) (BD Biosciences Clontech); conjugated Alexa-546 goat anti-rabbit secondary IgG was from Molecular Probes (Eugene, OR). The Alexa-568 protein labeling kit was also from Molecular Probes. All other reagents were of analytical grade.
2.2 Preparation of DNA constructs
cDNAs encoding the monomeric enhanced forms of CFP and YFP were a kind gift from Dr. R. Y. Tsien (U.C. San Diego). These mutated forms of CFP and YFP decrease the potential background of FRET due to their inability to form homodimers [34]. In addition, they have no internal targeting signals, reducing the possibility that they will affect SR-BI localization. Four plasmids encoding SR-BI fused with either CFP or YFP were generated. CFP or YFP fused to the N-terminal end of SR-BI were designated CFP-SR-BI and YFP-SR-BI, respectively; CFP or YFP fused to the C-terminal end of SR-BI were designated SR-BI-CFP and SR-BI-YFP, respectively (Figure 1A). The coding regions of CFP and YFP were amplified by PCR using the following primers: 5′-AGGCCAGGATCCACC-GGTCGCCACCATGGTGAGCAAGGGCGAGGAG-3′ (sense) and 5′-AGCCAGGATCCTCTAGAGGCCGCTTTACTTGTACAGCTCGTCCATGCCG-3′ (anti-sense). For construction of CFP-SR-BI and YFP-SR-BI, the resulting PCR products were digested with HindIII and KpnI, and ligated to similarly digested pcDNA3-SR-BI. For construction of SR-BI-CFP and SR-BI-YFP, the PCR products were digested with AgeI and PstI and ligated to similarly digested pGFPSRBI-N1, with CFP and YFP sequences replacing the GFP sequence.
Figure 1.
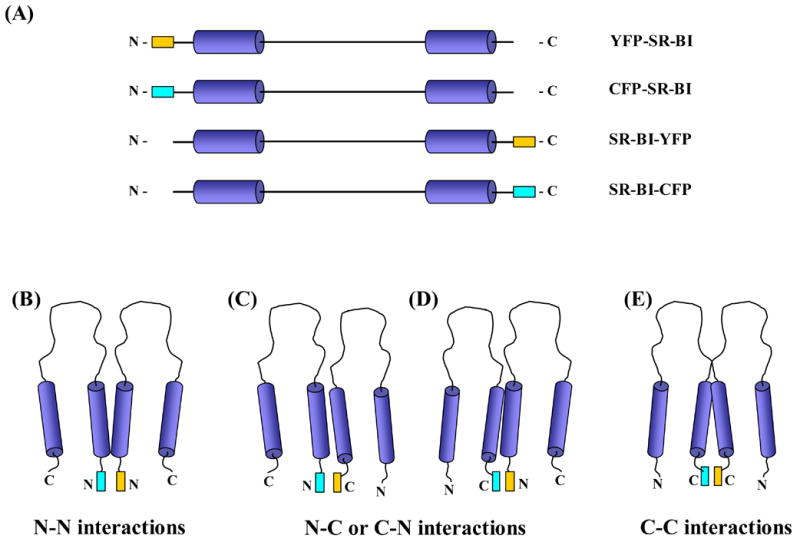
(A) SR-BI fusion proteins. Schematic illustrating linearized SR-BI proteins fused to a CFP or YFP protein either at the N-terminal or C-terminal end of the receptor. CFP protein is designated by the cyan rectangle; YFP protein is designated by the yellow rectangle. (B–E) Possible combinations of SR-BI monomers. COS-7 cells were co-transfected with CFP-SR-BI/YFP-SR-BI (B), CFP-SR-BI/SR-BI-YFP (C), SR-BI-CFP/YFP-SR-BI (D) or SR-BI-CFP/SR-BI-YFP (E).
All constructs were sequenced throughout the coding region to confirm the correct fluorescent protein addition and to ensure that no point mutations had been generated (automated sequencing facility at Stony Brook University).
2.3 Transient transfection of COS-7 cells
COS-7 cells were maintained and transfected as previously described [35]. The cells were assayed 48 h post-transfection unless otherwise indicated. Cell lysates were prepared [8, 36] and protein concentrations were determined by the Lowry method with modifications for use with detergents [37]. Protein lysates were resolved by SDS-PAGE [38], transferred onto nitrocellulose membranes and detected using a polyclonal anti-SR-BI C-terminal or anti-SR-BI extracellular antibody (1:5000) or anti-fluorescent protein antibody (A.v. peptide antibody) (1:100), followed by a horseradish peroxidase-conjugated anti-rabbit secondary antibody (1:10,000). Antigen-antibody complexes were detected with the SuperSignal West Pico reagent (Pierce).
2.4 Preparation of [125I]-DLT-[3H]COE-HDL, [3H]CE-HDL and fluorescent HDL
Human HDL3 (1.125 g/ml < ρ < 1.210 g/ml), herein referred to as HDL, was isolated by sequential ultracentrifugation [39]. The HDL was labeled with either non-hydrolysable [3H]-cholesteryl oleyl ether ([3H]-COE) (Amersham Life Sciences) or hydrolysable [3H]-cholesteryl oleate ([3H]-cholesteryl ester; [3H]-CE)) (Amersham Life Sciences) using recombinant cholesteryl ester transfer protein (Cardiovascular Targets, Inc.) as described [40] with modifications [41]. Labeled particles were reisolated by size exclusion chromatography and then labeled with [125I]-dilactitol tyramine (DLT) as previously described [35]. The average radiospecific activity of the [125I]-DLT-[3H]-COE-HDL was 330 dpm/ng protein for 125I and 4.0 dpm/ng protein (16.7 dpm/ng CE) for 3H. The average specific activity of the [3H]-CE-HDL was 13.5 dpm/ng protein (52.1 dpm/ng CE) for 3H. For fluorescent staining experiments, HDL was labeled with the Alexa-568 protein labeling kit according to manufacturer’s protocols.
2.5 HDL cell association, selective COE uptake and apolipoprotein degradation
Transiently transfected COS-7 cells (in 35 mm wells) were washed once with serum-free DMEM containing 0.5% BSA, and [125I]-DLT-[3H]-COE-HDL particles were added at a concentration of 10 μg protein/ml in serum-free DMEM containing 0.5% BSA. After incubation for 1.5 h at 37° C, the medium was removed and the cells were washed three times with PBS containing 0.1% BSA (pH 7.4) and one time with PBS (pH 7.4). The cells were lysed with 1.1 ml 0.1 N NaOH, and the lysate was processed to determine trichloroacetic acid soluble and insoluble 125I radioactivity and organic solvent-extractable 3H radioactivity. The values for cell-associated HDL apolipoprotein, total cell associated HDL-COE and the selective uptake of HDL-COE, were obtained as previously described [35]. Cholesteryl oleyl ether was used instead of cholesteryl ester because COE is non-hydrolysable, eliminating the complication of CE hydrolysis and flux of radioactive free cholesterol. The efficiency of HDL-COE selective uptake was determined by subtracting the values from vector-transfected cells and normalizing the amount of HDL-COE selective uptake to the amount of cell-associated HDL particles.
2.6 Cholesteryl ester hydrolysis assay
Assays for cholesteryl ester hydrolysis were performed as previously described using hydrolysable [3H]-CE-labeled HDL [41].
2.7 Fluorescence staining
Transiently-transfected cells were replated onto coverslips 24 h post-transfection and cells were processed 24 h after replating. For fluorescent HDL labeling experiments, cells were incubated with 20 μg/mL Alexa-568-HDL as previously described [15]. Alternatively, cells were stained with anti-SR-BI antibody (against the C-terminal domain), followed by Alexa-546 secondary antibody as described in [15].
2.8 Emission spectra of proteins tagged with fluorescent protein
Emission spectra were generated to confirm that all forms of fluorescent protein-tagged SR-BI emitted fluorescence at their expected wavelengths. COS-7 cells were transiently transfected with various constructs of fluorescent protein-tagged SR-BI. Cells were lysed 48 h post transfection by incubating with ice-cold cell lysis buffer (0.5% NP-40, 10 mM Tris-HCl pH 7.5, 1 mM MgCl2, 10 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 0.2 mM phenylmethylsulfonyl fluoride) on ice for 10 min. Cell lysates were collected and centrifuged at 12,000 rpm for 10 min to remove cellular debris. Emission spectra were collected on supernatants in a quartz cuvette using a Carey Eclipse Spectrometer. A band width of 5 nm was used for both excitation and emission monochromators. Fusion proteins tagged with CFP were excited at 435 nm and emission spectra were collected from 455 nm to 550 nm. Fusion proteins tagged with YFP were excited at 514 nm and emission spectra were collected from 520 nm to 600 nm. The results were analyzed using Carey Eclipse Scan software.
2.9 Live cell imaging and FRET
COS-7 cells were transiently transfected with various constructs of fluorescent protein-tagged SR-BI and plated onto glass bottom 35 mm dishes 24 h post-transfection. At 48 h post-transfection, live cells were illuminated with a Zeiss LSM510 laser module, and examined using a Zeiss Axiovert 200 microscope. Live cells were maintained at 37 °C in a temperature-controlled cell perfusion chamber. To minimize the contribution of background fluorescence prior to imaging, the phenol-red-containing DMEM was replaced with HEPES-modified phenol-red-free DMEM containing 1% BSA. Cells were examined with a 63 X 1.4 NA Zeiss oil immersion objective. To visualize CFP fluorescence, cells expressing fluorescent fusion proteins were excited at 458 nm and images were acquired using 470–500 nm band pass filters. To visualize YFP fluorescence, cells expressing fluorescent fusion proteins were excited at 514 nm and images were acquired using 530–600 nm band pass filters. To visualize possible FRET between tagged SR-BI constructs, cells were co-transfected with both C-terminally tagged SR-BI constructs, or both N-terminally tagged SR-BI constructs, or mixtures of C- and N-terminally tagged SR-BI constructs (Figure 1, B–E). A visual survey was used to identify cells with comparable CFP and YFP expression for further examination. A “timed bleach” protocol was employed to sequentially automate the acquisition of pre-bleach images, perform the acceptor photobleaching and acquire post-bleach images. A selected region of interest (ROI 1) within the live cell was scanned two times pre-bleaching. ROI 1 was either a region at the plasma membrane, or in regions within the cell that highly expressed SR-BI. A strong argon laser (514 nm), with 50% of 30 mW maximum laser power (100% transmission) for 100 iterations was used to photobleach YFP fluorescence in ROI 1. Four additional scans of CFP and YFP fluorescence of ROI 1 were acquired immediately following photobleaching. Two additional ROI’s were monitored as controls: (i) an area within the cell parallel to ROI 1 that was not bleached in order to detect fluorescence fluctuations independent of bleaching (ROI 2) and (ii) an area outside the cell to measure background fluorescence (ROI 3). In each time series, ROI 1 and ROI 2 intensities were background corrected using the fluorescence intensities of ROI 3. Fluorescence intensities of pre-photobleach and post-photobleach images, as well as percent FRET efficiencies within the region of interest were calculated by the “FRET macro” in the LSM 510 META software provided by Zeiss and based on calculations described in Karpova et al. [42]. An increase in CFP fluorescence intensity after photobleaching of YFP fluorescence indicated FRET. Average FRET efficiencies were calculated based on approximately 10–18 images taken from each experimental condition, where each experimental condition was performed at least 4 times.
RESULTS
Our laboratory has applied a variety of biochemical methods to show that SR-BI receptors may exist not only as monomers, but also as homo-dimers and homo-tetramers in the plasma membrane [19]. Most of the techniques we utilized to characterize potential SR-BI homo-oligomerization however, were wrought with the prospect of producing detergent-induced artifacts due to the nature of this cell surface receptor being a two-transmembrane domain protein. Therefore, we employed FRET to determine whether the monomeric subunits of SR-BI could be close enough in proximity to form oligomers through N-N, N-C or C-C terminal protein-protein interactions (Figure 1, A–E).
We generated four constructs for this study: CFP-SR-BI and YFP-SR-BI, where the fluorescent proteins were fused to the N-terminal domain of SR-BI, and SR-BI-CFP and SR-BI-YFP, where the fluorescent proteins were fused to the C-terminal domain of SR-BI (Figure 1, A–E). We used the monomeric forms of enhanced CFP and enhanced YFP to reduce the potential of causing artifactual dimerization of SR-BI [34]. We then performed several experiments to ensure that the fusion proteins generated from these constructs retained wild-type SR-BI cellular localization and cholesterol transport function.
Characterization of CFP and YFP fluorophores tagged to SR-BI
To confirm that fusion of SR-BI to CFP or YFP did not alter the emission spectra of these fluorescent proteins, we transiently transfected COS-7 cells with the various constructs. Cells expressing the CFP-tagged SR-BI fusion proteins were excited at 435 nm and generated the expected emission spectra with emission peaks at 478 and 500 nm (data not shown). In addition, cells expressing the YFP-tagged SR-BI fusion proteins were excited at 514 nm and generated expected emission spectra with emission peaks at 530 nm (data not shown).
Localization and oligomerization patterns of CFP- and YFP-tagged SR-BI
Wild-type SR-BI resides in irregularly-shaped clusters in COS-7 cells, including in patches and small extensions of the cell surface [15]. We confirmed the cellular localization of wild type SR-BI in COS-7 cells by confocal microscopy, either by staining with an antibody directed against the C-terminal end of SR-BI (Figure 2, panel E) or following incubation with 20 μg/mL Alexa-labeled HDL (Figure 2, panel F). To verify that our SR-BI fusion proteins retained wild-type SR-BI cellular localization, COS-7 cells were transiently transfected with various constructs of fluorescent protein-tagged SR-BI and viewed by confocal microscopy. Our data revealed that there were no visual changes, at the level of the light microscope, in the localization of SR-BI when the fluorescent proteins were fused to either its N- or C-terminal ends (Figure 2, panels A-D). To ensure that fluorescent protein-tagged SR-BI co-localized with wild-type SR-BI, COS-7 cells transiently expressing either of the fusion proteins along with wild-type SR-BI were incubated with antibody against the C-terminal cytoplasmic domain of SR-BI, and stained using Alexa-568 conjugated secondary antibody. Figure 3 shows that SR-BI-CFP (Panels A–C) and SR-BI-YFP (Panels D–F) co-localized with wild-type SR-BI. It is important to note that the anti-C-terminal SR-BI antibody does not interact with SR-BI-CFP or SR-BI-YFP due to steric hinderance by the fused fluorescent protein attached to SR-BI’s C-terminal end (data not shown). Therefore, this experiment could only be performed with the two C-terminally tagged, but not the two N-terminally tagged receptors.
Figure 2.
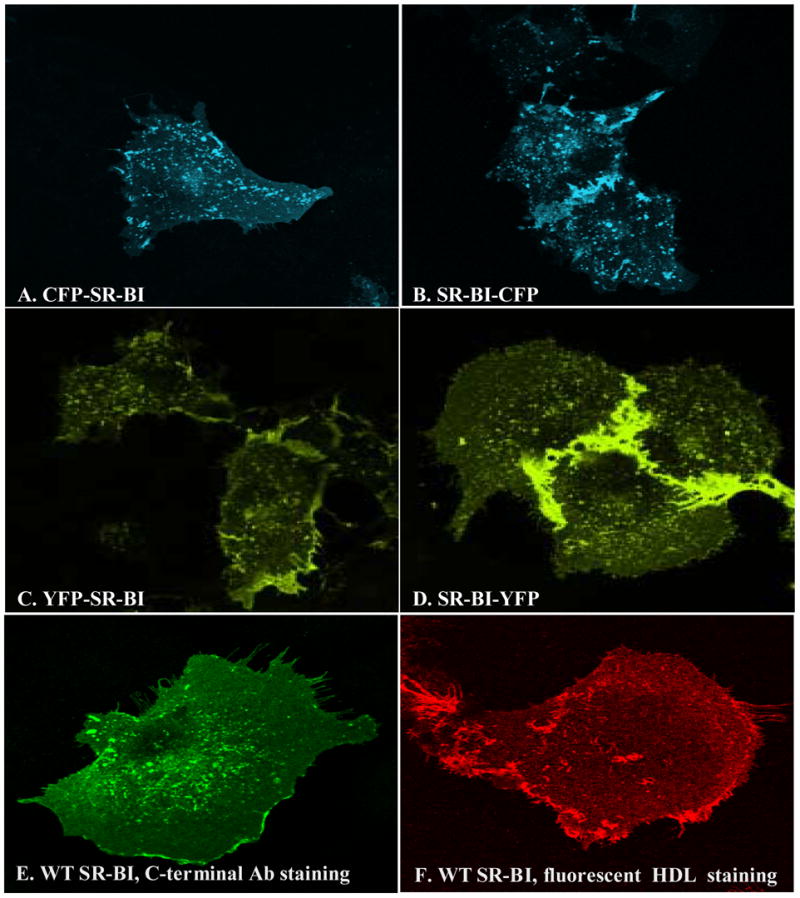
SR-BI/fluorescent protein fusion proteins retain wild-type SR-BI cellular localization. COS-7 cells were transiently transfected with various constructs of fluorescent protein-tagged SR-BI for 48 h, fixed with 4% paraformaldehyde, mounted and observed by confocal microscopy as described in Materials and Methods. CFP was excited at 458 nm and the images were collected from 475 nm to 525 nm; YFP was excited at 514 nm and the images were collected from 530 nm to 600 nm. Panel A: CFP-SR-BI; Panel B: SR-BI-CFP; Panel C: YFP-SR-BI; Panel D: SR-BI-YFP. Cellular localization of wild-type SR-BI transiently transfected into COS-7 cells is shown by staining with an anti-SR-BI antibody (anti- C-terminal domain), followed by Alexa-546 secondary antibody (panel E), or following incubation with 20 μg/mL Alexa-568-HDL (panel F) as described in Materials and Methods.
Figure 3.
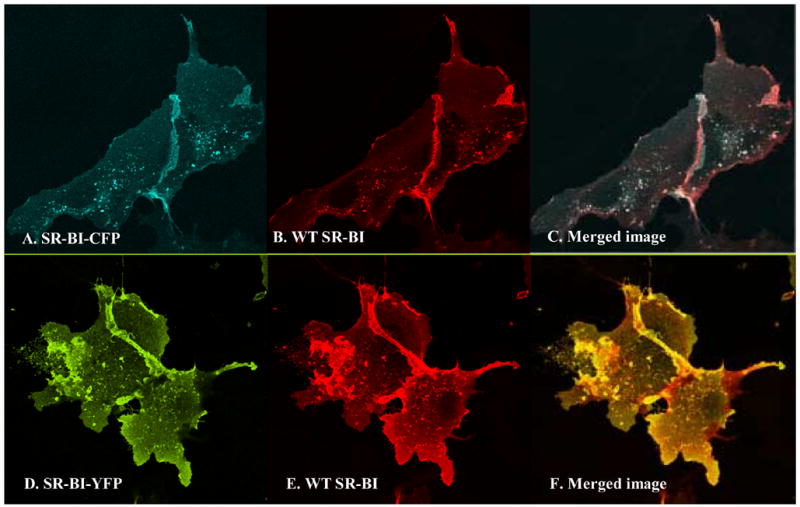
Fluorescent protein-tagged SR-BI co-localizes with wild-type SR-BI. COS-7 cells were co-transfected with fluorescent protein-tagged SR-BI and wild-type SR-BI for 48 h. Cells were fixed with 4% paraformaldehyde, permeabilized with 1% Triton X-100, and blocked with 3% BSA, 10 mM glycine in PBS. Panels A–C, cells co-transfected with SR-BI-CFP and wild-type SR-BI. Panels D–F, cells co-transfected with SR-BI-YFP and wild-type SR-BI. Panels A and D: fluorescent protein-tagged SR-BI; CFP was excited at 458 nm and images were acquired from 475 nm to 525 nm, whereas YFP was excited at 514 nm and images were acquired from 530 nm to 600 nm. Panels B and E: antibody staining using antibody directed against the C-terminal portion of SR-BI (primary antibody) and Alexa-546 anti-rabbit antibody (secondary antibody) as previously described [15]. Panels C and F: merged images.
In order to confirm that the SR-BI fusion proteins were properly expressed at the cell surface and functioned in their primary capacity as HDL receptors, we performed co-localization studies with the various SR-BI constructs and fluorescently-labeled HDL. The data clearly showed that the SR-BI fusion proteins bound HDL due to the extensive co-localization of Alexa-labeled HDL with CFP-tagged SR-BI or YFP-tagged SR-BI (data not shown). The co-localization between HDL labeling and fluorescent fusion protein expression was especially prominent on short cell surface extensions and on larger membrane extensions that extend between adjacent cells. These co-localization and cell surface expression patterns were very similar to those observed with wild-type SR-BI [15], confirming that all four fusion proteins retained their cell surface expression and HDL binding functions.
Finally, to confirm that fluorescent-tagged SR-BI multimerized similar to wild-type SR-BI, CFP- and YFP-tagged SR-BI were transiently transfected into COS-7 cells. Lysates were collected in dodecyl maltoside-containing lysis buffer and analyzed by blue native PAGE as described by Sahoo et al. [19]. The data revealed that all SR-BI fusion proteins existed as monomers, dimers and higher order oligomers, with patterns similar to those exhibited by wild-type receptor (data not shown).
CFP- and YFP-tagged SR-BI retain wild-type receptor functions
To ensure that the CFP- and YFP-tagged SR-BI proteins maintained wild-type cholesterol transport function, COS-7 cells transiently expressing wild-type SR-BI or the fusion proteins were incubated with radiolabeled HDL, after which cells were processed as previously described [35]. Figure 4 shows that cells expressing each of the SR-BI fusion proteins exhibited wild-type levels of cell-associated HDL (Figure 4, Panel A), selective uptake of HDL-COE (Figure 4, Panel B) and selective uptake efficiency (Figure 4, Panel C).
Figure 4.
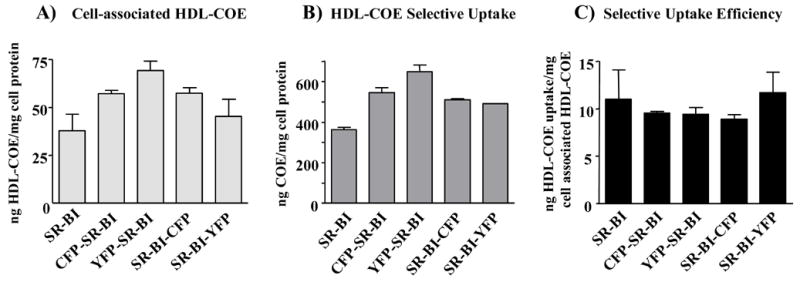
Cells expressing fluorescent protein-tagged SR-BI exhibit wild-type levels of cell-associated HDL, selective uptake of HDL-COE and selective uptake efficiency. COS-7 cells transiently expressing wild-type SR-BI or the fluorescent protein-tagged fusion proteins were incubated at 37° C for 1.5 h with [125I]DLT/[3H]COE-labeled HDL (10 μg of HDL protein/ml), after which cells were processed as previously described [35]. Cell-associated HDL-COE (Panel A), selective uptake of HDL-COE (Panel B), and selective uptake efficiency (Panel C) are shown. The efficiency of HDL-COE selective uptake was determined by subtracting the values from vector-transfected cells and normalizing the amount of HDL-COE selective uptake to the amount of cell-associated HDL particles. Values represent the mean ± SD of three replicates and are representative of three independent experiments.
SR-BI has also been shown to deliver HDL-CE into a metabolically active membrane pool where CE is efficiently hydrolyzed to FC [41]. To confirm that addition of CFP or YFP to SR-BI did not alter this receptor function, we tested the ability of the fusion proteins to facilitate CE hydrolysis. As shown in Figure 5, Panel A, levels of HDL-CE selective uptake were similar between cells transiently expressing wild-type SR-BI or the SR-BI fusion proteins. After incubating with radiolabeled HDL, similar percentages of total radioactive counts remained as CE in cells expressing wild-type SR-BI versus the SR-BI fusion proteins (Figure 5, Panel B), indicating that fluorescent protein-tagged SR-BI delivered HDL-CE to a metabolically active pool where CE could be efficiently hydrolyzed to FC. Immunoblot analysis of protein lysates from parallel wells of cells, detected with antibodies directed against SR-BI’s extracellular domain or the fused fluorescent protein, revealed that the SR-BI fusion proteins were expressed at similar levels within the cells (Figure 5, Panel C). However, due to differential detection with antibodies directed against different parts of the fusion proteins, HDL-COE selective uptake was normalized to HDL binding instead of SR-BI levels as revealed via immunoblot. In addition, the assays were repeated several times and each time they were repeated, all of the assays (Figures 4 and 5) were performed with the same transiently transfected cells. This way all of the cholesterol transport results were directly comparable to the same HDL binding data (Figure 4C), which should be a direct reflection of the cell surface expression of each of the fusion proteins.
Figure 5.
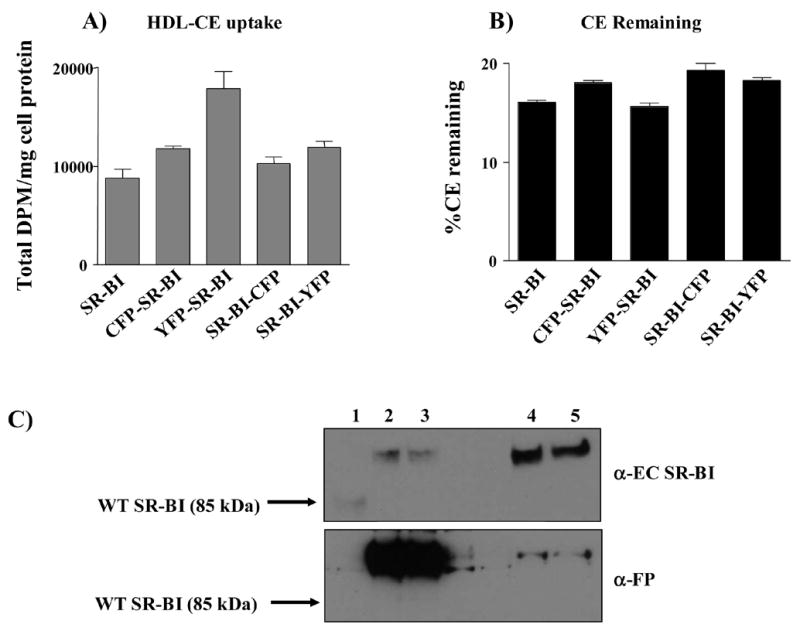
Cells expressing fluorescent protein-tagged SR-BI exhibit wild-type levels of HDL-CE hydrolysis. To measure CE hydrolysis, COS-7 cells transiently expressing wild-type SR-BI or fluorescent protein-tagged receptors were incubated at 37° C for 2 h with 10 μg/ml [3H]CE-labeled HDL in the presence of an acyl:CoA acyl transferase (ACAT) inhibitor at a final concentration of 2 μg/ml. After incubation, the cells were processed as previously described [41]. Total cell associated dpm/mg cell protein (Panel A) and the percentage of the total number of counts that remained as [3H]CE (Panel B) are shown. ACAT inhibitor was added to prevent re-esterification of radioactive FC generated by hydrolysis of radioactive CE. Values represent the mean ± SD of three replicates and are representative of three independent experiments. Panel C: Immunoblots showing expression of wild-type SR-BI (lane 1), SR-BI-CFP (lane 2), SR-BI-YFP (lane 3), CFP-SR-BI (lane 4) and YFP-SR-BI (lane 5) are shown using antibody directed against the extracellular domain of SR-BI (EC; top panel) or the fluorescent protein tag (FP; bottom panel). Immunoblot represents protein expression levels for experiments performed in Figures 4 and 5.
Taken together, analysis of the tagged SR-BI proteins revealed that the addition of CFP or YFP to the N- or C-termini did not affect SR-BI-mediated HDL binding or cholesterol transport, nor did we observe any changes in receptor oligomerization or cellular localization.
FRET analysis of SR-BI homo-oligomers by acceptor photobleaching
FRET is a very sensitive and powerful method for detecting and measuring changes in donor-acceptor fluorophore proximity. One of the most direct and widely-used approaches for measuring FRET is acceptor photobleaching [25, 42–46]. In this process, YFP fluorescence is photobleached at 514 nm, resulting in complete reduction of YFP fluorescence. If the CFP and YFP fluorophores are in close proximity (between 1–10 nm), then the destruction of YFP fluorescence releases the energy transferred from the donor, and causes an increase in CFP fluorescence. The changes in CFP intensity pre- and post-bleaching allow us to calculate the changes in FRET efficiency [32].
We used acceptor photobleaching to examine the various combinations of fluorescent protein-tagged SR-BI monomers to determine whether SR-BI homo-oligomerizes via N-N, C-C or N-C domain-domain interactions (Figure 1, B–E). We performed these experiments in living cells that were maintained at 37 °C in a temperature-controlled cell perfusion chamber so that we had the added benefit of examining the self-association of SR-BI intact in the plasma membrane, in the absence of detergents. Prior to initiation of our experiments, we tested whether photobleaching at 514 nm would affect the fluorescent intensity of the CFP fluorophore. We transiently transfected COS-7 cells with either CFP-SR-BI or SR-BI-CFP. Following photobleaching of the cells at 514 nm, the fluorescent images revealed that photobleaching did not affect the fluorescence intensities of CFP-tagged SR-BI (data not shown). Fluorescence quantification corroborated the lack of change in fluorescence intensities. To confirm that the CFP and YFP proteins themselves did not oligomerize and create artificial FRET signals, we co-expressed monomeric CFP and YFP in COS-7 cells. Cells expressing roughly equal amounts of CFP and YFP were chosen for examination. Following photobleaching of YFP fluorescence, there was a near absence of YFP fluorescence, yet there were no detectable changes in the fluorescence intensities of CFP (Figure 6). FRET efficiency between CFP and YFP was 0.6 %, strongly suggesting that monomeric CFP and YFP do not oligomerize under our experimental conditions. Therefore, tagging of either fluorophore to SR-BI should not artificially create FRET signals in our live cell system. Furthermore, co-transfection of CFP and YFP fluorophores into our cells served as an excellent negative control for all experimental conditions.
Figure 6.
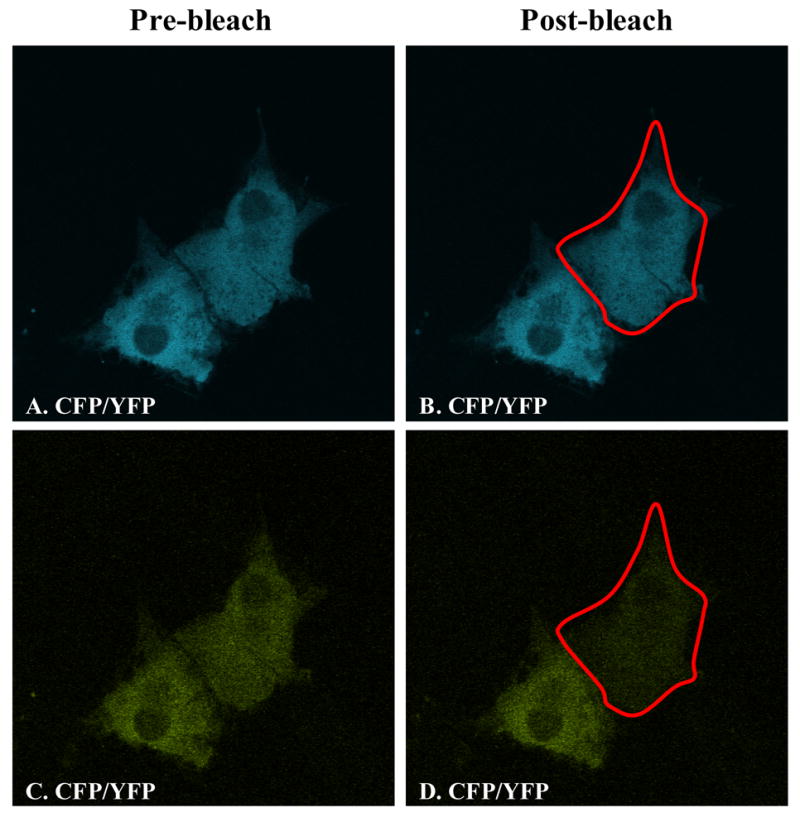
Monomeric forms of CFP and YFP do not generate FRET signals in live cells. COS-7 cells were transiently co-transfected with equal amounts of CFP and YFP proteins (non-tagged). Panels A, B show CFP fluorescence following excitation at 458 nm. Panels C, D show YFP fluorescence following excitation at 514 nm. Panels A, C: images taken prior to acceptor photobleaching; panels B, D: images taken after acceptor photobleaching. The red outline indicates the region of interest chosen for FRET measurements.
We chose to use the CFP-ApoE4-YFP construct as a positive control for our FRET experiments [31]. Apolipoprotein E4 is a major risk factor for Alzheimer’s disease, and it has been suggested that some of its detrimental effects in neurobiology may be the result of a domain-domain interaction mediated by a salt bridge between its N- and C-terminal domains [47, 48]. An ApoE4 construct with YFP fused at the N-terminus and CFP fused at the C-terminus of the protein was generated to show that intramolecular domain-domain interactions did take place based on a positive FRET signal that occurred upon expression of this construct in Neuro-2a cells [31]. We transiently expressed this fusion protein in COS-7 cells, photobleached the YFP fluorophore and measured FRET efficiency. Quantification of the fluorescent intensities revealed an increase in CFP fluorescence, resulting in a FRET efficiency of 6.4 %. Although the FRET efficiency was not calculated in the same fashion, these results were in line with those previously reported for this construct [31] and validated our FRET methodology.
With proper positive and negative controls in place, we proceeded to study the interactions between the SR-BI fusion proteins. COS-7 cells were transiently co-transfected with the tagged constructs in four different combinations: (i) CFP-SR-BI and YFP-SR-BI (Figure 1B) to examine the potential self-association between N-terminal domains; (ii) CFP-SR-BI and SR-BI-YFP (Figure 1C) or (iii) SR-BI-CFP and YFP-SR-BI (Figure 1D) to examine the potential self-association between the N- and C-terminal domains; and (iv) SR-BI-CFP and SR-BI-YFP (Figure 1E) to examine the potential self-association between C-terminal domains. COS-7 cells transiently co-expressing roughly equal amounts of these fusion proteins were photobleached at 514 nm and FRET efficiency was quantified. Figure 7 represents an example of images taken from cells co-expressing SR-BI-CFP and SR-BI-YFP to examine C-terminal domain-domain interactions. As summarized in Figure 8, only cells that were transfected with SR-BI-CFP/SR-BI-YFP produced a positive FRET efficiency of 12.3 %. The other combinations revealed near zero FRET efficiencies. Additionally, FRET measurements were also collected in transiently transfected HEK293 cells with similar results (data not shown). These data suggest that the C-terminal regions of SR-BI lie within 10 nm of each other in the plasma membrane. These observations are consistent with SR-BI forming homo-oligomeric protein-protein interactions through its C-terminal regions.
Figure 7.
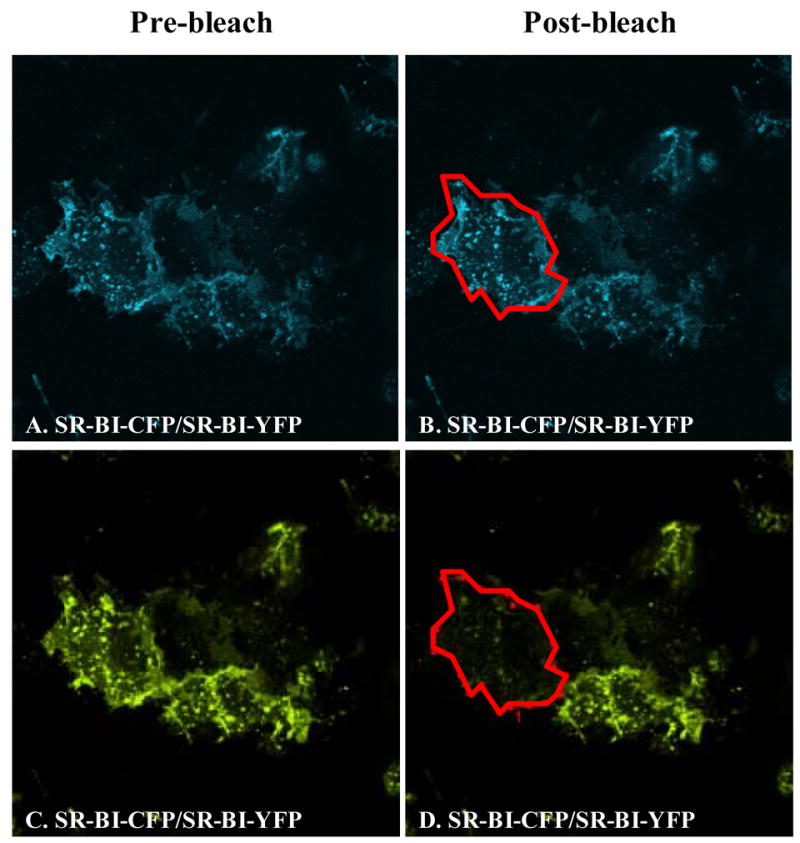
Photobleaching of YFP fluorescence in COS-7 cells expressing SR-BI-CFP/SR-BI-YFP constructs. COS-7 cells were transiently co-transfected with SR-BI-CFP and SR-BI-YFP proteins. Panels A, B show CFP fluorescence following excitation at 458 nm. Panels C, D show YFP fluorescence following excitation at 514 nm. Panels A, C: images taken prior to acceptor photobleaching; panels B, D: images taken after acceptor photobleaching. Images are representative of approximately 10–18 images taken for this particular experimental condition, which was performed at least 4 times. The red outline indicates the region of interest chosen for FRET measurements.
Figure 8.
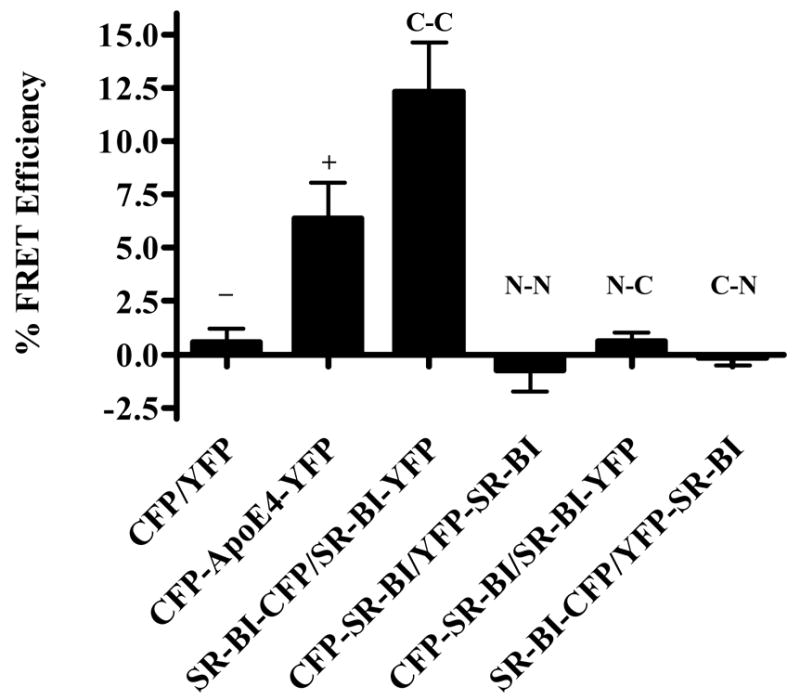
FRET efficiencies for all combinations of SR-BI monomers in COS-7 cells. FRET efficiencies were calculated using the “FRET macro” software provided by Zeiss. Domain-domain interactions are indicated above each bar, where “C” represents the C-terminal domain and “N” represents the N-terminal domain. “−“ and “+” indicate negative and positive controls, respectively. FRET efficiencies are the average of 16 images ± SD and are representative of 4 independent experiments.
DISCUSSION
In our previous study, we used a variety of biochemical techniques to show that SR-BI exists as an oligomer [19]. In the current report, we designed CFP- and YFP-fusion constructs of SR-BI for use in FRET studies to provide additional evidence for the oligomerization of SR-BI at the plasma membrane. Furthermore, we exploited the FRET technique to demonstrate that SR-BI self-associates via interactions within regions of its C-terminal domain (Figure 1E). These studies are of particular importance because they were performed in live cells, under non-invasive, physiological conditions, thus negating any suggestion that SR-BI oligomerization may be the result of detergent-induced aggregation.
We examined SR-BI oligomerization and spatial proximity relationships between SR-BI monomers using acceptor photobleaching [49]. The acceptor photobleaching method is rapidly increasing in popularity as a widely-used and trusted technique for analyzing FRET efficiencies [25, 42–44, 46] and is advantageous for several reasons: (i) it avoids the need for repeated imaging of fluorescence and spectral bleed-through corrections, both absolute requirements for the emission-sensitized imaging method [42, 50, 51]; (ii) use of strong lasers increases bleaching speeds, thus reducing the chance that live cells have moved; (iii) experiments can be performed without the need for additional specialized equipment; (iv) measurements are direct and quantitative and (v) bleaching is targeted, therefore ROI’s can be bleached and then compared to unbleached ROI’s within the same cell as a control [42].
Great care was taken to ensure that our observations were reliable: (i) we transfected cells with only CFP and photobleached the cells at the acceptor excitation wavelength to confirm that photobleaching of the acceptor will not photobleach donor fluorescence; (ii) prior to photobleaching, the cell media was changed to a colorless, pH-balanced HEPES-modified, phenol red-free DMEM that stabilizes the cells and produces less background fluorescence [52–54]; (iii) FRET efficiencies were calculated taking into account 3 different ROI’s from bleached and unbleached regions from at least 10 different cells from each of at least 4 independent transfections (see Materials and Methods); (iv) images that shifted during photobleaching of the live cells were not included in the calculations; (v) monomeric forms of CFP and YFP were used to prevent natural oligomerization [55] between fluorophores and served as our negative controls and (vi) the CFP-apoE4-YFP construct served as an excellent positive control, revealing a FRET efficiency of 6.4 % (this value compares well with an efficiency of 7.96 % obtained for a CFP-YFP fusion protein connected by a two amino acid linker by Karpova et al. [42]).
Based on the calculated FRET efficiencies, we concluded that SR-BI homo-oligomerized at the plasma membrane, and this self-association occurred via interactions between the C-terminal domains of SR-BI monomers (12.3 % FRET efficiency). The lack of observable FRET between two N-terminal domains or between N- and C- terminal domains of SR-BI revealed that, at least under the conditions of our assays, these domains were not in close proximity to each other (more than 10 nm apart). Furthermore, the negative results with the N-terminally tagged SR-BI proteins represented convenient internal controls, proving that the FRET signals we observed between two C-terminal domains of SR-BI were not due to overexpression of the receptors in the transiently-transfected cells.
Findings from our current FRET studies suggest that SR-BI self-association occurs via intermolecular C-terminal domain-domain interactions. In our previous study, however, we found that the last 47 amino acids of SR-BI were not responsible for oligomerization and that SR-BI oligomerization occurred in the absence of proteins that bind to the extreme C-terminal cytoplasmic tail of this receptor [19]. Therefore, it may be the C-terminal transmembrane domain and/or the extracellular region juxtaposed to it that mediate oligomerization. Intriguingly, these regions possess both “GXXXG” and leucine zipper motifs, which are known to be involved in the dimerization of transmembrane channel/transport proteins [56, 57]. Point mutations within the GXXXG motif, or the putative leucine zipper domain did not disrupt SR-BI oligomerization as measured by co-immunoprecipitation experiments [19]. However, the FRET data indicated that the C-terminal region of SR-BI was involved in homo-oligomerization. Therefore, the entire region of SR-BI spanning the GXXXG and leucine zipper motifs (L413-L455) may be required for self-association, and the mutations we created thus far may not have been disruptive enough to abrogate SR-BI oligomerization. Experiments are underway to test more mutations that will help us delineate the region(s) responsible for SR-BI multimerization.
These FRET studies were performed in the absence of ligand such as HDL. It is interesting to speculate that HDL binding might “trigger” SR-BI into an alternative oligomeric state, one that might facilitate CE uptake. For example, HDL may bind an already-existing oligomeric transport complex and induce a conformational change that would allow “opening” of a potential channel and selective HDL-CE uptake to occur. Such a conformational change has been observed with activation of integrin αIIβ3 [58, 59], where the transmembrane-cytoplasmic domains of integrin α and β subunits are likely to be closely aligned in the inactive state and separated in the activated state. It is also possible that HDL binding may alter the oligomeric structure so as to promote new N-N terminal or N-C terminal interactions between SR-BI monomers, and create a “channel” that would facilitate CE uptake from HDL into the plasma membrane for hydrolysis. Therefore, FRET studies in the presence and absence of SR-BI ligands such as HDL, LDL, and oxidized LDL are underway. The use of FRET in living cells to study possible ligand-induced conformational changes within SR-BI oligomers may provide valuable insight into the detailed mechanisms of HDL-CE delivery into the plasma membrane.
Acknowledgments
The authors thank Dr. Roger Y. Tsien (U.C. San Diego) for generously providing us with the cDNA constructs encoding monomeric CFP and YFP and Drs. Yadong Huang and Qin Xu (Gladstone Institute of Cardiovascular disease, U.C. San Francisco) for generously providing us with the CFP-ApoE4-YFP construct to use as a positive control for these FRET studies. We also thank Rhiannon Ledgerwood for excellent technical assistance and Dr. Victor Drover for preparing Figure 1. This work was supported by a National Institutes of Health Grant (HL58012) and by an American Heart Association National Scientist Development Grant, both awarded to D.S.
The abbreviations used are
- ACAT
acyl:CoA acyltransferase
- apoA-I
apolipoprotein A-I
- BSA
bovine serum albumin
- CE
cholesteryl ester
- CFP
monomeric enhanced cyan fluorescent protein
- COE
cholesteryl oleyl ether
- DLT
dilactitol tyramine
- DMEM
Dulbecco’s Minimal Essential media
- EC
extracellular
- EDTA
ethylenediamine-tetraacetic acid
- FC
free cholesterol
- FP
fluorescent protein
- FRET
fluorescence resonance energy transfer
- GFP
green fluorescent protein
- HDL
high density lipoprotein
- LDL
low density lipoprotein
- NP-40
nonidet P-40
- PBS
phosphate buffered saline
- RCT
reverse cholesterol transport
- ROI
region of interest
- SDS
sodium dodecyl sulfate
- SR-BI
scavenger receptor class B, type I
- YFP
monomeric enhanced yellow fluorescent protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gordon T, Castelli WP, Hjortland MC, Kannel WB, Dawber TR. High density lipoprotein as a protective factor against coronary heart disease. Am J Med. 1977;62:707–714. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- 2.Miller GJ, Miller NE. Plasma high density lipoprotein concentration and the development of ischaemic heart disease. Lancet. 1975;1:16–19. doi: 10.1016/s0140-6736(75)92376-4. [DOI] [PubMed] [Google Scholar]
- 3.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 4.Connelly MA, Williams DL. SR-BI and cholesterol uptake into steroidogenic cells. Trends Endocrinol Metab. 2003;14:467–472. doi: 10.1016/j.tem.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 5.Glass C, Pittman RC, Civen M, Steinberg D. Uptake of high-density lipoprotein-associated apoprotein A-I and cholesterol esters by 16 tissues of the rat in Vivo and by adrenal cells and hepatocytes in Vitro. J Biol Chem. 1985;260:744–750. [PubMed] [Google Scholar]
- 6.Pittman RC, Steinberg D. Sites and mechanisms of uptake and degradation of high density and low density lipoproteins. J Lipid Res. 1984;25:1577–1585. [PubMed] [Google Scholar]
- 7.Landschulz KT, Pathak RK, Rigotti A, Krieger M, Hobbs HH. Regulation of scavenger receptor, class B, type I, a high density lipoprotein receptor, in liver and steroidogenic tissues of the rat. J Clin Invest. 1996;98:984–995. doi: 10.1172/JCI118883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rigotti A, Edelman ER, Seifert P, Iqbal SN, DeMattos RB, Temel RE, Krieger M, Williams DL. Regulation by adrenocorticotropic hormone of the in vivo expression of scavenger receptor class B type I (SR-B1), a high density lipoprotein receptor, in steroidogenic cells of the murine adrenal gland. J Biol Chem. 1996;271:33545–33549. doi: 10.1074/jbc.271.52.33545. [DOI] [PubMed] [Google Scholar]
- 9.Ji Y, Jian B, Wang N, Sun Y, de la Llera Moya M, Phillips MC, Rothblat GH, Swaney JB, Tall AR. Scavenger receptor BI promotes high density lipoprotein-mediated cellular cholesterol efflux. J Biol Chem. 1997;272:20982–20985. doi: 10.1074/jbc.272.34.20982. [DOI] [PubMed] [Google Scholar]
- 10.Jian B, de la Llera-Moya M, Ji Y, Wang N, Phillips MC, Swaney JB, Tall AR, Rothblat GH. Scavenger receptor class B type I as a mediator of cellular cholesterol efflux to lipoproteins and phospholipid acceptors. J Biol Chem. 1998;273:5599–5606. doi: 10.1074/jbc.273.10.5599. [DOI] [PubMed] [Google Scholar]
- 11.de la Llera-Moya M, Connelly MA, Drazul D, Klein SM, Favari E, Yancey PG, Williams DL, Rothblat GH. Scavenger receptor, class B, type I, (SR-BI) affects cholesterol homeostasis by magnifying cholesterol flux between cells and HDL. J Lipid Res. 2001;42:1969–1978. [PubMed] [Google Scholar]
- 12.Kellner-Weibel G, de la Llera-Moya M, Connelly MA, Stoudt G, Christian AE, Haynes MP, Williams DL, Rothblat GH. Expression of scavenger receptor BI in COS-7 cells alters cholesterol content and distribution. Biochemistry. 2000;39:221–229. doi: 10.1021/bi991666c. [DOI] [PubMed] [Google Scholar]
- 13.Reaven E, Nomoto A, Leers-Sucheta S, Temel R, Williams DL, Azhar S. Expression and microvillar localization of scavenger receptor, class B, type I (a high density lipoprotein receptor) in luteinized and hormone-desensitized rat ovarian models. Endocrinology. 1998;139:2847–2856. doi: 10.1210/endo.139.6.6056. [DOI] [PubMed] [Google Scholar]
- 14.Reaven E, Spicher M, Azhar S. Microvillar channels: a unique plasma membrane compartment for concentrating lipoproteins on the surface of rat adrenal cortical cells. J Lipid Res. 1989;30:1551–1560. [PubMed] [Google Scholar]
- 15.Peng Y, Akmentin W, Connelly MA, Lund-Katz S, Phillips MC, Williams DL. Scavenger receptor BI (SR-BI) clustered on microvillar extensions suggests that this plasma membrane domain is a way station for cholesterol trafficking between cells and high-density lipoprotein. Mol Biol Cell. 2004;15:384–396. doi: 10.1091/mbc.E03-06-0445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krieger M. Charting the fate of the “good cholesterol”: identification and characterization of the high-density lipoprotein receptor SR-BI. Annu Rev Biochem. 1999;68:523–558. doi: 10.1146/annurev.biochem.68.1.523. [DOI] [PubMed] [Google Scholar]
- 17.Azhar S, Nomoto A, Reaven E. Hormonal regulation of adrenal microvillar channel formation. J Lipid Res. 2002;43:861–871. [PubMed] [Google Scholar]
- 18.Reaven E, Cortez Y, Leers-Sucheta S, Nomoto A, Azhar S. Dimerization of the scavenger receptor class B type I (SR-BI): formation, function and localization in diverse cells and tissues. J Lipid Res. 2004;45:513–528. doi: 10.1194/jlr.M300370-JLR200. [DOI] [PubMed] [Google Scholar]
- 19.Sahoo D, Darlington YF, Pop D, Williams DL, Connelly MA. Scavenger receptor class B Type I (SR-BI) assembles into detergent-sensitive dimers and tetramers. Biochim Biophys Acta. 2007;1771:807–17. doi: 10.1016/j.bbalip.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Rodrigueza WV, Thuahnai ST, Temel RE, Lund-Katz S, Phillips MC, Williams DL. Mechanism of scavenger receptor class B type I-mediated selective uptake of cholesteryl esters from high density lipoprotein to adrenal cells. J Biol Chem. 1999;274:20344–20350. doi: 10.1074/jbc.274.29.20344. [DOI] [PubMed] [Google Scholar]
- 21.Stryer L. Fluorescence energy transfer as a spectroscopic ruler. Annu Rev Biochem. 1978;47:819–846. doi: 10.1146/annurev.bi.47.070178.004131. [DOI] [PubMed] [Google Scholar]
- 22.Selvin PR. The renaissance of fluorescence resonance energy transfer. Nat Struct Biol. 2000;7:730–734. doi: 10.1038/78948. [DOI] [PubMed] [Google Scholar]
- 23.Tsien RY. Building and breeding molecules to spy on cells and tumors. FEBS Lett. 2005;579:927–932. doi: 10.1016/j.febslet.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 24.Vogel SS, Thaler C, Koushik SV. Fanciful FRET. Sci STKE 2006. 2006:re2. doi: 10.1126/stke.3312006re2. [DOI] [PubMed] [Google Scholar]
- 25.Zaccolo M. Use of chimeric fluorescent proteins and fluorescence resonance energy transfer to monitor cellular responses. Circ Res. 2004;94:866–873. doi: 10.1161/01.RES.0000123825.83803.CD. [DOI] [PubMed] [Google Scholar]
- 26.Forster T. Delocalized excitation and excitation transfer. In: Sinanoglu O, editor. Modern Quantum Chemistry. 3. Academic Press Inc; New York: 1965. pp. 93–137. [Google Scholar]
- 27.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2. Plenum Press; New York: 1999. [Google Scholar]
- 28.Giese B, Roderburg C, Sommerauer M, Wortmann SB, Metz S, Heinrich PC, Muller-Newen G. Dimerization of the cytokine receptors gp130 and LIFR analysed in single cells. J Cell Sci. 2005;118:5129–5140. doi: 10.1242/jcs.02628. [DOI] [PubMed] [Google Scholar]
- 29.Sekar RB, Periasamy A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J Cell Biol. 2003;160:629–633. doi: 10.1083/jcb.200210140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Storez H, Scott MG, Issafras H, Burtey A, Benmerah A, Muntaner O, Piolot T, Tramier M, Coppey-Moisan M, Bouvier M, Labbe-Jullie C, Marullo S. Homo- and hetero-oligomerization of beta-arrestins in living cells. J Biol Chem. 2005;280:40210–40215. doi: 10.1074/jbc.M508001200. [DOI] [PubMed] [Google Scholar]
- 31.Xu Q, Brecht WJ, Weisgraber KH, Mahley RW, Huang Y. Apolipoprotein E4 domain interaction occurs in living neuronal cells as determined by fluorescence resonance energy transfer. J Biol Chem. 2004;279:25511–25516. doi: 10.1074/jbc.M311256200. [DOI] [PubMed] [Google Scholar]
- 32.Zal T, Gascoigne NR. Using live FRET imaging to reveal early protein-protein interactions during T cell activation. Curr Opin Immunol. 2004;16:674–683. [PubMed] [Google Scholar]
- 33.Heim R. Green fluorescent protein forms for energy transfer. Methods Enzymol. 1999;302:408–423. doi: 10.1016/s0076-6879(99)02036-4. [DOI] [PubMed] [Google Scholar]
- 34.Truong K, Ikura M. The use of FRET imaging microscopy to detect protein-protein interactions and protein conformational changes in vivo. Curr Opin Struct Biol. 2001;11:573–578. doi: 10.1016/s0959-440x(00)00249-9. [DOI] [PubMed] [Google Scholar]
- 35.Connelly MA, Klein SM, Azhar S, Abumrad NA, Williams DL. Comparison of class B scavenger receptors, CD36 and SR-BI, shows that both receptors mediate HDL-cholesteryl ester selective uptake but SR-BI exhibits a unique enhancement of cholesteryl ester uptake. J Biol Chem. 1999;274:41–47. doi: 10.1074/jbc.274.1.41. [DOI] [PubMed] [Google Scholar]
- 36.Temel RE, Trigatti B, DeMattos RB, Azhar S, Krieger M, Williams DL. Scavenger receptor B, type I (SR-BI) is the major route for the delivery of high density lipoprotein cholesterol to the steroidogenic pathway in cultured mouse adrenocortical cells. Proc Natl Acad Sci USA. 1997;94:13600–13605. doi: 10.1073/pnas.94.25.13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 38.Laemmli UK. Cleavage of structural proteins during assembly of the bead of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 39.Havel RJ, Eder HA, Bragdon JH. The distribution and chemical composistion of ultracentrifugally separated lipoproteins in human serum. J Clin Invest. 1955;34:1345–1353. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Francone OL, Haghpassand M, Bennett JA, Royer L, McNeish J. Expression of human lecithin:cholesterol acyltransferase in transgenic mice: effects on cholesterol efflux, esterification, and transport. J Lipid Res. 1997;38:813–822. [PubMed] [Google Scholar]
- 41.Connelly MA, Kellner-Weiber G, Rothblat GH, Williams DL. Scavenger receptor, class B, type I (SR-BI)-directed HDL-cholesteryl ester hydrolysis. J Lipid Res. 2003;44:331–341. doi: 10.1194/jlr.M200186-JLR200. [DOI] [PubMed] [Google Scholar]
- 42.Karpova TS, Baumann CT, He L, Wu X, Grammer A, Lipsky P, Hager GL, McNally JG. Fluorescence resonance energy transfer from cyan to yellow fluorescent protein detected by acceptor photobleaching using confocal microscopy and a single laser. J Microsc. 2003;209:56–70. doi: 10.1046/j.1365-2818.2003.01100.x. [DOI] [PubMed] [Google Scholar]
- 43.Centonze VE, Firulli BA, Firulli AB. Fluorescence Resonance Energy Transfer (FRET) as a method to calculate the dimerization strength of basic Helix-Loop-Helix (bHLH) proteins. Biol Proced Online. 2004;6:78–82. doi: 10.1251/bpo75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goodwin JS, Kenworthy AK. Photobleaching approaches to investigate diffusional mobility and trafficking of Ras in living cells. Methods. 2005;37:154–164. doi: 10.1016/j.ymeth.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 45.Chen Z, Deddish PA, Minshall RD, Becker RP, Erdos EG, Tan F. Human ACE and bradykinin B2 receptors form a complex at the plasma membrane. Faseb J. 2006;20:2261–2270. doi: 10.1096/fj.06-6113com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sturmey RG, O’Toole J, Leese PHJ. Fluorescence resonance energy transfer analysis of mitochondrial:lipid association in the porcine oocyte. Reproduction. 2006;132:829–837. doi: 10.1530/REP-06-0073. [DOI] [PubMed] [Google Scholar]
- 47.Dong LM, Weisgraber KH. Human apolipoprotein E4 domain interaction. Arginine 61 and glutamic acid 255 interact to direct the preference for very low density lipoproteins. J Biol Chem. 1996;271:19053–19057. doi: 10.1074/jbc.271.32.19053. [DOI] [PubMed] [Google Scholar]
- 48.Dong LM, Wilson C, Wardell MR, Simmons T, Mahley RW, Weisgraber KH, Agard DA. Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms. J Biol Chem. 1994;269:22358–22365. [PubMed] [Google Scholar]
- 49.Kenworthy AK. Imaging protein-protein interactions using fluorescence resonance energy transfer microscopy. Methods. 2001;24:289–296. doi: 10.1006/meth.2001.1189. [DOI] [PubMed] [Google Scholar]
- 50.Wallrabe H, Periasamy A. Imaging protein molecules using FRET and FLIM microscopy. Curr Opin Biotechnol. 2005;16:19–27. doi: 10.1016/j.copbio.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 51.Zal T, Gascoigne NR. Photobleaching-corrected FRET efficiency imaging of live cells. Biophys J. 2004;86:3923–3939. doi: 10.1529/biophysj.103.022087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balish MF, Santurri RT, Ricci AM, Lee KK, Krause DC. Localization of Mycoplasma pneumoniae cytadherence-associated protein HMW2 by fusion with green fluorescent protein: implications for attachment organelle structure. Mol Microbiol. 2003;47:49–60. doi: 10.1046/j.1365-2958.2003.03282.x. [DOI] [PubMed] [Google Scholar]
- 53.Crowston JG, Chang LH, Daniels JT, Khaw PT, Akbar AN. T lymphocyte mediated lysis of mitomycin C treated Tenon’s capsule fibroblasts. Br J Ophthalmol. 2004;88:399–405. doi: 10.1136/bjo.2002.007708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maertens G, Vercammen J, Debyser Z, Engelborghs Y. Measuring protein-protein interactions inside living cells using single color fluorescence correlation spectroscopy. Application to human immunodeficiency virus type 1 integrase and LEDGF/p75. Faseb J. 2005;19:1039–1041. doi: 10.1096/fj.04-3373fje. [DOI] [PubMed] [Google Scholar]
- 55.Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 56.Curran AR, Engelman DM. Sequence motifs, polar interactions and confromational changes in helical membrane proteins. Curr Opin Struct Biol. 2003;13:412–417. doi: 10.1016/s0959-440x(03)00102-7. [DOI] [PubMed] [Google Scholar]
- 57.Russ WP, Engelman DM. The GxxxG motif: a framework for transmembrane helix-helix association. J Mol Biol. 2000;296:911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 58.Li R, Mitra N, Gratkoski H, Vilaire G, Litvinov R, Nagasami C, Weisel JW, Lear JD, DeGrado WF, Bennett JS. Activation of integrin αIIbβ3 by modulation of transmembrane helix associations. Science. 2003;300:795–798. doi: 10.1126/science.1079441. [DOI] [PubMed] [Google Scholar]
- 59.Tagaki J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110:599–611. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]