

Abstract
The effect of a sixth ligand in a series of low-spin thiocarbonyl-ligated iron(II)octaethylporphyrinates has been investigated. Six-coordinate complexes have been synthesized and characterized by Mössbauer and infrared spectroscopy and single-crystal X-ray structure determinations. The results are compared with the five-coordinate parent complex. The crystal structures of [Fe(OEP)(CS)(1-MeIm)] and [Fe(OEP)(CS)(Py)] are reported and discussed. The 1-methylimidazole and pyridine derivatives exhibit Fe–C(CS) bond distances of 1.703(4) and 1.706(2) Å that are significantly longer than the 1.662(3) Å reported for five-coordinate [Fe(OEP)(CS)] (Scheidt, W. R.; Geiger, D. K. Inorg. Chem. 1982, 21, 1208). The trans Fe-N(ligand) distances of 2.112(3) A and 2.1550(15) Å observed for the 1-methylimidazole and pyridine complex are ~0.13 Å longer than those observed for analogous bis-ligated complexes and are consistent with a significant structural trans effect for the CS ligand. Mössbauer investigations carried out for five- and six-coordinate thiocarbonyl derivatives with several different sixth axial ligands reveal interesting features. All derivatives exhibit very small isomer shift values, consistent with a very strong interaction between iron and CS. The five-coordinate derivative has δFe = 0.08 mm/s and the six-coordinate complexes exhibit δFe = 0.14 to 0.19 mm/s at 4.2 K. The five-coordinate complex shows a large quadrupole splitting (ΔEq = 1.93 mm/s at 4.2 K) which is reduced on coordination of the sixth ligand (ΔEq = 0.42 to 0.80 mm/s at 4.2 K). Addition of a sixth ligand also leads to a small decrease in the value of νCS. Correlations in structural, IR, and Mössbauer results suggest that the sixth ligand effect is primarily induced by changes in σ bonding. The structure of [Fe(OEP)(CS)(CH3OH)] is briefly reported. Crystal data: [Fe(OEP)(CS)(1-MeIm)] crystallizes in the monoclinic system, space group P21/n, Z = 4, a = 9.5906(5) Å, b = 16.704(4) Å, c = 23.1417(6) Å, β = 100.453(7)°. [Fe(OEP)(CS)(Py)] crystallizes in the triclinic system, space group P1̄, Z = 5, a = 13.9073(6) Å, b = 16.2624(7) Å, c = 22.0709(9) Å, α = 70.586(1)°, β = 77.242(1)°, γ = 77.959(1)°. [Fe(OEP)(CS)(CH3OH)] crystallizes in the triclinic system, space group P1̄, Z = 1, a = 9.0599(5) Å, b = 9.4389(5) Å, c = 11.0676(6) Å, α = 90.261(1)°, β = 100.362(1)°, γ = 114.664(1)°.
Introduction
Carbon monoxide has been used as ligand for exploring properties of iron porphyrins and heme proteins for over seventy years.1-11 As is well known, CO binds to the heme of the respiratory proteins hemoglobin and myoglobin more strongly than dioxygen. This tight binding of CO is the presumed result of a strong π-bond between CO and Fe(II). In part, this strong binding of CO has led to its use as a probe molecule in the study of iron porphyrinates and heme proteins.
Although the thiocarbonyl ligand (CS, also called carbon monosulfide) is isoelectronic with carbon monoxide, CS is considered to be both a better σ-donor and π-acceptor compared to the carbonyl ligand. This idea is supported by several early theoretical12-17 and spectroscopic studies.16-20 There are however only a relatively limited number of transition metal complexes reported,21 so that comparisons to transition metal carbonyl complexes are limited. Thiocar-bonyl derivatives of iron porphyrinates have been reported by the groups of Buchler22 and Mansuy.23, 24 The structure of one derivative, five-coordinate [Fe(OEP)(CS)],25, 26 has been reported. The very short Fe-C(CS) distance of 1.662(3) Å is consistent with the idea that CS is a strong π-accepting ligand as is the fact that the five-coordinate species is a low-spin derivative. In this regard CS appears to be very similar to the nitrite ligand. Five-coordinate [Fe(TpivPP)(NO2)]− is also a low-spin iron(II) derivative27 with a short axial Fe–N(NO2) distance. These properties for the nitrite derivative were ascribed to a very strong π-accepting character of N-bound NO2− in the iron(II) complex.
The nature of the strong π-accepting character of the nitrite ligand was found to be diminished substantially when an additional ligand was coordinated to form six-coordinate derivatives.28 This was seen in the Mössbauer spectra where a large quadrupole splitting was observed for five-coordinate [Fe(TpivPP)(NO2)]− and much smaller quadrupole splitting values for the six-coordinate derivatives [Fe(TpivPP)(NO2)(Py)] and [Fe(TpivPP)(NO2)(PMS)]. This was interpreted in terms of variable π-bonding exhibited by the nitrite ligand. The apparent differences in π-bonding were also supported by the structural data.28 The variation in π-bonding, as a function of coordination number was, in our view, unexpectedly large. This variation is distinctly different than that observed for another π-accepting ligand NO.29-37
These results have prompted us to investigate how the π-interaction in a series of thiocarbonyl-ligated iron porphyrinates varies as a function of an increase in coordination number from five to six. We have measured Mössbauer spectra for five-coordinate [Fe(OEP)(CS)] and a number of six-coordinate [Fe(OEP)(CS)(L)] derivatives (L = Py, 4-CNPy, 4-NMe2Py, 1-MeIm, and Pip) in zero and applied magnetic field. A pattern similar to that observed for nitrite is observed; a large quadruple splitting is shown by [Fe(OEP)(CS)] (ΔEQ = 1.93 mm/s at 4.2 K) with a large decrease in quadrupole splitting observed on coordination of a sixth ligand. Values of the quadrupole splitting are sensitive to the basicity of the sixth ligand. Another significant feature of the Mössbauer spectra of the thiocarbonyl porphyrinates is the unexpectedly low isomer shift values. Five-coordinate [Fe(OEP)(CS)] exhibits a low isomer shift value (δFe = 0.08 mm/s at 4.2 K); the six-coordinate species have higher values of isomer shift. The values of the isomer shifts are much lower than that observed for carbonyl derivatives.
The crystal structures of three derivatives, [Fe(OEP)(CS)(1-MeIm)], [Fe(OEP)(CS)(Py)] and [Fe(OEP)(CS)(CH3OH)] were solved during the course of the investigation. All show Fe–C(CS) distances that are longer than the 1.662 (2) Å-value of the five-coordinate species and thus suggest a weaker interaction of CS with the iron center in the six-coordinate complexes. The Fe–L bond distances trans to CS are all longer than expected for iron(II) species and show that the CS ligand exerts a strong trans effect.
Experimental Section
General Information
Tetrahydrofuran (THF) was dried and distilled over sodium/benzophenone. Chlorobenzene (C6H5Cl) was washed with concentrated sulphuric acid, aqueous NaHCO3, water till neutral, dried overnight over MgSO4 and distilled over P2O5. THF, C6H5Cl, 2,4,6-trimethylpyridine and CSCl2 were degassed before use by three freeze/pump/thaw cycles. Pyridine was dried over KOH, distilled under vacuum and used immediately. Piperidine was dried and distilled over calcium hydride. 1-Methylimidazole was dried and distilled over sodium. 4-Cyanopyridine was recrystallized from a dichloromethane/ethyl ether mixture. 4-Dimethylaminopyridine was recrystallized from toluene. 57Fe2O3 (95% enriched) was purchased from Cambridge Isotope Laboratories and used as received. Octaethylporphyrin was purchased from Midcentury Chemicals and was used as received. All other chemicals were used as received from Aldrich or Fisher. Most manipulations were carried out under argon using a double-manifold vacuum line, Schlenkware and cannula techniques. IR spectra were recorded on a Nicolet-Nexus 670 FT-IR spectrophotometer as KBr pellets. UV-vis spectra were recorded on a Perkin-Elmer Lambda 6 spectrophotometer. The solid-state Mössbauer samples were immobilized in Apiezon M grease. Spectra were obtained for [Fe(OEP)(CS)] using iron at normal and 95% enrichment in 57Fe while spectra for [Fe(OEP)(CS)(Py)], [Fe(OEP)(CS)(1-MeIm)], [Fe(OEP)(CS)(4-CNPy)], [Fe(OEP)(CS)(4-NMe2Py)] and [Fe(OEP)(CS)(Pip)], were obtained from enriched samples that had been prepared for another experiment. For the enriched iron derivatives, due to the small quantity of sample required, thin, dilute grease mulls were used to make the sample as uniform as possible. Mössbauer measurements were performed on a constant acceleration spectrometer at 4.2 K with zero field and up to 9 T in a magnetic field parallel to the γ-ray beam (Knox College).
Preparation of [Fe(OEP)(CS)]
[Fe(OEP)Cl] was prepared by a modified literature method.3,38 The five-coordinate thiocarbonyl complex was prepared according to the method reported by Buchler.22 Typically, 50 mg of [Fe(OEP)Cl] was dissolved in 25 mL of THF and degassed three times. The solution was transferred via cannula to 0.2 mL of sodium amalgam (1%). Immediate reduction occurred with a color change of the solution from brown to red. Thiophosgene (0.1 mL, degassed three times) was added over a period of 10 minutes and solution was stirred for 24 hours under argon at room temperature. The THF solution was decanted from the amalgam residue. The amalgam residue was washed twice with THF and the combined solution was evaporated to dryness under vacuum. The product was extracted with toluene and purified by column chromatography (neutral alumina, 50–200 μ, activity level 4). After evaporating the solvent in vacuum at 60 °C, the compound was washed with hexane (yield 33–40%). λmax(nm) in CH2Cl2 - 387, 516, 551 (reported, 388, 518, 551). IR (KBr), νCS- 1292 cm−1 (reported: 1292 cm−1).22
Preparation of [Fe(OEP)(CS)(L)] (L = Py, 1-MeIm, Pip, 4-CNPy and (4-NMe2Py)
The six-coordinate thiocarbonyl complexes were prepared by recrystallization (slow evaporation) from the CHCl3/CH3OH (3:1) solution of [Fe(OEP)(CS)](40 mg) containing 0.1 mL pyridine, 1-methylimidazole or piperidine. The 4-cyanopyridine and 4-dimethylaminopyridine derivatives were prepared by slow evaporation of the CHCl3/CH3OH solution containing five-coordinate [Fe(OEP)(CS)] and the respective ligand in 1:4 molar ratio. λmax (nm, log ε) in CH2Cl2: [Fe(OEP)(CS)(1-MeIm)] - 408, 531, 557 (reported, 410, 530, 560); [Fe(OEP)(CS)(Py)]- 407, 528, 559 (reported, 408, 529, 560); [Fe(OEP)(CS)(Pip)] - 409, 529, 559 (reported, 410, 529, 560);22 [Fe(OEP)(CS)(4-CNPy)] - 405, 5.190; 529, 4.148; 556, 4.285; [Fe(OEP)(CS)(4-NMe2Py) - 410, 5.195; 530, 4.122; 560, 4.170. νCS for [Fe(OEP)(CS)(Py)] - 1280 cm−1 (reported, 1282 cm−1); νCS for [Fe(OEP)(CS)(1-MeIm)] - 1272 cm−1 (reported, 1275 cm−1); νCS for [Fe(OEP)(CS)(Pip)] - 1280 cm−1 (reported, 1278);22 νCS for [Fe(OEP)(CS)(4-CNPy)] – 1284 cm−1, νCS for [Fe(OEP)(CS)(4-NMe2Py)] - 1279 cm−1.
Preparation of [57Fe(OEP)Cl] and [57Fe(OEP)(CS)]
The enriched iron porphyrin [57Fe(OEP)Cl] derivative was prepared following the reported method.39 95% enriched 57Fe2O3 and H2OEP were used in 1.8:1 molar ratio and the completion of metal insertion was monitored by UV-vis spectra. The reaction mixture was stirred at 80°; for 12 hours to ensure complete metallation. [57Fe(OEP)(CS)] was prepared by the same procedure as described above from57Fe(OEP)Cl. νCS - 1291 cm−1.
Preparation of [57Fe(OEP)(CS)(L)] (L = Py, 1-MeIm, Pip, 4-CNPy and (4-NMe2Py)
The iron-enriched six-coordinate derivatives were prepared by the same procedure as described above. The νCS values for the derivatives are: [57Fe(OEP)(CS)(Py)] -1280 cm−1, [57Fe(OEP)(CS)(1-MeIm)] - 1271 cm−1, [(57Fe(OEP)(CS)(4-CNPy)] - 1284 cm−1, [57Fe(OEP)(CS)(4-NMe2Py)] - 1279 cm−1 and [57Fe(OEP)(CS)(Pip)] - 1279 cm−1.
X-ray Structure Determinations. [Fe(OEP)(CS)(1-MeIm)]
A red block-shaped crystal of [Fe(OEP)(CS)(1-MeIm)] was used for data collection on an Enraf-Nonius FAST area detector diffractometer at 130 K using methods and procedures for small molecules standard in this lab.40 The structure was solved in the space group P21/n by direct methods with SHELXS-86.41 After all non-hydrogen atoms were refined anisotropically, most of the hydrogen atoms were located by the difference Fourier synthesis. However, in the least-squares refinement, all hydrogen atoms were put in idealized positions and refined with riding models (C-H = 0.95 Å for R2CH, 0.99 Å for R2CH2, 0.98 Å for RCH3). The structure was refined against F2 by the SHELXL-93 program.41 The structure was completely ordered.
[Fe(OEP)(CS)(Py)] and [Fe(OEP)(CS)(CH3OH)]
Crystalline samples of [Fe(OEP)(CS)(Py)] and [Fe(OEP)(CS)(CH3OH)] were placed in inert oil, mounted on glass pins, and transferred to the cold gas stream of the diffractometer. Crystal data were collected and integrated using a Bruker Apex system, with graphite monochromated Mo-Kα (λ = 0.71073 Å) radiation at 100 K. The structures were solved by direct methods using SHELXS-97 and refined using SHELXL-97 (Sheldrick, G.M., University of Göttingen). Non-hydrogen atoms were found by successive full matrix least squares refinement on F2 and refined with anisotropic thermal parameters. Hydrogen atom positions were located from difference Fourier maps and a riding model with fixed thermal parameters [uij = 1.2Uij(eq) for the atom to which they are bonded], was used for subsequent refinements. The asymmetric unit of [Fe(OEP)(CS)(Py)] contains two and one half iron-porphyrin units, with the Fe(II) ion of the half-unit located at a crystallographic inversion center and the corresponding axial ligands also disordered over two positions (50% occupancies). This porphyrin ring, containing Fe(1)–3, is also disordered due to a twofold disorder of two of the unique ethyl substituents, so that the two ethyl groups, one unique methylene bridge and four unique carbon atoms of the porphyrin backbone are present in 65%/35% relative occupancies. The porphyrin ring containing Fe(1)–1 includes two disordered sections, arising from two disordered ethyl groups apiece and including the corresponding β carbons, with site occupancies of 51%/49% and 65%/35%, respectively. The porphyrin ring containing Fe(1)–2 has one 73%/27% disorder of two ethyl groups and their β carbons, and one 50%/50% disorder of the methyl group of one ethyl substituent. The axial substituents of both of these porphyrin rings (attached to Fe(1)–1 and Fe(1)–2) are completely ordered. The asymmetric unit of [Fe(OEP)(CS)(CH3OH)] contains one half of an iron-porphyrin unit,42 with the position of the Fe(II) ion located at the crystallographic inversion center. Therefore, the methanol and CS ligands are disordered, and were refined with 50% site occupancies.
A brief description of crystallographic details for all three derivatives is given in Table 1 while complete information is given in the Supporting Information.
Table 1.
Crystallographic Details for [Fe(OEP)(CS)(L)] Derivatives
| [Fe(OEP)(CS)(1-MeIm)] | Fe(OEP)(CS)(Py) | [Fe(OEP)(CS)(CH3OH)] | |
|---|---|---|---|
| empirical formula | C41H50FeN6S | C42H49FeN5S | C38H52FeN4OS |
| formula weight | 714.78 | 711.77 | 668.75 |
| crystal system | monoclinic | triclinic | triclinic |
| space group | P21/n | P1̄ | P1̄ |
| a, Å | 9.5906(5) | 13.9073(6) | 9.0599(5) |
| b, Å | 16.704(4) | 16.2624(7) | 9.4389(5) |
| c, Å | 23.1417(6) | 22.0709(9) | 11.0676(6) |
| α(deg) | 90 | 70.5860(10) | 90.2610(10) |
| β(deg) | 100.453(7) | 77.2420(10) | 100.3620(10) |
| γ(deg) | 90 | 77.9590(10) | 114.6640(10) |
| V, (Å3) | 3645.9(8) | 4541.8(3) | 842.72(8) |
| Z | 4 | 5 | 1 |
| Dcalc, g/cm3 | 1.302 | 1.301 | 1.318 |
| wavelength, Å | 0.71073 | 0.71073 | 0.71073 |
| crystal size (mm) | 0.22 × 0.14 × 0.03 | 0.3 × 0.2 × 0.2 | 0.5 × 0.3 × 0.2 |
| temp, K | 130(2) | 100(2) | 100(2) |
| reflection collected | 22290 | 49146 | 9096 |
| independent reflections | 7882 | 22494 | 4178 |
| final R indices [I>2σ(I)] | R1 = 0.0735 | R1 = 0.0458 | R1 = 0.0587 |
| wR2 = 0.1742 | wR2 = 0.1106 | wR2 = 0.1395 | |
| R indices (all data) | R1 = 0.0987 | R1 = 0.0587 | R1 = 0.0608 |
| wR2 = 0.1902 | wR2 = 0.1185 | wR2 = 0.1403 |
Results
The synthesis, molecular structures, IR, UV-vis and Mössbauer spectra of several six-coordinate thiocarbonyl iron(II) porphyrinates are reported. The six-coordinate thiocarbonyl complexes exhibit UV-vis spectra with red-shifted Soret and visible absorption bands compared to the five-coordinate derivative. The CS stretching frequency is found to be lower in all the six-coordinate thiocarbonyl complexes (1272–1284 cm−1) compared to 1292 cm−1 observed for [Fe(OEP)(CS)]. Solid-state Mössbauer measurements were made on six samples, [Fe(OEP)(CS)] and [Fe(OEP)(CS)(L)] (L = 1-MeIm, Py, 4-CNPy, 4-NMe2Py, and Pip). The Mössbauer data will be discussed subsequently.
The molecular structures of three six-coordinate derivatives, [Fe(OEP)(CS)(1-MeIm)], [Fe(OEP)(CS)(Py)] and [Fe(OEP)(CS)(CH3OH)] have been determined. A labeled ORTEP diagram of [Fe(OEP)(CS)(1-MeIm)] is given in Figure 1. Crystals of [Fe(OEP)(CS)(Py)] contain two and one-half independent molecules per asymmetric unit,42 the axial ligands in the two full molecule in the unit are ordered while the half molecule in the unit has the axial ligands disordered over two positions. The three molecules are distinguished with atom labels followed by an underscore one(–1), two(–2), or three(–3) (half molecule); identical labeling schemes are used in all three. A labeled ORTEP diagram of molecule 2 of [Fe(OEP)(CS)(Py)] is given in Figure 2; ORTEP diagrams of the other two molecules are given in the Supporting Information, as is the diagram for [Fe(OEP)(CS)(CH3OH)]. A summary of selected bond distances and angles for the thiocarbonyl derivatives is given in Table 2.
Figure 1.
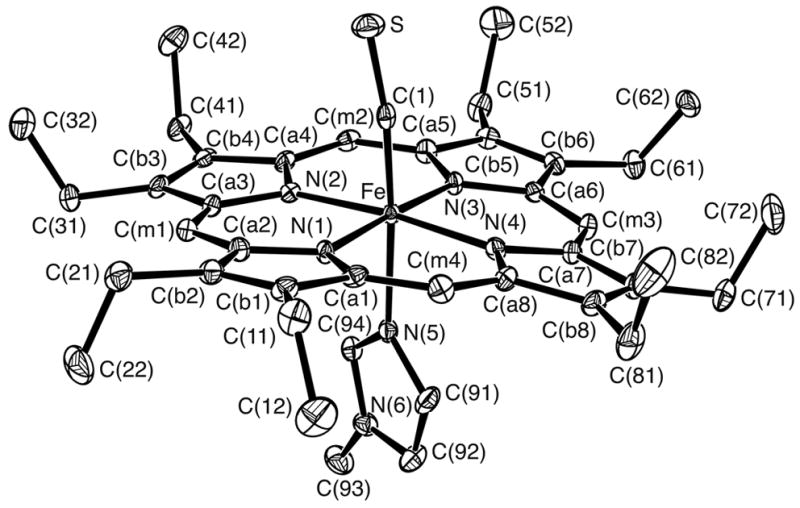
ORTEP diagram of [Fe(OEP)(CS)(1-MeIm)] displaying the atom labeling scheme. Thermal ellipsoids of all atoms are contoured at the 50% probability level. Hydrogen atoms have been omitted for clarity.
Figure 2.
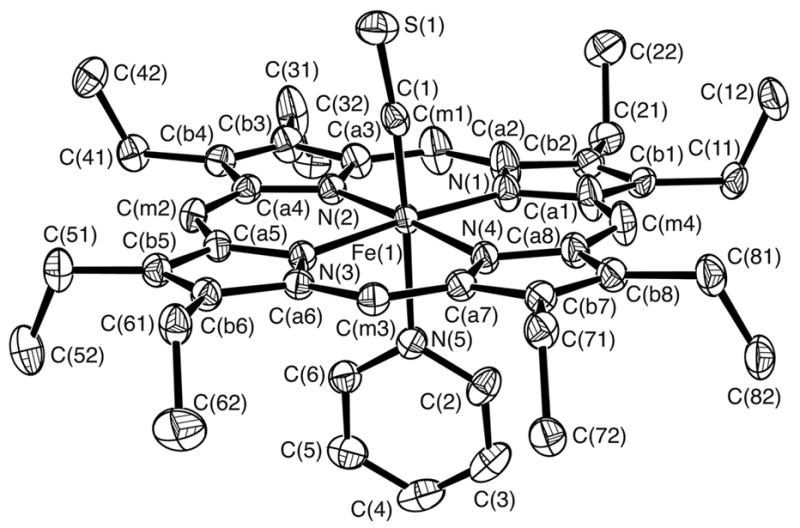
ORTEP diagram of molecule 2 of [Fe(OEP)(CS)(Py)] displaying the atom labeling scheme; this labeling scheme is used for all three molecules. Thermal ellipsoids of all atoms are contoured at the 50% probability level. Hydrogen atoms have been omitted for clarity.
Table 2.
Selected Bond Lengths, Bond Angles, and Iron Displacements for [Fe(OEP)(CS)(L)] Derivatives.
| [Fe(OEP)(CS)(L)]
|
||||||
|---|---|---|---|---|---|---|
| L = | —a | 1-MeIm | Py(1) | Py(2) | Py(3) | CH3OH |
| Fe–N(1)b | 1.981(3) | 2.002(3) | 2.0034(15) | 2.0048(8) | 2.000(2) | 1.992(2) |
| Fe–N(2)b | 1.989(3) | 1.996(3) | 2.0050(15) | 2.0043(14) | 1.998(2) | 1.994(2) |
| Fe–N(3)b | 1.979(3) | 2.000(3) | 2.0072(16) | 2.0039(14) | – | – |
| Fe–N(4)b | 1.980(2) | 2.006(3) | 2.0048(14) | 2.0108(14) | – | – |
| (Fe–Np)bav | 1.982(5) | 2.001(4) | 2.005(2) | 2.006(3) | 1.999(1) | 1.993(1) |
| Fe–C(CS)b | 1.662(3) | 1.703(4) | 1.707(2) | 1.7042(18) | 1.889(8) | 1.800(12) |
| Fe–Lb | – | 2.112(3) | 2.1469(18) | 2.1550(15) | 1.959(3) | 2.089(7) |
| C–Sb | 1.559(3) | 1.563(4) | 1.565(2) | 1.5626(18) | 1.603(8) | 1.576(12) |
| ΔFeb | 0.23 | 0.10 | 0.03–0.10c | 0.0–0.03c | 0.0d | 0.0d |
| Fe–C–Se | 176.3(2) | 172.2(2) | 176.24(13) | 174.57(12) | 175.4(5) | 175.4(6) |
| C–Fe–Le | 175.32(3) | 177.66(8) | 176.94(7) | 177.8(3) | 168.8(8) | |
| ref | 25 | tw | tw | tw | tw | tw |
Five-coordinate species.
Values in Å.
Range defined by choice of disordered atoms.
Value required by inversion symmetry.
Value in degrees.
A formal diagram showing the displacement of atoms (in unit of 0.01 Å) from the 24- atom mean plane of [Fe(OEP)(CS)(1-MeIm)] is given in Figure 3. The diagram also displays the average values for each chemically unique type of bond length and bond angle. Similar diagrams for the the pyridine and methanol derivatives are given in the Supporting Information. In contrast to the strongly ruffled core of [Fe(OEP)(CS)(1-MeIm)], the porphyrin cores in the remaining derivatives are more nearly planar.
Figure 3.
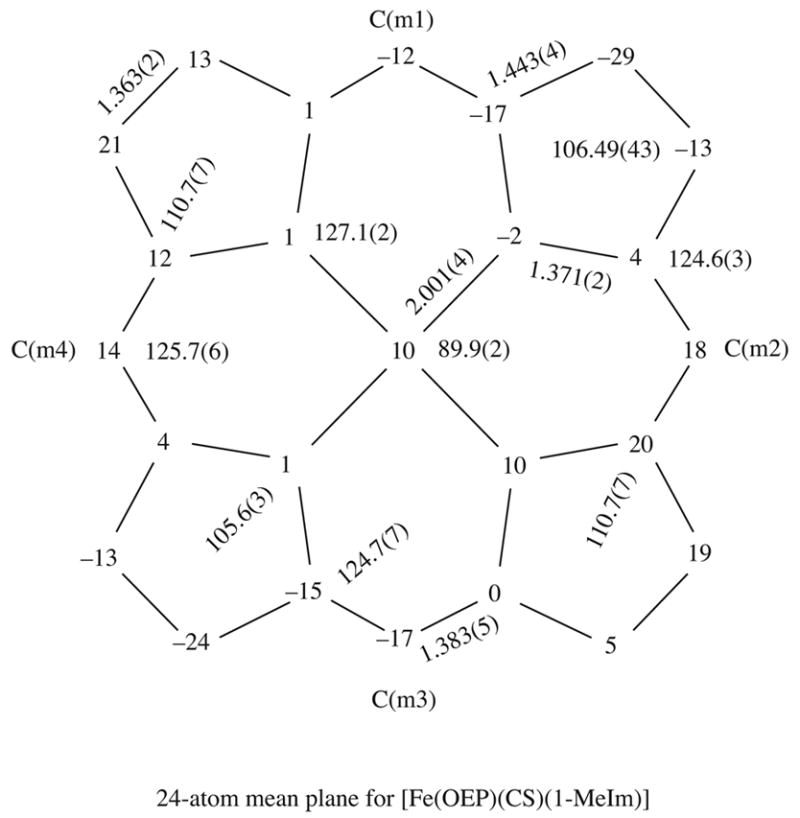
Formal diagram of the porphinato core displaying the perpendicular displacements, in units of 0.01 Å, of the core atoms from the 24-atom mean plane of [Fe(OEP)(CS)(1-MeIm)]. Positive values of displacments are towards the CS ligand. Also given on the diagram are the averaged values for the chemically unique bond distances and angles for the core.
The axial ligand plane in [Fe(OEP)(CS)(1-MeIm)] makes a dihedral angle of 21.9°; with the closest Fe–Np vector. The corresponding dihedral angles in [Fe(OEP)(CS)(Py)] are 42.93; (molecule 1), 39.1°; (molecule 2) and 35.0°; (molecule 3). The FeCS angles are close to linear with values of of 172.2(2)°; in the 1-methylimidazole complex and values of 176.2(1)°, 174.6(1)°; and 174.6(12)°; in the pyridine derivative. Complete listings of bond lengths and bond angles are included in the Supporting Information.
Discussion
A major objective of this investigation was to examine the effect of adding a sixth ligand on the Fe–C(CS) bonding in thiocarbonyl iron(II) porphyrinate derivatives. Accordingly, a number of [Fe(OEP)(CS)(L)] derivatives with neutral nitrogen donors (pyridine, substituted pyridines, piperidine and 1-methylimidazole) have been prepared. The vibrational spectra show that the addition of a sixth ligand has a small effect on the C–S stretching frequency. All six-coordinate derivatives have νCS at lower frequency than the 1292 cm−1 of five-coordinate [Fe(OEP)(CS)], with a maximum decrease of 20 cm−1.
The molecular structures of two nitrogen-ligated derivatives, [Fe(OEP)(CS)(1-MeIm)] and [Fe(OEP)(CS)(Py)], have been determined along with that of a methanol derivative [Fe(OEP)(CS)(CH3OH)]. This latter derivative, with the weakly binding methanol ligand, unfortunately has symmetry-required disorder of the axial ligands and provides no detailed metrical information about the axial ligand distances. The pyridine derivative has an unusual crystallographic feature with 2 and 1/2 molecules in the asymmetric unit.42 We first compare the new six-coordinate structures with the parent five-coordinate species.
As expected, the addition of a sixth ligand leads to a decrease in the out-of-plane displacement of the iron atom. Although the displacement of the iron atom from the 24-atom mean plane in five-coordinate [Fe(OEP)(CS)] is relatively small at 0.23 Å, owing to the low-spin state of iron(II), the displacement is decreased to 0.10 Å in [Fe(OEP)(CS)(1-MeIm)]. Similar or smaller values of the out-of-plane displacement are also found for the pyridine derivative; the precision of values is limited by slight disorder in the porphyrin cores. All displacements are on the thiocarbonyl side of the porphyrin plane. Concomitant with the decrease in the out-of-plane iron displacement, there is an increase of >0.02 Å in the average Fe–Np distance of the six-coordinate derivatives (See Table 2). The radius of the central hole, Ct···N, increases from 1.969 A in five-coordinate [Fe(OEP)(CS)] to 1.998 Å in the six-coordinate species.
The Fe–N bond distances trans to Fe–C(CS) are relatively long. The Fe–N(1-MeIm) distance is found to be 2.112(3) Å for [Fe(OEP)(CS)(1-MeIm)] and Fe-N(Py) distances of 2.1469(18) Å and 2.1550(15) Å are found for [Fe(OEP)(CS)(Py)]. Estimates of the magnitude of the increase in Fe–N(L) can be determined by comparison with a series of bis-ligated six-coordinate iron(II) porphyrinate complexes bonded to either imidazole43 or pyridine ligands.44 The bis-imidazole complexes43 display axial Fe–N(L) bonds ranging from 2.004(2) Å to 2.017(4) Å, a lengthening of ~0.10 Å is thus inferred. The Fe–N(L) bond in [Fe(Por)(L)2], where L is either pyridine or a substituted pyridine44 varies from 1.996(2) Å to 2.039(1) Å, with a median value of 2.019 Å. This gives a lengthening of 0.12 Å for the Fe–N(Py) bond. The bond lengthening is similar to that observed for the iron(II) nitrosyl porphyrinates where the Fe–N(L) distances of the ligand trans to nitrosyl is lengthened by > 0.2 Å. The thiocarbonyl ligand thus appears to have a significant structural trans effect similar to the nitrosyl ligand in iron(II) porphyrinates.35, 36, 45
The six-coordinate derivatives show a most significant structural feature: a general lengthening of the Fe–C(CS) bond on coordination of the sixth ligand. The 1-methylimidazole derivative exhibits an Fe–C(CS) distance of 1.703(4) Å, a distance significantly longer than the Fe–C(CS) distance of 1.662(3) Å observed for five-coordinate [Fe(OEP)(CS)].25 Similar values are seen in the [Fe(OEP)(CS)(Py)] structure where Fe–C(CS) distances of 1.707(2) Å and 1.704(2) Å are observed for the two molecules exhibiting ordered axial ligands. These values (average = 1.705(2) Å) appear to be reliable as can be attested by the constant value of the C–S bond distance of 1.56 Å. Thus there is a ~0.04 Å lengthening of Fe–C(CS) upon coordination of a trans neutral nitrogen ligand.
The Fe–C(CS) bond length of 1.703(4) Å observed for [Fe(OEP)(CS)(1-MeIm)] is much shorter than the Fe–C(CO) bond length of 1.744(5) Å found for [Fe(OEP)(CO)(1-MeIm)].11 Similarly, the Fe–C distances of 1.707(2) Å and 1.704(2) Å observed for the thiocarbonyl com- plex, [Fe(OEP)(CS)(Py)], are again shorter than the Fe–C distance of 1.77(2) Å observed for [Fe(TPP)(CO)(Py)].46 This 1.77- Å value is in the range of Fe–C(CO) distances for a number of other six-coordinate carbonyl complexes.47-53 The structure of a five-coordinate carbonyl derivative is unavailable. However, the Fe–C(CO) distance is 1.706(5) Å in [Fe(Deut)(CO)(THF)]54 where the trans ligand is the weakly binding tetrahydrofuran ligand.
The differences in the Fe–C(CS) and Fe–C(CO) bond distances immediately point towards a better bonding capability of the CS ligand compared to the CO ligand. The relatively short Fe–C and relatively long C–S bond distance point to significantly stronger Fe→C π-donation in the CS complexes relative to the CO complexes.55 The differences in Fe→C π-donation are also clearly reflected in the Mössbauer spectra of the thiocarbonyl and carbonyl iron porphyrinates.
Mössbauer spectra were obtained for five-coordinate [Fe(OEP)(CS)] and several six-coordinate complexes with neutral nitrogen donor ligands. Measurements were carried out in zero applied field at room temperature and 4.2 K and in a 9.0 T applied magnetic field at 4.2 K. The Mössbauer data for the thiocarbonyl complexes are summarized in Table 3 along with a number of related iron porphyrinate derivatives for comparison. Fits to the experimental data in the applied field show that five-coordinate [Fe(OEP)(CS)] and all its six-coordinate derivatives have the expected diamagnetic (low-spin) ground state.
Table 3.
Solid-State Mössbauer Parameters for Thiocarbonyl and Related Derivatives.
| complexa | Δ Eqb | δFeb | T(K) | νCSc | ref |
|---|---|---|---|---|---|
| Iron(II) Derivatives | |||||
| [Fe(OEP)(CS)] | 1.95 | −0.03 | 293 | 1292 | tw |
| 1.93 | 0.08 | 4.2 | |||
| [Fe(OEP)(CS)(4-CNPy)] | 0.90 | 0.03 | 293 | 1284 | tw |
| 0.80 | 0.19 | 4.2 | |||
| [Fe(OEP)(CS)(Py)] | 0.67 | 0.04 | 293 | 1280 | tw |
| 0.57 | 0.15 | 4.2 | |||
| [Fe(OEP)(CS)(Pip)] | 0.65 | 0.05 | 293 | 1279 | tw |
| 0.65 | 0.19 | 4.2 | |||
| [Fe(OEP)(CS)(4-NMe2Py)] | 0.497 | 0.06 | 293 | 1278 | tw |
| 0.44 | 0.19 | 4.2 | |||
| [Fe(OEP)(CS)(1-MeIm)] | 0.47 | 0.03 | 293 | 1271 | tw |
| 0.42 | 0.14 | 4.2 | |||
| [Fe(TPP)(CO)(Py)] | 0.57 | 0.28 | 293 | 56 | |
| [Fe(TPP)(CO)(1-MeIm)] | 0.35 | 0.20 | 293 | 56 | |
| [Fe(TPP)(CO)(Pip)] | 0.53 | 0.18 | 295 | 57 | |
| [Fe(TPP)(NO)] | 1.24 | 0.35 | 4.2 | 58 | |
| [Fe(OEP)(NO)] | 1.26 | 0.35 | 100 | 31 | |
| [Fe(TpivPP)(NO2)]− | 2.28 | 0.41 | 4.2 | 28 | |
| [Fe(TpivPP)(NO2)(PMS)]− | 1.18 | 0.42 | 4.2 | 28 | |
| [Fe(TpivPP)(NO2)(Py)]− | 0.93 | 0.41 | 4.2 | 28 | |
| Iron(III) Derivatives | |||||
| [Fe(TPP)(NO2)(NO)] | 1.37 | 0.02 | 293 | 59 | |
| 1.36 | 0.13 | 77 | 60 | ||
| 1.36 | 0.13 | 4.2 | 59 | ||
| [Fe(TpivPP)(NO2)(NO)] | 1.48 | 0.01 | 293 | 59 | |
| 1.43 | 0.09 | 4.2 | 59 | ||
| [Fe(OEP)(NO)(p-C6H4F)] | 0.56 | 0.05 | 293 | 61 | |
| 0.57 | 0.14 | 4.2 | 61 | ||
| [Fe(OEP)(NO)]ClO4 | 1.55 | 0.13 | 293 | 62 | |
| 1.64 | 0.20 | 4.2 | 62 | ||
| [Fe(OEP)(NO)]ClO4.CHCl3 | 1.63 | 0.12 | 293 | 62 | |
| 1.65 | 0.20 | 4.2 | 62 | ||
| [Fe(OEP)(NO)(Iz)]ClO4 | 1.99 | −0.07 | 293 | 62 | |
| 1.92 | 0.02 | 4.2 | 62 | ||
| [Fe(OEP)(t-BuNC)2]+ | 1.67 | 0.08 | 300 | 63 | |
| 1.98 | 0.16 | 120 | 63 | ||
| 2.06 | 0.18 | 4.2 | 63 | ||
| [Fe(TPP)(t-BuNC)2]+ | 2.12 | 0.13 | 120 | 63 | |
| Iron(III 1/2, IV) Derivatives | |||||
| {[Fe(TPP)]2N} | 1.08 | 0.10 | 300 | 64 | |
| 1.08 | 0.17 | 6 | |||
| {[Fe(TTP)]2N} | 1.15 | 0.04 | 293 | 65 | |
| {[Fe(TTP)]2N}SbCl6 | 2.04 | −0.13 | 293 | 65 | |
| {[Fe(TPP)]2C} | 1.88 | 0.10 | 131 | 66 | |
[Fe(OEP)(CS)] is found to exhibit a very small value for the isomer shift (δFe = 0.08 mm/s at 4.2 K and −0.03 mm/s at 293 K). The isomer shift is the smallest yet reported for a formally iron(II) porphyrinate complex. The isomer shift is comparable to values found for some iron porphyrin complexes with iron in formal oxidation state of +3 and +4, especially those with good π-accepting ligands.68 It is tempting to suggest that the extremely low value of the isomer shift is due solely to the π-accepting character of the axial thiocarbonyl ligand. However, decreases in the isomer shift in a similar series of complexes can be related to both increased σ-donating and π-accepting character of the ligands.69 Mössbauer spectra for the six-coordinate thiocarbonyl complexes provide evidence for the importance of σ-bonding as well as π-bonding for contributions to the extremely low isomer shift.
The axial ligands used include three pyridine derivatives, 4-cyanopyridine, pyridine, and 4-N-dimethylaminopyridine (these span the basicity range of pyridine derivatives), piperidine, a strong, pure σ-donor ligand and 1-methylimidazole, a strongly basic ligand that is also a modest π-donor. Mössbauer parameters of the six-coordinate thiocarbonyl complexes show increases in the isomer shift relative to the parent [Fe(OEP)(CS)]. Values for the five derivatives studied are given in Table 3. The increase in isomer shift is similar for all five complexes with dFe ranging from 0.14 to 0.19 mm/s at 4.2 K, values that are nonetheless still quite small for formally iron(II) species. Since the most basic and least basic sixth ligands give the same value of isomer shift (0.19 mm/s), we conclude that both σ and π effects must contribute to the changed isomer shift values in the six-coordinate complexes. As discussed below, a decreased σ-component of the Fe–C(CS) bond and not a changed π-component must be regarded as the primary cause for the decreased isomer shift in the six-coordinate species compared to the five-coordinate complex.
Mössbauer spectra have been measured at room temperature for several comparable carbonyl complexes: [Fe(TPP)(CO)(Py)],56 [Fe(TPP)(CO)(1-MeIm)],56 and [Fe(TPP)(CO)(Pip)].57 Significantly higher isomer shifts are found: δFe = 0.28 mm/s, 0.20 mm/s, and 0.18 mm/s at 293 K, respectively, for an increase of 0.15–0.20 mm/s compared to the range (0.03–0.06 mm/s) of the thiocarbonyl derivatives. The isomer shifts of the carbonyls themselves are also lower (by roughly 0.15–0.20 mm/s) compared to the value of δFe = 0.41–0.51 mm/s generally observed for typical low-spin iron(II) porphyrinates.68 The ~0.15 mm/s increases in isomer shift between members of the series CS,L > CO,L > bis neutral nitrogen donors are dominated by decreasing Fe→L(Axial) π-bonding in the order CS > CO > nitrogen donor.
The five-coordinate thiocarbonyl complex exhibits a large value of the Mössbauer quadrupole splitting constant, ΔEq = 1.93 mm/s at 4.2 K and 1.95 mm/s at 293 K, a relatively large value for a low-spin iron(II) species. A large decrease in quadrupole splitting is observed on coordination of a sixth ligand to [Fe(OEP)(CS)]; the quadrupole splitting values for the six-coordinate complexes range from 0.80 mm/s to 0.42 mm/s (Table 3). Moreover, all derivatives, except the piperidine derivative and the parent five-coordinate complex, show a substantial temperature dependence for the quadrupole splitting. The large temperature effect on the quadrupole splitting suggests the importance of a low-lying excited state for these complexes.
The large decrease in quadrupole splitting upon increase in coordination number is similar to that seen for iron(II) nitrite complexes. Five-coordinate [Fe(TpivPP)(NO2)]− exhibits a quadrupole splitting of 2.28 mm/s at 4.2 K while the six-coordinate complexes [Fe(TpivPP)(NO2)(PMS)]− and [Fe(TpivPP)(NO2)(Py)]− show smaller quadrupole splittings of 1.18 mm/s and 0.93 mm/s respectively at 4.2 K. The differences were attributed to a decrease in the importance of Fe donation to the π-acceptor nitrite upon addition of the sixth ligand.28 A comparable explanation for the changes in quadrupole splitting would appear applicable to the thiocarbonyls: a decrease in the importance of Fe donation to the π-acceptor CS upon addition of the sixth ligand. In the nitrite system, the rhombicity in the π interaction between nitrite and iron(II) was also considered to be an important factor in the large value of the quadrupole splitting. Clearly, rhombicity in π bonding for (linear) thiocarbonyl complexes is not applicable since both Fe dπ orbitals are involved in π bonding to CS. Thus, while the rhombic dπ donation in the Fe–N(NO2) π-interaction may have a significant effect on the value of the quadrupole splitting in five-coordinate [Fe(TpivPP)(NO2)] −, the thiocarbonyl results suggest that the rhombicity is neither the only, nor even the major, contributor to the large quadrupole splitting in these iron(II) porphyrinates. As shown in the next paragraphs, the variation in quadrupole splitting for the six-coordinate thiocarbonyl derivatives is not consistent with large changes in the character of the axial π-bonding and hence must primarily be due to cis effects.70
The six-coordinate thiocarbonyl complexes show substantial variation in quadrupole splitting with ΔEq values ranging from 0.80 to 0.42 mm/s at 4.2 K (Table 3). These variations in quadrupole splitting might be expected to reflect parallel differences in d-electron density on iron, differences caused by π bonding. However, the clear relationship between ligand basicity and quadrupole splitting is not consistent with axial ligand π effects. A linear relationship between ligand pKa and room temperature quadrupole splitting for the three pyridine derivatives studied, shows the most basic pyridine derivative has the lowest value of quadrupole splitting, not the highest, the opposite of that expected for π effects. The three derivatives encompass the entire pKa range of pyridines.71 This relationship is by no means an exact one; the ligands piperidine and 1-methylimidazole only fit the correlation in a general way. Piperidine, the most basic of the ligands studied, does not have the smallest value of quadrupole splitting, the 1-methylimidazole derivative does. Figure 4 gives a plot of quadrupole splitting vs. ligand pKa; the line shown is the best fit to the three pyridine values.
Figure 4.
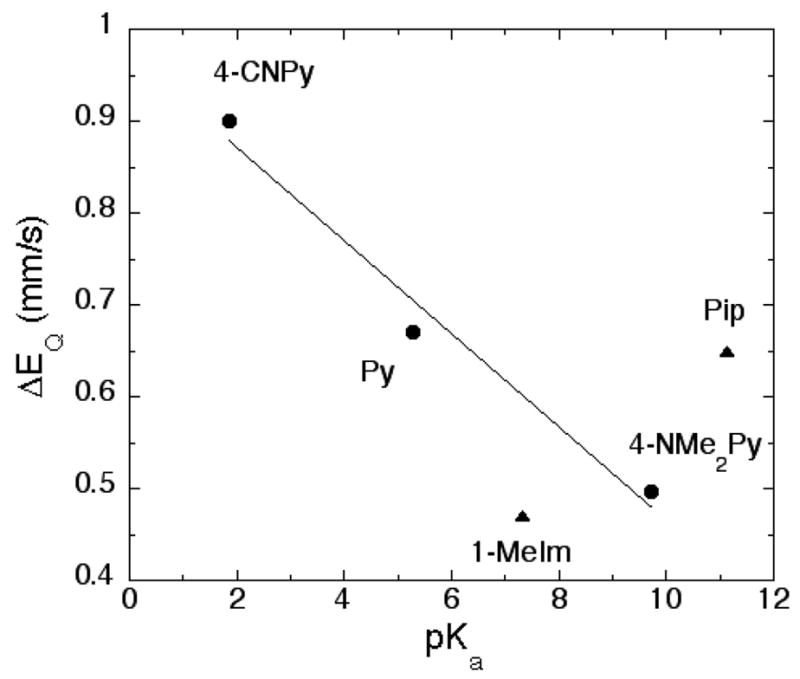
Plot displaying the relationship between ligand pKa and Mössbauer quadrupole splitting constant for the six-coordinate [Fe(OEP)(CS)(L)]. The line is the best fit to the three pyridine derivatives.
The quadrupole splitting values are also correlated with the CS stretching frequency. Values of νCS are given in Table 3; stretching frequencies decrease with increasing pKa of the added ligand.72 Figure 5 displays the correlation: a decreased value of the quadrupole splitting is related to a decreased value of νCS. Any decrease in νCS is expected to arise from an increased population of the CS π* orbitals, which in turn derives from increased Fe→C π-donation. But the small changes in values of νCS suggests only a very modest decrease in the C–S bond strength for the set of compounds studied, even when the five-coordinate complex is included. Thus, we can conclude that the change in Fe→C π-donation upon addition of a sixth ligand must also be very small.
Figure 5.
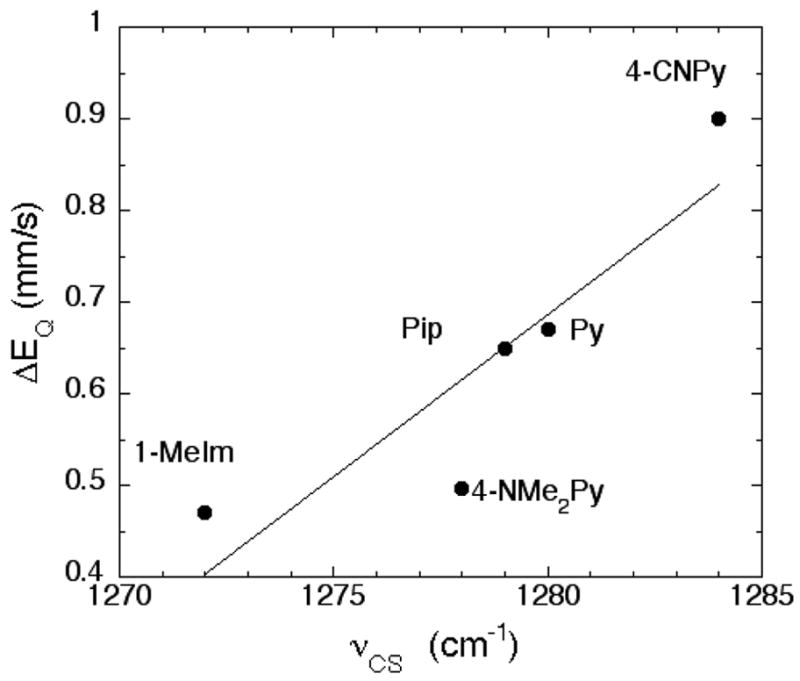
Plot displaying the relationship between νCS and Mössbauer quadrupole splitting constant for the six-coordinate [Fe(OEP)(CS)(L)]. The line is the best fit to all derivatives.
The variation in quadrupole splitting can be analyzed in terms of the Vzz component of the electric field gradient. Vzz is approximately given by the expression
| (1) |
where k is the scaling factor and the ni values are the effective populations of the 3d orbitals.74 Values of Vzz are related to the measured 57Fe quadrupole splitting constants by the expression
| (2) |
where e is the charge on the electron, η is the asymmetry parameter (Vxx−Vyy/Vzz), and Q is the quadrupole moment of the 57Fe nucleus in its excited state. Measurements in applied magnetic field show that the asymmetry parameter γ is effectively zero for all CS complexes studied and that the sign of Vzz is always positive. Expression (1) shows that increased populations of the dz2, dxy, and dxz orbitals and a decreased population of the dx2−y2 orbital lead to decreased values of Vzz and hence decreased quadrupole splitting (expression (2)). Since an increase in the population of dxy and dxz from decreased axial π-bonding has already been shown to be unlikely, as demonstrated by the νCS values, any increase in these populations must come from porphyrin→Fe π-bonding. A decrease in the dx2−y2 population must come from decreased σ-bonding between iron and porphyrin, probably in response to the increased σ-bonding from the sixth ligand. The porphyrin–iron σ- and π-effects are counter to each other and must be be approximately equivalent. The involvement of dz2 is consistent with the σ-donation and the general pKa dependence of the sixth ligand, while the changes in iron–porphyrin bonding are cis effects induced by addition of the sixth ligand.
These observations allow two important conclusions. First, the small changes in axial Fe→C π-bonding on addition of a sixth ligand means that the ~0.04 Å increase in Fe–C bond length in the six-coordinate species can only be the result of decreased σ-donation by the CS ligand. Clearly the strong structural trans effect of CS must result from σ-donation competition and CS remains a strong σ-donor. Second, the large d-electron distribution changes consistent with the large change in the quadrupole splitting must arise, in large part, from cis effects induced by binding the ligand trans to CS. The cis effects are both an increase in the porphyrin→Fe π donation and a decrease in the σ bonding.
Conclusion
The structural and Mössbauer studies of these iron(II)octaethylporphyrinate thiocarbonyl complexes show the effects of adding the sixth ligand are primarily the result of changes induced by the σ bonding of the axial ligand. The large quadrupole splitting and exceptionally small isomer shifts observed for the [Fe(OEP)(CS)] derivatives indicates that CS must be an exceptionally good π-bonding ligand and that the character of the axial Fe→C(CS) π-bonding is little affected by the sixth ligand.
Supplementary Material
Figures S1-S7 giving ORTEP diagrams and mean plane drawings for [Fe(OEP)(CS)(Py)] and [Fe(OEP)(CS)(CH3OH)]. Tables S1-S18, giving complete crystallographic details, atomic coordinates, bond distances and angles, anisotropic temperature factors, and fixed hydrogen atom positions for [Fe(OEP)(CS)(1-MeIm)], [Fe(OEP)(CS)(Py)], and [Fe(OEP)(CS)(CH3OH)]. (Available as a PDF file.) X-ray crystallographic information (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
Figure S1. ORTEP diagram of [Fe(OEP)(CS)(Py1)] displaying the atom labeling scheme. Thermal ellipsoids of all atoms are contoured at the 50% probability level.
Figure S2. ORTEP diagram of [Fe(OEP)(CS)(Py3)] displaying the atom labeling scheme. Thermal ellipsoids of all atoms are contoured at the 50% probability level.
Figure S3. ORTEP diagram of [Fe(OEP)(CS)(CH3OH)] displaying the atom labeling scheme. Thermal ellipsoids of all atoms are contoured at the 50% probability level.
Figure S4. Formal diagrams of the porphinato core displaying the perpendicular displacements, in units of 0.01 Å, of the core atoms from the 24-atom mean plane of [Fe(OEP)(CS)(Py)] (molecule 1).
Figure S5. Formal diagrams of the porphinato core displaying the perpendicular displacements, in units of 0.01 Å, of the core atoms from the 24-atom mean plane of [Fe(OEP)(CS)(Py)] (molecule 2).
Figure S6. Formal diagrams of the porphinato core displaying the perpendicular displacements, in units of 0.01 Å, of the core atoms from the 24-atom mean plane of [Fe(OEP)(CS)(Py)] (molecule 3). Centrosymmetrically related atoms in the diagram have equal magnitude displacements but are of opposite sign.
Figure S7. Formal diagrams of the porphinato core displaying the perpendicular displacements, in units of 0.01Å, of the core atoms from the 24-atom mean plane of [Fe(OEP)(CS)(CH3OH)]. Centrosymmetrically related atoms in the diagram have equal magnitude displacements but of are opposite sign.
Table S1. Complete Crystallographic Details for [Fe(OEP)(CS)(1-MeIm)].
Table S2. Atomic Coordinates and Equivalent Isotropic Displacement Parameters for [Fe(OEP)(CS)(1-MeIm)].
Table S3. Bond Lengths for [Fe(OEP)(CS)(1-MeIm)].
Table S4. Bond Angles for [Fe(OEP)(CS)(1-MeIm)].
Table S5. Anisotropic Displacement Parameters for [Fe(OEP)(CS)(1-MeIm)].
Table S6. Hydrogen Coordinates and Isotropic Displacement Parameters for [Fe(OEP)(CS)(1-MeIm)].
Table S7. Complete Crystallographic Details for [Fe(OEP)(CS)(CH3OH)].
Table S8. Atomic Coordinates and Equivalent Isotropic Displacement Parameters for [Fe(OEP)(CS)(CH3OH)]
Table S9. Bond Lengths for [Fe(OEP)(CS)(CH3OH)].
Table S10. Bond Angles for [Fe(OEP)(CS)(CH3OH)].
Table S11. Anisotropic Displacement Parameters for [Fe(OEP)(CS)(CH3OH)].
Table S12. Hydrogen Coordinates and Isotropic Displacement Parameters for [Fe(OEP)(CS)(CH3OH)].
Table S13. Complete Crystallographic Details for [Fe(OEP)(CS)(Py)].
Table S14. Atomic Coordinates and Equivalent Isotropic Displacement Parameters for [Fe(OEP)(CS)(Py)]
Table S15. Bond Lengths for [Fe(OEP)(CS)(Py)].
Table S16. Bond Angles for [Fe(OEP)(CS)(Py)].
Table S17. Anisotropic Displacement Parameters for [Fe(OEP)(CS)(Py)].
Table S18. Hydrogen Coordinates and Isotropic Displacement Parameters for [Fe(OEP)-(CS)(Py)].
Acknowledgments
We thank the National Institutes of Health for the support of this research under Grant GM-38401 to W.R.S. Funds for the purchase of the FAST area detector diffractometer was provided through NIH Grant RR-06709 to the University of Notre Dame.
Footnotes
Contribution from The Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, Indiana 46556 and Department of Physics, Knox College, Galesburg, Illinois 61401
References and Notes
- 1.Keilin D. Roy Soc Proc B. 1925;98:312. [Google Scholar]
- 2.Pauling L, Coryell CD. Proc Natl Acad Sci USA. 1936;22:210. doi: 10.1073/pnas.22.4.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buchler JW. In: Porphyrins and Metalloporphyrins. Smith KM, editor. Chapter 5 Elsevier Scientific Publishing; Amsterdam, The Netherlands: 1975. [Google Scholar]
- 4.Collman JP, Gagne RR, Reed CA, Robinson WT, Rodley GA. Proc Natl Acad Sci USA. 1974;71:1326. doi: 10.1073/pnas.71.4.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Traylor TG, Koga N, Deardruff LA. J Am Chem Soc. 1985;107:6504. [Google Scholar]
- 6.Springer BA, Sligar SG, Olson JS, Philips GN., Jr Chem Rev. 1994;94:699. [Google Scholar]
- 7.Olson JS, Phillips GN., Jr J Biol Inorg Chem. 1997;2:544. [Google Scholar]
- 8.Slebodnick C, Ibers JA. J Biol Inorg Chem. 1997;2:521. [Google Scholar]
- 9.Sage JT. J Biol Inorg Chem. 1997;2:537. [Google Scholar]
- 10.Spiro TG, Kozlowski PM. J Am Chem Soc. 1998;120:4524. [Google Scholar]
- 11.Salzmann R, McMahon MT, Godbout N, Sanders LK, Wojdelski M, Oldfield E. J Am Chem Soc. 1999;121:3818. [Google Scholar]
- 12.Richards WG. Faraday Soc Trans. 1967;63:257. [Google Scholar]
- 13.Ziegler T, Rauk A. Inorg Chem. 1979;18:1755. [Google Scholar]
- 14.Hubbard JL, Lichtenberger DL. Inorg Chem. 1980;19:3865. [Google Scholar]
- 15.Ziegler T. Inorg Chem. 1986;25:2721. [Google Scholar]
- 16.Lichtenberger DL, Fenske RF. Inorg Chem. 1976;15:2015. [Google Scholar]
- 17.Saillard JY, Grandjean D, Caillet P, le Beuze AJ. J Organomet Chem. 1980;190:371. [Google Scholar]
- 18.Butler IS. Acc Chem Res. 1977;10:359. [Google Scholar]
- 19.Andrews MA. Inorg Chem. 1977;16:496. [Google Scholar]
- 20.Woodward SS, Angelici RJ, Dombek BD. Inorg Chem. 1978;17:1634. [Google Scholar]
- 21.Moltzen EK, Klabunde KJ. Chem Rev. 1988;88:391. [Google Scholar]
- 22.Buchler JW, Kokisch W, Smith PD, Tonn B. Z Naturforsch. 1978;33b:1371. [Google Scholar]
- 23.Mansuy D, Battioni JP, Chottard JC. J Am Chem Soc. 1978;100:4311. doi: 10.1021/ja00461a046. [DOI] [PubMed] [Google Scholar]
- 24.Battioni JP, Chottard JC, Mansuy D. Inorg Chem. 1982;21:2056. [Google Scholar]
- 25.Scheidt WR, Geiger DK. Inorg Chem. 1982;21:1208. [Google Scholar]
- 26.Abbreviations: OEP, dianion of octaethylporphyrin; TPP, dianion of tetraphenylporphyrin; TpivPP, dianion of α, α, α, α -tetrakis(o-pivalamidophenyl)porphyrin; TTP, dianion of tetratolylporphyrin; Py, pyridine; 4-CNPy, 4-cyanopyridine; 4-NMe2Py, 4-N-dimethylaminopyridine; 1-MeIm, 1-methylimidazole; Pip, piperidine; Np, porphyrinato nitrogen; PMS, pentamethylene sulfide; Iz, indazole; t-BuCN t-butyl isocyanide.
- 27.Nasri H, Wang Y, Huynh BH, Scheidt WR. J Am Chem Soc. 1991;113:717. [Google Scholar]
- 28.Nasri H, Ellison MK, Krebs C, Huynh BH, Scheidt WR. J Am Chem Soc. 2000;122:10795. [Google Scholar]
- 29.Scheidt WR, Frisse ME. J Am Chem Soc. 1975;97:17. doi: 10.1021/ja00834a005. [DOI] [PubMed] [Google Scholar]
- 30.Bohle DS, Hung CH. J Am Chem Soc. 1995;117:9584. [Google Scholar]
- 31.Bohle DS, Debrunner PG, Fitzgerald J, Hansert B, Hung CH, Thompson AJ. J Chem Soc, Chem Commun. 1997:91. [Google Scholar]
- 32.Ellison MK, Scheidt WR. J Am Chem Soc. 1997;119:7404. [Google Scholar]
- 33.Scheidt WR, Duval HF, Neal TJ, Ellison MK. J Am Chem Soc. 2000;122:4651. [Google Scholar]
- 34.Scheidt WR, Lee YJ, Hatano K. J Am Chem Soc. 1984;106:3191. [Google Scholar]
- 35.Scheidt WR, Piciulo PL. J Am Chem Soc. 1976;98:1913. doi: 10.1021/ja00423a044. [DOI] [PubMed] [Google Scholar]
- 36.Scheidt WR, Brinegar AC, Ferro EB, Kirner JF. J Am Chem Soc. 1977;99:7315. [Google Scholar]
- 37.Ellison MK, Scheidt WR. J Am Chem Soc. 1999;121:5210. [Google Scholar]
- 38.Adler AD, Longo FR, Kampas F, Kim J. J Inorg Nucl Chem. 1970;32:2443. [Google Scholar]
- 39.Landergren M, Baltzer L. Inorg Chem. 1990;29:556. [Google Scholar]
- 40.Scheidt WR, Turowska-Tyrk I. Inorg Chem. 1994;33:1314. [Google Scholar]
- 41.Programs used in this study included SHELXS-86 (Sheldrick GM. Acta Crystallogr, Sect A. 1990;A46:467.Sheldrick GM. SHELXL-93. local modifications of Johnson’s ORTEP2. Scattering factors were taken from International Tables for Crystallography. In: Wilson AJC, editor. J Appl Cryst. C. Kluwer Academic Publishers; Dordrecht: 1992.
- 42.For these triclinic systems, there are two asymmetric units per unit cell.
- 43.Several bis-ligated iron(II) complexes of general formula [Fe(Por)(L)2] where L is an imidazole derivative have been structurally characterized. The values of Fe–N(L) are as follows: (a) [Fe(TPP)(1-MeIm)2], 2.014(5) Å, Hoard, J. L. personal communication to WRS; (b) [Fe(TPP)(1-VinIm)2], 2.004(2) Å; [Fe(TPP)(1-BzIm)2], 2.017(4), Safo MK, Scheidt WR, Gupta GP. Inorg Chem. 1990;29:626.
- 44.Several bis-ligated iron(II) complexes of general formula [Fe(Por)(L)2] where L is either pyridine or a substituted pyridine have been structurally characterized. The values of Fe–N(L) are as follows: (a) [Fe(TPP)(Py)2], 2.037(1) Å, Li N, Coppens P, Landrum J. Inorg Chem. 1988;27:482.; (b) [Fe(TPP)(Py)2]·2Py, 2.039(1) Å, Ni L, Petricek V, Coppens P, Landrum J. Acta Crystallogr, Sect C. 1985;C41:902.; (c) [Fe(TMP)(4-MePy)2], 2.010(2) Å; [Fe(TMP)(4-CNPy)2], 1.996(2) Å; [Fe(TMP)(3-CNPy)2], 2.026(2) Å, Safo MK, Nesset MJM, Walker FA, Debrunner PG, Scheidt WR. J Am Chem Soc. 1997;119:9438.
- 45.Wyllie GRA, Schulz CE, Scheidt WR. submitted for publication. [Google Scholar]
- 46.Peng SM, Ibers JA. J Am Chem Soc. 1976;98:8032. doi: 10.1021/ja00441a025. [DOI] [PubMed] [Google Scholar]
- 47.Caron C, Mitschler A, Rivère G, Ricard L, Schappacher M, Weiss R. J Am Chem Soc. 1979;101:7401. [Google Scholar]
- 48.Ricard L, Weiss R, Momenteau M. J Chem Soc, Chem Commun. 1986:818. [Google Scholar]
- 49.Kim K, Fettinger J, Sessler JL, Cyr M, Hugdahl J, Collman JP, Ibers JA. J Am Chem Soc. 1989;111:403. [Google Scholar]
- 50.Kim K, Ibers JA. J Am Chem Soc. 1991;113:6077. [Google Scholar]
- 51.Slebodnick C, Duval ML, Ibers JA. Inorg Chem. 1996;35:3607. [Google Scholar]
- 52.Slebodnick C, Fettinger JC, Peterson HB, Ibers JA. J Am Chem Soc. 1996;118:3216. [Google Scholar]
- 53.Salzmann R, Ziegler CJ, Godbout N, McMahon MT, Suslick KS, Oldfield E. J Am Chem Soc. 1998;120:11323. [Google Scholar]
- 54.Scheidt WR, Haller KJ, Fons M, Mashiko T, Reed CA. Biochemistry. 1981;20:3653. doi: 10.1021/bi00515a054. [DOI] [PubMed] [Google Scholar]
- 55.Richardson JW, Jr, Angelici RJ, Jacobson RA. Inorg Chem. 1987;26:452. [Google Scholar]
- 56.Havlin RH, Godbout N, Salzmann R, Wojdelski M, Arnold W, Schulz CE, Oldfield E. J Am Chem Soc. 1998;120:3144. [Google Scholar]
- 57.James BR, Sams JR, Tsin TB, Reimer KJ. J Chem Soc, Chem Commun. 1978:746. [Google Scholar]
- 58.Nasri H, Ellison MK, Chen S, Hyunh BH, Scheidt WR. J Am Chem Soc. 1997;119:6274. [Google Scholar]
- 59.Ellison MK, Schulz CE, Scheidt WR. Inorg Chem. 1999;38:100. doi: 10.1021/ic000789e. [DOI] [PubMed] [Google Scholar]
- 60.Settin MF, Fanning JC. Inorg Chem. 1988;27:1431. [Google Scholar]
- 61.Richter-Addo GB, Wheeler RA, Hixon CA, Chen Li, Khan MA, Ellison MK, Schulz CE, Scheidt WR. J Am Chem Soc. 2001;123:6314. doi: 10.1021/ja010276m. [DOI] [PubMed] [Google Scholar]
- 62.Ellison MK, Schulz CE, Scheidt WR. Inorg Chem. 2000;39:5102. doi: 10.1021/ic000789e. [DOI] [PubMed] [Google Scholar]
- 63.Walker FA, Nasri H, Turowska-Tyrk I, Mohanrao K, Watson CT, Shokhirev NV, Debrunner PG, Scheidt WR. J Am Chem Soc. 1996;118:12109. [Google Scholar]
- 64.Summerville DA, Cohen IA. J Am Chem Soc. 1976;98:1747. doi: 10.1021/ja00423a019. [DOI] [PubMed] [Google Scholar]
- 65.Li M, Shang M, Ehlinger N, Schulz CE, Scheidt WR. Inorg Chem. 2000;39:580. doi: 10.1021/ic990900k. [DOI] [PubMed] [Google Scholar]
- 66.English DR, Hendrickson DN, Suslick KS. Inorg Chem. 1983;23:367. [Google Scholar]
- 67.Rhynard D, Lang G, Spartalian K, Yonetani TJ. J Chem Phys. 1979;71:3715. [Google Scholar]
- 68.Debrunner PG. In: Iron Porphyrins. Lever ABP, Gray HB, editors. Part 3, Chapter 2 VCH Publishers Inc; New York: 1983. [Google Scholar]
- 69.Shenoy GK. Mössbauer E3ect Isomer Shifts. In: Long GJ, editor. Mössbauer Spectroscopy Applied to Inorganic Chemistry. Plenum Press; NY: 1984. pp. 57–76. [Google Scholar]
- 70.Buchler JW, Kokisch W, Smith PD. Struct Bonding (Berlin) 1978;34:79. [Google Scholar]
- 71.Schoefield KS. Hetero-Aromatic Nitrogen Compounds. Plenum Press; New York, N. Y: 1967. p. 146. [Google Scholar]
- 72.The variation of νCS with the basicity of the trans ligand has been noted in earlier studies.23, 24 A similar pattern has been noted for [Fe(por)(CO)(L)] systems.73
- 73.Alben JO, Caughey WS. Biochemistry. 1968;7:175. doi: 10.1021/bi00841a022. [DOI] [PubMed] [Google Scholar]
- 74.Bancroft JM, Platt RH. Adv Inorg Chem Radiochem. 1972;15:59. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1-S7 giving ORTEP diagrams and mean plane drawings for [Fe(OEP)(CS)(Py)] and [Fe(OEP)(CS)(CH3OH)]. Tables S1-S18, giving complete crystallographic details, atomic coordinates, bond distances and angles, anisotropic temperature factors, and fixed hydrogen atom positions for [Fe(OEP)(CS)(1-MeIm)], [Fe(OEP)(CS)(Py)], and [Fe(OEP)(CS)(CH3OH)]. (Available as a PDF file.) X-ray crystallographic information (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
Figure S1. ORTEP diagram of [Fe(OEP)(CS)(Py1)] displaying the atom labeling scheme. Thermal ellipsoids of all atoms are contoured at the 50% probability level.
Figure S2. ORTEP diagram of [Fe(OEP)(CS)(Py3)] displaying the atom labeling scheme. Thermal ellipsoids of all atoms are contoured at the 50% probability level.
Figure S3. ORTEP diagram of [Fe(OEP)(CS)(CH3OH)] displaying the atom labeling scheme. Thermal ellipsoids of all atoms are contoured at the 50% probability level.
Figure S4. Formal diagrams of the porphinato core displaying the perpendicular displacements, in units of 0.01 Å, of the core atoms from the 24-atom mean plane of [Fe(OEP)(CS)(Py)] (molecule 1).
Figure S5. Formal diagrams of the porphinato core displaying the perpendicular displacements, in units of 0.01 Å, of the core atoms from the 24-atom mean plane of [Fe(OEP)(CS)(Py)] (molecule 2).
Figure S6. Formal diagrams of the porphinato core displaying the perpendicular displacements, in units of 0.01 Å, of the core atoms from the 24-atom mean plane of [Fe(OEP)(CS)(Py)] (molecule 3). Centrosymmetrically related atoms in the diagram have equal magnitude displacements but are of opposite sign.
Figure S7. Formal diagrams of the porphinato core displaying the perpendicular displacements, in units of 0.01Å, of the core atoms from the 24-atom mean plane of [Fe(OEP)(CS)(CH3OH)]. Centrosymmetrically related atoms in the diagram have equal magnitude displacements but of are opposite sign.
Table S1. Complete Crystallographic Details for [Fe(OEP)(CS)(1-MeIm)].
Table S2. Atomic Coordinates and Equivalent Isotropic Displacement Parameters for [Fe(OEP)(CS)(1-MeIm)].
Table S3. Bond Lengths for [Fe(OEP)(CS)(1-MeIm)].
Table S4. Bond Angles for [Fe(OEP)(CS)(1-MeIm)].
Table S5. Anisotropic Displacement Parameters for [Fe(OEP)(CS)(1-MeIm)].
Table S6. Hydrogen Coordinates and Isotropic Displacement Parameters for [Fe(OEP)(CS)(1-MeIm)].
Table S7. Complete Crystallographic Details for [Fe(OEP)(CS)(CH3OH)].
Table S8. Atomic Coordinates and Equivalent Isotropic Displacement Parameters for [Fe(OEP)(CS)(CH3OH)]
Table S9. Bond Lengths for [Fe(OEP)(CS)(CH3OH)].
Table S10. Bond Angles for [Fe(OEP)(CS)(CH3OH)].
Table S11. Anisotropic Displacement Parameters for [Fe(OEP)(CS)(CH3OH)].
Table S12. Hydrogen Coordinates and Isotropic Displacement Parameters for [Fe(OEP)(CS)(CH3OH)].
Table S13. Complete Crystallographic Details for [Fe(OEP)(CS)(Py)].
Table S14. Atomic Coordinates and Equivalent Isotropic Displacement Parameters for [Fe(OEP)(CS)(Py)]
Table S15. Bond Lengths for [Fe(OEP)(CS)(Py)].
Table S16. Bond Angles for [Fe(OEP)(CS)(Py)].
Table S17. Anisotropic Displacement Parameters for [Fe(OEP)(CS)(Py)].
Table S18. Hydrogen Coordinates and Isotropic Displacement Parameters for [Fe(OEP)-(CS)(Py)].