

Abstract
Heparinase I from Flavobacterium heparinum, a source of diverse polysaccharidases, suffers from low yields, insufficient purity for structural studies and insolubility when expressed as a recombinant product in Escherichia coli that is devoid of glycosaminoglycan polysaccharidases. In this study, cDNA coding for the orthologue of F. heparinum heparinase I was constructed from genomic information from the mammalian gut symbiont Bacteroides thetaiotaomicron and expressed in E. coli as a fusion protein with GST at the N-terminus. This resulted in high yield (30 mg/g dry bacteria) of soluble product and facilitated one-step affinity purification to homogeneity. Purified heparinase I bearing the GST fusion exhibited a Km of 2.3 μM and Vmax of 42.7 μmol/min with a specific activity of 164 units/mg with heparin (average 12000 Da) as substrate. The results indicate a 2-fold improvement in yield, specific activity and affinity for heparin as substrate over previous reports. The data suggest that the heparinase I from the gut symbiont exhibits a higher intrinsic affinity for heparin than that from F. heparinum. The purified GST fusion enzyme exhibited a requirement for Ca2++ and a pH optimum between 6.7 to 7.3 that was similar to the enzyme freed of the N-terminal GST portion. Our study revealed that catalytic activity of heparinase I requires a reducing environment. The GST facilitated immobilization of heparinase I in solid phase either for clinical purposes or for structural studies in absence of interference by contaminating polysaccharidases.
Keywords: bacterial symbionts, carbohydrate microsequencing, chimeric tagged enzymes, enzyme immobilization, extracellular matrices, FGF signaling, glycobiology, heparan sulfate, heparin, oligosaccharides, polysaccharidases
Heparan sulfate (HS) glycosaminoglycan (GAG) is a cell surface and extracellular matrix component in vertebrates and invertebrates. In addition to roles in tissue organization, HS binds and impacts biological activities of numerous proteins that regulate diverse physiological and pathological processes that span cell growth, differentiation, migration and defense against infection [1–3]. HS and heparin GAG polysaccharides are comprised of structurally related uronate-glucosamine (UA-GlcN) disaccharide repeats that vary in length and degree and disposition of epimerized, acetylated or sulfated side groups that impart to HS strong anionic polyelectrolyte character. Variations in sulfation, size and topography of HS chains impacts function of diverse proteins [3–6]. However, it remains unclear whether the regulation is mediated by relatively non-specific variations in overall charge density or a specific arrangement of modified residues for a structurally specific motif within the HS chains [3,4,7]. To resolve this question requires isolation and characterization of potentially rare and specific oligosaccharide motifs from complex mixtures with highly specific bioprobes followed by microsequence analysis using group specific HS degradative enzymes and gel electrophoresis or mass spectrometry. Recently we have used members of the FGF polypeptide family as bioaffinity reagents to extract extremely rare oligosaccharide motifs that exhibit FGF-specific activity in support of FGFR signal transduction [5, 6]. An indirect deduction of structure indicated that such motifs may be undersulfated and exhibit a unique distribution of sulfated and non-sulfated sidegroups.
In experiments to demonstrate FGF-specific oligosaccharides by fragmentation of long chain heparin and heparan sulfates, we found that carefully controlled enzymatic reduction by heparinase I was the most promising compared to other methods [5]. However, currently available heparinase preparations yielded equivocal results and hampered generation of yields sufficient for routine structural analyses. Heparinase 1 (EC 4.2.2.7) cleaves primarily at α-D-GlcNS(±6S) (1→4) α-L-IdoA/GlcA2S linkage in the highly N-sulfated domains. Commercial heparinase I is a heparin-induced product derived from the soil bacterium F. heparinum that was selected for use of heparin as sole carbon, nitrogen and sulfur source [8]. F. heparinum is a natural source of diverse polysaccharidases including heparinases 1, 2, and 3 and sulfatases that degrade heparin and heparan sulfates at distinct positions [9]. More recently other bacteria strains have been examined as sources of heparinases [10–13]. However, the variability in inductive yields, complexity of purification procedures and potential co-purification with multiple heparin lyases of distinct specific activities have generally hampered both analytical and commercial applications of heparinase I and made them costly. The presence of trace amounts of other enzymes with different specificity compromises accurate microsequencing of rare oligosaccharide motifs [8, 9]. To bypass these limits, attempts have been made to express and recover recombinant heparinases in E. coli that do not express GAG polysaccharidases [14,15]. However, active soluble product was in low yield and solubilization and refolding was required [14,15,16]. Sequencing of the complete genomes of new bacterial strains has revealed homologues to F. heparinum polysaccharidases. One of the first to emerge was the genome of the human and mouse gut symbiotic Bacteroides thetaiotaomicron that exhibits one of the richest adaptive proteomes of polysaccharidases of any bacterium to date [17]. These include a variety of enzymes that target host-derived glycans such as chondroitin sulfates, mucins, hyaluronate and heparin/heparan sulfate. The B. thetaiotaomicron polysaccharidase repertoire is thought to be part of a finely tuned evolutionary niche that is symbiotic with humans and mice [18,19]. We posited that B. thetaiotaomicron gene products might be a superior source of conventional and potentially novel variants of lyases for heparin and heparan sulfate, as well as other glycosaminoglycans.
In this report in the first of a series to express, recover in high yield and purity and characterize B. thetaiotaomicron heparan sulfate lyases, we describe the cloning, recombinant expression and high yield recovery from E. coli and enzymatic characterization of B. thetaiotaomicron heparinase I, and compare the data to that of heparinase I encoded by F. heparinum. Both enzymes were soluble when expressed in E. coli with GST fused at the N-terminus. The GST fusion protein facilitated one-step affinity purification and exhibited similar kinetic parameters and co-factor requirements to heparinase I alone after removal of the GST. The approach represented improvements in yield and activity of purified heparinase I preparations. The GST-fused enzyme with high purity and specific activity may be useful in solid phase clinical applications and sequence characterizations of structurally specific heparin and heparan sulfate motifs.
Experimental procedures
Materials
Heparin (porcine intestinal mucosa, 170 USP units/mg), heparan sulfate (bovine kidney), chondroitin sulfate (bovine cartilage), dithiothreitol (DTT) and reduced glutathione were from Sigma (St. Louis, MO). Thrombin, HiTrap Glutathione-Sepharose, Benzamidine-Sepharose and Superose 12 columns were from GE HealthCare Bio-Sciences Corp (Piscataway, NJ). B. thetaiotaomicron (#29148) and F. heparinum (#13125) were from the American Type Culture Collection (Manassas, VA).
Culture of bacteria and recovery of DNA
B. thetaiotaomicron was cultured anaerobically in airtight tubes wrapped with aluminum foil containing chopped meat glucose broth medium (Becton Dickinson, Sparks, MD) at 25°C for 2 days. Bacteria was collected from 1.5 ml of culture by centrifugation at 5000 rpm for 2 min and bacterial genomic DNA extracted by modification of a previously described method [20]. Briefly, the bacterial pellet was re-suspended in 567 μl TE buffer with addition of 30 μl 10% SDS and 3 μl of 20 mg/ml proteinase K. After incubation at 37°C for 1 hr, 100 μl of 5 M NaCl was added and mixed thoroughly, and then 80 μl CTAB/NaCl solution was added followed by incubation at 65°C for 10 min. DNA was extracted with an equal volume of chloroform/isoamyl alcohol and then phenol/chloroform/isoamyl alcohol. After centrifugation, DNA in the aqueous phase was precipitated with 0.6 volumes of isopropanol, washed with 70% ethanol, and re-suspended in 100 μl TE buffer.
F. heparinum was cultured in medium comprised of 1% tryptone, 0.2% soya peptone, 0.5% NaCl and 0.03% heparin sodium salt in phosphate buffer containing additional 1 mM L-Histidine. Bacteria were collected and DNA prepared as described above for B. thetaiotaomicron.
Construction of recombinant plasmids and expression of GST-heparinase I in E. coli
DNA encoding B. Thetaiotaomicron heparinase I (cDNA) was prepared using the PCR with primers 5′-GGGGATCCATGCTGACTGCTCAGACT-3′ and 5′-GGGAATTCTTATCTTTCCGAATATCC-3′ and purified genomic DNA from B. thetaiotaomicron as template. The PCR product was digested and ligated into pGEX-2T vector at BamHI and EcoRI sites in frame with coding sequence for GST at the 5′ end. A thrombin cut site was introduced between the GST and heparinase I. Sequence of the heparinase I cDNA insert was confirmed. F. heparinium heparinase I cDNA was cloned by the same approach using primers 5′-GCGGATCCCAGCAAAAAAAATCCGGT-3′, 5′-GCGAATTCCTATCTGGCAATTTCGCT-3′ and F. heparinium genomic DNA template, and inserted similarly into pGEX-2T. Plasmids were introduced into E. coli BL21(DE3) for expression. Single colonies were grown overnight and diluted 50 times in 600 ml of LB medium containing 100 μg/ml ampicillin. Heparinase I expression was induced with 0.5 mM isopropyl β-D-thiogalactopyranoside (IPTG; Gold Bio Technology, St. Louis, MO) for 3 hr at 30°C when cultures reached a density of 1.0 at A600. Bacteria were harvested by centrifugation at 5000 rpm for 5 min and the cell pellet stored at −20°C until use.
Purification of recombinant heparinase I
Frozen pellets from 300 ml culture were re-suspended in 30 ml cold buffer A (20 mM Tris-HCl, pH 7.0; 0.5% Triton X-100; 1 mM CaCl2, 0.5 M NaCl, 0.1 mM DTT, and 0.1 mM PMSF). The bacterial suspension was sonicated and then centrifuged at 16000 g at 4°C for 10 min. Soluble GST-heparinase I in the supernatant was captured with Glutathione-Sepharose 4B (GE HealthCare BioSciences Corp, Piscataway, NJ). Beads were washed several times with buffer A and then twice with buffer B (20 mM Tris-HCl, pH 7.0; 0.15 M NaCl; 1 mM CaCl2; and 0.1 mM DTT). Bound GST-Heparinase I was eluted with 10 mM GSH (reduced glutathione) in buffer B. Free GSH was removed by HiTrap Desalt column (GE HealthCare Bio-Sciences Corp, Piscataway, NJ). The GST fusion partner was removed from GST-heparinase I while it was immobilized on Gluthathione-Sepharose beads. After multiple washes with buffer B as described above, beads were incubated with 10 units thrombin (GE HealthCare BioSciences Corp, Piscataway, NJ) at ambient temperature for 4 hr to release heparinase I from the GST. The thrombin was removed by passage of the released product over Benzamidine-Sepharose (GE HealthCare Bio-Sciences Corp, Piscataway, NJ). If necessary, the heparinase I devoid of GST can be further purified by a Superose 12 column in buffer B. The size and purity of heparinase I was confirmed by 10% SDS-PAGE. Protein concentrations in the range of 25 to 400 μg/ml were determined by the Micro BCATM Protein Assay (Pierce, Rockford, IL) using BSA as a standard.
The binding of GST-heparinase I or heparinase I to heparin in the absence of catalytic activity was tested in the absence of calcium and DTT. Purified heparinase I was loaded on a 1 ml HiTrap Heparin-Sepharose column (GE HealthCare Bio-Sciences Corp, Piscataway, NJ) and eluted by linear gradient concentration of NaCl from 0 to 1 M generated by an AKTApurifier unit with binary pumps of solution A (10 mM Tris-HCl; pH 7.0 and 5 mM EDTA) and B (1 M NaCl in pump A buffer). Eluted protein was detected by UV absorbance at 280 nm.
Analysis of enzymatic activity
Heparinase I activity was determined by ability to cleave heparin that yields an unsaturated double bond at C4-C5 position at the non-reducing terminus of the resulting saccharide with a peak absorbance at about 230 nm (226 was used in the current study). The initial reaction rate was determined by the increase in absorbance for cleaved heparin for the first 1 min at 37°C with heparin concentrations ranging from 0.03125 to 1 mg/ml and 1 μg/ml GST-heparinase I in 1 ml buffer B. Unless otherwise noted, all enzyme assays consisted of buffer B containing 1 mM freshly prepared DTT (buffer B-DTT). Samples were boiled for 2 min and the absorbance was determined in an Ultraspec II 4050 spectrophotometer (LKB, Cambridge, England) at 226 nm. One unit of activity was defined as the amount of heparinase I that catalyses the formation of 1 μM of unsaturated uronic acid per minute at 37°C using heparin as substrate. Absorption units were converted to concentration of uronic acid liberated based on a molar absorption coefficient at 226 nm for the unsaturated C4-C5 bond of uronate of 4433 cm−1M−1. Kinetic parameters were derived from nonlinear least-squares regression analysis of initial velocity versus heparin concentration using the Michaelis-Menten equation. Substrate specificity was assessed at 0.5 mg/ml heparin, heparan sulfate or chondroitin sulfate. Effect of DTT and divalent cations at concentrations in the text were determined at 0.50 mg/ml heparin in buffer B-DTT missing DTT or CaCl2, respectively. For assessment of pH effects from 5.7 to 7.9, Tris-HCl in buffer B-DTT was replaced with phosphate buffer adjusted for the ratio of Na2HPO4 and NaH2PO4. Analyses were performed at least three times with independent preparations of GST-heparinase I. Unless otherwise indicated, a single representative experiment showing the mean of triplicate assays ± SD is presented in the text.
Neutralization of anticoagulant activity
Tritium-labeled dodecasaccharide with anticoagulant activity that had never been exposed to heparinase was prepared by modified method of partial nitrous acid deaminative cleavage of crude heparin [21]. Briefly, about 600 mg porcine intestinal mucosa heparin (170 USP unit/mg) (Sigma, St. Louis, MA) in 50 mM NaNO2 was acidified to pH 1.5 with 6 N HCl. After 6 min at room temperature, the solution was raised to pH 9 with 2 M Na2CO3. The reducing end aldehyde groups were reduced to alcohol by 10 mg [3H]-NaBH4 at room temperature for 3 hr. Free tritium was removed by adjustment to pH 4 with 4 M acetic acid followed by neutralization with 4 M NaOH. Oligosaccharides in the cleavage mixture were traced by tritium, separated by tandem Bio-Gel P-10 (Bio-Rad, Hercules, CA) XK26/100 columns (26 mm × 200 cm, Amersham Pharmacia Biotech, Piscataway, NJ) and then size purified as described [5].
Dodecasaccharide from the partial nitrous acid cleavage or synthetic anticoagulant pentasaccharide (a gift from Dr. R. Linhardt) was reacted with 1 μg/ml GST-heparinase I in buffer B-DTT at 37°C for the times indicated in the text. Reactions were stopped by boiling for 2 min. Oligosaccharide at 0.3 μM was added to antithrombin-mediated anti-Factor Xa assays as described [22].
Results
Comparison of sequences of B. thetaiotaomicron and F. heparinum heparinase I
A systematic search of sequence databanks for enzymes with homology to heparin and heparan sulfate lyases from F. heparinum revealed homologues in a variety of organisms whose genomes have recently become available. These included B. thetaiotaomicron and subsequently other organisms as Mycobacterium tuberculosis, Streptomyces coelicolor, Mycobacterium vanbaalenii and Cellulophaga agricola. Homologues with heparinase I as the initial prototype was chosen from the human and mouse gut symbiont B. thetaiotaomicron for initial studies because of the highly evolved and diverse inducible repertoire of polysaccharidases indicated by the genome sequence [17]. The coding sequence for B. thetaiotaomicron heparinase I predicts a 376 amino acid protein with a molecular mass of 42 kDa (Fig. 1). B. thetaiotaomicron heparinase I exhibits about a 70 percent homology to F. heparinum heparinase I. B. thetaiotaomicron heparinase I differs from F. heparinum heparinase I by the absence of an apparent signal peptide at the N-terminus predicated by the signal peptide predication program SignalP and an 8 amino acid insert in the putative heparin-binding domain [23] and calcium co-coordinating motif [24, 25] deduced from structure-function analysis of F. heparinum heparinase I. Cysteine 121 and histidine 189 (residues 135 and 203 in F. heparinum heparinase I) [26,27] that are thought to be essential catalytic site residues within the heparin-binding and calcium-binding pockets are conserved.
Fig. 1.

Amino acid sequences of B. thetaiotaomicron and F. heparinum heparinase I. Identical and similar residues are highlighted in black and gray, respectively. A closed rectangle indicates the putative signal peptide cleavage site at alanine-21 in F. heparinum heparinase I. Closed and open circles indicate conserved active site cysteine and histidine, respectively [26, 27]. The solid overline indicates the putative calcium-coordinating domain [25]. The putative heparin-binding domain is indicated by the hatched underline. FH = F. heparinum; BT = B. thetaiotaomicron.
High yield expression and recovery of soluble recombinant GST-heparinase I
Preliminary experiments to express and recover both F. heparinum and B. thetaiotaomicron heparinase I from the predicted mature N-terminus (Fig. 1) in a variety of E. coli expression hosts and conditions resulted in product directed to inclusion bodies as reported for F. heparinum heparinase I by others [14]. To potentially improve solubility, facilitate immobilization and affinity purification under mild non-denaturing conditions, both B. thetaiotaomicron and F. heparinum heparinase I were expressed as a fusion protein with glutathione-S-transferase (GST) at the N-terminus. Over 85 percent of the expected GST fusion product that could be captured by a GSH affinity column was in the soluble fraction of the bacterial lysate (Fig. 2). Elution from the GSH affinity columns with GSH yielded about 39 mg per liter per A600 unit of bacteria, which is about 30 mg per gram of dried bacteria. If necessary, the GST-heparinase I can be further purified by size-exclusion to remove minor products revealed by high load analysis on SDS-PAGE (Fig. 2A, lane 5). The GST portion (26 kDa) was removed by incubation of the immobilized GST-heparinase I with thrombin. The treatment released an apparently homogenous 42 kDa product consistent with the molecular mass of B. thetaiotaomicron heparinase I deduced from sequence and that of recombinant heparinase from F. heparinum [14]. In the absence of calcium and DTT that is essential for catalytic activity, B. thetaiotaomicron heparinase I bound transiently and weakly to immobilized heparin eluting at 0.25 to 0.3 M NaCl (Fig. 2B).
Fig. 2.

Expression and recovery of soluble recombinant B. thetaiotaomicron GST-heparinase I from E. coli and binding to immobilized heparin. (A) SDS-PAGE analysis at different steps. Std, protein standards; lanes 1, crude E. coli lysate; 2, soluble fraction of crude lysate; 3, GST-heparinase I captured on GSH-Sepharose beads; 4, material released from beads by GSH; 5, heparinase I devoid of GST after thrombin treatment. About 10 to 30 μg of the indicated samples prepared as described in Experimental Procedures was applied to each lane. Gels were stained with Coomassie Brilliant Blue R-250. (B) Binding of GST-heparinase I to immobilized heparin. B. thetaiotaomicron GST-heparinase I purified by GSH-Sepharose was applied and eluted by the indicated salt gradient as described in Experimental Procedures. The data is representative of 3 experiments from three independent preparations or more.
Parallel experiments indicated that solubility, yields and purity of F. heparinum heparinase I was similar to that of B. thetaiotaomicron heparinase I when expressed as a GST fusion protein by the same methods.
Co-factor requirements of B. thetaiotaomicron heparinase I activity
Activity of both the recombinant GST fusion and heparinase I after removal of the GST was monitored by increase in absorbance at 226 nm of the unsaturated double bond at the C4-C5 position at the non-reducing terminus that results from cleavage at the α-1,4 glycosidic linkage between disaccharide repeats of heparin. Conservation of the essential surface-exposed cysteine in the active site of F. heparinum heparinase I [27] suggested that heparinase I may have a requirement for the presence of reducing agent. Fig. 3A indicates that B. thetaiotaomicron GST-heparinase I activity is stimulated over 8 fold by the presence of an optimum 1 mM DTT and tolerates up to 4 mM before a detrimental effect was observed. The requirement could not be alleviated by increasing levels of substrate heparin indicating a near absolute requirement of the reducing environment for activity (Fig. 3B). Separate experiments indicated that F. heparinum heparinase I exhibited a similar requirement for reducing agent (results not shown). Both B. thetaiotaomicron (Fig. 4A) and F. heparinum (not shown) GST-heparinase I activity was optimum between pH 6.7 and 7.2.
Fig. 3.
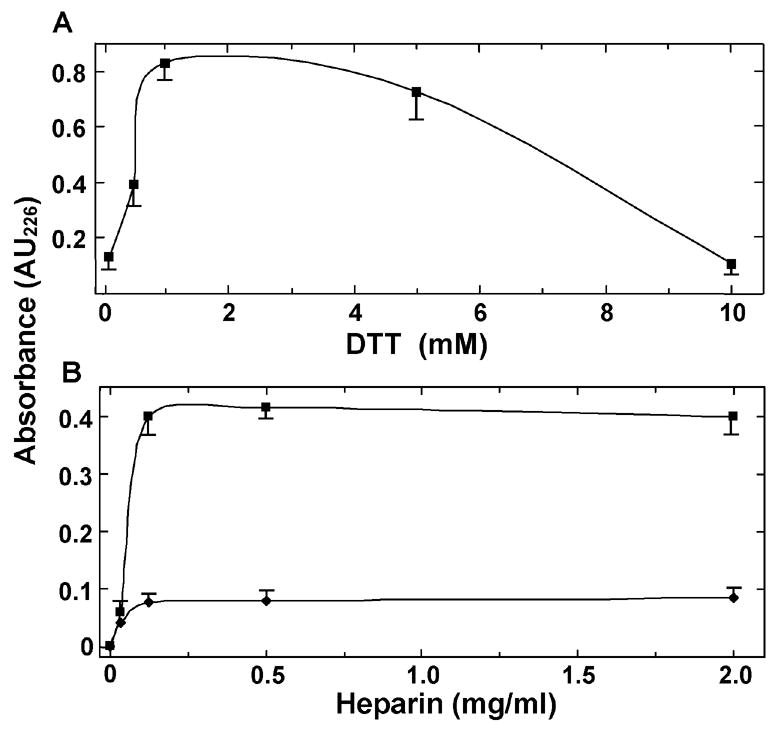
Dependence of heparinase I activity on reducing agent. Effect of the indicated concentrations of DTT on activity of B. thetaiotaomicron GST-heparinase I in the presence of 0.5 mg/ml heparin (A) and of 1 mM DTT at the indicated concentrations of heparin (B) was determined as described in Experimental Procedures. Squares indicate presence and diamonds indicate absence of DTT. Data are the mean ± S.E. (n=3) from three experiments performed with three independent enzyme and DTT preparations.
Fig. 4.

pH optimum and dependence on divalent cation. Effect of pH was determined as described in Experimental Procedures (A). The indicated divalent cations were added as the chloride salt at 1 mM (B). Data are the mean ± S.E. (n=3) from three experiments performed with three independent enzyme and reagents preparations.
Although B. thetaiotaomicron heparinase I exhibits an 8 amino acid insert in the domain relative to the enzyme from F. heparinum, the proposed Ca++-binding residues in F. heparinum heparinase I are conserved in the B. thetaiotaomicron enzyme (Fig. 1). An analysis of divalent ion requirements revealed that similar to results reported for native F. heparinum heparinase I [24, 25], activity of B. thetaiotaomicron GST-heparinase I was stimulated 7 fold by 1 mM Ca++ (Fig. 4B). Mg++ and Mn++ were much less effective at the same concentrations while Cu++ and Zn++ were ineffective. This indicates that similar to F. heparinum heparinase I [24, 25], B. thetaiotaomicron heparinase I also requires Ca++ as a co-factor for optimal enzymatic activity. This requirement differs from native B. stercoris heparinase I that was reported to prefer Co2+ as a co-factor [13].
Recombinant F. heparinum GST-heparinase I and both recombinant B. thetaiotaomicron and F. heparinum heparinase I free of the GST N-terminus exhibited similar co-factor requirements to those of B. thetaiotaomicron GST-heparinase I (not shown).
Substrate specificity and kinetic analysis
A screen of different glycosaminoglycan substrates for lyase activity of purified B. thetaiotaomicron GST-heparinase I revealed specificity for heparin (Fig. 5A) similar to that reported for native F. heparinum heparinase I [23, 28]. No significant activity was observed with heparan sulfate, chondroitin sulfate (Fig. 5A) or dermatan or keratan sulfate (not shown). Examination of initial rates of B. thetaiotaomicron GST-heparinase I activity over a range of substrate concentrations from 15.5 μg to 1 mg per ml (1.3 μM to 83.3 μM based on an average molecular mass of 12000 Da) revealed a simple hyperbolic response curve. The calculated specific activity of the purified enzyme was 164 units/mg. A double-reciprocal plot of the data conformed to a simple linear plot with a regression coefficient of 0.9967 (Fig. 5B). Nonlinear least-squares regression analysis of the data fit to the Michealis-Menten equation indicated an apparent Km of 2.3±0.41 μM and Vmax of 42.7±3.55 μmol/min (Fig. 5B inset). B. thetaiotaomicron heparinase I after enzymatic removal of the GST exhibited similar kinetics (data not shown). Recombinant F. heparinum GST-heparinase I exhibited a Km of 3.3±0.39 μM and Vmax of 40.8±3.86 μmol/min with a specific activity of 154 units/mg.
Fig. 5.

Substrate specificity and kinetic analysis of B. thetaiotaomicron GST-heparinase I. (A) Comparison of lyase activity on porcine intestinal mucosa heparin (diamonds), bovine cartilage heparan sulfate (squares) and chondroitin sulfate (triangles). (B) Kinetic analysis. The initial rates of B. thetaiotaomicron heparinase I were determined in the presence of various concentrations of heparin (average MW 12000 Da) as described in Experimental Procedures. Data from the inset (triplicates of each heparin concentration) was fit to the Michaelis-Menten equation and kinetic parameters determined by nonlinear least-squares regression analysis. A double reciprocal plot of the data is also shown. The indicate data is representative of three experiments with three independent heparinase I preparations.
Neutralization of heparin-dependent anticoagulant activity
The endolytic activity of heparinases is of utility for controlling the hemorrhagic complications of extracorporeal heparin administered as a clinical anticoagulant [29]. Inhibition by B. thetaiotaomicron GST-heparinase I of heparin-dependent anticoagulant activity was assessed by activity of Factor Xa in the presence of antithrombin (Fig. 6). Within 5 min, 1 μg B. thetaiotaomicron GST-heparinase I resulted in restoration of 78 percent of Factor Xa activity that was inhibited by 0.3 μM of a heparin dodecasaccharide in the presence of antithrombin. Near complete restoration was observed after 20 min. The GST-heparinase I also neutralized a chemically-defined antithrombin-binding pentasaccharide, but at a markedly reduced rate (Fig. 6). Separate experiments not shown here indicated that B. thetaiotaomicron heparinase I devoid of GST and both recombinant F. heparinum heparinase I and GST-heparinase I exhibited a similar neutralization activity.
Fig. 6.

Neutralization of anticoagulant activity by purified recombinant B. thetaiotaomicron GST-heparinase I. Purified heparin dodecasaccharide from partial nitrous acid cleavage (closed diamond) or synthetic anticoagulant pentasaccharide (closed triangle) was reacted with 1 μg/ml GST-heparinase I for the indicated times and remaining anticoagulant activity reported by anti-factor Xa activity. Data is expressed as the mean ± S.E. (n=3) from three experiments performed with three independent enzyme preparations.
Discussion
Ultrapure, reproducible and economical GAG polysaccharidases of defined specificity are essential for enzyme-based degradation strategies for structural determination and sequencing as well as clinical applications. Heparan sulfate and its highly sulfated prototype heparin are the most widely studied of the sulfated glycosaminoglycans [2]. An increasing amount of evidence suggests that oligosaccharide motifs within heparan sulfates have biospecific effects due to distribution of sidechains within the uronate-glucosamine disaccharide repeats that define the highly charged polyanion [3]. Therefore, general and group-specific lyases of high quality for heparin and heparan sulfate are of particular utility.
Bacterial heparinases cleaving at the glucosamine-uronate bond between disaccharides are of utility in reduction of long chain heparin and heparan sulfates for use as clinical anticoagulants [30], in de-heparinization to control anticoagulation, in treatments for other diseases as cancer and inflammation [31–33] and in generation of oligosaccharides [5, 34] for structural and functional studies [4, 5, 35, 36]. Native heparinases have been traditionally prepared by conventional multi-step purification from lysates of F. heparinum after induction by heparin that has suffered from variability, low yield and purity [8, 9]. In addition lysates of F. heparinum contain three isozymes of heparinases 1, 2 and 3, multiple group-specific heparan sulfate sulfatases and other GAG-degrading enzymes [9, 28, 37] that may contaminate individual lyase preparations.
The current study was precipitated by the failure of commercially available heparinases to reproducibly generate in sufficient yields oligosaccharides of 4 to 14 monosaccharide units containing FGF-specific motifs by controlled reduction of heparin (or heparan sulfate). Other experiments also indicated the lack of absolute specificity of commercially available group-specific sulfatases for heparan sulfate [38]. Here we report the heterologous expression and high yield recovery by one step affinity purification of soluble recombinant B. thetaiotaomicron heparinase I expressed in E. coli. The human and mouse gut symbiont B. thetaiotaomicron was chosen because of its highly evolved diverse repertoire of polysaccharidases indicated by the complete genome sequence [17]. E. coli are devoid of similar GAG polysaccharidases. Recombinant heparinase I from F. heparinum has been expressed in E. coli, however, the product was insoluble and required renaturation from inclusion bodies for activity [14]. Fusion with GST at the N-terminus resulted in highly soluble recombinant B. thetaiotaomicron or F. heparinum heparinase I in lysates of E. coli without detrimental effect on the enzymatic activity. The GST tag facilitated one-step purification to homogeneity and also solid phase immobilization. In contrast to commercial preparations of heparinase I, application of this material for generation of size-defined heparin oligosaccharides from high molecular weight heparin facilitated the reproducible isolation of a novel undersulfated octasaccharide with specific distribution of side groups required for high affinity binding of FGF7 to FGFR2IIIb and formation of an active signaling complex [5, 6].
Table 1 compares the preparation and properties of recombinant GST-heparinase I from B. thetaiotaomicron and F. heparinum to previous reports of native enzymes from F. heparinum and B. stercoris, or refolded recombinant enzyme from F. heparinum expressed in E. coli. The approach reported here represents a 8 to 100-fold improvement in yield from that reported for the recovery of purified native enzyme from F. heparinum [28] and B. stercoris [13] and an over 2-fold improvement over that reported for the refolded recombinant enzyme expressed in and recovered from E. coli [14]. The specific activity of recombinant B. thetaiotaomicron heparinase I was 2 to 10-fold that of previously reported recombinant and native F. heparinum enzymes [14, 28] and B. stercoris [13]. Notably, the purified recombinant B. thetaiotaomicron GST-heparinase I exhibited a higher apparent affinity (p<0.01) for heparin than those of F. heparinum prepared the same way and native B. stercoris heparinase I [13]. This improvement in affinity and catalytic activity may reflect the 8 amino acid insert in the putative heparin-binding domain [23] and calcium co-coordinating motif [24, 25]. Our method also provides cost-effective one-step purification. Otherwise the B. thetaiotaomicron enzyme exhibited similar properties to the F. heparinum enzyme in respect to calcium requirement, pH optimum and preference for more highly sulfated heparin over heparan sulfate. The simple kinetic response of the recombinant heparinase I to long chain heparin and the lack of high affinity for immobilized heparin in absence of catalytic activity indicate that the catalytic site is likely the single high affinity interaction site in the enzyme. We report here for the first time the requirement for reducing agent as DTT for heparinase I activity. This may indicate that the reduced ionizable sulfhydral group in residues cysteine-121 and cysteine-135 in the B. thetaiotaomicron and F. heparinum enzymes, respectively, may be essential for activity. Structural evidence indicates that cysteine-135 of F. heparinum heparinase I is in the active site of the enzyme [27].
Table 1.
Comparison of the efficiencies of expression, purification and activity of native and recombinant heparinase I from F. heparinum, B. stercoris and B. thetaiotaomicron
| Enzyme | Reference | Purified heparinase I (mg/g bacteria) | Yield (%) | SA (units/mg) | Km (μM) | Vmax (μmol/min) |
|---|---|---|---|---|---|---|
| nFH | [28] | NP | 0.8–1.2 | 27–130 | 0.3–42 | 19.7–219 |
| rFH | [14] | 12 | 43 | 117 | 3.9a | 37.4b |
| nBS | [13] | NP | 1.1 | 16 | 13 | 8.8 |
| rGST-BT | This work | 39 | 91 | 164 | 2.3±0.41 | 42.7±3.55 |
| rBT | This work | 22 | 82 | 228 | 2.5±0.34 | 41.8±3.73 |
| rGST-FH | This work | 34 | 88 | 154 | 3.3±0.39 | 40.8±3.86 |
| rFH | This work | 19 | 79 | 213 | 3.5±0.47 | 39.9±2.87 |
Value was derived from 47 μg/ml [14] with an average molecular weight of heparin of 12000 Da.
Value was derived from 2.37 milli-absorbance units per second [14] with the average molecular weight of heparin of 12000 Da and a molar absorbance coefficient at 232 nm for the unsaturated C4-C5 bond of uronate of 3800 cm−1.M−1.
SA = specific activity; n = native; r = recombinant; FH = F. heparinum; BS= B. stercoris; BT = B. thetaiotaomicron; NP = not reported.
Heparin is best known for the prophylaxis of thrombosis and blood clotting related disorders [4]. A variety of heparin derivatives have been developed as improved heparin-based anticoagulants, including several low molecular weight heparins (LMWHs) generated by heparinase I [30]. Oligosaccharides generated from heparin are the most widely available source of heparan sulfate oligosaccharides for structure-function studies in a number of biological systems [2]. Our method for efficient expression, recovery and optimization of enzymatic conditions of highly active recombinant heparinase I from B. thetaiotaomicron and other organisms in E. coli free of other heparin/heparan sulfate lyases may be useful in these applications. Heparinase I has also been applied for extracorporeal deheparinization of blood [29]. GST-heparinase I is amenable to solid phase immobilization via the GST portion without interference with its catalytic activity. This may be useful for extracorporeal flow-through columns or filters for heparinized blood, or for general removal of the enzyme from the soluble phase by affinity absorption. Utility of the immobilized GST-heparinase I of high specific activity free of other lyases has been demonstrated in identification and retention of motif specificity of rare FGF7-FGFR2IIIb specific octasaccharides in heparin [5, 6, 29]. Preliminary studies indicate that the described heterologous expression approach for recombinant N-terminal GST-fusion protein can be applied to a variety of glycosaminoglycan lyases including heparinase 3 and multiple heparin/heparan sulfate-specific sulfatases that are otherwise insoluble from both B. thetaiotaomicron and other novel bacterial genomes that have recently emerged (Luo and McKeehan, manuscript in preparation). Such a repertoire of immobilized ultrapure degrading enzymes with high specificity and specific activity are essential for miniaturization of solid state microsequencing of rare natural oligosaccharide motifs as the 7,8-S-OctaF7 [5, 6] involved in the FGF7-FGFR2IIIb signaling complex.
Acknowledgments
This work was supported by Public Health Service Grants DK35310 from the National Institute of Diabetes and Digestive Diseases and CA59971 from the National Cancer Institute.
Abbreviations used
- B. thetaiotaomicron
Bacteroides thetaiotaomicron
- DTT
dithiothreitol
- FGF
fibroblast growth factor
- FGFR
fibroblast growth factor receptor
- F. heparinum
Flavobacterium heparinum
- GAG
glycosaminoglycan
- GlcN
glycosamine residue that can be N-acetylated (Ac) or multiple sulfated (S)
- GST
glutathione-S-transferase
- HS
heparan sulfate
- UA
uronic acid residue including glucuronic acid (GlcA) and Iduronic acid (IdoA) that can be sulfated at 2-OH (2S)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sasisekharan R, Venkataraman G. Curr Opin Chem Biol. 2000;4:626–631. doi: 10.1016/s1367-5931(00)00145-9. [DOI] [PubMed] [Google Scholar]
- 2.Linhardt RJ. J Med Chem. 2003;46:2551–2564. doi: 10.1021/jm030176m. [DOI] [PubMed] [Google Scholar]
- 3.Capila I, Linhardt RJ. Angew Chem Int Ed Engl. 2002;41:391–412. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 4.Petitou M, Casu B, Lindahl U. Biochimie. 2003;85:83–89. doi: 10.1016/s0300-9084(03)00078-6. [DOI] [PubMed] [Google Scholar]
- 5.Luo Y, Ye S, Kan M, McKeehan WL. J Cell Biochem. 2006;97:1241–1258. doi: 10.1002/jcb.20724. [DOI] [PubMed] [Google Scholar]
- 6.Luo Y, Ye S, Kan M, McKeehan WL. J Biol Chem. 2006;281:21052–21061. doi: 10.1074/jbc.M601559200. [DOI] [PubMed] [Google Scholar]
- 7.Kreuger J, Jemth P, Sanders-Lindberg E, Eliahu L, Ron D, Basilico C, Salmivirta M, Lindahl U. Biochem J. 2005;389:145–150. doi: 10.1042/BJ20042129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galliher PM, Cooney CL, Langer R, Linhardt RJ. Appl Environ Microbiol. 1981;41:360–365. doi: 10.1128/aem.41.2.360-365.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ototani N, Kikuchi M, Yosizawa Z. Carbohydr Res. 1981;88:291–303. doi: 10.1016/s0008-6215(00)85542-1. [DOI] [PubMed] [Google Scholar]
- 10.Watanabe M, Tsuda H, Yamada S, Shibata Y, Nakamura T, Sugahara K. J Biochem (Tokyo) 1998;123:283–288. doi: 10.1093/oxfordjournals.jbchem.a021934. [DOI] [PubMed] [Google Scholar]
- 11.Yoshida E, Sakai K, Tokuyama S, Miyazono H, Maruyama H, Morikawa K, Yoshida K, Tahara Y. Biosci Biotechnol Biochem. 2002;66:1181–1184. doi: 10.1271/bbb.66.1181. [DOI] [PubMed] [Google Scholar]
- 12.Kim BT, Kim WS, Kim YS, Linhardt RJ, Kim DH. J Biochem (Tokyo) 2000;128:323–328. doi: 10.1093/oxfordjournals.jbchem.a022756. [DOI] [PubMed] [Google Scholar]
- 13.Kim WS, Kim BT, Kim DH, Kim YS. J Biochem Mol Biol. 2004;37:684–690. doi: 10.5483/bmbrep.2004.37.6.684. [DOI] [PubMed] [Google Scholar]
- 14.Ernst S, Venkataraman G, Winkler S, Godavarti R, Langer R, Cooney CL, Sasisekharan R. Biochem J. 1996;315( Pt 2):589–597. doi: 10.1042/bj3150589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoshida E, Arakawa S, Matsunaga T, Toriumi S, Tokuyama S, Morikawa K, Tahara Y. Biosci Biotechnol Biochem. 2002;66:1873–1879. doi: 10.1271/bbb.66.1873. [DOI] [PubMed] [Google Scholar]
- 16.Shaya D, Tocilj A, Li Y, Myette J, Venkataraman G, Sasisekharan R, Cygler M. J Biol Chem. 2006;281:15525–15535. doi: 10.1074/jbc.M512055200. [DOI] [PubMed] [Google Scholar]
- 17.Xu J, Bjursell MK, Himrod J, Deng S, Carmichael LK, Chiang HC, Hooper LV, Gordon JI. Science. 2003;299:2074–2076. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- 18.Hooper LV, Gordon JI. Glycobiology. 2001;11:1R–10R. doi: 10.1093/glycob/11.2.1r. [DOI] [PubMed] [Google Scholar]
- 19.Hooper LV, Midtvedt T, Gordon JI. Annu Rev Nutr. 2002;22:283–307. doi: 10.1146/annurev.nutr.22.011602.092259. [DOI] [PubMed] [Google Scholar]
- 20.Ausubel RBFM, Kingston RE, Moore DD, Smith JA, Seidman JG, Struhl K. Current Protocols in Molecular Biology. Greene Publishing Associates and Wiley-Interscience; New York: 1987. Preparation and Analysis of DNA. [Google Scholar]
- 21.Shively JE, Conrad HE. Biochemistry. 1976;15:3932–3942. doi: 10.1021/bi00663a005. [DOI] [PubMed] [Google Scholar]
- 22.Luo Y, Cho HH, McKeehan WL. J Pharm Sci. 2003;92:2117–2127. doi: 10.1002/jps.10472. [DOI] [PubMed] [Google Scholar]
- 23.Sasisekharan R, Venkataraman G, Godavarti R, Ernst S, Cooney CL, Langer R. J Biol Chem. 1996;271:3124–3131. doi: 10.1074/jbc.271.6.3124. [DOI] [PubMed] [Google Scholar]
- 24.Shriver Z, Liu D, Hu Y, Sasisekharan R. J Biol Chem. 1999;274:4082–4088. doi: 10.1074/jbc.274.7.4082. [DOI] [PubMed] [Google Scholar]
- 25.Liu D, Shriver Z, Godavarti R, Venkataraman G, Sasisekharan R. J Biol Chem. 1999;274:4089–4095. doi: 10.1074/jbc.274.7.4089. [DOI] [PubMed] [Google Scholar]
- 26.Godavarti R, Cooney CL, Langer R, Sasisekharan R. Biochemistry. 1996;35:6846–6852. doi: 10.1021/bi960356g. [DOI] [PubMed] [Google Scholar]
- 27.Sasisekharan R, Leckband D, Godavarti R, Venkataraman G, Cooney CL, Langer R. Biochemistry. 1995;34:14441–14448. doi: 10.1021/bi00044a022. [DOI] [PubMed] [Google Scholar]
- 28.Lohse DL, Linhardt RJ. J Biol Chem. 1992;267:24347–24355. [PubMed] [Google Scholar]
- 29.Heres EK, Horrow JC, Gravlee GP, Tardiff BE, Luber J, Jr, Schneider J, Barragry T, Broughton R. Anesth Analg. 2001;93:1446–1452. doi: 10.1097/00000539-200112000-00019. [DOI] [PubMed] [Google Scholar]
- 30.Mousa SA. Cardiovasc Drug Rev. 2002;20:199–216. doi: 10.1111/j.1527-3466.2002.tb00087.x. [DOI] [PubMed] [Google Scholar]
- 31.Sasisekharan R, Moses MA, Nugent MA, Cooney CL, Langer R. Proc Natl Acad Sci U S A. 1994;91:1524–1528. doi: 10.1073/pnas.91.4.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Silver PJ, Moreau JP, Denholm E, Lin YQ, Nguyen L, Bennett C, Recktenwald A, DeBlois D, Baker S, Ranger S. Eur J Pharmacol. 1998;351:79–83. doi: 10.1016/s0014-2999(98)00293-3. [DOI] [PubMed] [Google Scholar]
- 33.Temkin V, Aingorn H, Puxeddu I, Goldshmidt O, Zcharia E, Gleich GJ, Vlodavsky I, Levi-Schaffer F. J Allergy Clin Immunol. 2004;113:703–709. doi: 10.1016/j.jaci.2003.11.038. [DOI] [PubMed] [Google Scholar]
- 34.Ernst S, Langer R, Cooney CL, Sasisekharan R. Crit Rev Biochem Mol Biol. 1995;30:387–444. doi: 10.3109/10409239509083490. [DOI] [PubMed] [Google Scholar]
- 35.Turnbull JE. Methods Mol Biol. 2001;171:129–139. doi: 10.1385/1-59259-209-0:129. [DOI] [PubMed] [Google Scholar]
- 36.Venkataraman G, Shriver Z, Raman R, Sasisekharan R. Science. 1999;286:537–542. doi: 10.1126/science.286.5439.537. [DOI] [PubMed] [Google Scholar]
- 37.Yang VC, Linhardt RJ, Bernstein H, Cooney CL, Langer R. J Biol Chem. 1985;260:1849–1857. [PubMed] [Google Scholar]
- 38.Turnbull JE, Hopwood JJ, Gallagher JT. Proc Natl Acad Sci U S A. 1999;96:2698–2703. doi: 10.1073/pnas.96.6.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]