

Abstract
A previous analysis of serum insulin-like growth factor I (IGF-I) levels in a mouse population (n = 961) derived from a cross of (BALB/cJ × C57BL/6J) F1 females and (C3H/HeJ × DBA/2J) F1 males documented quantitative trait loci (QTL) on chromosomes 1, 10, and 17. We employed a newly developed, random walk-based method to search for three- and four-way allelic combinations that might influence IGF-I levels through nonadditive (conditional or epistatic) interactions among 185 genotyped biallelic loci and with significance defined by experiment-wide permutation (P < 0.05). We documented a three-locus combination in which an epistatic interaction between QTL on paternal-derived chromosomes 5 and 18 had an opposite effect on the phenotype based on the allele inherited at a third locus on maternal-derived chromosome 17. The search also revealed three four-locus combinations that influence IGF-I levels through nonadditive genetic interactions. In two cases, the four-allele combinations were associated with animals having high levels of IGF-I, and, in the third case, a four-allele combination was associated with animals having low IGF-I levels. The multiple-locus genome scan algorithm revealed new IGF-I QTL on chromosomes 2, 4, 5, 7, 8, and 12 that had not been detected in the single-locus genome search and showed that levels of this hormone can be regulated by complex, nonadditive interactions among multiple loci. The analysis method can detect multilocus interactions in a genome scan experiment and may provide new ways to explore the genetic architecture of complex physiological phenotypes.
Keywords: quantitative trait loci, epistasis, gene interactions
MANY TRAITS OF INTEREST to biological and medical science are determined by the interaction of multiple factors. The genetic and environmental variation among individuals in a population results in a broadened phenotype range and, in many cases, obscures the relationships connecting the causative factors. Although the paradigm of “one gene-one phenotype” has been successfully exploited in experimental biology, the multiple genes that underlie interindividual variation in most traits remain unresolved. Consequently, a significant challenge remains for biomedical research to develop the tools for the deconstruction and understanding of the genetic network, or architecture, of complex traits (16, 21, 25, 33, 41).
In experimental organisms, the individual causative genes that underlie a phenotype can be identified through conventional linkage studies, targeted mutational analysis, or quantitative trait locus (QTL) analysis (16, 20). After identification, the single genes can be shown, by experiment, to interact within a more complex functional pathway or network. Alternatively, interconnected genetic factors can be identified by searches for second-site modifier genes. In model organisms such as yeast, Caenorhabditis elegans, and Drosophila melanogaster, the modifier gene strategy has been exceptionally valuable (1, 35). The mapping and molecular cloning of second-site modifiers of specific phenotypes has also been successful in the mouse, to a lesser degree (9, 11).
Practical and theoretical challenges face genetic searches for the interacting causative factors in mammalian complex traits. First, the genome is large, in both the number of nucleotides and number of genes, and comprehensive genome-wide genotyping remains costly (36). Second, the numbers of individuals that can be obtained and phenotyped are often constrained. Breeding and housing are limiting in laboratory animal studies, whereas, in human studies, challenges are faced in the assessment of large populations or families. Finally, traditional statistical analysis methods are primarily suited to finding single effects rather than components in complex networks (13, 14, 25). In human populations, the reality of genetic and environmental heterogeneity and the lack of models for their interactions remain a major obstacle to identifying genetic causes of complex phenotypes (6, 8).
Interactions among genes remain largely unexplored, yet may contribute significantly to the heritable component of phenotype variation (5, 10, 14). The detection of multigene interaction effects is difficult, for several reasons: 1) the low frequency of individuals in any two (or more) gene combination class necessitates a large sample population; 2) multiple models of dominance and recessive interactions must be accommodated; 3) the appropriate measurement scale for the phenotype may be unknown; and 4) the multiple statistical tests that must be employed result in high threshold levels for statistical significance, leaving only extremely strong interactions for detection. To date, several analytical strategies have been developed for identifying pairs of interacting loci in mammalian systems (4, 38), including ANOVA analysis for all possible two-locus effects. Detection of interactions among greater than two loci are typically based on previously identified single, or paired, loci (12, 19, 32, 38).
We reported an experimental and analytical strategy that can simultaneously identify additive and nonadditive interactions of up to four separate loci. The experimental system measures phenotype and whole genome genotype in a genetically heterogeneous mouse population. The mouse model allows for a controlled environment, uniform breeding structure, and the availability of extensive genetic resources as well as the measurement of tissue-specific phenotypes. The genetic variability of our study population is constructed in a reproducible manner, because the source alleles are derived from four common inbred strains: BALB/cJ (C), C57BL/6J (B6), C3H/HeJ (C3), and DBA/2J (D2). The four progenitor strains are crossed as (C × B6) F1 females and (C3 × D2) F1 males, yielding a heterogeneous study population, called UM-HET3. All members of the UM-HET3 study population are genetically equivalent to full siblings derived from heterozygous parents, with known linkage phase. Each individual in the population represents a unique combination of alleles from the inbred grandparents, yet at any locus there are only two possible maternal genetic contributions (C or B6) and two possible paternal alleles (C3 or D2). In the UM-HET3 population, the four genotype classes for a locus (C/C3, C/D2, B6/C3, and B6/D2) appear in approximately equal numbers. No segregation distortion has been detected in any genotyped region of the genome. Genome-wide genotyping is readily performed on all members of the population, and the breeding structure allows replicate populations (18, 22, 26, 27, 37).
In a previous report (18) searching for QTL influencing serum insulin-like growth factor (IGF)-I levels (18), a conventional ANOVA-based genome scan was performed in a population of genetically heterogeneous mice (UM-HET3) measured for serum IGF-I and genotyped across the genome. The genome scan identified three QTL using an experiment-wide significance criterion of P < 0.05 calculated with reference to 1,000 permutation distributions of the data. The QTL were linked to genotyped loci on maternal-derived chromosomes 1 and 17 (D1Mit206 and D17Mit185) and a locus on paternal-derived chromosome 10 (D10Mit230).
A newly developed multilocus search algorithm described by Hanlon and Lorenz (17) uses a series of random walks to efficiently explore the large set of gene interactions and phenotype distributions obtained in the population. The random walk process is reiterated, with each subsequent search using information obtained in earlier walks. Allelic combinations that contribute to the phenotype variance in the population are identified and assessed for significance by genome-wide permutation testing. We report here on the use of the Hanlon-Lorenz (H-L) random walk method to search for loci that regulate the level of the serum hormone IGF-I. The search algorithm reveals loci on six additional chromosomes that had not previously been shown, in a conventional single-locus search, to influence IGF-I levels. Polymorphisms at these loci are involved in a three-way interaction and three different four-way interactions, each revealing nonadditive interactions among three or more alleles. Thus the search method provides new insights into the complexity of the genetic networks that influence endocrine levels in a segregating mouse population.
MATERIALS AND METHODS
Mice and Husbandry
Briefly, the mouse population UM-HET3 was derived from a four-way cross among four inbred strains: C, B6, C3, and D2. The experimental animals (n = 961) were the progeny of (C × B6) F1 females and (C3 × D2) F1 males. The F1 breeding animals were purchased from Jackson Laboratories (Bar Harbor, ME). Animals were housed segregated by sex in a single suite of specific pathogen-free rooms and were exposed to identical environmental conditions (12:12-h light-dark cycle, 23°C). Mice were given ad libitum access to water and laboratory mouse chow. The cages were covered with microisolator tops to control the spread of infectious agents. Sentinel mice were tested every 3 mo to verify the pathogen-free status. All such tests were negative throughout the course of the study. The work was approved by the Animal Care and Use Committee of the University of Michigan.
Genotyping Assays
Genomic DNA was prepared from 1-cm sections of the tail from 4-wk-old animals and tested for concentration, ability to sustain PCR amplification under standard conditions, and electrophoretic size distribution. Genomic DNA was PCR amplified and detected using an ALFexpress automated sequence analyzer (GE Healthcare, Piscataway, NJ); the details of this genotyping method have been described previously (22). Oligodeoxynucleotide primer pairs were purchased from MWG Biotech (High Point, NC). In total, 185 biallelic informative loci were examined using 99 simple sequence-repeat marker PCRs. Of the 99 reactions, 86 loci were informative for both the maternal- and paternal-derived alleles and 13 loci were only informative for maternal- or paternal-derived alleles. The selection of genetic loci has been described previously in detail (37). Loci were chosen distributed across the genome at 15- to 20-cM intervals, yielding a genotyping density with ~95% of the autosomal genome within 30 cM of a marker.
For clarity, in the UM-HET3 population, maternal-derived alleles at a genomic locus (i.e., C or B6) were given the appended designation “m” and paternal-derived alleles (i.e., C3 or D2) were appended with “p.” The chromosomal localization and order of markers were calculated using the Map Manager QTX program package (http://www.mapmanager.org/mmQTX.html). Genomic sequence locations were based on the Mouse Genome Sequencing Consortium Version 3 Whole Genome Shotgun Assembly (http://www.ncbi.nih.gov/genome/guide/mouse/).
Phenotype Measurement of IGF-I
IGF-I levels were evaluated by immunoassay of serum drawn from each animal at 4 mo of age, as previously described (18). Briefly, serum samples were taken by tail venipuncture between 7 and 11 AM and stored at −70°C. Levels of IGF-I were quantified by a double-antibody radioimmunoassay kit (Diagnostic Systems Laboratories; Webster, TX) run at one-quarter volume according to the manufacturer’s instructions. All samples were assayed in duplicate for each animal.
Genome-Wide Search Method for Detecting QTL Pairs and Triplets
A two-step process was used to identify statistically significant single- and multiple-locus effects that contributed to the phenotype of IGF-I levels. The first step used a random walk-based algorithm method (17) with permutation testing to identify QTL interaction effects and to give an estimate of statistical significance. As a second step, we applied ANOVA tests to the locus groups to determine which triplets and quadruplets showed additive effects and which showed evidence for nonadditive or conditional effects. We describe these steps separately below.
Step 1: application of the Hanlon-Lorenz random walk method with permutation testing
This method is a computational approach to determining the genetic architecture of a single quantitative trait. A technical description of the algorithm, including analyses of instructive synthetic data sets, is available (17). The following description provides a less technical summary of the approach. The method proceeds by a series of random walks, each of which searches for a close approximation of the phenotype as a sum of genetic effects, including either additive or nonadditive effects of multiple loci. Incorporated into the random walk process are constraints that prevent the algorithm from overfitting the data from the sample. The algorithm can be viewed as a walk through a “space” consisting of sets of genetic loci and the phenotypic trait value associated with each locus set. Within this space, there is a notion of adjacency. Namely, two sets of genetic effects are “adjacent” if the two effects differ by only one allele at one genotyped locus. The score of a set of genetic effects is its residual sum of squares for a least-squares fit to the phenotype. The random walk that underlies the algorithms proceeds as follows: from a particular set, the algorithm chooses an adjacent set and moves to that set if the score of the new set is better than that of the current set. In part, the power of the algorithm comes from the way in which potential steps are chosen. The algorithm “learns” how to make that choice wisely based on earlier experiences in the random walk process. As steps are repeated in these walks, loci whose choice tends to bring about a large improvement in score are given greater probability to be chosen at later steps.
In our analysis, each random walk consisted of 5,000 steps, and we conducted 400 such random walks. The output of each random walk was a locus or combination of loci that was associated with a relatively extreme value of the phenotype. Because the walks were random, outputs of successive runs were seldom identical, but if a particular locus combination showed up a large fraction of the time in the output of these runs, this showed that the algorithm had difficulty finding an approximation of the phenotype that did not include that particular combination of loci. Hence, the locus combination was likely to represent a genetic effect that modulated the phenotype.
Each locus or locus combination received a score corresponding to the number of times, out of 400 walks, that the combination effect was included in the terminal state of the walk. Significance levels were estimated by comparing the score for each combination with an empirical distribution produced by permutations of the same data set. To produce this distribution, we held the genotype database constant and permuted the values of the phenotype across individuals and applied the algorithm to this permuted dataset, again taking 400 walks and recording the score of the locus combinations that appeared most frequently at the terminus of the walks. A second permuted dataset was then examined, and so forth, until 1,000 such datasets had been evaluated. Significance levels for the scores observed from the actual, nonpermuted data set were then assigned with respect to the highest scores seen in the series of 1,000 permuted datasets. Locus combinations can consist of a single locus, pairs of loci, triplets, or quadruplets, and the significance tests for each combination were evaluated with respect to the scores of similar order in the set of permuted datasets. This procedure resulted in a listing of loci and locus combinations for which the experiment-wide P values, evaluated by permutation, were P < 0.05.
Step 2: post hoc ANOVA
Pairs and triplets of genetic loci found by the genome-wide search process to influence traits of interest were then evaluated further by ANOVA methods. For pairs of loci, the equation took the following form:
where T indicates the level of the trait, A and B indicate the effects of the two marker loci of interest, and AB indicates the interaction term. The significance of the AB interaction term (at P < 0.05) was interpreted as evidence that the effects of the A locus allele were conditional on the allele inherited at locus B and vice versa. Similarly, tests of three-way interactions were evaluated by ANOVA using models of the following form:
where C is the effect of a third locus of interest. In this case, significance of any two-way interaction term (AB, AC, or BC) was interpreted as an indication of epistatic interaction between the pair of alleles involved, and significance of the ABC term was taken as evidence that the strength of the epistatic interaction depended on the allele inherited at the third locus. Analyses of four-locus interactions were conducted using a similar strategy with terms for each of the four individual loci, the six pairs, the four triplets, and the single quadruplet.
RESULTS
In the initial description of the H-L QTL search algorithm (17), the method was tested extensively on simulated genome scan data to explore the power of the method to uncover novel multilocus interactions. The simulated analyses clearly showed the computational efficiency of the method; however, the simulated data did not provide insight into the extent to which multiple genes might interact to modulate measured traits in real populations. We therefore applied this new search algorithm to measures of serum IGF-I levels in a population of 961 UM-HET3 mice that had been genotyped at 185 biallelic loci across the genome.
The goal of the present analysis was to determine whether the genome-wide search method could detect groups of QTL that modulate the levels of serum IGF-I with additive and nonadditive interaction effects. In the H-L method, QTL interaction effects are not searched for by evaluating the effects of each locus after conditioning on previously identified loci. Rather, the single- and multiple-locus models are examined concurrently. As a consequence, it is possible to detect nonadditive (e.g., conditional) interactions among loci. The conditional interactions, in particular, are of biological interest, because they may directly expose interacting (epistatic) factors in quantitative phenotypes. Experiment-wide significance was determined by a permutation analysis in which the complete random walk search process was repeated with 1,000 permuted shuffles of the phenotype and genotype data, yielding an empirical null distribution. For those multigene combinations meeting the empirical significance level, we performed a post hoc analysis to determine whether the loci interacted additively or nonadditively.
Three-Way Interaction
One three-way interaction was detected (experiment-wide P = 0.01) using the H-L random walk approach, involving alleles linked to D5Mit95p, D17Mit185m, and D18Mit55p. The three-locus combination was then further evaluated by post hoc ANOVA using models that incorporated all three single alleles, all three two-way combinations, and the three-way interaction as independent predictors. We note that of the three loci, only the one linked to D17Mit185m had been detected previously using the conventional single-locus ANOVA method (18). Figure 1A shows the interaction between D5Mit95p and D18Mit55p for the half of the mice that inherit the D17Mit185m C allele. For this subset of mice, the quantitative value and direction of the effect of the D5Mit95p allele depended on the allele of D18Mit55p received. For mice with the C3 allele of D18Mit55p, high levels of IGF-I were associated with the C3 allele of D5Mit95p. In contrast, for mice with the D2 allele at D18Mit55p, high levels of IGF-I were associated with the alternative D2 allele of D5Mit95p. Thus the effects of D5Mit95p and D18Mit55p cannot be explained by an additive interaction. The loci have their significant effect on IGF-I conditionally, an effect likely to reflect an epistatic interaction between the loci. The display of Fig. 1 allows a straightforward visualization of additive and nonadditive effects. Had the effects been additive, the two lines in Fig. 1A would have been approximately parallel. The lines are, in fact, not parallel, a graphical indication of the conditional interaction between the effects of D5Mit95p and D18Mit55p alleles.
Fig. 1.
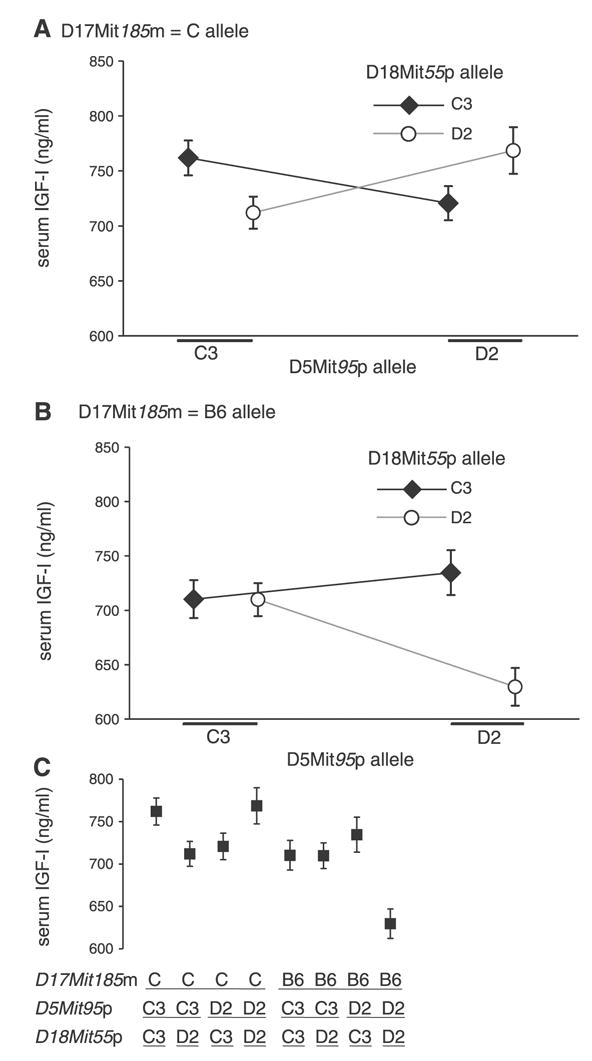
A conditional quantitative trait loci (QTL) triplet combination observed for serum insulin-like growth factor (IGF)-I levels in the UM-HET3 population. A: interactions between alleles at D5Mit95p and D18Mit55p for the mice with the BALB/cJ (C) allele at D17Mit185m. B: similar display for mice with the C57BL/6J (B6) allele at D17Mit185m. C: values for the means of the 8 genotypic groups defined by alleles at the three loci. The nonadditive interactions among the three QTL are observed as nonparallel lines (A and B) and by the outlying position of the single three-allele group D17Mit185m, allele B6; D5Mit95p, allele DBA/2J (D2); and D18Mit55p, allele D2, in C. C3, C3H/HeJ allele. Values are means ± SE. Each of the 8 genotypic groups contained between 97 and 120 mice.
Similarly, Fig. 1B documents conditional effects between the two interacting loci (D5Mit95p and D18Mit55p) for the other half of the population of mice, i.e., those that inherit the B6 allele at D17Mit185m. In this case, too, the lines are not parallel, suggesting a conditional interaction between D5Mit95p and D18Mit55p. In contrast to Fig. 1A, however, the mice that have inherited the B6 allele at D17Mit185m showed a qualitatively different interaction: in this group, the line slopes upward for the C3 allele at D18Mit55p and slope downward for the D2 allele at D18Mit55p. For example, mice that inherited the D2 allele at both D5Mit95p and D18Mit55p showed the highest levels of IGF-I when they received the C allele at D17Mit185m but showed the lowest IGF-I levels when they received the B6 allele at D17Mit185m. Thus the interaction between D5Mit95p and D18Mit55p depends on the allele of D17Mit185m present.
These graphical impressions were evaluated by a formal, post hoc ANOVA calculation. This analysis showed a significant two-way interaction between D17Mit185m and D18Mit55p (P = 0.047). In addition, the three-way interaction was significant at P = 0.0004, confirming the impression that the degree of interaction between D17Mit185m and D18Mit55p depended on the allele inherited at D5Mit95p.
The nonadditive interaction among the three loci can be clearly seen when all eight possible three-locus combinations are examined together in Fig. 1C. Seven of the eight subgroups determined by three-locus genotype have group means between 710 and 770 ng/ml. In contrast, the single three-locus allele combination, D17Mit185m (B6), D5Mit95p (D2), and D18Mit55p (D2), showed a group mean of 630 ng/ml.
Four-Way Interactions
The genome-wide search identified significant four-locus effects on the IGF-I phenotype in the population. Three quadruplets were found: 1) D8Mit51m, D2Mit285p, D5Mit205p, and D10Mit230p; 2) D4Mit170m, D12Mit167m, D7Mit91p, and D10Mit230p; and 3) D8Mit51m, D17Mit185m, D5Mit95p, and D7Mit25p. In each case, the experiment-wide significance was obtained by permutation testing, giving P = 0.002, 0.028, and 0.002, respectively. Post hoc analysis identified the alleles of each locus that mediate the phenotype effect. Figure 2, A–C, provides a straightforward view of the interactions of the loci and is similar in concept to Fig. 1C, although in this case the mice are divided into 16 subpopulations depending of the combination of alleles inherited at the four loci indicated.
Fig. 2.
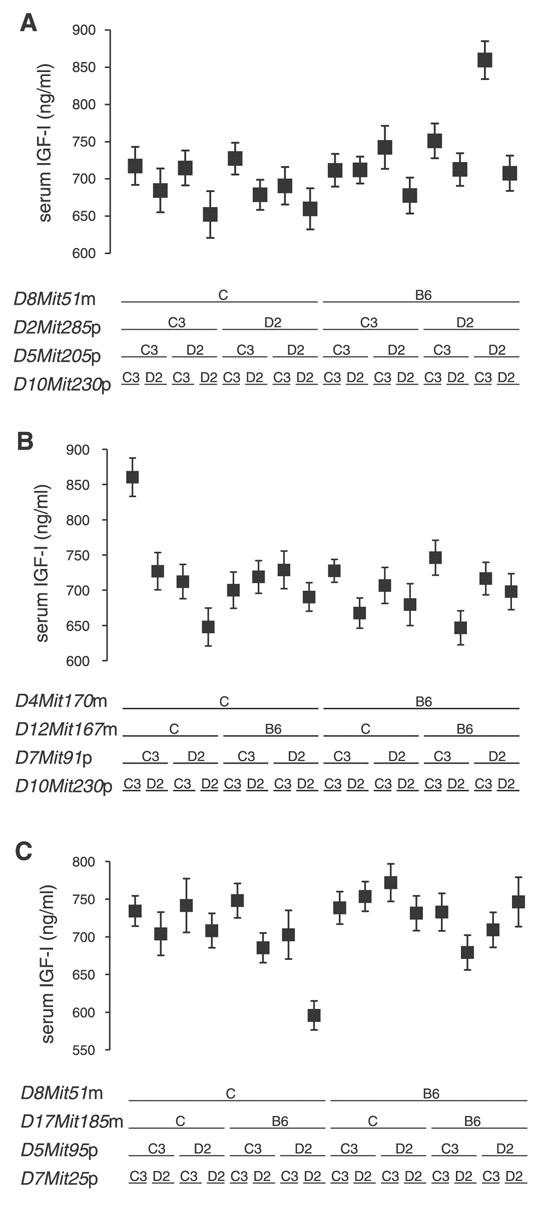
Three QTL quadruplet combinations observed for serum IGF-I levels. A–C: phenotype mean ± SE values for the 16 groups defined by the two alleles at four loci. In each case, a single four-locus combination yielded an extreme phenotype group mean, whereas the 15 alternative combinations were clustered near the overall population mean value (718 ng/ml). A: four-locus effect of the QTL D8Mit51m, D2Mit285p, D5Mit205p, and D10Mit230p. The experiment-wide significance for detection of the quadruplet was P = 0.002. Each of the 16 genotypic groups contained between 36 and 63 mice; the total number of mice with complete genotypes was 805. B: four-locus effect of the QTL D4Mit170m, D12Mit167m, D7Mit91p, and D10Mit230p. The experiment-wide significance for detection of the quadruplet was P = 0.028. Each of the 16 genotypic groups contained between 40 and 64 mice; the total number of mice with complete genotypes was 822. C: four-locus effect of the QTL D8Mit51m, D17Mit185m, D5Mit95p, and D7Mit25p. The experiment-wide P value for detection of the quadruplet was P = 0.002. Each of the 16 genotypic groups contained between 41 and 68 mice; the total number of mice with complete genotypes was 856.
In each case, the nonadditive interactions among the QTL can be observed as the outlying position of a single four-locus combination. Two of the quadruplets were associated with four-locus groups with exceptionally high IGF-I serum values (Fig. 2, A and B) and one four-locus group associated with exceptionally low IGF-I serum values (Fig. 2C). The two quadruplets with a high-phenotype group shared one QTL, D10Mit230p, which had been identified in the previous single-locus genome search (18) but did not share any other loci in common. The low-IGF-I phenotype quadruplet (Fig. 2C) shared three chromosomal regions with the high-phenotype quadruplets, paternal-derived chromosomes 5 and 7, and maternal-derived chromosome 8. The QTL locus on chromosome 8 (D8Mit51m) had the B6-derived allele associated with the high-phenotype group and the alternative allele (C) associated with the low-genotype quadruplet. Similarly, the identified loci on chromosome 7, D7Mit91p and D7Mit25p, had alternative alleles associated with high- and low-phenotype groups. The two chromosome 7 loci were adjacent in the mapping panel (9 cM) and may reflect the effect of a single common underlying QTL. In contrast, the identified chromosome 5 loci, D5Mit205p and D5Mit95p, had the D2 allele associated with both the high- and low-phenotype group. The two loci were adjacent in the genotyping panel (17 cM) and may represent detection of the same effective QTL, but having opposite phenotypic effects in different genetic interactions.
The phenotype distributions of the individual animals identified as members of the four-locus groups are given in Fig. 3 and are shown relative to the population distribution as a whole. The mean serum IGF-I value for the entire UM-HET3 population was 718 ng/ml, whereas the two high-phenotype quadruplet groups had mean level values of 859 and 860 ng/ml (Fig. 3A, triangles). The low-phenotype quadruplet group had a mean level of 595 ng/ml (Fig. 3B, triangles). Individual animals with any specific four-locus genotype should represent ∼1/16 (6.25%) of the population with genotype information available at all 4 loci. This expectation was confirmed for each quadruplet: 1) D8Mit51m, D2Mit285p, D5Mit205p, and D10Mit230p, 56 of 805 animals; 2) D4Mit170m, D12Mit167m, D7Mit91p, and D10Mit230p, 58 of 822 animals; and 3) D8Mit51m, D17Mit185m, D5Mit95p, and D7Mit25p, 54 of 856 animals.
Fig. 3.
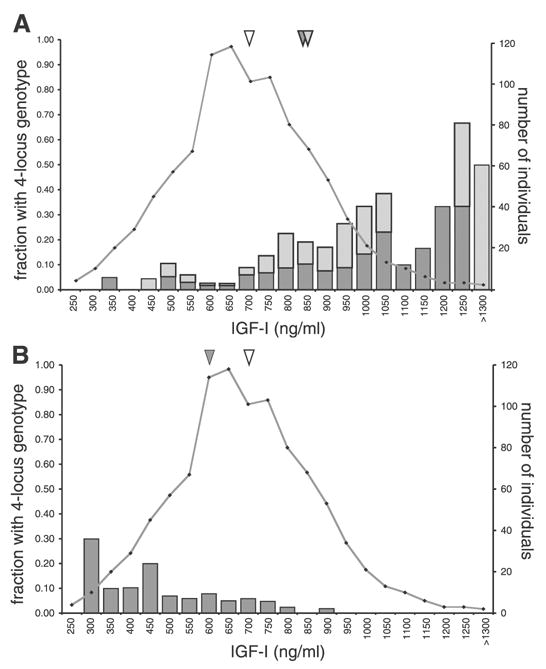
Serum IGF-I phenotype distributions of individuals with the detected four-locus genotype combinations. In both A and B, the UM-HET3 population distribution is given as the number of individuals within each IGF-I phenotype interval (connected points, right vertical scale). The full population distribution represents 961 animals, with a mean value of 718 ng/ml (open inverted triangle). A: individuals with the two four-locus genotype combinations [D8Mit51m, allele B6; D2Mit285p, allele D2; D5Mit205p, allele D2; and D10Mit230p, allele C3 (dark shaded bars); and D4Mit170m, allele C; D12Mit167m, allele C; D7Mit91p, allele C3; and D10Mit230p, allele C3 (light shaded bars)] are presented for each IGF-I value interval. The distribution is given as the fraction of individuals with the four-locus combination in each phenotype interval (left vertical scale). The genotype distributions represent 56 animals (mean 859 ng/ml, dark shaded inverted triangle) and 58 animals (mean 860 ng/ml, light shaded inverted triangle), respectively. B: individuals with the four-locus genotype combination (D8Mit51m, allele C; D17Mit185m, allele B6; D5Mit95p, allele D2; and D7Mit25p, allele D2) are presented for each IGF-I value interval. The distribution is given as the fraction of individuals with the four-locus combination in each phenotype interval (shaded bars, left vertical scale). Fifty-four animals were identified with the four-locus genotype, with a mean value of 595 ng/ml (shaded inverted triangle).
The individuals with extreme high or low phenotypes were strongly represented by members of the quadruplet genotype groups (Fig. 3, bars). For example, of the eight individuals having serum IGF-I values of >1,200 ng/ml, four have the D8Mit51m, D2Mit285p, D5Mit205p, and D10Mit230p quadruplet genotype or the D4Mit170m, D12Mit167m, D7Mit91p, and D10Mit230p quadruplet genotype. This represents 50% of the animals over 1,200 ng/ml, rather than the simple expectation of ∼13%. Finally, of the 56 mice with the quadruplet allele combination D8Mit51m, D2Mit285p, D5Mit205p, and D10Mit230p, 9 also shared the allele pattern of the other quadruplet associated with high IGF-I levels: D4Mit170m, D12Mit167m, D7Mit91p, and D10Mit230p. The phenotype values for these nine animals yielded an average of 866 ng/ml, which was only slightly greater than the means for the two underlying quadruplets (859 and 860 ng/ml).
DISCUSSION
The analytical strategy presented here provides a framework for uncovering multilocus additive and nonadditive genetic interactions in an experimentally accessible mammalian genetic system. Although conventional QTL genome scan approaches have been successful in providing locations for loci that influence traits of interest in a variety of segregating populations, the computational challenges of identifying three-and four-locus interactive effects have proven more difficult. It is straightforward to evaluate interactions among known loci that affect a specific trait, to determine whether the allelic effects at these loci are additive or conditional. A related strategy begins with a single locus with significant effects and then scans among remaining loci for those that add a significant degree of correlation with the trait of interest. Such post hoc analyses, however, do not consider interactions among loci whose individual effects were too small to reach the chosen significance criterion of the initial scan. In addition, interaction effects of the sort shown in Figs. 1 and 2 can result in a situation in which no one locus has a large net effect on the phenotype, yet each is strongly influential when considered in combination with the others.
The mouse UM-HET3 genetic system has several advantages as a test bed for analytical algorithms. First, phenotypes and genotypes were measured on a large number of animals (in the present study, up to 961 animals); therefore, for a single biallelic locus, ∼480 siblings could be be assigned to each allelic class. Under conditions of complete genotype data, each of the 4 two-locus groups contained ≈240 siblings, the 8 three-locus groups ≈120 siblings, and the 16 four-locus groups ≈60 siblings. This was confirmed in practice, because none of the four-locus genotype groups shown in Fig. 2 had <36 members. As a consequence, statistically useful subpopulations for each of the multiple-locus genotype combinations were available. Second, knowledge of the maternal-derived and paternal-derived alleles at the genotyped loci yielded unambiguous, phase-known linkage information. Genetic recombination between genotyped loci and QTL was limited to a single meiotic generation. Third, unlike recombinant inbred strains, the study animals were not the product of extensive inbreeding; consequently, genotypes that may be infertile have not been lost by selection. Fourth, the extensive heterozygosity of the UM-HET3 animals is likely to provide reduced phenotype variability arising from environmental and stochastic processes (30, 40). In contrast, breeding F2 populations from inbred lines can yield extensive regions of homozygosity in each individual. Finallly, the population could be readily expanded or duplicated for future confirmation studies or for refinement of the QTL map locations.
To evaluate the recently developed H-L random walk approach as a means of finding examples of conditional gene effects (i.e., genetic effects whose strength or direction depended on the allele inherited at one or more other loci), we applied the algorithm to a phenotype data set of quantitative serum IGF-I levels. The experiment-wide significance level was set at P < 0.05 to focus attention on triplets and quadruplets of loci whose combined effects were unlikely to reflect chance alone. The strength of each association between trait and locus combination was evaluated with respect to a set of 1,000 permuted datasets to provide an experiment-wide confidence level adjusted for the multiple sets of loci tested simultaneously. The search method successfully identified locus triplets and quadruplets in this experimental population but did not provide information as to which combination(s) of alleles showed conditional effects. Post hoc ANOVA calculations were used to show significant two-factor interactions in all QTL triplets and quadruplets observed in this study.
The genome search method, as presently implemented, does not discriminate between loci whose individual effects are strictly additive and those in which the effect of alleles at one locus is conditional on the presence of a given allele at additional loci (i.e., nonadditive or epistatic effects). From this perspective, it is notable that all of the triplet and quadruplets combinations identified in this study were found to show evidence for conditional interlocus effects. For example, the interaction illustrated in Fig. 1 is one in which the strength and direction of the two-way interaction between D5Mit95p and D18Mit55p is itself dependent on the allele inherited at D17Mit185m. These findings suggest that interaction effects of this kind may be quite common, at least among loci segregating within common laboratory mouse populations.
Conventional ANOVA searches for interactions between pairs of loci, although computationally demanding, are technically feasible, using searches across all possible pairs of loci to produce an empirical distribution of the test statistic across permuted datasets. Such an approach using a group of 200 genetic markers would involve systematic evaluation of 200 × 199/2 locus pairs (about 20,000 combinations) for association with the trait of interest, accompanied by a similar evaluation of 20,000 pairs in each of the permuted dataset analyses. In our UM-HET3 population data, using Solaris 2.9 on a Sun Blade 1000 computer system, analyses of this kind required ∼15 h of computer time for each trait under study and are thus impractical for more than a small number of traits. Searches for three-way or higher-order interactions, moreover, would require >200-fold more computational time and are thus undesirable. The search method examined in this report, in contrast, can generate lists of multilocus interactions, complete with permutations, in a comparatively short time. Each series of 400 random walks takes ∼5 min, and thus an analysis of a complete set of 1,000 permuted data sets takes ∼4 days for a single trait. Notably, the approach yields information on QTL for the high-order multiples simultaneously. In this study, we limited the analysis to interactions of four loci or less, so that genotype groups remained of reasonable size. However, the computational method can search for higher-order interactions of five or more loci, with no change in the process algorithm.
Additional analyses using other segregating populations and other sets of traits will be needed to determine how often the H-L genome search approach succeeds in detecting examples of conditional interaction of three way or higher orders. In a simulation study (17), synthetic datasets were constructed, each containing a specific number of individuals with unique combinations of biallelic markers and a phenotype determined as an additive combination of single-gene and multiple-gene effects plus a noise component. The algorithm was then run multiple times to determine its effectiveness at uncovering the genetic architecture of the phenotype (i.e., at determining the single gene and epistatic effects from which the phenotype was constructed). Parameters of the system, such as the number of steps in each walk, the number of walks conducted, the number of loci and the relative strength of the epistatic effects, and the number of individuals tested, were then varied to determine their effects on the detection power of the algorithm. The simulation system also allowed for variation of the genetic effects relative to the level of noise, corresponding to a mixture of uncertainty in the phenotype measure and the unknown effects of genes that influence the phenotype but are not modeled in the simulation. The H-L simulation model did not explore alternative genotype-phenotype data structures, such as restricted genotyping to individuals at phenotypic extremes.
Extensive tests were run on a simulation dataset with 140 biallelic marker loci and walks of length 5,000, roughly identical to the parameter values used in the experimental results reported here. First, the ability to detect a locus-pair with moderate effects on the phenotype was strongly influenced by population size. Second, increasing the random walk length improved sensitivity, particularly at high experimental noise levels. Finallly, the simulations (and the present experimental study) showed that multiple-locus effects are detected by the H-L algorithm even when the effects of individual loci were not detected separately.
Interactions between loci having statistically significant marginal effects have been reported by other investigators (2, 3, 19, 25, 31). Our analysis algorithm, however, also detects QTL combinations that are likely to have remained hidden if only statistically significant marginal effects were examined (5, 10). Previous work on defining the multiple genetic components in complex phenotypes, including additive and nonadditive effects, has been extensively reviewed (8, 15, 34). Genetic interaction effects have been examined with statistical methods and include combinatorial partitioning methods (28), orthogonal variance components models (7, 23), and physiological epistasis parameterization (5, 29). However, the challenge of extensive computation and determination of significance thresholds have generally limited searches for interactions to studies of loci each of which achieved significance on their own or, alternately, to detection of loci whose effects are significant after conditioning for previously detected loci. A growing set of analysis methods can model both main and pairwise epistatic effects independently, using Bayesian methods for reduction of the complex, multilocus model space (39) or multiple interval mapping strategies (24).
In this study, significant nonadditive (or epistatic) QTL interactions were readily identified, suggesting that such complex interactions may underlie many complex traits. In each of the four-locus interactions detected, 1 of the 16 genotype classes showed an extreme phenotype mean (Fig. 2). In each case, the alternative 15 genotype classes showed mean values clustered near the overall population mean. Our results also indicated that nonadditive, multilocus interaction effects may underlie a significant proportion of the individuals that display extreme phenotypes with a population. As shown in Fig. 3, the group of individuals with extreme phenotypes consisted to a large extent of mice with the detected quadruplet allele combinations. The extreme phenotype groups highlighted in Fig. 3 contained few individuals (n < 10) and will require confirmation within a replicate experiment to assess biological significance.
Although alternative explanations may be invoked, it is likely that the nonadditive locus interactions represent genes whose products interact in the same molecular pathway to elaborate the measured phenotype. In the future, given rapid improvements in genomic analysis, it may be possible to identify the individual genes (and variants) that encode these QTL, leading to understanding of the network of interacting factors that yield the phenotype. Conventional genetic search strategies for polymorphic loci influencing quantitative traits that rely on independent marginal effects are likely to underestimate the complexity of the genetic architecture. Clearly, complex gene-by-gene interactions are occurring in mammalian systems and can be detected by experimental systems and algorithms of sufficient power.
Acknowledgments
We thank Emily Gray and Dana Knutzen for the assistance with genotyping; Steve Pinkosky, Gretchen Buehner, and Maggie Vergera for the technical and husbandry assistance; and Shu Chen for the assistance with the statistical analyses.
Footnotes
GRANTS
This work was supported by National Institutes of Health (NIH) Grants AG-16699, AG-11687, AR-46024, AG-08808, and HG-01984 and National Science Foundation Grants DMS-9977371 and DMS-0073785. J. M. Harper was supported by NIH Training Grant T32-AG-00114.
References
- 1.Avery L, Wasserman S. Ordering gene function: the interpretation of epistasis in regulatory hierarchies. Trends Genet. 1992;8:312–316. doi: 10.1016/0168-9525(92)90263-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badano JL, Kim JC, Hoskins BE, Lewis RA, Ansley SJ, Cutler DJ, Castellan C, Beales PL, Leroux MR, Katsanis N. Heterozygous mutations in BBS1, BBS2 and BBS6 have a potential epistatic effect on Bardet-Biedl patients with two mutations at a second BBS locus. Hum Mol Genet. 2003;12:1651–1659. doi: 10.1093/hmg/ddg188. [DOI] [PubMed] [Google Scholar]
- 3.Blangero J, Williams JT, Almasy L. Quantitative trait locus mapping using human pedigrees. Hum Biol. 2000;72:35–62. [PubMed] [Google Scholar]
- 4.Carlborg O, Andersson L, Kinghorn B. The use of a genetic algorithm for simultaneous mapping of multiple interacting quantitative trait loci. Genetics. 2000;155:2003–2010. doi: 10.1093/genetics/155.4.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheverud JM, Routman EJ. Epistasis and its contribution to genetic variance components. Genetics. 1995;139:1455–1461. doi: 10.1093/genetics/139.3.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 7.Cockerham CC, Zeng ZB. Design III with marker loci. Genetics. 1996;143:1437–1456. doi: 10.1093/genetics/143.3.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cordell HJ. Epistasis: what it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum Mol Genet. 2002;11:2463–2468. doi: 10.1093/hmg/11.20.2463. [DOI] [PubMed] [Google Scholar]
- 9.Cormier RT, Hong KH, Halberg RB, Hawkins TL, Richardson P, Mulherkar R, Dove WF, Lander ES. Secretory phospholipase Pla2g2a confers resistance to intestinal tumorigenesis. Nat Genet. 1997;17:88–91. doi: 10.1038/ng0997-88. [DOI] [PubMed] [Google Scholar]
- 10.Culverhouse R, Suarez BK, Lin J, Reich T. A perspective on epistasis: limits of models displaying no main effect. Am J Hum Genet. 2002;70:461–471. doi: 10.1086/338759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dietrich WF, Lander ES, Smith JS, Moser AR, Gould KA, Luongo C, Borenstein N, Dove WF. Genetic identification of Mom-1, a major modifier locus affecting Min-induced intestinal neoplasia in the mouse. Cell. 1993;75:631–639. doi: 10.1016/0092-8674(93)90484-8. [DOI] [PubMed] [Google Scholar]
- 12.Doerge RW. Mapping and analysis of quantitative trait loci in experimental populations. Nat Rev Genet. 2002;3:43–52. doi: 10.1038/nrg703. [DOI] [PubMed] [Google Scholar]
- 13.Falconer DS, Mackay TFC. Introduction to Quantitative Genetics. Essex, UK: Addison Wesley Longman; 1996. [Google Scholar]
- 14.Frankel WN, Schork NJ. Who’s afraid of epistasis? Nat Genet. 1996;14:371–373. doi: 10.1038/ng1296-371. [DOI] [PubMed] [Google Scholar]
- 15.Franklin I, Lewontin RC. Is the gene the unit of selection? Genetics. 1970;65:707–734. doi: 10.1093/genetics/65.4.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glazier AM, Nadeau JH, Aitman TJ. Finding genes that underlie complex traits. Science. 2002;298:2345–2349. doi: 10.1126/science.1076641. [DOI] [PubMed] [Google Scholar]
- 17.Hanlon P, Lorenz WA. A computational method to detect epistatic effects contributing to a quantitative trait. J Theor Biol. 2005;235:350–364. doi: 10.1016/j.jtbi.2005.01.015. [DOI] [PubMed] [Google Scholar]
- 18.Harper JM, Galecki AT, Burke DT, Pinkosky SL, Miller RA. Quantitative trait loci for insulin-like growth factor-I, leptin, thyroxine, and corticosterone in genetically heterogeneous mice. Physiol Genomics. 2003;15:44–51. doi: 10.1152/physiolgenomics.00063.2003. [DOI] [PubMed] [Google Scholar]
- 19.Hoh J, Ott J. Mathematical multi-locus approaches to localizing complex human trait genes. Nat Rev Genet. 2003;4:701–709. doi: 10.1038/nrg1155. [DOI] [PubMed] [Google Scholar]
- 20.Hrabe de Angelis MH, Flaswinkel H, Fuchs H, Rathkolb B, Soewarto D, Marschall S, Heffner S, Pargent W, Wuensch K, Jung M, Reis A, Richter T, Alessandrini F, Jakob T, Fuchs E, Kolb H, Kremmer E, Schaeble K, Rollinski B, Roscher A, Peters C, Meitinger T, Strom T, Steckler T, Holsboer F, Klopstock T, Gekeler F, Schindewolf C, Jung T, Avraham K, Behrendt H, Ring J, Zimmer A, Schughart K, Pfeffer K, Wolf E, Balling R. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat Genet. 2000;25:444–447. doi: 10.1038/78146. [DOI] [PubMed] [Google Scholar]
- 21.Ideker T, Galitski T, Hood L. A new approach to decoding life: systems biology. Annu Rev Genomics Hum Genet. 2001;2:343–372. doi: 10.1146/annurev.genom.2.1.343. [DOI] [PubMed] [Google Scholar]
- 22.Jackson AU, Fornes A, Galecki AT, Miller RA, Burke DT. Multiple-trait quantitative trait loci analysis using a large mouse sibship. Genetics. 1999;151:785–795. doi: 10.1093/genetics/151.2.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kao CH, Zeng ZB. Modeling epistasis of quantitative trait loci using Cockerham’s model. Genetics. 2002;160:1243–1261. doi: 10.1093/genetics/160.3.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kao CH, Zeng ZB, Teasdale RD. Multiple interval mapping for quantitative trait loci. Genetics. 1999;152:1203–1216. doi: 10.1093/genetics/152.3.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mackay TFC. The genetic architecture of quantitative traits. Annu Rev Genet. 2001;35:303–339. doi: 10.1146/annurev.genet.35.102401.090633. [DOI] [PubMed] [Google Scholar]
- 26.Miller RA, Chrisp C, Jackson AU, Burke DT. Marker loci associated with lifespan in genetically heterogeneous mice. J Gerontol Med Sci. 1998;53A:M257–M263. doi: 10.1093/gerona/53a.4.m257. [DOI] [PubMed] [Google Scholar]
- 27.Miller RA, Jackson AU, Galecki AT, Burke DT. Genetic polymorphisms in mouse genes regulating age-sensitive and age-stable T cell subsets in mice. Genes Immunity. 2003;4:30–39. doi: 10.1038/sj.gene.6363895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nelson MR, Kardia SL, Ferrell RE, Sing CF. A combinatorial partitioning method to identify multilocus genotypic partitions that predict quantitative trait variation. Genome Res. 2000;11:458–470. doi: 10.1101/gr.172901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peripato AC, De Brito RA, Vaughn TT, Pletscher LS, Matioli SR, Cheverud JM. Quantitative trait loci for maternal performance for offspring survival in mice. Genetics. 2002;162:1341–1353. doi: 10.1093/genetics/162.3.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phelan JP, Austad SN. Selecting animal models of human aging: inbred strains often exhibit less biological uniformity than F1 hybrids. J Gerontol Biol Sci. 1994;49:B1–B11. doi: 10.1093/geronj/49.1.b1. [DOI] [PubMed] [Google Scholar]
- 31.Routman EJ, Cheverud JM. Gene effects on a quantitative trait: two-locus epistatic effects measured at microsatellite markers and at estimated QTL. Evolution. 1997;51:1654–1662. doi: 10.1111/j.1558-5646.1997.tb01488.x. [DOI] [PubMed] [Google Scholar]
- 32.Sen S, Churchill GA. A statistical framework for quantitative trait mapping. Genetics. 2001;159:371–387. doi: 10.1093/genetics/159.1.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sing CF, Stengard JH, Kardia SL. Genes, environment, and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2003;23:1190–1196. doi: 10.1161/01.ATV.0000075081.51227.86. [DOI] [PubMed] [Google Scholar]
- 34.Templeton AR. Epistasis and Complex Traits, in Epistasis and the Evolutionary Process. New York: Oxford Univ. Press; 2000. pp. 41–57. [Google Scholar]
- 35.Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, Chen Y, Cheng X, Chua G, Friesen H, Goldberg DS, Haynes J, Humphries C, He G, Hussein S, Ke L, Krogan N, Li Z, Levinson JN, Lu H, Menard P, Munyana C, Parsons AB, Ryan O, Tonikian R, Roberts T, Sdicu AM, Shapiro J, Sheikh B, Suter B, Wong SL, Zhang LV, Zhu H, Burd CG, Munro S, Sander C, Rine J, Greenblatt J, Peter M, Bretscher A, Bell G, Roth FP, Brown GW, Andrews B, Bussey H, Boone C. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 36.Tsuchihashi Z, Dracopoli NC. Progress in high throughput SNP genotyping methods. Pharmacogenomics J. 2002;2:103–110. doi: 10.1038/sj.tpj.6500094. [DOI] [PubMed] [Google Scholar]
- 37.Volkman SK, Galecki AT, Burke DT, Paczas MR, Moalli MR, Miller RA, Goldstein SA. Quantitative trait loci for femoral size and shape in a genetically heterogeneous mouse population. J Bone Miner Res. 2003;18:1497–1505. doi: 10.1359/jbmr.2003.18.8.1497. [DOI] [PubMed] [Google Scholar]
- 38.Yi N, Xu S, Allison DB. Bayesian model choice and search strategies for mapping interacting quantitative trait loci. Genetics. 2003;165:867–883. doi: 10.1093/genetics/165.2.867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yi N, Yandell BS, Churchill GA, Allison DB, Eisen EJ, Pomp D. Bayesian model selection for genome-wide epistatic quantitative trait loci analysis. Genetics. 2005;170:1333–1344. doi: 10.1534/genetics.104.040386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu SB, Li JX, Xu CG, Tan YF, Gao YJ, Li XH, Zhang Q, Maroof MA. Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid. Proc Natl Acad Sci USA. 1997;94:9226–9231. doi: 10.1073/pnas.94.17.9226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zwick ME, Cutler DJ, Chakravarti A. Patterns of genetic variation in Mendelian and complex traits. Annu Rev Genomics Hum Genet. 2000;1:387–407. doi: 10.1146/annurev.genom.1.1.387. [DOI] [PubMed] [Google Scholar]