

Abstract
Diabetes-associated vascular complications are collectively the major clinical problems facing patients with diabetes and lead to the considerably higher mortality rate than that of the general population. People with diabetes have a much higher incidence of coronary artery disease as well as peripheral vascular diseases in part because of accelerated atherogenesis. Despite the introduction of new therapies, it has not been possible to effectively reduce the high cardiovascular morbidity and mortality associated with diabetes. Of additional concern is the recognition by the World Health Organization that we are facing a global epidemic of type 2 diabetes. Endothelial dysfunction is an early indicator of cardiovascular disease, including that seen in type 2 diabetes. A healthy endothelium, as defined in terms of the vasodilator/blood flow response to an endothelium-dependent vasodilator, is an important indicator of cardiovascular health and, therefore, a goal for corrective interventions. In this review we explore the cellular basis for endothelial dysfunction in an attempt to identify appropriate new targets and strategies for the treatment of diabetes. In addition, we consider the question of biomarkers for vascular disease and evaluate their usefulness for the early detection of and their role as contributors to vascular dysfunction.
Keywords: endothelium, diabetes, nitric oxide (NO), C-reactive protein (CRP), asymmetric dimethylarginine (ADMA), endothelial progenitor cells (EPCs)
Diabetes – the problem
“Diabetes is a growing and massive silent epidemic that has the potential to cripple health services in all parts of the world”, quote from Dr Robert Beaglehole, Director Chronic Diseases and Health Promotion, speaking at the launch of Diabetes Action Now, 5 May, 2004. Diabetes Action Now is a joint program of the World Health Organization and the International Diabetes Federation; further information can be found on the World Health Organization website.
The incidence of type 2 diabetes (characterized initially by insulin resistance, whereas type 1 is characterized by insulin deficiency) has already reached epidemic proportions. To date, epidemics have been associated with communicable diseases; however, type 2 diabetes is the only non-infectious condition that has been classified as an epidemic. According to the World Health Organization, there are some 150 million diagnosed cases of type 2 diabetes in the world, and current estimates predict that this will increase to 300 million by 2025. An unofficial estimate, given the suspected high number of undiagnosed cases, boosts the current number of people with diabetes to 1.2 billion. Regardless of the accuracy of these estimates, there are severe global and societal implications resulting from this emerging epidemic in diabetes (Zimmet et al 2001). The increase in the incidence of type 2 diabetes is mainly the result of genetic predisposition and a higher standard of living that has changed eating habits and led to a more sedentary lifestyle. The link between genetically determined susceptibility and “lifestyle” is particularly intriguing. The incidence of type 2 diabetes in North Americans and individuals of European origin has risen from 2% in 1981 to 8% in 2002; the increase in “westernized” indigenous people is even more dramatic, from essentially 0% to as high as 50% in some societies (Diamond 2003). This increase in the incidence of type 2 diabetes has already placed a huge burden on healthcare, and the early detection, treatment, and preferably prevention of type 2 diabetes are significant challenges for this century. With an alarming increase in the number of overweight adults and children in the Western world, the need for better treatment regimens and early diagnosis of vascular disease has never been greater.
Another concern for the global impact of type 2 diabetes is an apparent association between the use of certain drugs for the treatment of AIDS and an increase in the incidence of diabetes in people with AIDS. Concurrent with the improvement in life expectancy and quality of life of AIDS patients by the increased availability of HIV-1 protease inhibitors and highly active antiretroviral therapy (HAART) is evidence that long-term HAART leads to insulin resistance, impaired glucose tolerance, and also dyslipidemia and thus an increase in the prevalence of diabetes and cardiovascular disease in people with AIDS (Mondal et al 2001; Tershakovec et al 2004). It was reported in the 2004 Report on the Global AIDS Epidemic (UNAIDS 2004) that there are approximately 38 (range 34.6–42.3) million people worldwide living with HIV and this number is increasing by about 3 million a year – an additional frightening statistic to the worldwide problem of diabetes.
Diabetes – a vascular disease
Diabetes-associated vascular complications are a major clinical problem, and diabetic patients have a mortality rate 3–4 times that of the general population (Haffner et al 1998). Diabetic patients have a 2- to 4-fold increase in the incidence of coronary artery disease (CAD) and 10-fold increase in peripheral vascular diseases (Laakso 1999), in part because of accelerated atherogenesis.
Cardiovascular disease and the endothelium
Communication between endothelial and vascular smooth muscle cells and vice versa plays an important role in the regulation of vascular tone. Impaired endothelial function, characterized by nitric oxide (NO) scavenging due to increased superoxide () production, is a hallmark of vascular disease states. Endothelial dysfunction can result in a number of pathophysiological complications such as enhanced inflammatory responses, ie, increased leukocyteendothelial cell adhesions (Pieper 1999), possibly due to the increased expression of adhesion molecules (Li and Forstermann 2000), enhanced activation of platelets and clotting factors resulting in a procoagulant state, and ultimately structural changes resulting from defective modulation of vascular growth and remodeling by the endothelium (for review see McLeod et al 1995). Up to 75% of patients with diabetes die from vascular disease and dysfunction of the endothelium. Endothelial dysfunction plays an early (possibly preceding frank symptoms of diabetes itself) and prominent role in this process (Andrews et al 1987; Cohen et al 1988). In addition, vascular dysfunction can continue to progress after a hyperglycemic insult despite the subsequent maintenance of normal blood glucose levels (so-called hyperglycemic memory) (Engerman and Kern 1987; Fioretto et al 1998).
Endothelial dysfunction can be defined as a reduced endothelium-dependent vasodilator response to acetylcholine1 and, although the endothelium has many additional functions beyond the control of vascular tone, this reduced response to acetylcholine serves as an important indicator of vascular dysfunction (De Vriese et al 2000; Pannirselvam et al 2003; Triggle et al 2003; Andrews et al in press).
Endothelial function can be readily measured using isolated blood vessels and studying the ability of endothelium-dependent vasodilators, such as acetylcholine, to relax vessels with pre-existing tone either under “organ bath” (for macrovessels) or “wire myograph” (for microvessels) conditions (Mulvany and Halpern 1977). Endothelial function can also be assessed in microvessels, under pressurized conditions wherein either the effects of endothelium-dependent vasodilator-mediated or flow-mediated dilations can be assessed (for review see Davis and Hill 1999). Endothelium function can be assessed using in vitro techniques with isolated vessels from humans; however, the data generated, by virtue of the limitations in the sources of the tissues, often reflect function in diseased vessels only, and a reference to a normal non-diseased control is usually absent. Thus in humans both invasive and non-invasive in vivo techniques are employed and include direct intracoronary measurements, positron emission tomography, impedance plethysmography, brachial ultrasound, and, for venous studies, dorsal hand vein compliance. The utility of these techniques has been reviewed by Anderson (1999), with intracoronary measures providing the gold standard. In humans the status of the endothelium (Anderson 1999; Verma and Anderson 2002; Verma, Szmitko, and Anderson 2004) is an important surrogate marker of atherosclerosis activity. In addition, attenuation of endothelium-dependent vasodilation is associated with an increased risk of cardiovascular complications during long-term follow-up (Halcox et al 2002). It is therefore not surprising that endothelium dysfunction is seen as a key risk factor and the endothelium as a target for correctional intervention (De Vriese et al 2000; Pannirselvam et al 2003; Triggle et al 2003; Aird 2004; Andrews et al in press), although outcomes may be complicated by the increasing evidence that the endothelium represents a heterogeneous population of cells with, probably, considerable vessel specialization (Andrews et al in press).
Several clinical studies have specifically addressed the question as to whether improving glycemic control in diabetes also improves endothelial function, and the results and significance of some of these studies are discussed below.
Mather et al (2001) measured blood flow responses to intra-arterial administration of the endothelium-dependent (acetylcholine) and endothelium-independent (sodium nitroprusside) agonists as well as nitrate-independent (verapamil) vasodilators using forearm plethysmography. Data from their study provided the first in vivo evidence that, in type 2 diabetic patients, metformin therapy for 12 weeks significantly improved acetylcholine-stimulated, endothelium-dependent vasodilation compared with placebo. The improvement in endothelial function probably reflects the beneficial influence of metformin on insulin resistance and glucose-lowering and antioxidant effects, and possibly favorable effects on lipids and free fatty acids as well as direct vasodilating actions. Peroxisome proliferator-activated receptor-gamma (PPARγ) agonists, notably the thiazolidinediones (glitazones), such as rosiglitazone, are being used as insulin-sensitizers in the treatment of type 2 diabetes and, in some studies, endothelial function has been assessed. Thus, Caballero et al (2003) used high-resolution ultrasound images to determine the effects of troglitazone treatment on endothelium-dependent flow-mediated dilation and endothelium-independent nitroglycerin-induced dilation in the brachial artery, as well as laser Doppler perfusion imaging to measure acetylcholine- and sodium nitroprusside-mediated vasodilation in the forearm skin. Their results indicate that 12 weeks of troglitazone treatment improved endothelial function in the macrocirculation of patients with recently diagnosed type 2 diabetes, and improvement was strongly associated with the improvement of fasting plasma insulin. Sidhu et al (2004) studied another glitazone, rosiglitazone, and demonstrated reduced markers of inflammation and endothelial activation. Despite rosiglitazone treatment significantly reducing C-reactive protein (CRP; the significance of which is discussed later in this review), the circulating endothelial cell marker von Willebrand factor, insulin resistance index, and mean low-density lipoprotein (LDL), compared with placebo, there was no improvement in flow-mediated dilation in diabetic patients (Sidhu et al 2004). We await additional results from other clinical trials that are investigating the effects of glitazones on the endothelium and cardiovascular morbidity (eg, the GATE – Glitazones And The Endothelium – Study).
Exercise alone can improve endothelial function in patients with stable CAD. A study by Hambrecht et al (2003) demonstrated that exercise training in stable CAD improved agonist-mediated, acetylcholine-mediated, endothelium-dependent vasodilation as assessed invasively in the left internal mammary artery before and after 4 weeks of exercise training on bicycle and row ergometers. The change in acetylcholine-induced vasodilation was linked to a shear stress-induced/Akt (protein kinase B)-dependent phosphorylation of endothelial nitric oxide synthase (eNOS) on Ser1177; shear stress activation of eNOS had been previously reported by Dimmeler et al (1999).
Yeh (2004) credits Dr William Osler with stating in his Principles and Practice of Medicine: “Longevity is a vascular question. A man is only as old as his arteries”. Today we might change that to “a man is only as old as his endothelium”. We therefore need to investigate and assess techniques whereby the health of the endothelium can be accurately monitored.
Endothelium-derived vasoactive factors
Vascular tone is intricately regulated by a number of vasorelaxant and contractile factors synthesized and released from endothelial cells: vasodilators (NO, prostacyclin, endothelium-derived hyperpolarizing factor, bradykinin, adrenomedullin, C-natriuretic peptide) and vasoconstrictors (endothelin-1, angiotensin-II, thromboxane A2, prostaglandins, hydrogen peroxide (H2O2), reactive oxygen species (ROS)) (McGuire et al 2001; Triggle et al 2003). The L-arginine-NO pathway is, however, thought to be the most important vasodilator source. In addition to its function as a vasodilator, NO, released from endothelial cells, reduces vascular permeability, is a potent inhibitor of platelet aggregation and adhesion to the vascular wall, controls the expression of proteins involved in atheroma formation, and decreases the expression of the chemoattractant protein monocyte chemoattractant protein (commonly known as MCP-1) and of surface adhesion molecules such as CD11/CD18, P-selectin, vascular cell adhesion molecule-1 (VCAM-1), and intercellular adhesion molecule (ICAM-1). In addition, NO decreases oxidation of LDL and inhibits proliferation of vascular smooth muscle cells (Li and Forstermann 2000), producing antiatherogenic effects.
NO is synthesized by a family of oxidoreductases, called NO synthases (NOS), that bear close sequence homology to cytochrome P450 (CYP) enzymes. Three isoforms are known to exist: endothelial (eNOS), inducible (iNOS), and neuronal (nNOS). In the endothelium eNOS is activated by a number of agonists acting on G protein-coupled receptors and by physical stimuli such as shear stress and changes in oxygen levels that lead to the activation of stretch-operated nonselective cation channels. NO is cleaved from L-arginine and this process requires a number of cofactors: nicotinamide adenine dinucleotide phosphate (NADPH), flavin adenine dinucleotide (FAD), heme, flavin adenine mononucleotide (FMN), calmodulin, and tetrahydrobiopterin (BH4).
Of relevance to diabetes is the link between the availability of BH4 and cardiovascular disease (Alp and Channon 2004). Numerous studies, in both humans and animals, have demonstrated the beneficial effects of BH4 on endothelial function. The heterogeneity of the human populations (as well as the animal models) wherein beneficial effects of BH4 have been reported indicates that a common mechanism probably exists whereby endothelial function is affected in disease states and is linked to oxidative stress and the reduced bioavailability of NO (Katusic 2001). However, a report from Nyström et al (2004) indicated that BH4 infusion in a random crossover study in diabetic versus nondiabetic patients improved insulin sensitivity, as assessed by hyperinsulinemic isoglycemic clamp, but not endothelial function, as assessed with brachial artery ultrasonography. Ihlemann et al (2003) reported that BH4 corrects endothelial dysfunction induced by an oral glucose challenge in healthy subjects and that this was specific for the 6R and not the 6S stereoisomer of BH4. Since both 6R and 6S are equally effective as antioxidants but only 6R is effective as a cofactor for eNOS, one can conclude that increasing the synthesis of NO is likely to be more important than a simple antioxidant action of BH4.
These data enforce the notion that nonspecific antioxidant treatment may not prove effective in reducing endothelial dysfunction and vascular disease (see Oxidative stress, below). Data from animal studies have also been illuminating as, for instance, Yang et al (2003) also used R and S BH4, but in isolated aortae from spontaneously hypertensive and control rats, and their data indicate that superoxide generated from the autoxidation of BH4 enhances endothelium-dependent contractions in tissues from the hypertensive but not control animals. Thus, supplementation with BH4 may also result in detrimental effects on vascular functions resulting from the enhanced synthesis and/or release of a putative endothelium-derived contracting factor(s) (EDCF).
Overall we can conclude that cardiovascular function will be compromised when the level of NO and/or bioavailability is reduced. However, other endothelium-derived factors may compensate for the loss of NO, and one such endothelial cell-derived relaxing factor, prostacyclin, can also be released via the activation of G protein-coupled receptors and nonselective (stretch-operated) cation channels on endothelial cells. Activation of cyclooxygenase (COX), the synthesis of prostacyclin, and activation of the prostacyclin receptor on vascular smooth muscle cells increases cyclic adenosine monophosphate levels and induces vasorelaxation.
Although it has been reported that COX-2 blockade does not impair endothelium-dependent function in healthy individuals (Verma et al 2001), changes in the ratio of COX-derived vasodilator versus vasoconstrictor products, most likely prostaglandin endoperoxides, may contribute to endothelial-vascular dysfunction in cardiovascular disease states including diabetes (Tesfamariam et al 1989, 1990; Shimizu et al 1993; De Vriese et al 2000; Lagaud et al 2001; Pannirselvam et al in press).
Role of endothelium-derived hyperpolarizing factor
In many vessels, inhibition of endothelial NO and prostacyclin synthesis does not completely inhibit endothelium-dependent vasodilation. This NOS/COX-insensitive relaxation has been linked to vascular smooth muscle cell hyperpolarization, which has given rise to the postulation of the “third mediator pathway” (that functions in addition to the pathways regulated by NO and prostacyclin) (McGuire et al 2001; Vanhoutte 2004). Rather than a true chemical mediator, endothelium-derived hyperpolarizing factor (EDHF) may reflect a purely electrical coupling process between cells mediated by myoendothelial gap junctions that are made up of the connexion proteins 37, 40, 43, and 45 (Griffith 2004; Sandow 2004). Considerable heterogeneity has been reported with respect to the pharmacological properties of EDHF, which has given rise to the hypothesis that multiple EDHFs may exist (Ding and Triggle 2001a; McGuire et al 2001; Triggle and Ding 2002). There are several putative candidates for EDHF: potassium ions, epoxyeicotetranoic acid (often simply abbreviated as EETs), and H2O2. H2O2 is discussed below (see Oxidative stress). The EETs, which are products of CYP enzymic metabolism of arachidonic acid, are attractive candidates for EDHF because they hyperpolarize and relax vascular smooth muscle cells by activating calcium-sensitive potassium (KCa) channels. In situations of reduced NO availability in eNOS knock-out mice there is an up-regulation of EDHF-mediated relaxation in resistance arteries (Waldron et al 1999; Ding et al 2000). If multiple EDHFs exist, either as chemical mediators or as distinct cell-cell (gap junctions) communication pathways, there is the exciting prospect that novel therapeutic agents could, in a vascular bed-dependent manner, be developed to selectively modulate blood flow to a specific organ (Ding and Triggle 2001b; Feletou and Vanhoutte 2004). Conceivably, in disease states, the availability of EDHF (or expression of gap junctions) may, like NO, be compromised. Alternatively, EDHF may not simply act as a substitute for NO, but may function in a pathophysiological manner and have both short- and long-term detrimental effects on vascular function. Regardless, a better understanding of the cellular mechanisms that mediate EDHF may allow for the therapeutic manipulation of this cell-cell communication pathway (Ding and Triggle 2001b).
A number of studies have linked EETs to the EDHF phenomenon and also to a protective role in cardiovascular function. Provocatively, Fisslthaler et al (1999), on the basis of their studies with native porcine coronary artery cells, concluded that CYP 2C is an EDHF synthase. Krötz et al (2004) demonstrated that EETs, in addition to hyperpolarizing platelets, inhibited platelet P-selectin expression in response to a procoagulation stimulus. Thum and Borlak (2004) reported that oxidized LDL repressed the synthesis of EETs in primary cultures of human coronary arterial endothelial cells at the transcriptional level, possibly via oxidized LDL, and enhanced ROS production and the regulation of CYP genes by the transcription factor nuclear factor 1 (NF-1). A polymorphism in the enzyme that metabolizes the EETs, soluble epoxide hydrolase, has also been associated with coronary artery calcification, and a defect in this enzyme has been linked to the development of atherosclerosis (Fornage et al 2004). Conversely, inhibition of CYP 2C9 with sulfaphenazole has been shown to improve endothelium-dependent vasodilation in patients with CAD via, it is argued, the repression of ROS generation in endothelial cells by CYP 2C9 (Fichtlscherer, Dimmeler, et al 2004). Conceivably these data reflect that CYP 2C9 can (in a similar manner to that in which eNOS generates both NO and ROS) generate both EETs and ROS and that an imbalance in the generation of ROS exacerbates vascular disease.
Thus, therapeutic advantages could result from increased knowledge of the cellular basis of how the endothelium modulates vascular tone and blood flow. The development of novel therapeutic agents, as well as both diet and exercise interventions, to enhance the bioavailability of NO as well as enhance or inhibit the synthesis or action of EDHF-mediated cell–cell communication pathways may have benefits for the treatment of a variety of cardiovascular diseases (Ding and Triggle 2001b; Feletou and Vanhoutte 2004).
Type 2 diabetes is frequently associated with obesity, and of potential interest to the question of endothelial dysfunction are a limited number of reports that describe an adipocyte-derived vascular relaxing factor (ADRF) that appears to be of periadventitial origin (Löhn et al 2002; Dubrovska et al 2004; Fernández-Alfonso 2004; Verlohren et al 2004). To date, the data available on ADRF are from nondiabetic subjects and the nature of ADRF remains unknown (other than that it is a hyperpolarizing factor) (Dubrovska et al 2004). But what if adipocytes from diabetics do not produce ADRF or the relaxing versus contracting profile is changed and a contracting factor, ADCF, is produced?
Biomarkers for cardiovascular dysfunction
Although endothelial function can be readily assessed in humans (Anderson 1999), it does require a level of technical sophistication that may not be readily available to all clinics, and thus the question arises as to whether there is another readily accessible surrogate marker for the health of the endothelium.
Figure 1 provides an overview of some of the potential pathways involved in linking several potential biomarkers to endothelial dysfunction. Such a biomarker could provide a very useful and, it is hoped, early indicator of vascular disease and would provide important prognostic information concerning the risk of future cardiovascular events (Suwaidi et al 2000; Heitzer et al 2001). There is thus an urgent need to identify non-invasive surrogate measurements of early stage vascular disease in patients with diabetes; monitoring the levels of such a biomarker(s) would be of obvious benefit as a measure of the effectiveness of interventions. Indeed, there has been considerable interest in identifying and developing appropriate assays for both plasma biomarkers (Ridker, Brown, et al 2004) for risk assessment of future vascular events and genetic markers for predicting risk and the progress of the disease (Gibbons et al 2004). Although hyperlipidemia has long been used as a routine measure, it is also apparent that the greater percentage of vascular events occurs in individuals whose lipid profile is within the normal range. Thus, given our increasing awareness that so-called cholesterol-lowering drugs, such as the statins, have a considerable repertoire of pleiotropic actions that involve direct actions on the vasculature, cholesterol levels may not directly reflect the health of the vascular system (Liao and Laufs 2004). Since the topics of biomarkers and genetic markers for cardiovascular disease have been recently reviewed, by Ridker, Brown, et al (2004) and Gibbons et al (2004), respectively, we focus in the current review on the appropriateness of two intensely studied biomarkers that have been considered to be not only indicators of vascular disease but, perhaps, also mediators of endothelial dysfunction.
Figure 1.
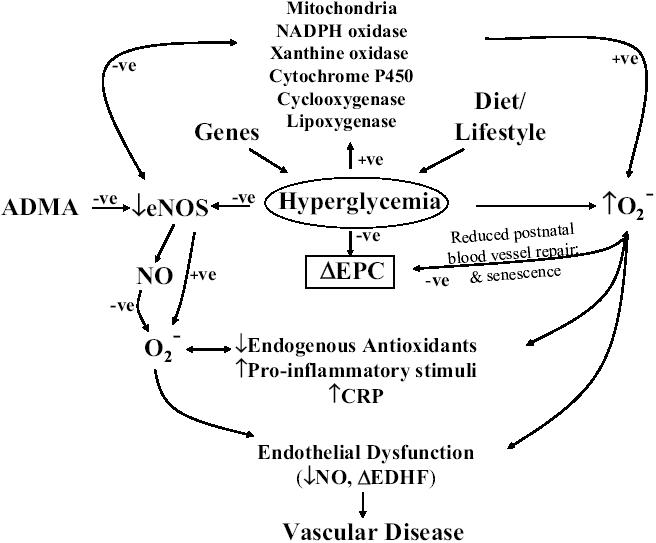
An elevation in plasma glucose brought on by the combined effects of diet, lifestyle, and genes leads to hyperglycemia. Hyperglycemia decreases endothelial nitric oxide synthase (eNOS) activity and thus reduces the bioavailability of nitric oxide (NO) and also increases oxidative stress (enhanced production of ) via, potentially, a multitude of pathways that may include the involvement of mitochondria, NADPH oxidase, xanthine oxidase, cytochrome P450, cyclooxygenase, and lipoxygenase. Oxidative stress also further reduces the bioavailability of NO, thus reducing the inhibitory actions of NO on cytochrome P450 and other enzymes that are involved in the elevation of . Hyperglycemia may have direct or indirect effects to decrease the numbers and/or viability of endothelial progenitor cells (EPCs) and thus reduce postnatal blood vessel repair and enhance the rate of endothelial cell senescence. Asymmetric dimethylmethylarginine (ADMA) is an endogenous inhibitor of eNOS, and the metabolism of ADMA is decreased when oxidative stress is high and thus elevated ADMA will further decrease the bioavailability of NO. Reduced availability of NO, elevated oxidative stress, and reduced levels of endogenous antioxidants add to the proinflammatory stimuli, and plasma and possibly vascular levels of C-reactive protein (CRP) are elevated. CRP has been reported to decrease the stability of mRNA for eNOS, thus further reducing the synthesis of NO. Endothelial dysfunction results from the reduction in the local production/bioavailability of NO and, possibly, changes in the contribution of endothelium-derived hyperpolarizing factor (EDHF) to the regulation of vascular tone.
C-reactive protein
Studies of CRP have now gone full circle. It has been known for almost 75 years that CRP is an acute-phase protein and blood CRP rises from trace levels to high micrograms per milliliter during inflammatory diseases (Tillett and Francis 1930; Gabay and Kushner 1999; Szalai and McCrory 2002). The acute-phase response is a fundamental host defense response, and CRP synthesis is rapidly elevated by the liver as a result of an increase in interleukin (IL)-6 levels, with serum concentrations of CRP rising from less than 1 μg/mL to a thousand times higher within 48 hours of the inflammatory stimulus (Gabay and Kushner 1999). It was recognized as early as 1941 that CRP plays a potentially important antimicrobial role by virtue of the ability of CRP to bind to bacterial wall phosphocholine and precipitate the collapse of the cell wall of Streptococcus pneumoniae (Abernathy and Avery 1941). CRP has also been used as a prognostic marker in several malignancies, and Wieland et al (2003) concluded that CRP was a promising diagnostic and prognostic index and follow-up monitor for both pediatric and adolescent patients with Hodgkin's disease. Of particular interest is that Wieland et al (2003) reported plasma CRP levels as high as 211 μg/mL in the plasma of Hodgkin's disease patients, far exceeding the levels reported in patients with CAD. As discussed later in this review, Szalai (2004) also presents compelling evidence that CRP may play a protective role in autoimmune diseases and thus it should not be surprising that plasma CRP levels are raised in inflammatory diseases.
Considerable attention has been placed on CRP as a powerful indicator of cardiovascular risk (Libby et al 2002; Ridker et al 2002; Taubes 2002). Ridker et al (2002) published the results of a large epidemiological study of 27 939 women and concluded that circulating CRP levels were a better indicator of CAD than cholesterol. Similarly, it was concluded that elevated levels of CRP and IL-6 predict the development of type 2 diabetes and support a possible role for inflammation in diabetogenesis (Pradhan et al 2001). CRP is now accepted as a valuable prognostic marker – blood levels are routinely measured – and by inference, has come to be considered as an actual mediator/modulator, rather than merely a corollary marker, of the inflammation that contributes to CAD (Yeh 2004). These conclusions also seem to have correlates in animal research, as studies with the Israel sand gerbil (Psammomys obesus), an excellent polygenic animal model of type 2 diabetes, provide a similar correlation between acute-phase proteins (Tanis and serum amyloid A protein), inflammation, and a mechanistic link to type 2 diabetes and cardiovascular disease (Walder et al 2002). Furthermore, a mechanistic argument has been presented that CRP mediates inhibitory actions on eNOS and angiogenesis via a posttranscriptional effect on eNOS mRNA stability (Verma et al 2002). CRP levels in patients with CAD may also predict the bioavailability of NO (Fichtlscherer, Breuer, et al 2004). Complexities in interpretation of some of the clinical data, however, may relate to the fact that CRP exists as both pentameric CRP and monomeric CRP (mCRP) and risk may be greater for patients with more of the mCRP (Khreiss et al 2004; Verma, Szmitko, Yeh, et al 2004). Nonetheless, collectively these reports provide both epidemiological and molecular support for a direct role for CRP in diabetogenesis and endothelial dysfunction. A CRP promoter polymorphism has also been associated with type 2 diabetes in Pima Indians (Wolford et al 2003). However, for reasons discussed below, we argue that it is premature to conclude that such proteins are mechanistically directly linked to the pathophysiology of type 2 diabetes-related vascular diseases.
Should the above data that seem to strongly link plasma levels of CRP to the incidence and severity of cardiovascular disease be re-interpreted in terms of the long-established primary function for CRP (and other acute-phase proteins) as a host defense molecule? Certainly the results of another large and very comprehensive study by Danesh, Wheeler, et al (2004) challenge the utility of routine CRP measurements as an indicator of CAD, questioning, in particular, the magnitude of the association between CRP and cardiovascular risk. These data have been the subject of follow-up letters to the Editor of the New England Journal of Medicine (Danesh, Pepys, et al 2004; Foody et al 2004; Glynn and Cook 2004; Libby et al 2004; Ridker, Koenig, et al 2004) and considerable debate. Should we reconsider the use of CRP levels as an exact predictor of the severity of CAD and possibly negate the contribution of CRP as a direct mediator of endothelial dysfunction and CAD? We need to closely follow this debate before reaching a final conclusion. Caution must also be applied to in vitro studies with human recombinant CRP, as it has been reported that commercial samples contain high levels of sodium azide, and the activation of guanylyl cyclase by azide, as well as direct toxic effects of azide, may explain at least some of the reported in vitro effects of CRP (Van Den Berg et al 2004).
Studies with transgenic mice that express human CRP have helped elucidate the biological functions of CRP in humans. In the mouse, unique among mammals, CRP is not an acute-phase protein and blood levels do not change appreciably during inflammation, but rather this function is served by mouse serum amyloid P protein. Ciliberto et al (1987) generated CRP transgenic mice (CRPtg) that, with an appropriate stimulus, express human CRP as an acute-phase protein with blood levels comparable to those reported for humans. Thus, CRPtg mice constitutively produce human CRP such that serum levels are 10–20 μg/mL and in the range seen in patients at high risk of CAD (Ridker et al 1997; Szalai and McCrory 2002). Acute-phase levels of human CRP in CRPtg mice also dramatically rise with an endotoxin challenge (based on studies by Murphy et al 1995 that indicate that a sequence downstream of the human CRP coding sequence regulates induction by endotoxin), closely resembling that seen in humans, and this increase in CRP provides protection against bacterial infection (Szalai et al 1995, 2000; Szalai 2002). Studies by Szalai and colleagues (Szalai and McCrory 2002; Szalai 2004) have been interpreted to reflect a role for CRP in CRPtg mice to act as an important host defense molecule. Szalai (2004) also argues that CRP serves to bind autoantigens and protect against autoimmunity as well as promote clearance of apoptotic cells. In contrast, Hirschfield et al (2003) reported that human CRP did not protect against lipopolysaccharide challenge in CRPtg mice. This is perhaps not surprising, since human CRP does not bind lipopolysaccharide and the clinical data indicate that CRP levels remain in patients with septic shock, suggesting that CRP is not protective, at least in such extreme conditions. Danenberg et al (2003) reported that arterial injury in CRPtg mice results in a faster rate of arterial thrombosis than in control mice. Furthermore, human CRPtg expression into the apolipoprotein knock-out (ApoE−/−) mouse to produce the CRPtg+/0/ApoE−/− resulted in significantly larger atherosclerotic lesions, but only at 29 weeks and not at 15 weeks (Paul et al 2004). The data from Paul et al (2004) suggest that whereas CRP may contribute to the chronic progression of the atherosclerotic process, it may not be involved in the initiation of atherosclerosis; however, data from the study by Danenberg et al (2003) imply a role for CRP as an important causal agent in acute cardiovascular events. Thus, quite differing conclusions have been reached from studies of CRPtg mice, and this may reflect the different contributions of CRP in acute versus chronic pathophysiological states. Clearly further studies must be pursued in this area.
Thus, as clearly stated by Goodman (2004), CRP is a marker of inflammation, and serum levels will rise in almost any condition when an individual is sick, reemphasizing the long-standing contribution of Mark Pepys to our knowledge of CRP. Since CRP is involved in host defense mechanisms, should it also be considered as a contributor to the pathophysiology and a specific marker for cardiovascular disease? Perhaps the persistent increase in liver-derived CRP, as seen in advanced cancer, AIDS, and cardiovascular disease, is a key factor that contributes, in a variable manner, to the development of endothelial dysfunction. Further studies are justified concerning the direct contribution of CRP to vascular pathophysiology, and a point for consideration is the question: Does locally produced (not of liver origin) CRP in the vasculature contribute to the development of atherosclerosis (Yasojima et al 2001)?
Asymmetric dimethylarginine
Asymmetric dimethylarginine (ADMA) is described in the literature as an “endogenous inhibitor of NO synthase” and as an “uber marker”, a biochemical factor that mediates the adverse effects of a plethora of other risk factors and biochemical markers (Böger 2003; Cooke 2004). Vallance et al (1992a, 1992b) demonstrated that both Nw-monomethyl-L-arginine (L-NMMA) and ADMA were endogenous inhibitors of NOS, with ADMA being produced in sufficient amounts that it could be considered to be a potentially important endogenous modulator of eNOS activity. Numerous studies provide data that seem to back this pronouncement. The “normal” plasma levels of ADMA have been reported to be 1.0 ± 0.1 μmol/L (Böger et al 1998). Elevated plasma ADMA levels have been associated with many cardiovascular risk factors including age, diabetes, hyperlipidemias, hypertension, and insulin resistance (see recent review by Cooke 2004). Plasma levels have been reported ranging from 2.2 μmol/L in young hyper-cholesterolemic, but otherwise healthy, individuals (Böger et al 1997) to 2.5–3.5 μmol/L in elderly patients with atherosclerosis and peripheral vascular disease (Böger et al 1998). The highest levels, however, are reported in renal failure (Vallance et al 1992a; Kielstein et al 1999; Zoccali et al 2001). ADMA is excreted in the urine either unchanged or as the dimethylarginine dimethylaminohydrolase (DDAH) metabolite and may rise as high as 8.7±0.7μmol/L in end-stage renal failure (Vallance et al 1992a). The calculated 50% inhibitory concentration (IC50) for ADMA against NOS is 1.8 ± 0.1 μmol/L (Faraci et al 1995); it is therefore not surprising that considerable attention has been placed on ADMA as a diagnostic marker and as a mediator of endothelial dysfunction. Interestingly, ADMA levels are doubled and endothelium function decreased in diabetic patients after a high-fat meal (Achan et al 2003), and correlations have been made between insulin resistance and ADMA levels (Stuhlinger et al 2002). Furthermore, LDL has been shown to enhance ADMA synthesis in human endothelial cells (Böger et al 2000).
Many of the above studies are, however, based on correlations between plasma levels of ADMA and normal versus disease states in patients. Although ADMA may prove to be an appropriate “diagnostic marker” for vascular disease, and there is a considerable data bank to back up this assumption, it is unlikely that the pathophysiological actions of ADMA are entirely mediated via the inhibition of eNOS. This conclusion is reached by examining the data and conclusions reported by Suda et al (2004) with eNOS knock-out mice. The long-term treatment of mice with ADMA induced coronary microvascular lesions to the same extent in both wild-type and eNOS knock-out animals, and the induction of these lesions was not prevented by the administration of L-arginine. Furthermore, the treatment also caused an up-regulation of vascular angiotensin-converting enzyme and an increase in superoxide production to a comparable extent in both strains. Finally, neither systemic nor vascular NO production was attenuated by treatment with ADMA. These data may reflect differences observed when comparing acute with chronic effects of ADMA, as the acute administration of ADMA in humans and animals clearly leads to inhibition of endothelial NO synthesis (Vallance et al 1992b), whereas the chronic effects of long-term administration are not associated with reductions in NO production (Suda et al 2004). Similar data have been reported for short-term versus long-term administration of the NOS inhibitor NG-nitro-L-arginine methyl ester (Bryant et al 1995). These data indicate that the simple inhibition of endothelial NO synthesis cannot be the primary mechanism whereby the long-term vascular effect of ADMA leads to endothelial dysfunction in vivo. The data also stress the value of studies with knock-out mice for the elucidation of pathophysiological mechanisms in humans. The data from Suda et al (2004) indicate that ADMA enhances superoxide production, via an angiotensin-I receptor-mediated process, and also increases angiotensin-converting enzyme activity. ADMA may therefore prove to be a good marker for oxidative stress, as elevated oxidative stress inhibits dimethylarginine dimethylaminohydrolase, the enzyme responsible for degrading ADMA, and thus raises plasma ADMA levels (Leiper et al 2002; Sydow and Munzel 2003).
In conclusion, since elevated oxidative stress is associated with endothelial dysfunction, plasma ADMA levels may prove to be a good indicator of the health of the endothelium (Sydow and Munzel 2003), but there may not be any direct correlation between chronic plasma levels of ADMA and eNOS activity.
Additional measures of endothelial dysfunction
If CRP and ADMA do not necessarily reflect a direct cause-effect relationship to endothelial dysfunction (at least via inhibition of eNOS), what other measures could also be considered as potential biomarkers? Elevated oxidative stress, although not specific for diabetes, would provide an indication of the health of the endothelium and, in addition, endothelial progenitor cells (EPCs) provide a measure of the repair/regenerative capability of the cardiovascular system.
Oxidative stress
Oxidative stress plays an important role in the pathogenesis of cardiovascular disease, including atherosclerosis, hypertension, and the macro- and microvascular diseases associated with diabetes. Oxidative stress is defined as an increase in ROS and/or a decrease in the antioxidant defense mechanisms such as catalase, glutathione, superoxide dismutase (SOD), thioredoxin, and selenoproteins, as well as certain vitamins, notably vitamins C and E (Halliwell 1999; Halliwell and Whiteman 2004). It should also be noted that the antioxidant roles of beta-carotene and ascorbic acid have been questioned (Halliwell 1999).
We have recently reviewed the literature related to oxidative stress and the endothelium and discussed the nature of ROS and possible cellular sources (Ellis and Triggle 2003). Touyz and Schiffrin (2004) have also recently provided an excellent review of ROS in vascular biology, and thus only a brief overview of this field follows.
ROS are a family of molecules, including molecular oxygen and its derivatives (, •OH, and H2O2), which are produced in all aerobic cells as a result of reduction-oxidation (or redox processes) in cells. The free radical form of NO, NO•, is also a “reactive oxygen species”. , having an unpaired electron, is quite unstable and short-lived; however, although cell membranes are not permeable to , it can enter cells via anion channels. is readily reduced to H2O2, which (like NO•) can cross cell membranes, but compared with is not a free radical and is comparatively stable; it is produced from the dismutation of via a SOD-mediated or spontaneous process and is scavenged by catalase and glutathione peroxidase. Since the formation of H2O2 is favored when levels are low (or SOD high), there has been speculation that H2O2 may have important physiological functions that include a role as an EDHF in humans and mice (Matoba and Shimokawa 2003), but see Ellis et al (2003). •OH is the highly reactive reduction product of H2O2 and is produced in the presence of ferric ions (Fe2+) via the Fenton reaction. In situations where high levels of are produced and in the presence of NO, NO is oxidized to produce the highly reactive and cytotoxic peroxynitrite anion, ONOO−. There is a considerable literature that supports the argument that ONOO−, as a reactive nitrogen species and via the formation of nitrotyrosine species, results in tissue damage that includes the oxidation of eNOS and the subsequent dissociation of dimeric eNOS into the inactive monomeric form, as well as the inactivation of prostacyclin synthase (Zou et al 2004). Alternatively, it has also been argued that the cytotoxicity of ONOO− has been overestimated and that it may simply serve as an intermediate, by virtue of its rapid reaction with antioxidants, in the use of NO to dispose of excess , or in the use of to dispose of excess NO (Halliwell et al 1999).
Multifaceted sources of ROS in type 1 and type 2 diabetes include auto-oxidation of glucose, increased substrate flux and decreased levels of NADPH through the polyol pathway, formation of advanced glycation end products and interaction with cellular targets, such as endothelial cells, which may lead to oxidative stress and oxidized LDL. Several enzymes are reported to be a source of ROS, including xanthine oxidase (XO), NADH/NADPH oxidase, COX, CYP, and uncoupled eNOS (Ellis and Triggle 2003; Pannirselvam et al 2003). Brownlee and colleagues (Nishikawa, Edelstein, Brownlee 2000; Nishikawa, Edelstein, Du, et al 2000) have argued that mitochondria are the source of ROS and that uncoupling, for instance, oxidative phosphorylation in endothelial cells that have been treated with high glucose, prevents the sequelae of hyperglycemia. Furthermore, NO is an important tonic inhibitory factor for controlling mitochondrial respiration, and thus a decrease in eNOS activity (or NO bioavailability) will result in an increase in superoxide () production by mitochondria. In particular, overproduction of may be detrimental because of its rapid interaction with NO, which leads to the loss of NO bioavailability and a reduction in the antiatherogenic effects of NO.
Elevated production of may also be linked to plaque instability (Cai and Harrison 2000; De Meyer et al 2003) with the shoulder region of the plaque being a particularly active area for production (Sorescu et al 2002). Patients with endothelial dysfunction, and in whom arterial superoxide production is also elevated, are at highest risk for vascular morbidity and mortality (Guzik et al 2000; Heitzer et al 2001; reviewed by Channon and Guzik 2002). The NAD(P)H oxidase family has been identified as a major enzyme system involved in the generation of vascular oxidative stress, and the molecular composition of vascular NAD(P)H oxidases appears to vary in different cell types (Paravicini et al 2002; Bengtsson et al 2003). The expression of the membrane-bound cytochrome reductase domain α-subunit, p22phox, and the larger flavin-containing β-subunit called gp91phox has been reported to be elevated in human sclerotic lesions (Azumi et al 1999; Sorescu et al 2002). It is likely that the molecular regulation of the NAD(P)H oxidases within atherosclerotic plaques may be highly relevant to plaque events, such as rupture. Further investigations of the relevance of the NAD(P)H oxidases versus other cellular sources of to human vascular disease pathogenesis are therefore justified and are required particularly with respect to the contribution of other systemic factors, such as oxidized LDL, angiotensin-II, and proinflammatory cytokines, to atherosclerotic risk and the progression of vascular disease in diabetes. However, changes in the expression of -producing enzymes may prove to be an important predictor of the severity and potential morbidity associated with vascular disease as well as the enzymes serving as potential targets for corrective interventions.
Hyperglycemia impairs endothelium-dependent relaxation and this effect can be reversed with SOD, an endogenous scavenger of superoxide anions (Tesfamariam and Cohen 1992). Gene transfer of CuZn SOD and Mn SOD reduced both production and endothelial dysfunction in experimental diabetes, thus supporting the argument that oxidative stress is an important target for the correction of diabetes-related vascular disease (Zanetti et al 2001). Clinical studies have also demonstrated that normal endothelium function can be impaired by a 6-hour exposure to high glucose (Williams et al 1998) and this impairment can be prevented by vitamin C and vitamin E. Furthermore, flow-mediated vasodilation is differentially affected by a glucose load in normal versus impaired glucose tolerance and type 2 diabetic patients (Kawano et al 1999). Endothelium function in those with impaired glucose tolerance and type 2 diabetes took considerably longer to recover and was associated with a glucose load-induced increase in oxidative stress (Kawano et al 1999). One can speculate that endothelium function can be rapidly affected by changes in blood glucose levels and that susceptibility to long-term dysfunction may be determined as a result of a failure of antioxidant protective mechanisms linked, at least in part, to genetically determined susceptibility.
Hyperglycemia may competitively inhibit the transport of ascorbic acid through facilitative glucose transporters, thus promoting oxidative stress. There is also direct evidence in humans that patients with type 2 diabetes have enhanced oxidative stress as measured by lipid peroxidation (Pinkney et al 1999). Unfortunately, clinical intervention studies with antioxidants (notably vitamin C and/or E) have provided confusing and conflicting data (Antoniades et al 2003), as the interventions produced only variable reductions in oxidative stress. Two large prospective intervention studies with vitamin E and ramipril (the HOPE Study; Lonn et al 2002) and with vitamin C, vitamin E, and beta-carotene (the MRC/BHF Heart Protection Study; Heart Protection Study Collaborative Group 2002) reported no benefit for patients with diabetes and cardiovascular disease. In both these studies plasma concentrations of the vitamins as well as the lipid profile were measured and reported but, surprisingly, no measure was made of oxidative stress. Thus, in neither study was it possible to conclude whether the interventions were modifying oxidative stress in the patients – somewhat akin, as noted by Halliwell (2000), to conducting a trial with antihypertensive drugs but not monitoring blood pressure. Although it can be argued that there is no clear agreement as to which biomarkers best monitor oxidative stress, the measurement of, for instance, the isoprostanes as an indicator of lipid peroxidation would have provided one reference set of data (Halliwell and Whiteman 2004). In addition, the choice of the interventions may be questioned, as vitamins C and E do not greatly affect isoprostane levels (Levine et al 2001; Meagher et al 2001), although their inclusion was probably based on their link to dietary sources. In addition, it may well be that as yet undefined “coantioxidants” are essential and present in dietary sources of vitamin E but missing in the supplements used in the studies (Stocker 1999). It is also doubtful whether any of the “classical antioxidants” are capable of preventing the reaction of superoxide with NO, as this is one of the fastest known biological reactions. Furthermore, concerns over the measurement of ROS as well as the interpretation of antioxidant intervention studies have been detailed by Halliwell and Whiteman (2004) and Halliwell (1999, 2000a, 2000b).
As also noted by Kritharides and Stocker (2002), it is important to determine and to then target the oxidative processes that are operative within the artery wall. It is well established that vitamin E (α-tocopherol) can inhibit the oxidation of LDL in vitro, and each particle of LDL contains between 6 and 10 molecules of α-tocopherol; however, via the formation of α-tocopheroxyl, lipid peroxidation of LDL can also be enhanced under certain conditions (Stocker 1999). Another consideration is that vitamin E is associated with the lipophilic/hydrophobic domains of lipoproteins and cell membranes and ROS are generated in the cytosolic and extracellular compartments; it is conceivable that such compartments are just far too separate. Touyz (2004) reached similar conclusions in her review of why antioxidant supplements have failed to benefit patients with elevated blood pressure and, in addition, pointed out that many patients would have been taking aspirin prophylactically, and aspirin has antioxidant activity (Wu et al 2002). In addition, non-oxidant actions of vitamin E may also be important considerations in determining the effectiveness of vitamin E use in clinical situations (Munteanu et al 2004).
Endothelial cell turnover and bone marrow-derived endothelial progenitor cells
The endothelial dysfunction that is commonly seen in cardiovascular disease states may be linked to accelerated endothelial cell death and reduced regeneration of endothelial cells, as many of the changes reported in endothelial cell function in cardiovascular disease are also seen in senescent cells (Minamino et al 2004). Endothelial cells normally turn over quite slowly, but nonetheless have a finite lifetime. Thus, cardiovascular disease may induce premature aging of the endothelium. Premature senescence and a reduced ability of endothelial cells to be replaced by progenitor cells may therefore be an important component of the poor cardiovascular health of people with diabetes (Minamino et al 2004; Voghel et al 2004). In the Zucker diabetic rat, a model of insulin resistance and obesity, Brodsky, Gealekman, et al (2004) reported that endothelial cells undergo premature senescence in the vasculature, and this senescence is associated with signs of vasculopathy. Furthermore, endothelium-derived microparticles accelerate endothelial dysfunction in cardiovascular disease states, including diabetes (Tepper et al 2002; Brodsky, Zhang, et al 2004). There are numerous descriptions of eukaryotic cells shedding components of their plasma membrane expressing the same antigenic determinants as the corresponding cell surface and, in the case of endothelial cells, such microparticles have been reported to have procoagulant capability and express CD31, CD54, and CD62E etc (Combes et al 1999; Brodsky, Zhang, et al 2004).
Voghel et al (2004) have explicitly stated that chronic exposure to risk factors for cardiovascular disease leads to telomere-associated premature senescence of the vascular endothelium and may explain the early endothelial dysfunction in patients with risk factors for cardiovascular disease. The telomere, a cap at the end of each chromosome, is very important for the chromosome replication process, and with each replication cycle the telomere shortens. A link between telomere shortening, cellular senescence (Karlseder et al 2002) and human vascular disease has been reported (Minamino and Komuro 2002). Acceleration of telomere shortening, perhaps linked directly or indirectly to endothelial dysfunction (ie, hyperglycemia and oxidative stress), would also lead to the reduced viability of endothelial progenitor cells (EPCs). EPCs are a subset of progenitor cells that can be found in bone marrow and are important for postnatal neovascularization and vascular repair, and thus a decrease in the numbers and/or viability of EPCs would exacerbate vascular disease (Hristov et al 2003a, 2003b). Of relevance to this issue is our increasing awareness that circulating endothelial cells (CECs) may reflect the functional state of the endothelium. Koc et al (2003) recently reported increased CECs in hemodialysis and hypertensive patients and patients with diabetes. Their criterion for identifying CECs was positive immunostaining with the endothelial cell marker von Willebrand factor. Their study, however, did not attempt to distinguish CECs from EPCs. EPCs are usually identified by the expression of the stem cell markers CD133, CD34, and vascular endothelial growth factor receptor 2. As EPCs differentiate into endothelial cells they lose CD133 and later CD34 expression and then start to express von Willebrand factor as well as VE-cadherin and, finally, eNOS (Hristov et al 2003a, 2003b). Asahara et al (1997) first described the isolation of putative progenitor CD34+ cells with the use of magnetic microbeads from human blood. Although the percentage of EPCs in the peripheral blood is normally very low (0.002%), numbers can be dramatically increased by, for instance, granulocyte colony-stimulating factor.
Rauscher et al (2003) reported that the defective vascular repair observed in the ApoE-deficient mouse is associated with an age-dependent reduction of EPCs from these animals thus suggesting an important role for EPC senescence in the progression of vascular disease. Of interest too was the ability of EPCs from young ApoE-deficient mice to suppress plasma levels of the inflammatory cytokine IL-6 (Rauscher et al 2003). Raised plasma IL-6 is reported to be reflective of both aging and atherosclerosis and is argued to be also a predictive marker for future CAD (Ferrucci et al 1999; Huber et al 1999). The classification of IL-6 solely as an inflammatory cytokine has been challenged (Carey and Febbraio 2004), and indeed IL-6 may play an important role as an exercise factor or myokine in humans (Pedersen et al 2004). IL-6 is also involved in the stimulation of CRP production by the liver (Gabay and Kushner 1999) and this, again, raises the issue of whether CRP and IL-6 are “friends or foes”. Quite possibly such distinctions may relate to both the absolute level (ie, concentration) and the regulation of plasma levels in acute versus chronic conditions.
In people with diabetes, EPCs demonstrate impaired activity and these changes may contribute to the high vascular morbidity that is inherent in diabetics (Tepper et al 2002). Little is known, however, about the processes that regulate EPC numbers and function, but of importance for people with diabetes is that exercise-induced myocardial ischemia in patients with symptomatic CAD results in an increase in circulating EPCs (Adams et al 2004). Additional studies to explore how exercise, diet, and drug interventions may interact and affect EPC numbers and function would be of obvious interest.
Hyperglycemia, hyperglycemic memory, and endothelial dysfunction
In type 1 diabetes it is not until at least 5 years after pancreas transplantation that reversal of diabetic nephropathy lesions is observed (Fioretto et al 1998). Therefore, it has been suggested that hyperglycemia induces phenotypic changes in cells that persist despite a return to normoglycemia (Fioretto et al 1998). Roy et al (1990) have also suggested that hyperglycemia is responsible for the “induction of self-perpetuating changes in gene expression”. The concept of hyperglycemic memory (and the ability to reverse it) may be an important factor in determining the success of interventions directed towards improving vascular function in diabetic patients. It is therefore worthy of speculation that the premature senescence of endothelial cells linked to a decreased potential for EPCs as contributors to endothelial regeneration are key events in the deterioration of endothelial function that is seen in diabetes and other vascular diseases. It is clearly important to investigate the effects of diabetes on endothelial cell senescence and the viability of EPCs and whether controlling insulin sensitivity and blood glucose levels with drug, diet, and/or exercise interventions affects EPC levels and function. In this regard, Loomans et al (2004) reported that EPCs from type 1 patients are dysfunctional as illustrated in an in vitro angiogenesis assay and, despite culturing these EPCs under normoglycemic conditions, such functional differences between diabetes patients and controls were maintained.
Concluding comments
Diabetes-associated vascular complications are a growing concern worldwide, a major clinical problem, and there is thus an urgent need to characterize appropriate diagnostic markers that can provide an early prognostic indicator of developing vascular disease. Endothelial dysfunction is an early indicator of vascular disease and may directly or indirectly be associated with increases in plasma ADMA, CRP, IL-6, ROS, endothelial cell senescence, and decreases in the numbers and viability of EPCs. Recent data from both human and animal studies question a direct association between an increase in plasma CRP and endothelial dysfunction and also between ADMA and a reduction in NO production, and further studies are needed to answer the questions:
To what extent do CRP and ADMA serve as specific biomarkers of vascular disease?
Is there a cause and effect relationship between CRP and ADMA plasma/tissue levels in the development of vascular disease?
Assays of the status of EPCs are likely to prove to be critical for assessing the health of the vascular system, and interventions that enhance EPC numbers and restore angiogenic activity in diabetes may prove to be particularly beneficial.
Acknowledgments
Supported by an internal Research Infrastructure Funding grant from RMIT University, Melbourne, Australia to HD and CRT and operating grants from the Canadian Diabetes Association, Canadian Institutes of Health Research, and the Heart & Stroke Foundation of Alberta to CRT.
Notes
We note that whereas acetylcholine (and in some clinical studies methacholine) is employed it is unlikely that acetylcholine is the physiological mediator of endothelial cell-dependent relaxation; it is the most commonly used agent for probing endothelial function/dysfunction.
References
- Abernathy TJ, Avery OT. The occurrence during acute infections of a protein not normally present in the blood. I. Distribution of the reactive protein in patient's sera and the effects of calcium on the flocculation reaction with C-polysaccharide of pneumococcus. J Exp Med. 1941;73:173–82. doi: 10.1084/jem.73.2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achan V, Broadhead M, Malaki M, et al. Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolized by dimethylarginine dimethylaminohydrolase. Arterioscler Thromb Vasc Biol. 2003;23:1455–9. doi: 10.1161/01.ATV.0000081742.92006.59. [DOI] [PubMed] [Google Scholar]
- Adams V, Lenk K, Linke A, et al. Increase of circulating endothelial progenitor cells in patients with coronary artery disease after exercise-induced ischemia. Arterioscler Thromb Vasc Biol. 2004;24:684–90. doi: 10.1161/01.ATV.0000124104.23702.a0. [DOI] [PubMed] [Google Scholar]
- Aird WC. Endothelium as an organ system. Crit Care Med. 2004;32:S271–9. doi: 10.1097/01.ccm.0000129669.21649.40. [DOI] [PubMed] [Google Scholar]
- Alp NJ, Channon KM. Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler Thromb Vasc Biol. 2004;24:413–20. doi: 10.1161/01.ATV.0000110785.96039.f6. [DOI] [PubMed] [Google Scholar]
- Anderson TJ. Assessment and treatment of endothelial dysfunction in humans. J Am Coll Cardiol. 1999;34:631–8. doi: 10.1016/s0735-1097(99)00259-4. [DOI] [PubMed] [Google Scholar]
- Andrews HE, Bruckdorfer KR, Dunn RC, et al. Low-density lipoproteins inhibit endothelium-dependent relaxation in rabbit aorta. Nature. 1987;327:237–9. doi: 10.1038/327237a0. [DOI] [PubMed] [Google Scholar]
- Andrews K, Pannirselvam M, Anderson TJ, et al. The vascular endothelium in diabetes: a practical target for drug treatment? Expert Opin Ther Targets. doi: 10.1517/14728222.9.1.101. In press. [DOI] [PubMed] [Google Scholar]
- Antoniades C, Tousoulis D, Tentolouris C, et al. Oxidative stress, antioxidant vitamins, and atherosclerosis. From basic research to clinical practice. Herz. 2003;28:628–38. doi: 10.1007/s00059-003-2417-8. [DOI] [PubMed] [Google Scholar]
- Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–7. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- Azumi H, Inoue N, Takeshita S, et al. Expression of NADH/NADPH oxidase p22phox in human coronary arteries. Circulation. 1999;100:1494–8. doi: 10.1161/01.cir.100.14.1494. [DOI] [PubMed] [Google Scholar]
- Bengtsson SH, Gulluyan LM, Dusting GJ, et al. Novel isoforms of NADPH oxidase in vascular physiology and pathophysiology. Clin Exp Pharmacol Physiol. 2003;11:849–54. doi: 10.1046/j.1440-1681.2003.03929.x. [DOI] [PubMed] [Google Scholar]
- Böger RH. Asymmetric dimethylarginine (ADMA) modulates endothelial function – therapeutic implications. Vasc Med. 2003;8:149–51. doi: 10.1191/1358863x03vm501ed. [DOI] [PubMed] [Google Scholar]
- Böger RH, Bode-Boger SM, Szuba A, et al. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation. 1998;98:1842–7. doi: 10.1161/01.cir.98.18.1842. [DOI] [PubMed] [Google Scholar]
- Böger RH, Bode-Boger SM, Thiele W, et al. Biochemical evidence for impaired nitric oxide synthesis in patients with peripheral arterial occlusive disease. Circulation. 1997;95:2068–74. doi: 10.1161/01.cir.95.8.2068. [DOI] [PubMed] [Google Scholar]
- Böger RH, Sydow K, Borlak J, et al. LDL cholesterol upregulates synthesis of asymmetrical dimethylarginine in human endothelial cells:involvement of S-adenosylmethionine-dependent methyltransferases. Circ Res. 2000;87:99–105. doi: 10.1161/01.res.87.2.99. [DOI] [PubMed] [Google Scholar]
- Brodsky SV, Gealekman O, Chen J, et al. Prevention and reversal of premature endothelial cell senescence and vasculopathy in obesity-induced diabetes by ebselen. Circ Res. 2004;94:377–84. doi: 10.1161/01.RES.0000111802.09964.EF. [DOI] [PubMed] [Google Scholar]
- Brodsky SV, Zhang F, Nasjletti A, et al. Endothelium-derived microparticles impair endothelial function in vitro. Am J Physiol Heart Circ Physiol. 2004;286:H1910–15. doi: 10.1152/ajpheart.01172.2003. [DOI] [PubMed] [Google Scholar]
- Bryant CE, Allcock GH, Warner TD. Comparison of effects of chronic and acute administration of NG-nitro-L-arginine methyl ester to the rat on inhibition of nitric oxide-mediated responses. Br J Pharmacol. 1995;114:1673–9. doi: 10.1111/j.1476-5381.1995.tb14956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero AE, Saouaf R, Lim SC, et al. The effects of troglitazone, an insulin-sensitizing agent, on the endothelial function in early and late type 2 diabetes:a placebo-controlled randomized clinical trial. Metabolism. 2003;52:173–80. doi: 10.1053/meta.2003.50023. [DOI] [PubMed] [Google Scholar]
- Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases:the role of oxidant stress. Circ Res. 2000;87:840–4. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- Carey AL, Febbraio MA. Interleukin-6 and insulin sensitivity:friend or foe? Diabetologia. 2004;47:1135–42. doi: 10.1007/s00125-004-1447-y. [DOI] [PubMed] [Google Scholar]
- Channon KM, Guzik TJ. Mechanisms of superoxide production in human blood vessels:relationship to endothelial dysfunction, clinical and genetic risk factors. J Physiol Pharmacol. 2002;53:515–24. [PubMed] [Google Scholar]
- Ciliberto G, Arcone R, Wagner EF, et al. Inducible and tissue-specific expression of human C-reactive protein in transgenic mice. EMBO J. 1987;6:4017–22. doi: 10.1002/j.1460-2075.1987.tb02745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen RA, Zitnay KM, Haudenschild CC, et al. Loss of selective endothelial cell vasoactive functions caused by hypercholesterolemia in pig coronary arteries. Circ Res. 1988;63:903–10. doi: 10.1161/01.res.63.5.903. [DOI] [PubMed] [Google Scholar]
- Combes V, Simon AC, Grau GE, et al. In vitro generation of endothelial microparticles and possible prothrombotic activity in patients with lupus anticoagulant. J Clin Invest. 1999;104:93–102. doi: 10.1172/JCI4985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke JP. Asymmetrical dimethylarginine:the uber marker? Circulation. 2004;109:1813–18. doi: 10.1161/01.CIR.0000126823.07732.D5. [DOI] [PubMed] [Google Scholar]
- Danenberg HD, Szalai AJ, Swaminathan RV, et al. Increased thrombosis after arterial injury in human C-reactive protein-transgenic mice. Circulation. 2003;108:512–15. doi: 10.1161/01.CIR.0000085568.13915.1E. [DOI] [PubMed] [Google Scholar]
- Danesh J, Pepys MB, Gudnason V. C-reactive protein and coronary heart disease [letter] N Engl J Med. 2004;351:297. doi: 10.1056/NEJMoa032804. [DOI] [PubMed] [Google Scholar]
- Danesh J, Wheeler JG, Hirschfield GM, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med. 2004;350:1387–97. doi: 10.1056/NEJMoa032804. [DOI] [PubMed] [Google Scholar]
- Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. doi: 10.1152/physrev.1999.79.2.387. [DOI] [PubMed] [Google Scholar]
- De Meyer GR, Kockx MM, Knaapen MW, et al. Nitric oxide donor molsidomine favors features of atherosclerotic plaque stability during cholesterol lowering in rabbits. J Cardiovasc Pharmacol. 2003;41:970–8. doi: 10.1097/00005344-200306000-00021. [DOI] [PubMed] [Google Scholar]
- De Vriese AS, Verbeuren TJ, Van de Voorde J, et al. Endothelial dysfunction in diabetes. Br J Pharmacol. 2000;130:963–74. doi: 10.1038/sj.bjp.0703393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond J. The double puzzle of diabetes. Nature. 2003;423:599–602. doi: 10.1038/423599a. [DOI] [PubMed] [Google Scholar]
- Dimmeler S, Fleming I, Fisslthaler B, et al. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–5. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- Ding H, Kubes P, Triggle CR. Potassium and acetylcholine-induced vasorelaxation in mice lacking endothelial nitric oxide synthase. Br J Pharmacol. 2000;129:1194–200. doi: 10.1038/sj.bjp.0703144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Triggle CR. Novel endothelium-derived relaxing factors:identification of factors and cellular targets. J Pharmacol Toxicol Methods. 2001a;44:441–52. doi: 10.1016/s1056-8719(00)00127-1. [DOI] [PubMed] [Google Scholar]
- Ding H, Triggle CR. Relaxing blood vessels:are there novel endothelium-derived mediators to be found and can their discovery lead to the development of new therapeutic agents? Pharm News. 2001b;8:42–9. [Google Scholar]
- Dubrovska G, Verlohren S, Luft FC, et al. Mechanisms of ADRF release from rat aortic adventitial adipose tissue. Am J Physiol Heart Circ Physiol. 2004;286:H1107–13. doi: 10.1152/ajpheart.00656.2003. [DOI] [PubMed] [Google Scholar]
- Ellis A, Pannirselvam M, Anderson TJ, et al. Catalase has negligible inhibitory effects on endothelium-dependent relaxations in mouse isolated aorta and small mesenteric artery. Br J Pharmacol. 2003;140:1193–200. doi: 10.1038/sj.bjp.0705549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis A, Triggle CR. Involvement of endothelium-derived reactive oxygen species in the regulation of vascular tone (a survey review) Can J Physiol Pharmacol. 2003;81:1013–28. doi: 10.1139/y03-106. [DOI] [PubMed] [Google Scholar]
- Engerman RL, Kern TS. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes. 1987;36:808–12. doi: 10.2337/diab.36.7.808. [DOI] [PubMed] [Google Scholar]
- Faraci FM, Brian JE, Jr, Heistad DD. Response of cerebral blood vessels to an endogenous inhibitor of nitric oxide synthase. Am J Physiol. 1995;269:H1522–7. doi: 10.1152/ajpheart.1995.269.5.H1522. [DOI] [PubMed] [Google Scholar]
- Feletou M, Vanhoutte PM. EDHF:new therapeutic targets? Pharmacol Res. 2004;49:565–80. doi: 10.1016/j.phrs.2003.10.017. [DOI] [PubMed] [Google Scholar]
- Fernández-Alfonso MS. Regulation of vascular tone:the fat connection. Hypertension. 2004;44:255–6. doi: 10.1161/01.HYP.0000140056.64321.f9. [DOI] [PubMed] [Google Scholar]
- Ferrucci L, Harris TB, Guralnik JM, et al. Serum IL-6 level and the development of disability in older persons. J Am Geriatr Soc. 1999;47:639–46. doi: 10.1111/j.1532-5415.1999.tb01583.x. [DOI] [PubMed] [Google Scholar]
- Fichtlscherer S, Breuer S, Schachinger V, et al. C-reactive protein levels determine systemic nitric oxide bioavailability in patients with coronary artery disease. Eur Heart J. 2004;25:1412–18. doi: 10.1016/j.ehj.2004.06.026. [DOI] [PubMed] [Google Scholar]
- Fichtlscherer S, Dimmeler S, Breuer S, et al. Inhibition of cytochrome P450 2C9 improves endothelium-dependent, nitric oxide-mediated vasodilatation in patients with coronary artery disease. Circulation. 2004;109:178–83. doi: 10.1161/01.CIR.0000105763.51286.7F. [DOI] [PubMed] [Google Scholar]
- Fioretto P, Steffes MW, Sutherland DE, et al. Reversal of lesions of diabetic nephropathy after pancreas transplantation. N Engl J Med. 1998;339:69–75. doi: 10.1056/NEJM199807093390202. [DOI] [PubMed] [Google Scholar]
- Fisslthaler B, Popp R, Kiss L, et al. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature. 1999;401:493–7. doi: 10.1038/46816. [DOI] [PubMed] [Google Scholar]
- Foody JM, Gotto AM, Wenger N. C-reactive protein and coronary heart disease [letter] N Engl J Med. 2004;351:296. [PubMed] [Google Scholar]
- Fornage M, Boerwinkle E, Doris PA, et al. Polymorphism of the soluble epoxide hydrolase is associated with coronary artery calcification in African-American subjects:the Coronary Artery Risk Development in Young Adults (CARDIA) study. Circulation. 2004;109:335–9. doi: 10.1161/01.CIR.0000109487.46725.02. [DOI] [PubMed] [Google Scholar]
- Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. N Engl J Med. 1999;340:448–54. doi: 10.1056/NEJM199902113400607. [Erratum:N Engl J Med, 1999, 340:1376.] [DOI] [PubMed] [Google Scholar]
- Gibbons GH, Liew CC, Goodarzi MO, et al. Genetic markers:progress and potential for cardiovascular disease. Circulation. 2004;109:IV47–58. doi: 10.1161/01.CIR.0000133440.86427.26. [DOI] [PubMed] [Google Scholar]
- Glynn RJ, Cook NR. C-reactive protein and coronary heart disease [letter] N Engl J Med. 2004;351:295. doi: 10.1056/NEJM200407153510318. [DOI] [PubMed] [Google Scholar]
- Goodman L. Hard-hearted CRP. J Clin Invest. 2004;113:1244–5. doi: 10.1172/JCI21836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith TM. Endothelium-dependent smooth muscle hyperpolarization:do gap junctions provide a unifying hypothesis? Br J Pharmacol. 2004;141:881–903. doi: 10.1038/sj.bjp.0705698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik TJ, West NE, Black E, et al. Vascular superoxide production by NAD(P)H oxidase:association with endothelial dysfunction and clinical risk factors. Circ Res. 2000;86:E85–90. doi: 10.1161/01.res.86.9.e85. [DOI] [PubMed] [Google Scholar]
- Haffner SM, Lehto S, Ronnemaa T, et al. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. 1998;339:229–34. doi: 10.1056/NEJM199807233390404. [DOI] [PubMed] [Google Scholar]
- Halcox JP, Schenke WH, Zalos G, et al. Prognostic value of coronary vascular endothelial dysfunction. Circulation. 2002;106:653–8. doi: 10.1161/01.cir.0000025404.78001.d8. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Antioxidant defence mechanisms:from the beginning to the end (of the beginning) Free Radic Res. 1999;31:261–72. doi: 10.1080/10715769900300841. [DOI] [PubMed] [Google Scholar]
- Halliwell B. The antioxidant paradox. Lancet. 2000a;355:1179–80. doi: 10.1016/S0140-6736(00)02075-4. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Why and how should we measure oxidative DNA damage in nutritional studies? How far have we come? Am J Clin Nutr. 2000b;72:1082–7. doi: 10.1093/ajcn/72.5.1082. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Whiteman M. Measuring reactive species and oxidative damage in vivo and in cell culture:how should you do it and what do the results mean? Br J Pharmacol. 2004;142:231–55. doi: 10.1038/sj.bjp.0705776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B, Zhao K, Whiteman M. Nitric oxide and peroxynitrite. The ugly, the uglier and the not so good:a personal view of recent controversies. Free Radic Res. 1999;31:651–69. doi: 10.1080/10715769900301221. [DOI] [PubMed] [Google Scholar]
- Hambrecht R, Adams V, Erbs S, et al. Regular physical activity improves endothelial function in patients with coronary artery disease by increasing phosphorylation of endothelial nitric oxide synthase. Circulation. 2003;107:3152–8. doi: 10.1161/01.CIR.0000074229.93804.5C. [DOI] [PubMed] [Google Scholar]
- Heart Protection Study Collaborative Group MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20 536 high-risk individuals:a randomised placebo-controlled trial. Lancet. 2002;360:23–33. [Google Scholar]
- Heitzer T, Schlinzig T, Krohn K, et al. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation. 2001;104:2673–8. doi: 10.1161/hc4601.099485. [Erratum:Circulation, 2003, 108:500.] [DOI] [PubMed] [Google Scholar]
- Hirschfield GM, Herbert J, Kahan MC, et al. Human C-reactive protein does not protect against acute lipopolysaccharide challenge in mice. J Immunol. 2003;171:6046–51. doi: 10.4049/jimmunol.171.11.6046. [DOI] [PubMed] [Google Scholar]
- Hristov M, Erl W, Weber PC. Endothelial progenitor cells:mobilization, differentiation, and homing. Arterioscler Thromb Vasc Biol. 2003a;23:1185–9. doi: 10.1161/01.ATV.0000073832.49290.B5. [DOI] [PubMed] [Google Scholar]
- Hristov M, Erl W, Weber PC. Endothelial progenitor cells:isolation and characterization. Trends Cardiovasc Med. 2003b;13:201–6. doi: 10.1016/s1050-1738(03)00077-x. [DOI] [PubMed] [Google Scholar]
- Huber SA, Sakkinen P, Conze D, et al. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 1999;19:2364–7. doi: 10.1161/01.atv.19.10.2364. [DOI] [PubMed] [Google Scholar]
- Ihlemann N, Rask-Madsen C, Perner A, et al. Tetrahydrobiopterin restores endothelial dysfunction induced by an oral glucose challenge in healthy subjects. Am J Physiol Heart Circ Physiol. 2003;285:H875–82. doi: 10.1152/ajpheart.00008.2003. [DOI] [PubMed] [Google Scholar]
- Karlseder J, Smogorzewska A, de Lange T. Senescence induced by altered telomere state, not telomere loss. Science. 2002;295:2446–9. doi: 10.1126/science.1069523. [DOI] [PubMed] [Google Scholar]
- Katusic ZS. Vascular endothelial dysfunction:does tetrahydrobiopterin play a role? Am J Physiol Heart Circ Physiol. 2001;281:H981–6. doi: 10.1152/ajpheart.2001.281.3.H981. [DOI] [PubMed] [Google Scholar]
- Kawano H, Motoyama T, Hirashima O, et al. Hyperglycemia rapidly suppresses flow-mediated endothelium-dependent vasodilation of brachial artery. J Am Coll Cardiol. 1999;34:146–54. doi: 10.1016/s0735-1097(99)00168-0. [DOI] [PubMed] [Google Scholar]
- Khreiss T, Jozsef L, Potempa LA, et al. Conformational rearrangement in C-reactive protein is required for proinflammatory actions on human endothelial cells. Circulation. 2004;109:2016–22. doi: 10.1161/01.CIR.0000125527.41598.68. [DOI] [PubMed] [Google Scholar]
- Kielstein JT, Boger RH, Bode-Boger SM, et al. Asymmetric dimethylarginine plasma concentrations differ in patients with end-stage renal disease:relationship to treatment method and atherosclerotic disease. J Am Soc Nephrol. 1999;10:594–600. doi: 10.1681/ASN.V103594. [DOI] [PubMed] [Google Scholar]
- Koc M, Bihorac A, Segal MS. Circulating endothelial cells as potential markers of the state of the endothelium in hemodialysis patients. Am J Kidney Dis. 2003;42:704–12. doi: 10.1016/s0272-6386(03)00906-5. [DOI] [PubMed] [Google Scholar]
- Kritharides L, Stocker R. The use of antioxidant supplements in coronary heart disease. Atherosclerosis. 2002;164:211–19. doi: 10.1016/s0021-9150(02)00011-4. [DOI] [PubMed] [Google Scholar]
- Krötz F, Riexinger T, Buerkle MA, et al. Membrane potential-dependent inhibition of platelet adhesion to endothelial cells by epoxyeicosatrienoic acids. Arterioscler Thromb Vasc Biol. 2004;24:595–600. doi: 10.1161/01.ATV.0000116219.09040.8c. [DOI] [PubMed] [Google Scholar]
- Laakso M. Hyperglycemia as a risk factor for cardiovascular disease in type 2 diabetes. Prim Care. 1999;26:829–39. doi: 10.1016/s0095-4543(05)70133-0. [DOI] [PubMed] [Google Scholar]
- Lagaud GJL, Mash-Khan E, Kai S, et al. Influence of type II diabetes on arterial tone and endothelium function in murine resistance arteries. J Vasc Res. 2001;38:578–89. doi: 10.1159/000051094. [DOI] [PubMed] [Google Scholar]
- Leiper J, Murray-Rust J, McDonald N, et al. S-nitrosylation of dimethylarginine dimethylaminohydrolase regulates enzyme activity:further interactions between nitric oxide synthase and dimethylarginine dimethylaminohydrolase. Proc Natl Acad Sci U S A. 2002;99:13527–32. doi: 10.1073/pnas.212269799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Wang Y, Padayatty SJ, et al. A new recommended dietary allowance of vitamin C for healthy young women. Proc Natl Acad Sci U S A. 2001;98:9842–6. doi: 10.1073/pnas.171318198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Forstermann U. Nitric oxide in the pathogenesis of vascular disease. J Pathol. 2000;190:244–54. doi: 10.1002/(SICI)1096-9896(200002)190:3<244::AID-PATH575>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2004;45 doi: 10.1146/annurev.pharmtox.45.120403.095748. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–43. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- Libby P, Willerson JT, Braunwald E. C-reactive protein and coronary heart disease [letter] N Engl J Med. 2004;351:295–6. [PubMed] [Google Scholar]
- Löhn M, Dubrovska G, Lauterbach B, et al. Periadventitial fat releases a vascular relaxing factor. FASEB J. 2002;16:1057–63. doi: 10.1096/fj.02-0024com. [DOI] [PubMed] [Google Scholar]
- Lonn E, Yusuf S, Hoogwerf B, et al. Effects of vitamin E on cardiovascular and microvascular outcomes in high-risk patients with diabetes:results of the HOPE study and MICRO-HOPE substudy. Diabetes Care. 2002;25:1919–27. doi: 10.2337/diacare.25.11.1919. [DOI] [PubMed] [Google Scholar]
- Loomans CJ, de Koning EJ, Staal FJ, et al. Endothelial progenitor cell dysfunction:a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes. 2004;53:195–9. doi: 10.2337/diabetes.53.1.195. [DOI] [PubMed] [Google Scholar]
- Mather KJ, Verma S, Anderson TJ. Improved endothelial function with metformin in type 2 diabetes mellitus. J Am Coll Cardiol. 2001;37:1344–50. doi: 10.1016/s0735-1097(01)01129-9. [DOI] [PubMed] [Google Scholar]
- Matoba T, Shimokawa H. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in animals and humans. J Pharmacol Sci. 2003;92:1–6. doi: 10.1254/jphs.92.1. [DOI] [PubMed] [Google Scholar]
- McGuire JJ, Ding H, Triggle CR. Endothelium-derived relaxing factors:a focus on endothelium-derived hyperpolarizing factor(s) Can J Physiol Pharmacol. 2001;79:443–70. [PubMed] [Google Scholar]
- McLeod DS, Lefer DJ, Merges C, et al. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol. 1995;147:642–53. [PMC free article] [PubMed] [Google Scholar]
- Meagher EA, Barry OP, Lawson JA, et al. Effects of vitamin E on lipid peroxidation in healthy persons. JAMA. 2001;285:1178–82. doi: 10.1001/jama.285.9.1178. [DOI] [PubMed] [Google Scholar]
- Minamino T, Komuro I. Role of telomere in endothelial dysfunction in atherosclerosis. Curr Opin Lipidol. 2002;13:537–43. doi: 10.1097/00041433-200210000-00010. [DOI] [PubMed] [Google Scholar]
- Minamino T, Miyauchi H, Yoshida T, et al. The role of vascular cell senescence in atherosclerosis:antisenescence as a novel therapeutic strategy for vascular aging. Curr Vasc Pharmacol. 2004;2:141–8. doi: 10.2174/1570161043476393. [DOI] [PubMed] [Google Scholar]
- Mondal M, Larussa VF, Agrawal KC. Synergistic antiadipogenic effects of HIV type 1 protease inhibitors with tumor necrosis factor α:suppression of extracellular insulin action mediated by extracellular matrix-degrading proteases. AIDS Res Hum Retroviruses. 2001;17:1569–84. doi: 10.1089/088922201753341988. [DOI] [PubMed] [Google Scholar]
- Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circ Res. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- Munteanu A, Zingg JM, Azzi A. Anti-atherosclerotic effects of vitamin E – myth or reality? J Cell Mol Med. 2004;8:59–76. doi: 10.1111/j.1582-4934.2004.tb00260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy C, Beckers J, Ruther Ü. Regulation of the human C-reactive protein gene in transgenic mice. J Biol Chem. 1995;270:704–8. doi: 10.1074/jbc.270.2.704. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Edelstein D, Brownlee M. The missing link:a single unifying mechanism for diabetic complications. Kidney Int Suppl. 2000;77:S26–30. doi: 10.1046/j.1523-1755.2000.07705.x. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–90. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- Nyström T, Nygren A, Sjohölm Å. Tetrahydrobiopterin increases insulin sensitivity in patients with type 2 diabetes and coronary heart disease. Am J Physiol Endocrinol Metab. 2004;287:E919–25. doi: 10.1152/ajpendo.00046.2004. [DOI] [PubMed] [Google Scholar]
- Pannirselvam M, Anderson TJ, Triggle CR. Endothelial cell dysfunction in type I and II diabetes – the cellular basis for dysfunction. Drug Dev Res. 2003;58:28–41. [Google Scholar]
- Pannirselvam M, Wiehler WB, Anderson TJ, et al. Enhanced vascular reactivity of small mesenteric arteries from diabetic mice is associated with enhanced oxidative stress and cyclooxygenase products. Br J Pharmacol. doi: 10.1038/sj.bjp.0706121. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paravicini TM, Gulluyan LM, Dusting GJ, et al. Increased NADPH oxidase activity, gp91phox expression, and endothelium-dependent vasorelaxation during neointima formation in rabbits. Circ Res. 2002;91:54–61. doi: 10.1161/01.res.0000024106.81401.95. [DOI] [PubMed] [Google Scholar]
- Paul A, Ko KW, Li L, et al. C-reactive protein accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109:647–55. doi: 10.1161/01.CIR.0000114526.50618.24. [DOI] [PubMed] [Google Scholar]
- Pedersen BK, Steensberg A, Fischer C, et al. The metabolic role of IL-6 produced during exercise:is IL-6 an exercise factor? Proc Nutr Soc. 2004;63:263–7. doi: 10.1079/PNS2004338. [DOI] [PubMed] [Google Scholar]
- Pieper GM. Enhanced, unaltered and impaired nitric oxide-mediated endothelium-dependent relaxation in experimental diabetes mellitus:importance of disease duration. Diabetologia. 1999;42:204–13. doi: 10.1007/s001250051140. [DOI] [PubMed] [Google Scholar]
- Pinkney JH, Downs L, Hopton M, et al. Endothelial dysfunction in type 1 diabetes mellitus:relationship with LDL oxidation and the effects of vitamin E. Diabet Med. 1999;16:993–9. doi: 10.1046/j.1464-5491.1999.00191.x. [DOI] [PubMed] [Google Scholar]
- Pradhan AD, Manson JE, Rifai N, et al. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–34. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- Rauscher FM, Goldschmidt-Clermont PJ, Davis BH, et al. Aging, progenitor cell exhaustion, and atherosclerosis. Circulation. 2003;108:457–63. doi: 10.1161/01.CIR.0000082924.75945.48. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Brown NJ, Vaughan DE, et al. Established and emerging plasma biomarkers in the prediction of first atherothrombotic events. Circulation. 2004;109(Suppl 1):IV6–19. doi: 10.1161/01.CIR.0000133444.17867.56. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Cushman M, Stampfer MJ, et al. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336:973–9. doi: 10.1056/NEJM199704033361401. [Erratum:N Engl J Med, 1997, 337:356.] [DOI] [PubMed] [Google Scholar]
- Ridker PM, Koenig W, Fuster V. C-reactive protein and coronary heart disease [letter] N Engl J Med. 2004;351:296–7. [PubMed] [Google Scholar]
- Ridker PM, Rifai N, Rose L, et al. Comparison of C-reactive protein and low-density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med. 2002;34:1557–65. doi: 10.1056/NEJMoa021993. [DOI] [PubMed] [Google Scholar]
- Roy S, Sala R, Cagliero E, et al. Expression of fibronectin induced by diabetes or high glucose:phenomenon with a memory. Proc Natl Acad Sci U S A. 1990;87:404–8. doi: 10.1073/pnas.87.1.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandow SL. Factors, fiction and endothelium-derived hyperpolarizing factor. Clin Exp Pharmacol Physiol. 2004;31:563–70. doi: 10.1111/j.1440-1681.2004.04048.x. [DOI] [PubMed] [Google Scholar]
- Shimizu K, Muramatsu M, Kakegawa Y, et al. Role of prostaglandin H2 as an endothelium-derived contracting factor in diabetic state. Diabetes. 1993;42:1246–52. doi: 10.2337/diab.42.9.1246. [DOI] [PubMed] [Google Scholar]
- Sidhu JS, Cowan D, Kaski JC. Effects of rosiglitazone on endothelial function in men with coronary artery disease without diabetes mellitus. Am J Cardiol. 2004;94:151–6. doi: 10.1016/j.amjcard.2004.03.051. [DOI] [PubMed] [Google Scholar]
- Sorescu D, Weiss D, Lassegue B, et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation. 2002;105:1429–35. doi: 10.1161/01.cir.0000012917.74432.66. [DOI] [PubMed] [Google Scholar]
- Stocker R. The ambivalence of vitamin E in atherogenesis. Trends Biochem Sci. 1999;24:219–23. doi: 10.1016/s0968-0004(99)01404-8. [DOI] [PubMed] [Google Scholar]
- Stuhlinger MC, Abbasi F, Chu JW, et al. Relationship between insulin resistance and an endogenous nitric oxide synthase inhibitor. JAMA. 2002;287:1420–6. doi: 10.1001/jama.287.11.1420. [DOI] [PubMed] [Google Scholar]
- Suda O, Tsutsui M, Morishita T, et al. Asymmetric dimethylarginine produces vascular lesions in endothelial nitric oxide synthase-deficient mice:involvement of renin-angiotensin system and oxidative stress. Arterioscler Thromb Vasc Biol. 2004;24:1682–8. doi: 10.1161/01.ATV.0000136656.26019.6e. [DOI] [PubMed] [Google Scholar]
- Suwaidi JA, Hamasaki S, Higano ST, et al. Long-term follow-up of patients with mild coronary artery disease and endothelial dysfunction. Circulation. 2000;101:948–54. doi: 10.1161/01.cir.101.9.948. [DOI] [PubMed] [Google Scholar]
- Sydow K, Munzel T. ADMA and oxidative stress. Atherosclerosis Suppl. 2003;4:41–51. doi: 10.1016/s1567-5688(03)00033-3. [DOI] [PubMed] [Google Scholar]
- Szalai AJ. The antimicrobial activity of C-reactive protein. Microbes Infect. 2002;4:201–15. doi: 10.1016/s1286-4579(01)01528-3. [DOI] [PubMed] [Google Scholar]
- Szalai AJ. C-reactive protein (CRP) and autoimmune disease:facts and conjectures. Clin Dev Immunol. 2004;11:221–6. doi: 10.1080/17402520400001751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szalai AJ, Briles DE, Volanakis JE. Human C-reactive protein is protective against fatal Streptococcus pneumoniae infection in transgenic mice. J Immunol. 1995;155:2557–63. [PubMed] [Google Scholar]
- Szalai AJ, McCrory MA. Varied biologic functions of C-reactive protein:lessons learned from transgenic mice. Immunol Res. 2002;26:279–87. doi: 10.1385/IR:26:1-3:279. [DOI] [PubMed] [Google Scholar]
- Szalai AJ, VanCott JL, McGhee JR, et al. Human C-reactive protein is protective against fatal Salmonella enterica serovar typhimurium infection in transgenic mice. Infect Immun. 2000;68:5652–6. doi: 10.1128/iai.68.10.5652-5656.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubes G. Cardiovascular disease. Does inflammation cut to the heart of the matter? Science. 2002;296:242–5. doi: 10.1126/science.296.5566.242. [DOI] [PubMed] [Google Scholar]
- Tepper OM, Galiano RD, Capla JM, et al. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106:2781–6. doi: 10.1161/01.cir.0000039526.42991.93. [DOI] [PubMed] [Google Scholar]
- Tershakovec AM, Frank I, Rader D. HIV-related lipodystrophy and related factors. Atherosclerosis. 2004;174:1–10. doi: 10.1016/S0021-9150(03)00246-6. [DOI] [PubMed] [Google Scholar]
- Tesfamariam B, Brown ML, Deykin D, et al. Elevated glucose promotes generation of endothelium-derived vasoconstrictor prostanoids in rabbit aorta. J Clin Invest. 1990;85:929–32. doi: 10.1172/JCI114521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesfamariam B, Cohen RA. Free radicals mediate endothelial cell dysfunction caused by elevated glucose. Am J Physiol. 1992;263:H321–6. doi: 10.1152/ajpheart.1992.263.2.H321. [DOI] [PubMed] [Google Scholar]
- Tesfamariam B, Jakubowski JA, Cohen RA. Contraction of diabetic rabbit aorta caused by endothelium-derived PGH2-TxA2. Am J Physiol. 1989;257:H1327–33. doi: 10.1152/ajpheart.1989.257.5.H1327. [DOI] [PubMed] [Google Scholar]
- Thum T, Borlak J. Mechanistic role of cytochrome P450 monooxygenases in oxidized low-density lipoprotein-induced vascular injury:therapy through LOX-1 receptor antagonism? Circ Res. 2004;94:e1–13. doi: 10.1161/01.RES.0000110081.03480.E9. [DOI] [PubMed] [Google Scholar]
- Tillett WS, Francis T., Jr Serological reactions in pneumonia with non-protein somatic fraction of pneumococcus. J Exp Med. 1930;52:561–71. doi: 10.1084/jem.52.4.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension:what is the clinical significance? Hypertension. 2004;44:248–52. doi: 10.1161/01.HYP.0000138070.47616.9d. [DOI] [PubMed] [Google Scholar]
- Touyz RM, Schiffrin EL. Reactive oxygen species in vascular biology:implications in hypertension. Histochem Cell Biol. 2004;122:339–52. doi: 10.1007/s00418-004-0696-7. [DOI] [PubMed] [Google Scholar]
- Triggle CR, Ding H. Endothelium-dependent hyperpolarizing factor:is there a novel chemical mediator? Clin Exp Pharmacol Physiol. 2002;129:153–60. doi: 10.1046/j.1440-1681.2002.03632.x. [DOI] [PubMed] [Google Scholar]
- Triggle CR, Hollenberg M, Anderson TJ, et al. The endothelium in health and disease – a target for therapeutic intervention. J Smooth Musc Res. 2003;39:249–67. doi: 10.1540/jsmr.39.249. [DOI] [PubMed] [Google Scholar]
- UNAIDS International AIDS Conference, Bangkok, 11–16 July 2004. 2004. Accessed 14 Jan 2005. URL: http://www.unaids.org/bangkok2004/report_pdf.html.
- Vallance P, Leone A, Calver A, et al. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet. 1992a;339:572–5. doi: 10.1016/0140-6736(92)90865-z. [DOI] [PubMed] [Google Scholar]
- Vallance P, Leone A, Calver A, et al. Endogenous dimethylarginine as an inhibitor of nitric oxide synthesis. J Cardiovasc Pharmacol. 1992b;20(Suppl 12):S60–2. doi: 10.1097/00005344-199204002-00018. [DOI] [PubMed] [Google Scholar]
- Van Den Berg CW, Taylor KE, Lang D. C-reactive protein-induced in vitro vasorelaxation is an artefact caused by the presence of sodium azide in commercial preparations. Arterioscler Thromb Vasc Biol. 2004;24:E168–71. doi: 10.1161/01.ATV.0000142807.92781.d9. [DOI] [PubMed] [Google Scholar]
- Vanhoutte PM. Endothelium-dependent hyperpolarizations:the history. Pharmacol Res. 2004;49:503–8. doi: 10.1016/j.phrs.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Verlohren S, Dubrovska G, Tsang SY, et al. Visceral periadventitial adipose tissue regulates arterial tone of mesenteric arteries. Hypertension. 2004;44:271–6. doi: 10.1161/01.HYP.0000140058.28994.ec. [DOI] [PubMed] [Google Scholar]
- Verma S, Anderson TJ. Fundamentals of endothelial function for the clinical cardiologist. Circulation. 2002;105:546–9. doi: 10.1161/hc0502.104540. [DOI] [PubMed] [Google Scholar]
- Verma S, Raj SR, Shewchuk L, et al. Cyclooxygenase-2 blockade does not impair endothelial vasodilator function in healthy volunteers:randomized evaluation of rofecoxib versus naproxen on endothelium-dependent vasodilatation. Circulation. 2001;104:2879–82. doi: 10.1161/hc4901.101350. [DOI] [PubMed] [Google Scholar]
- Verma S, Szmitko PE, Anderson TJ. Endothelial function:ready for prime time? Can J Cardiol. 2004;20:1335–9. [PubMed] [Google Scholar]
- Verma S, Szmitko PE, Yeh ET. C-reactive protein:structure affects function. Circulation. 2004;109:1914–17. doi: 10.1161/01.CIR.0000127085.32999.64. [DOI] [PubMed] [Google Scholar]
- Verma S, Wang CH, Li SH, et al. A self-fulfilling prophecy:C-reactive protein attenuates nitric oxide production and inhibits angiogenesis. Circulation. 2002;106:913–19. doi: 10.1161/01.cir.0000029802.88087.5e. [DOI] [PubMed] [Google Scholar]
- Voghel G, Thorin-Trescases N, Perrault LP, et al. Telomere length predicts premature senescence of the vascular endothelium in patients with risk factors for cardiovascular diseases. Circulation. 2004;110(17):368. [Google Scholar]
- Walder K, Kantham L, McMillan JS, et al. Tanis:a link between type 2 diabetes and inflammation? Diabetes. 2002;51:1859–66. doi: 10.2337/diabetes.51.6.1859. [DOI] [PubMed] [Google Scholar]
- Waldron GJ, Ding H, Lovren F, et al. Acetylcholine-induced relaxation of arteries isolated from mice lacking endothelial nitric oxide synthase. Br J Pharmacol. 1999;128:653–8. doi: 10.1038/sj.bjp.0702858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland A, Kerbl R, Berghold A, et al. C-reactive protein (CRP) as tumor marker in pediatric and adolescent patients with Hodgkin disease. Med Pediatr Oncol. 2003;41:1–5. doi: 10.1002/mpo.10286. [DOI] [PubMed] [Google Scholar]
- Williams SB, Goldfine AB, Timimi FK, et al. Acute hyperglycemia attenuates endothelium-dependent vasodilation in humans in vivo. Circulation. 1998;97:1695–701. doi: 10.1161/01.cir.97.17.1695. [DOI] [PubMed] [Google Scholar]
- Wolford JK, Gruber JD, Ossowski VM, et al. A C-reactive protein promoter polymorphism is associated with type 2 diabetes mellitus in Pima Indians. Mol Genet Metab. 2003;78:136–44. doi: 10.1016/s1096-7192(02)00230-5. [DOI] [PubMed] [Google Scholar]
- Wu R, Lamontagne D, de Champlain J. Antioxidative properties of acetylsalicylic acid on vascular tissues from normotensive and spontaneously hypertensive rats. Circulation. 2002;105:387–92. doi: 10.1161/hc0302.102609. [DOI] [PubMed] [Google Scholar]
- Yang D, Levens N, Zhang JN, et al. Specific potentiation of endothelium-dependent contractions in SHR by tetrahydrobiopterin. Hypertension. 2003;41:136–42. doi: 10.1161/01.hyp.0000047669.93078.a7. [DOI] [PubMed] [Google Scholar]
- Yasojima K, Schwab C, McGeer EG, et al. Generation of C-reactive protein and complement components in atherosclerotic plaques. Am J Pathol. 2001;158:1039–51. doi: 10.1016/S0002-9440(10)64051-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh ETH. CRP as a mediator of disease. Circulation. 2004;109(Suppl):II:II11–14. doi: 10.1161/01.CIR.0000129507.12719.80. [DOI] [PubMed] [Google Scholar]
- Zanetti M, Sato J, Katusic ZS, et al. Gene transfer of superoxide dismutase isoforms reverses endothelial dysfunction in diabetic rabbit aorta. Am J Physiol Heart Circ Physiol. 2001;280:H2516–23. doi: 10.1152/ajpheart.2001.280.6.H2516. [DOI] [PubMed] [Google Scholar]
- Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001;414:782–7. doi: 10.1038/414782a. [DOI] [PubMed] [Google Scholar]
- Zoccali C, Bode-Boger S, Mallamaci F, et al. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease:a prospective study. Lancet. 2001;358:2113–17. doi: 10.1016/s0140-6736(01)07217-8. [DOI] [PubMed] [Google Scholar]
- Zou MH, Cohen R, Ullrich V. Peroxynitrite and vascular endothelial dysfunction in diabetes mellitus. Endothelium. 2004;11:89–97. doi: 10.1080/10623320490482619. [DOI] [PubMed] [Google Scholar]