

Abstract
The renin-angiotensin-system (RAS) is a cascade of enzymatic reactions resulting ultimately in the formation of angiotensin II. Recent research has expanded the knowledge about the RAS by adding new components to the pathways: angiotensin-(1–5) [Ang-1–5], angiotensin-(1–7) [Ang-(1–7)], angiotensin-(1–9) [Ang-(1–9)], an ACE homologous enzyme, ACE2, and the G-protein-coupled receptor mas as a molecular receptor for Ang-(1–7). Although previous studies provided some conflicting evidence about the relevance of Ang-(1–7) in the regulation of vascular and renal function, data now demonstrate that Ang-(1–7) contributes to the cardiovascular effects of ACE-inhibitors (ACE-I) and AT1-receptor-blockers (ARBs) both in experimental conditions and in humans. This review summarizes and critically discusses the currently available experimental and clinical study evidence for the role of Ang-(1–7) as a vasodilator and anti-trophic peptide in cardiovascular drug therapy. In addition, the potential therapeutic impact of currently available RAS blocking agents (ACE-I and ARBs) and new agents still under development (renin-inhibitors) on the RAS-effector peptides is highlighted.
Keywords: renin-angiotensin-system, cardiovascular drug therapy, Angiotensin-(1–7)
Introduction
The importance of the renin-angiotensin-system (RAS) in the development of hypertension and cardiovascular disease is well established (Unger 2002). Chronic RAS activation has been identified as a major factor contributing to progressive dysfunction of end organs including blood vessels, the kidneys and the heart (Unger 2002). This prompted the development of agents, capable of blocking the RAS and reversing the associated pathologies.
The first group of drugs targeting the RAS which became available in the late 1970-ies were ACE-inhibitors (ACE-I) (Brunner et al 1978). Captopril was the first ACE-I which was and still is clinically used for the treatment of hypertension. The first–although peptide – AT1-receptor-blocker (ARB) saralasin was identified in 1971 (Pals et al 1971) and the antihypertensive effect demonstrated in patients in 1979 (Case et al 1979). Due to the lack of oral bioavailability this agent did not achieve wider clinical use. More than ten years elapsed before the first selective synthetic AT1-receptor antagonist (Losartan) became available for the treatment of hypertension (Johnston 1995). It soon became clear that the reduction in cardiovascular events demonstrated in clinical trials with ACE-I (Yusuf et al 2000) and ARBs (Pitt et al 1997) was not only related to their blood pressure lowering capacity (Burnier and Brunner 2000; Hayoz 2002; Cipollone et al 2004) but to a more complex effect in which reversal of target organ damage might be explained by inhibition of synthesis or activity of angiotensin II (Ang II) or transformation to a metabolite with pleiotropic activity.
The understanding of the RAS has been extended in the last few years with respect to the recognition that angiotensin-(1–7) [Ang-(1–7)] – a metabolite cleaved from Ang I and Ang II – is a biologically active product of the renin-angiotensin cascade (Carey and Siragy 2003). This heptapeptide has been recognized to counterbalance the effects of Ang II by stimulating the activity of vasodilator autocoids and nitric oxide (NO) (Ferrario et al 1997). The characterization of Ang-(1–7) (Schiavone et al 1988; Campagnole-Santos et al 1989; Ferrario et al 1991) as an amino-terminal angiotensin peptide product generated from either angiotensin I (Ang I) or Ang II provided a foundation for the pursuit of a new concept regarding the regulation of cardiovascular function by the RAS. While prostacyclin, bradykinin and NO act as vasodilator hormones limiting the pressor and proliferative actions of Ang II, it had not been considered that products derived independently from either angiotensin I (Ang I) or Ang II could also function to counterbalance the actions of Ang II.
This review focuses on the currently available evidence for the contribution of the vasodilator peptide Ang-(1–7) to the beneficial effects of cardiovascular drugs blocking the RAS and discusses the different pharmacological actions of ACE-I and ARBs on the balance of different RAS-effectors and the potential of new concepts like vasopeptidase and renin inhibition.
Ang-(1–7)
Ang-(1–7) as an antagonist of angiotensin II
Eighteen years ago the presence of the heptapeptide Ang-(1–7) as a product of the metabolism of Ang I found in brain homogenates (Santos et al 1988) led to the later demonstration of its action in counteracting the pressor and baroreflex effects of Ang II (Campagnole-Santos et al 1989; Benter et al 1993; Benter et al 1995a). The existence of biologically active fragments, other than Ang II, suggests a process of biotransformation involving a cascade of multiple upstream enzymes but also raises the problem that the organization of the physiological network determining the actions of Ang II needs revision to account for the actions of the additional peptides. Figure 1 gives an overview about the pathways for the formation of biologically active angiotensin peptides. Two important and recently published findings provide strong evidence for an extended understanding of the RAS as a regulatory system which does not necessarily have to be pharmacologically inhibited in order to treat hypertension and RAS-mediated end-organ damage but which can be pharmacologically modified by amplifying pathways generating Ang-(1–7). This has the potential to counterbalance the detrimental effects of Ang II: First, identification of the ACE-homologue ACE2 which forms Ang-(1–7) from Ang II (Donoghue et al 2000; Tipnis et al 2000; Crackower et al 2002) and second the identification of the mas-receptor (Santos et al 2003) as molecular target structure for the Ang-(1–7)-molecule mediating its beneficial effects.
Figure 1.
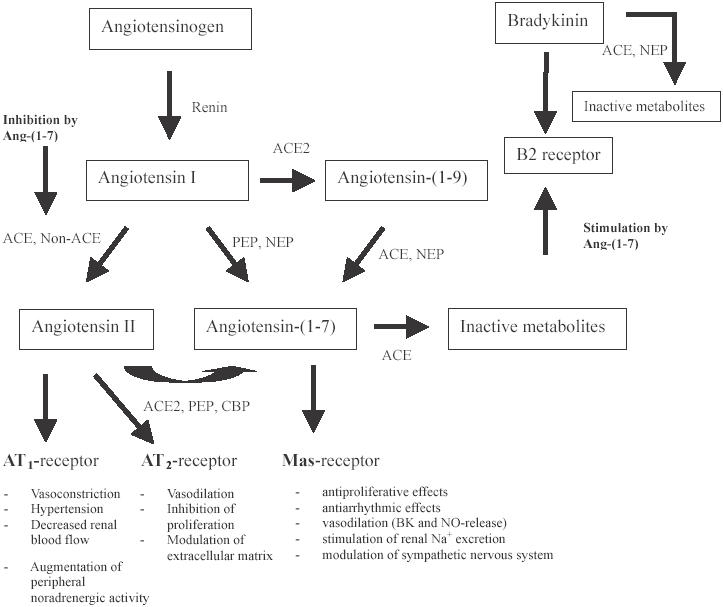
Pathways for the formation of biologically active angiotensin peptides.
Abbreviations: ACE, angiotensin-converting enzyme; ACE2, ACE-related carboxypeptidase; CBP, carboxypeptidase; BK, bradykinine; CBP, carboxypeptidase; EDRF, endothelium derived relaxing factor; NEP, neutral endopeptidase; NO, nitric oxide; PEP, prolylendopetidase; PKG, proteine kinase G.
Figure 2.
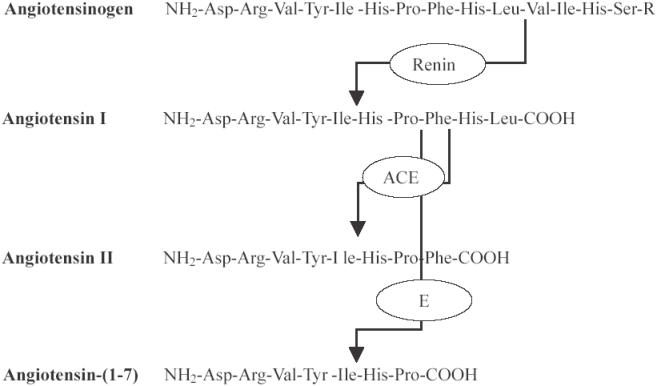
Systematic overview of angiotensinogen (its crucial n-terminal part), angiotensin I, angiotensin II, and angiotensin-(1-7) with sites of enzymatic cleavage.
Origin of Ang-(1–7)
Ang-(1–7) is a fragment of Ang I which can be cleaved by endopeptidases through the removal of the last three amino acids of the Ang I precursor molecule. The relative importance of the endopeptidases, prolyl endopeptidase 24.26 (PEP), neutral endopeptidases (NEP) 24.11 (neprilysin), and thimet oligopeptidase 24.15 in the conversion of Ang I into Ang-(1–7) appears to depend on both tissue enzyme distribution and substrate availability since neprilysin is highly active in the circulation and vascular endothelium whereas in vascular smooth muscle Ang-(1–7) formation is dependent on the hydrolytic activity of thimet oligopeptidase 24.15 (Welches et al 1993; Chappell et al 1994).
More recently, however, a pathway for Ang-(1–7) production has been demonstrated through the cloning and characterization of an ACE homologue–termed ACE2. This enzyme, shown to convert Ang II into Ang-(1–7) established a new pathway by which the trophic, vasoconstrictor, and pro-fibrotic effects of Ang II would be mitigated by this alternate processing pathway (see Figure 1).
ACE2 was identified as a new homologue of ACE (Donoghue et al 2000; Tipnis et al 2000; Crackower et al 2002) which – in contrast to ACE – is not inhibited by ACE-I nor does it share the same catalytic properties. The catalytic activity of ACE2 on Ang II as a substrate is much higher than its ability to cleave Ang-(1–9) from Ang I. ACE2 exhibits the highest efficacy (kcat/km) among Ang-(1–7)-forming enzymes and a 500-fold greater kcat/Km for Ang II compared with Ang I. Determination of the kcat/Km ratio gives a measurement of the substrate specificity. Thus, ACE2 appears to function to decrease Ang II concentration. ACE2 exists in both soluble and membrane-bound forms with high expression in the kidney, heart, cardiovascular tissues, brain and testes (Harmer et al 2002). Animal studies in the ACE2 knockout model demonstrated higher circulating and tissue levels of Ang II suggesting that reductions in ACE2 expression may lead to higher endogenous levels of Ang II and contribute to cardiac and renal pathologies associated with this model (Crackower et al 2002). Therefore, ACE2 might have an important function as a counter-regulatory enzyme to decrease local cardiac Ang II concentrations.
A way to degrade Ang-(1–7) is ACE which hydrolyses Ang-(1–7) to Ang-(1–5), thus regulating/limiting the physiological effects of Ang-(1–7) (Chappell et al 1998; Deddish et al 1998).
Ang-(1–7) receptor
Several studies gave evidence for the existence of a non-AT1/AT2-receptor that mediates the effects of Ang-(1–7) (Tallant et al 1991; Campagnole-Santos et al 1992; Diz and Pirro, 1992; Jaiswal et al 1992). This was obtained using the selective Ang-(1–7)-antagonist A-779 (Ambuhl et al 1994; Santos et al 1994). In addition, studies in mas-recep-tor transfected cells using Ang-(1–7) or the non-peptide Ang-(1–7)-agonist AVE-0991 showed that the mas-receptor mediates the effect of Ang-(1–7) on prostaglandins and NO release (Santos et al 2003; Pinheiro et al 2004). Recent studies using antisense probes directed to the cardiac mas receptor further showed abolition of the anti-hypertrophic effects of Ang-(1–7) on cardiac myocytes (Tallant et al 2005). These effects were not blocked by specific AT1–or AT2-receptor-blockers. The mas proto-oncogene encodes a seven-transmembrane – domain G-protein-coupled orphan receptor that was erroneously identified as an Ang II receptor in the late 1980ies. mas mRNA has been detected in the heart, testes, kidney, and the brain (Metzger et al 1995). Isolated hearts of mas-deficient mice (see (Walther et al 1998) for details about the phenotype of mas-deficient mice) showed marked changes in cardiac function. The interaction of Ang-(1–7) with its mas-receptor may have an important role in the regulation of cardiac function (Castro et al 2005). Today it is known that the mas-receptor mediates antiproliferative and antiarrhythmic effects, leads to vasodilation via bradykinin (BK) and NO-release, and stimulates renal sodium excretion and the sympathetic nervous system function.
Ang-(1–7) actions in preclinical studies
Renal actions of Ang-(1–7)
The RAS is a key regulator of kidney function, playing an essential role in the homeostasis of blood volume and hydro-electrolyte balance (Hall, 1991). Evidence suggests that not only Ang II but also Ang-(1–7) plays a significant role in renal function. Ang-(1–7) has been described as a potent diuretic and natriuretic agent (Andreatta-van Leyen et al 1993; DelliPizzi et al 1994; Handa et al 1996). It increases the renal blood flow in anesthetized rats (Sampaio et al 2003) and produces afferent arteriolar relaxation through specific receptor-mediated NO-release in isolated kidneys of rabbits (Ren et al 2002). In humans, the concentration of Ang-(1–7) in renal veins is several times higher than in the systemic circulation (Admiraal et al 1990). In addition, Ang-(1–7) is excreted into the urine of normal healthy volunteers in amounts 2.5 fold higher than measured in the plasma (Ferrario et al 1998). Control studies in untreated hypertensive patients showed a significantly reduced excretion of Ang-(1–7). Importantly, urinary concentrations of Ang-(1–7) showed an inverse correlation with blood pressure and were suggestive for the association with hypertension. The relatively higher concentrations of Ang-(1–7) in urine compared with plasma provide evidence that locally produced Ang-(1–7) may contribute to the regulation of renal function.
Cardiovascular actions of Ang-(1–7)
Ang-(1–7) is formed (Santos et al 1992) and metabolized (Chappell et al 1998) in endothelial cells. Vasorelaxant effects of the peptide have been demonstrated in animals in several vascular beds (see Table 1). Ang-(1–7)-induced vasorelaxation mainly results from amplification of bradykinin-induced dilation, by stimulation of vasodilator prostaglandins and by mediation of NO-release. In some vascular beds data suggest a role for Ang-(1–7) in mediating EDRF – vasodilation. The biological actions of Ang-(1–7) are both activation of peripheral vasodilatory mechanisms and antitrophic effects mediated by the inhibition of protein synthesis (see Table 2). Ang-(1–7) exerts biological effects on three critical organ systems regulating blood pressure (brain, blood vessels, and kidney). The most important studies providing evidence for active vascular effects of Ang-(1–7) are summarized in Table 3. Preclinical studies demonstrate an important action of Ang-(1–7) in potentiation of the vasodilator actions of bradykinin. Roks et al (1999) showed in human internal mammary arteries that contractions induced by Ang I and Ang II were antagonized by Ang-(1–7) in a non-competitive way, with a 60% inhibition of the maximal response to Ang II. The data further revealed an ACE-inhibiting effect by Ang-(1–7) in plasma and atrial tissue up to 100%. At supraphysiologic concentrations a vasoconstrictive effect of Ang-(1–7) has also been postulated – most likely through a weak AT1-receptor agonist action (Santos et al 2000). The observation that Ang-(1–7) increases following long-term-administration of ACE-I and ARBs (Santos et al 2000) raises the possibility that Ang-(1–7) might contribute to the pharmacological effects of both ACE-I and ARBs.
Table 1.
Cardiovascular actions of Ang-(1–7)
| Ang-(1–7) — effect | Model | Study |
|---|---|---|
| Amplification of vasodilation mediated by bradykinin | Conscious rats | (Paula et al 1995) |
| Reduction of NE-release acting through a BK/NO-mediated mechanism stimulating cGMP/PKG-signaling | Hypertensive rat | (Gironacci et al 2004) |
| Coronary vasodilation mediated by NO | Canine coronary arteries in vitro | (Brosnihan et al 1996) |
| Release of vasodilator prostaglandins | Sprague-Dawley rats | (Benter et al 1993) |
| Reduction in plasma vasopressin concentration, blood pressure reduction | Hypertensive rats | (Benter et al 1995b) |
| Active vasodilation | Isolated rabbit afferent arterioles | (Ren et al 2002) |
| Bradykinin potentiation | Arterioles hypertensive rats in vivo | (Fernandes et al 2001) |
| Release of NO by Ang-(1–7) | Porcine coronary endothelium | (Porsti et al 1994) |
| Stimulation and release of vasodilator prostaglandins | Porcine aortic endothelial cells | (Jaiswal et al 1992) |
| Induction of bradykinin-mediated hypotensive responses | Anesthetized rat | (Abbas et al 1997) |
| Augmentation of bradykinin-induced vasodilation | Canine coronary artery rings | (Li et al 1997) |
| Endothelium-dependent relaxation | Canine middle cerebral artery | (Feterik et al 2000) |
| Cerebral vasodilation mediated by prostaglandins | Piglet pial arterioles | (Meng and Busija 1993) |
| Vasodilation mediated by EDRF | Feline mesenteric vascular bed | (Osei et al 1993) |
| Relaxation potentiated by losartan | Rat aorta | (le Tran and Forster 1997) |
| Vasodilation in the cutaneous and implant vasculature | Newly formed vasculatures (sponge implants) | (Machado et al 2002) |
| Bradykinin-induced vasodilation by Ang-(1–7) | Anestethized Wistar Rats | (Oliveira et al 1999) |
| Unmasking of a bradykinin mediated potentiation of ACE-inhibitors | Spontaneously hypertensive rats | (Fernandes et al 2001) |
Abbreviations: See Figure 1.
Table 2.
Biological actions of Ang-(1–7)
| Organ or system | Biological action | Reference |
|---|---|---|
| Cellular actions | • Stimulation of release of PGE2 and 6-keto-PGF1α | (Trachte et al 1990; Jaiswal et al 1992, 1993a, b; Benter et al 1995b) |
| • Augmentation of the vasodilator action of bradykinin | (Porsti et al 1994; Santos et al 1994; Paula et al 1995; Abbas et al 1997; Li et al 1997; Lima et al 1997) | |
| • Increased release of NO | (Osei et al 1993; Brosnihan et al 1996; Abbas et al 1997; Li et al 1997) | |
| • Antiproliferative actions in vascular smooth muscle | (Freeman et al 1996) | |
| Brain | • Stimulation of vasopressin release | (Schiavone et al 1988; Baracho et al 1995; Santos et al 1996) |
| • Facilitation of baroreflexes | (Campagnole-Santos et al 1989; Campagnole-Santos et al 1992; Silva et al 1993) | |
| Blood vessels | • Vasodilation and antihypertensive actions | (Benter et al 1993, 1995a, b) |
| Kidney | • Diuresis and natriuresis | (DelliPizzi et al 1994; Handa et al 1996; Andreatta-van Leyen et al 1993; Hilchey and Bell-Quilley 1995) |
| • Inhibition of tubular sodium and bicarbonate transport | (Handa et al 1994, 1996) |
Abbreviations: See Figure 1.
Table 3.
Interactions between Ang-(1–7), kinins, and Ang II in kidney and blood vessels
| Action | Model | Reference |
|---|---|---|
| Potentiation of bradykinin by Ang-(1–7) by kinins | • Normo- and hypertensive rats | • (Santos et al 2000) |
| • (Paula et al 1995) | ||
| • (Fernandes et al 2001) | ||
| • Canine coronary arteries | • (Brosnihan et al 1996; Li et al 1997) | |
| • Isolated rat hearts | • (Almeida et al 2000) | |
| • Conscious male Wistar rats | • (Bomtempo et al 1998) | |
| • Chinese hamster ovary cells transfected with human cDNA for BK-B2receptors and ACE | • (Deddish et al 1998) | |
| Mediation of Ang-(1–7) actions by kinins | • Canine coronary arteries | • (Brosnihan et al 1996; Li et al 1997) |
| • Conscious male Wistar rats | • (Bomtempo et al 1998) | |
| • Bovine aortic endothelial cells | • (Heitsch et al 2001) | |
| • Anesthetized rats | • (Abbas et al 1997) |
Interaction with bradykinin
Most of the interactions between Ang-(1–7) and bradykinin (BK) have been reported to occur in blood vessels (Paula et al 1995; Brosnihan et al 1996; Hecker et al 1997; Li et al 1997; Santos et al 2000; Almeida et al 2000; Fernandes et al 2001). Two major types of interactions are postulated: 1. potentiation of BK by Ang-(1–7) and 2. mediation of the vascular actions of Ang-(1–7) by kinins. An overview about the currently available study evidence demonstrating interactions between bradykinin and Ang-(1–7) is summarized in Table 3. The mechanism underlying the BK potentiating activity of Ang-(1–7) is complex: There is considerable evidence for mas-receptor-mediated facilitation of NO release (Li et al 1997; Almeida et al 2000; Heitsch et al 2001) and/or prostaglandins (Paula et al 1995; Almeida et al 2000; Fernandes et al 2001), endothelium-derived hyperpolarizing factor (Fernandes et al 2001), binding to ACE and because of that facilitation of the crosstalk between ACE and BK-B2 receptors (Deddish et al 1998) and ACE-inhibition (Li et al 1997; Chappell et al 1998). It is assumed that the relative contribution of each of these mechanisms changes from vascular bed to vascular bed, with species and probably with vessel diameter (Santos et al 2000).
Role of Ang-(1–7) as physiologic antagonist of angiotensin II
The first study describing an interaction between Ang-(1–7) and Ang II was published by Bovy et al (1989) who described inhibition of the contractile effect of Ang II in the rabbit aorta by the Ang-(1–7) analogue Sar1-Ang-(1–7). Several studies confirmed the ability of Ang-(1–7) to antagonize the vascular effects of Ang II (Mahon et al 1994; Roks et al 1999; Ueda et al 2000). Most evidence about the physiological actions of Ang-(1–7) has been demonstrated in animal models. However, there is also evidence from human studies: In a study performed in the arterial vascular bed of healthy young men, Ang-(1–7) was able to antagonize Ang II-induced vasoconstriction (Ueda et al 2000) by shifting the dose-response-curve of Ang II to the right. Several mechanisms have been suggested for this interaction of Ang-(1–7) and Ang II in blood vessels:
The weak constrictive effect described in the study by Ueda (2000) (humans) and in the studies of Abbas (1997) and Benter (1995b) (animals) is mediated by low binding of Ang-(1–7) to the AT1-receptor causing an agonistic effect.
Interaction of Ang-(1–7) with extracellular Ca2+ influx into smooth muscle cells might also contribute to this weak constrictive effect (Chansel et al 2001).
Because Ang-(1–7) does not antagonize the vasoconstrictor action of α-adrenergic drugs in vitro (Mahon et al 1994) or in human forearm (Ueda et al 2000) it seems unlikely that these effects contribute to its antagonistic effects of Ang II-actions.
An influence of Ang-(1–7) on the synthesis of Ang II at the mRNA-level has been described (Moritz et al 2001).
ACE-inhibiting properties of Ang-(1–7) have been described (Li et al 1997, Tom et al 2001) based on a crosstalk between ACE and the BK-B2 receptor and mas-receptor-mediated changes in the coupling and/or signalling of bradykinin.
A crosstalk between the mas-receptor and other receptors such as AT1-and AT2-receptors may counterbalance constrictive pathways towards pathways resulting in vasodilation.
Ang-(1–7) potentiates the vasodilatory and hypotensive effects of bradykinin (Paula et al 1995; Oliveira et al 1999; Almeida et al 2000; Tom et al 2001).
Interpretation of currently available study evidence with Ang-(1–7) in humans
All human trials investigating the cardiovascular effects of Ang-(1–7) are summarized in Table 4. Sasaki and co-workers (Sasaki et al 2001) reported vasodilation in the human forearm whereas Davie and McMurray (Davie and McMurray 1999) did not observe an acute hemodynamic short-term effect of Ang-(1–7) in ACE-I treated patients. Failure to obtain a vasodilator response in patients given an ACE-I is not surprising as this treatment is associated with increased Ang-(1–7) levels due to both prevention of peptide metabolism by ACE and increased production from elevated levels of Ang I. In keeping with this interpretation, increased circulating Ang-(1–7)-levels have been clearly demonstrated in humans after therapy with RAS-inhibitors (Chappell et al 1998; Iyer et al 1998b; Davie and McMurray 1999). Even if we assume that there is only a weak or even no agonistic short-term effect of Ang-(1–7) – infusion favoring vasodilation in the human forearm vascular bed, this does not exclude hemodynamic effects in other vascular beds (eg, veins) or beneficial long-term-effects after drug therapy with ACE-I or ARBs mediated by increases in Ang-(1-7)-peptide levels. It has to be noted that the long-term effects of ACE-I and ARBs on Ang-(1–7)-metabolism have not been investigated in humans so far.
Table 4.
Studies investigating the effects of Ang-(1–7) in humans
| Author | Subjects | Methods | Results | Interpretation |
|---|---|---|---|---|
| (Roks et al 1999) | Human arteries in vivo | Internal mammary arteries; Incubation with Ang-(1–7) IC 50: 3 and 4 mmol/L | • ACE activity in plasma and atrial tissue was inhibited by Ang-(1–7) up to 100% | • Ang-(1–7) blocks Ang II induced vasoconstriction and inhibits ACE in human cardiovascular tissues |
| • Ang-(1–7) may be an important modulator of the human RAS. | ||||
| (Davie and McMurray 1999) | Patients with heart failure on treatment with an ACE-inhibitor | Brachial artery infusions of Ang-(1–7), venous occlusion plethys-mography; 5–50.000 pmol/min. | • No effect of Ang-(1–7) on its own or any effect of Ang-(1–7) on the response to bradykinin | • Ang-(1–7) is biologically inactive in the forearm circulation of patients with heart failure treated with an ACE-inhibitor |
| (Ueda et al 2000) | Healthy normotensive men, forearm arteries | Brachial artery infusions of Ang-(1–7) 0.1–2000 pmol/min. | • Ang-(1–7) at 0.5–40 nmol/min caused weak but significant vasoconstriction | • Ang-(1–7) antagonizes vasoconstriction by Ang II in human resistant vessels and might act as an endogenous Ang II antagonist |
| • Ang-(1–7) at 100 pmol/min, but not at 10 pmol/min, significantly shifted the Ang II DRC towards the right | ||||
| • Ang-(1–7) did not affect the DRC of NA | ||||
| (Ueda et al 2001) | Normotensive healthy men, forearm arteries | Coinfusion of bradykinin with placebo, Ang-(1–7) 1000 pmol/min, Angiotensin II and L-NMMA | • Ang-(1–7) potentiated bradykinine-induced vasodilation | • Ang-(1–7) potentiates the vasodilating effect of bradykinin through mechanisms involving nitric oxide release in human forearm resistance vessels. |
| • Dilation was completely abolished by L-NMMA | ||||
| • Ang-(1–7) did not affect the vasodilating effects of Acetylcholine or nitro-prusside | ||||
| (Wilsdorf et al 2001) | Normotensive healthy men Human forearm resistant vessels | Intraarterial infusion of Ang-(1–7) (10, 100, 300 pmol/min), bradykinin (47, 94, 189 pmol/min and Ang I (1, 10, 30 pmol/min.) | • No effect of Ang-(1–7) on forearm blood flow | • Does not support a role of Ang-(1–7) given at supraphysiological doses in the regulation of human peripheral vascular resistance |
| (Sasaki et al 2001) | Patients with essential hypertension and normo-tensive control subjects | Intraarterial infusion of Ang-(1–7) 10−10, 10−9, 10−8 mol/min | • Ang-(1–7) infusion significantly increased forearm bloodflow dose-dependently in normo- and hypertensive patients | • Ang-(1–7) causes vasodilation in forearm circulation of normotensive subjects and patients with essential hypertension through a pathway independent of NO-synthesis |
| • Ang-(1–7)-induced dilation was similar in both groups | ||||
| • L-NMMA coinfusion did not alter the forearm blood flow response to Ang-(1–7) |
Abbreviations: NA, noradrenaline.
Although it is not completely clear from human studies if exogenously infused Ang-(1–7) acts as a vasodilator (Wilsdorf et al 2001), the available experimental evidence suggests that Ang-(1–7) contributes to the cardiovascular effects of ACE-I and ARBs by directly acting as an ACE-I or by an interaction with ACE favoring a crosstalk between the ACE-, the BK-B2- and the mas-receptor by mediating changes in coupling and signalling of bradykinin. However, randomized controlled studies systematically investigating the effects of RAS-blockade with ACE-I and ARBs on Ang-(1–7)-biology and catabolism have not been conducted. The main problem with the results obtained in human studies might be related to the extremely low case numbers included in clinical studies (usually n = 8) and to methodological differences in the study protocols. Therefore, the initiation of large randomized controlled clinical studies systematically investigating long-term-effects of pharmacological RAS-blockade on Ang-(1–7)-metabolism seems to be highly desirable.
We have recently demonstrated in healthy male subjects that a 4 week treatment period with 150 mg of the AT1-receptor-antagonist irbesartan results in significant increases of Ang-(1–7) peptide levels which points towards a contribution of Ang-(1–7) to the antihypertensive and beneficial vascular effects of AT1-receptor blockade (Schindler et al 2007). This study provides evidence that Ang-(1–7)-biol-ogy might also be involved in humans when inhibitors of the RAS are being used.
In addition it has to be noted that the majority of published human studies points towards a RAS-modulatory role of Ang-(1–7) in humans (Roks et al 1999; Ueda et al 2000; Sasaki et al 2001; Ueda et al 2001) supporting vasodilation. Especially the recent data showing a role for ACE2 in cardiopulmonary pathophysiology (Reudelhuber 2006) and clinical data from healthy human subjects (Schindler et al 2007) suggest that the effects of drug treatment with ACE-I and ARBs merit another look with regard to Ang-(1–7)-biology and its relevant signalling pathways.
Pharmacologic RAS inhibitors and Ang-(1–7)
Different actions of ACE-I and ARBs on the RAS
The general ability of ACE-I and of ARBs to attenuate virtually all of the cardiovascular actions of the RAS is commonly accepted (for review see (Dendorfer et al 2005)). Although ACE-I and ARB block the same system, they have important mechanistic differences because they act on different sites of the RAS (Burnier and Brunner 2000). The differences between the two drug classes have a differential impact on the affected angiotensin receptor subtypes, a different involvement of peptide hormones other than Ang II, and possible differences in the efficacy of RAS suppression. ACE-I reduce the stimulation of AT1 and AT2 receptors, but inhibition may be overcome by activation of renin, by induction of ACE, and by alternative Ang II forming enzymes such as chymase. Inhibition of ACE blocks the degradation of various peptides, especially the vasodilator bradykinin. ACE-I potentiate the effects of bradykinin by 3–50-fold (Bonner et al 1990). Bradykinin stimulates NO-release from endothelial cells via B2-receptors (Unger, 2002). These effects might contribute to the antihypertensive effects of ACE-I.
Effect of ACE-I and ARBs on Ang-(1–7) generation
Drug therapy with an ACE-I compared with ARB-therapy exerts different effects on the RAS-balance. Pharmacologically blocking ACE results in lower circulating Ang II peptide levels which means that less substrate is available for ACE2 and for Ang-(1–7)-generation. The catabolism of Ang-(1–7) and Ang II-levels is decreased by ACE-I-treatment, whereas the level of Ang I which is a substrate for Ang-(1–7)-forming enzymes to generate Ang-(1–7) is increased. Ang-(1–7) can then act via the mas-receptor (see also Figure 1). The observation that the Ang-(1–7) antagonist D-Ala7-Ang-(1–7) reverts the potentiation of bradykinin by enalapril or enalaprilat in mesenteric microvessels (Fernandes et al 2001), or attenuates the potential hypotensive response to BK in captopril-treated rats (Maia et al 2004) indicates that an Ang-(1–7)-related mechanism significantly contributes to the hypotensive and beneficial effects of ACE-I.
ARBs completely block all effects mediated via the AT1-receptor which results in increased Ang II levels. This increase in circulating Ang II levels causes a shunting of Ang II to the unopposed AT2-receptor. It has been demonstrated experimentally that AT2-receptor stimulation enhances bradykinin-generation, so that some vasodilator or diuretic actions of ARB may arise through this pathway (Carey et al 2001). However, bradykinin potentiation by ARBs is less to that of ACE-I which might have impact on the therapeutically desired actions. On the other hand, AT1-receptor blockade prevents the AT1-mediated actions of Ang II and might hence be capable of potentiating its effects via AT2-receptors. However, work by Ferrario and colleagues found no evidence for a role of AT2-receptors in mediating the vasodilator response produce by long-term administration of lisinopril and losartan (Iyer et al 1998a, b, 2000). In addition, increased levels of both Ang I and Ang II change the metabolic cascade through ACE2 towards Ang-(1–7)-generation as recent work demonstrated that administration of either losartan or olmesartan upregulated cardiac and renal ACE2 mRNA (Ishiyama et al 2004; Ferrario et al 2005a, b; Igase et al 2005; Jessup et al 2006). ACE-I and ARBs have important similarities because they block the same system but they also have several differences in their mechanisms of action, eg, regarding their effects on kinins, Ang-(1–7) and the AT2-receptor. The clinical relevance of these different therapeutic principles of RAS-blockade requires further study. Although beyond the scope of the present article there is also increasing interest and evidence to support the combined use of ACE-I and ARB in various clinical settings such as heart failure (Burnier and Brunner 2000). The Candesartan in Heart Failure Assessment of Reduction in Morbidity and mortality (CHARM) trial revealed that candesartan treatment significantly reduced cardiovascular deaths and hospital admissions for heart failure (Pfeffer et al 2003). In addition, the COOPERATE-trial highlighted that combination treatment of an ACE-I with an ARB safely retards progression of non-diabetic renal disease compared with monotherapy (Nakao et al 2003).
What this means in terms of the regulations and interplay of the components of the RAS is still widely unknown.
Is there scientific evidence for therapeutic superiority of either ARBs or ACE-I for any medical indication?
ACE-I and ARBs are successfully applied in the treatment of hypertension, heart failure, diabetes and coronary heart disease (Dendorfer et al 2005). Although ACE-I and ARBs block the same system, they may have important differences because they act on different sites of the RAS. However, therapeutic superiority of ARBs over ACE-I has never been proven. International treatment guidelines for hypertension (Chobanian et al 2003) cite ACE-I and ARBs as equally effective in reducing blood pressure but explicitly recommend ARBs only in case of ACE-I induced cough (Stergiou and Skeva 2004). Although clinical trials with head to head comparison of ACE-I with ARBs are few, there is some evidence suggesting differences in their effect on insulin sensitivity (Moan et al 1996) and pulse pressure (Stergiou et al 2002). In addition, selective ARBs have consistently been shown to inhibit several physiologic mechanisms that are involved in the development of in-stent restenosis: neointimal formation, vascular smooth cell migration, oxidative stress, anti-inflammatory effects and suppression of smooth muscle cell differentiation. There is increasing evidence from clinical studies that ARBs reduce restenosis rates after coronary (Peters et al 2001; Yoshida et al 2005) or superficial femoral artery stenting (Schindler et al 2005) whereas ACE-I do not have these beneficial effects (Faxon 1995; Cashin-Hemphill et al 1999; MacMahon et al 2000). Considering the pharmacological differences between ACE-I and ARBs it has to be pointed out that treatment with an ARB results in significantly increased Ang-(1–7)-peptide levels (Schindler et al 2007) whereas treatment with an ACE-I does not. Together with the experimental results of Langeveld (Langeveld et al 2005) who described a significant reduction in neointimal thickness after Ang-(1–7) infusions in an experimental model after stent implantation one might speculate that increased Ang-(1–7)-levels might causally contribute to the improved outcomes documented with ARBs after vascular interventions whereas increased bradykinin-levels after treatment with an ACE-I do not seem to have beneficial vascular effects. This hypothesis should be confirmed in large, randomized controlled clinical studies.
However, the significance of most of the reported differences between the two drug classes remains largely unknown and in clinical practice the only clearly established advantage of ARBs over ACE-I is the absence of cough as a side effect.
Other cardiovascular drugs with therapeutic implications on the RAS
Renin-inhibitors
Experimental and clinical studies have indicated that blockade of the RAS is an important therapeutic strategy in reducing cardiovascular and renal disease. However, the therapeutic response achieved with currently available blockers of the RAS – angiotensin-converting-enzyme-inhibitors and angiotensin receptor blockers – although efficacious, is limited. This may be partly because of the reactive rise in renin induced by these agents with the resultant increase in angiotensin peptides. Therefore, other more effective strategies to block the RAS have been sought. Just recently a new renin inhibitor (aliskiren) was FDA-approved for the treatment of hypertension in April 2007 (for review see (Azizi et al 2006)). Renin inhibitors prevent the formation of Ang I and Ang II and so may act differently from ARBs and ACE-I. Currently there is no published data from preclinical or clinical studies investigating the influence of pharmacological renin-inhibition on either urinary or serum peptide levels of Ang-(1–7).
Vasopeptidase-inhibitors
Drugs that possess the ability to inhibit simultaneously ACE and the neutral endopeptidase 24.11 (NEP) represent another development of compounds affecting the RAS. These dual inhibitors, also named vasopeptidase inhibitors, decrease Ang II generation by inhibiting ACE activity, and reduce the metabolic degradation of natriuretic peptides by inhibiting NEP. Natriuretic peptides represent a family of peptides that include the atrial natriuretic peptide (ANP), the BNP, and the C-type natriuretic peptide (CNP). Bradykinin and substance P are two other peptides metabolized by ACE and NEP, the accumulation of which may contribute to the vasodilatory effects of dual inhibitors.
Ferrario et al (Ferrario et al 2002a) found a strong correlation between the antihypertensive response to omapatrilat and increases in urinary excretion rates of Ang I and Ang-(1–7) in spontaneously hypertensive rats and also in salt-sensitive hypertensive subjects (Ferrario et al 2002b) suggesting a contribution of Ang-(1–7) to the vasodilator response mediated by this agent. This important association between increases in both plasma and urinary Ang-(1–7) and the antihypertensive effect of omapatrilat suggests, even if it does not prove, that Ang-(1–7) as humoral regulator may play a contributing role in the mechanisms that account for the control of blood pressure.
However, omapatrilat as the prototype of drugs with this pharmacological principle of dual inhibition was clinically developed but never got final approval by the health authorities due to unforeseen side-effects.
Perspectives
Ang-(1–7) as potential cardiovascular drug target
Besides its direct vascular effects which are not completely clear in humans, Ang-(1–7) can act as an ACE-I and as an Ang II antagonist and facilitates the vascular and cardiac effects of bradykinin, making it an attractive target for the development of new cardiovascular drugs. The growing evidence that at least part of the beneficial effects of ACE-I and ARBs are mediated by Ang-(1–7) further strengthens this view. The recently described model compound AVE-0991 is the first available Ang-(1–7) receptor agonist. A recently published study (Wiemer et al 2002) demonstrated that the new nonpeptide compound AVE 0991 is able to evoke effects on endothelial cells similar to that observed for heptapeptide Ang-(1–7). The amount of AVE 0991-stimulated NO production was about five times higher than that stimulated by Ang-(1–7) (Wiemer et al 2002). In addition it was experimentally shown that AVE is an Ang-(1–7) receptor mas-receptor agonist (Pinheiro et al 2004). This promising drug characteristic makes the orally active Ang-(1–7)-agonist AVE-0991, as a potent mimic of the unique NO/O2-releasing profile of Ang-(1–7), an attractive target for further future development for patients, eg, for indications such as endothelial dysfunction and stable coronary heart disease. Besides their potential as vascular drugs Ang-(1–7) or other mas-receptor-agonists might also exert beneficial effects on structural organ disease such as myocardial fibrosis which is a key pathological process in left ventricular hypertrophy. The optimal treatment of hypertensive patients should target a parallel decrease in cardiac mass and fibrosis. Preliminary evidence suggests that not all antihypertensive agents affect fibrosis to the same extent. However, agents directly blocking the RAS such as ACE-I and ARBs appear particularly effective (Cuspidi et al 2006) which underlines that mas-receptor-agonists might have a therapeutic potential and should be systematically investigated for this indication.
In addition, the presence of polymorphisms in the Ang-(1–7) forming enzyme genes and ACE2 need further exploration as we showed that men with the T allele showed higher Ang-(1–7) levels compared with those with the MM genotype (Reyes-Engel et al 2006). Furthermore, additional studies should be directed to determine whether the efficacy of long-term effects of ACE-I or ARB on cardiac remodeling and preservation of renal function correlates with the effect of these drugs on plasma levels of Ang-(1–7).
Conclusion
Ang-(1–7) as the most pleiotropic angiotensin peptide can act at several levels on the RAS, counterbalancing the detrimental vascular effects consequent to Ang I and Ang II formation and is therefore an attractive candidate as a therapeutic drug target. The majority of clinical studies investigating cardiovascular effects of Ang-(1–7) are in favor of beneficial RAS-modulating effects. Conflicting evidence about the RAS-modulating role of Ang-(1–7) reported from human studies might be related to extremely low case numbers investigated and due to different study protocols. The relevance of beneficial effects of Ang-(1–7) for patients on therapy with ACE-I or ARBs should therefore be systematically studied in randomized controlled trials in humans.
References
- Abbas A, Gorelik G, Carbini LA, et al. Angiotensin-(1-7) induces bradykinin-mediated hypotensive responses in anesthetized rats. Hypertension. 1997;30:217–21. doi: 10.1161/01.hyp.30.2.217. [DOI] [PubMed] [Google Scholar]
- Admiraal PJ, Derkx FH, Danser AH, et al. Metabolism and production of angiotensin I in different vascular beds in subjects with hypertension. Hypertension. 1990;15:44–55. doi: 10.1161/01.hyp.15.1.44. [DOI] [PubMed] [Google Scholar]
- Almeida AP, Frabregas BC, Madureira MM, et al. Angiotensin-(1–7) potentiates the coronary vasodilatatory effect of bradykinin in the isolated rat heart. Braz J Med Biol Res. 2000;33:709–13. doi: 10.1590/s0100-879x2000000600012. [DOI] [PubMed] [Google Scholar]
- Ambuhl P, Felix D, Khosla MC. [7-D-ALA]-angiotensin-(1–7): selective antagonism of angiotensin-(1–7) in the rat paraventricular nucleus. Brain Res Bull. 1994;35:289–91. doi: 10.1016/0361-9230(94)90103-1. [DOI] [PubMed] [Google Scholar]
- Andreatta-Van Leyen S, Romero MF, Khosla MC, et al. Modulation of phospholipase A2-activity and sodium transport by angiotensin-(1–7) Kidney Int. 1993;44:932–6. doi: 10.1038/ki.1993.334. [DOI] [PubMed] [Google Scholar]
- Azizi M, Webb R, Nussberger J, et al. Renin inhibition with aliskiren: where are we now, and where are we going. J Hypertens. 2006;24:243–56. doi: 10.1097/01.hjh.0000202812.72341.99. [DOI] [PubMed] [Google Scholar]
- Baracho NCV, Silva ACS, Khosla M, et al. Characterization of the antidiuretic action of angiotensin-(1–7) in water-loaded rats. Hypertension. 1995;25:1408. [Google Scholar]
- Benter IF, Diz DI, Ferrario CM. Cardiovascular actions of angioten-sin(1–7) Peptides. 1993;14:679–84. doi: 10.1016/0196-9781(93)90097-z. [DOI] [PubMed] [Google Scholar]
- Benter IF, Diz DI, Ferrario CM. Pressor and reflex sensitivity is altered in spontaneously hypertensive rats treated with angiotensin-(1–7) Hypertension. 1995a;26:1138–44. doi: 10.1161/01.hyp.26.6.1138. [DOI] [PubMed] [Google Scholar]
- Benter IF, Ferrario CM, Morris M, et al. Antihypertensive actions of angiotensin-(1–7) in spontaneously hypertensive rats. Am J Physiol. 1995b;269:H313–9. doi: 10.1152/ajpheart.1995.269.1.H313. [DOI] [PubMed] [Google Scholar]
- Bomtempo CA, Santos GF, Santos RA, et al. Interaction of bradykinin and angiotensin-(1–7) in the central modulation of the baroreflex control of the heart rate. J Hypertens. 1998;16:1797–804. doi: 10.1097/00004872-199816120-00013. [DOI] [PubMed] [Google Scholar]
- Bonner G, Preis S, Schunk U, et al. Hemodynamic effects of bradykinin on systemic and pulmonary circulation in healthy and hypertensive humans. J Cardiovasc Pharmacol. 1990;15(Suppl 6):S46–56. [PubMed] [Google Scholar]
- Bovy PR, Trapani AJ, McMahon EG, et al. A carboxy-terminus truncated analogue of angiotensin II, [Sar1]angiotensin II-(1–7)-amide, provides an entry to a new class of angiotensin II antagonists. J Med Chem. 1989;32:520–2. doi: 10.1021/jm00123a002. [DOI] [PubMed] [Google Scholar]
- Brosnihan KB, Li P, Ferrario CM. Angiotensin-(1–7) dilates canine coronary arteries through kinins and nitric oxide. Hypertension. 1996;27:523–8. doi: 10.1161/01.hyp.27.3.523. [DOI] [PubMed] [Google Scholar]
- Brunner HR, Gavras H, Turini GA, et al. Long-term treatment of hypertension in man by an orally active angiotensin-converting enzyme inhibitor. Clin Sci Mol Med Suppl. 1978;4:293s–295s. doi: 10.1042/cs055293s. [DOI] [PubMed] [Google Scholar]
- Burnier M, Brunner HR. Angiotensin II receptor antagonists. Lancet. 2000;355:637–45. doi: 10.1016/s0140-6736(99)10365-9. [DOI] [PubMed] [Google Scholar]
- Campagnole-Santos MJ, Diz DI, Santos RA, et al. Cardiovascular effects of angiotensin-(1–7) injected into the dorsal medulla of rats. Am J Physiol. 1989;257:H324–9. doi: 10.1152/ajpheart.1989.257.1.H324. [DOI] [PubMed] [Google Scholar]
- Campagnole-Santos MJ, Heringer SB, Batista EN, et al. Differential baroreceptor reflex modulation by centrally infused angiotensin peptides. Am J Physiol. 1992;263:R89–94. doi: 10.1152/ajpregu.1992.263.1.R89. [DOI] [PubMed] [Google Scholar]
- Carey RM, Howell NL, Jin XH, et al. Angiotensin type 2 receptor-mediated hypotension in angiotensin type-1 receptor-blocked rats. Hypertension. 2001;38:1272–7. doi: 10.1161/hy1201.096576. [DOI] [PubMed] [Google Scholar]
- Carey RM, Siragy HM. Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocr Rev. 2003;24:261–71. doi: 10.1210/er.2003-0001. [DOI] [PubMed] [Google Scholar]
- Case DB, Wallace JM, Laragh JH. Comparison between saralsin and converting enzyme inhibitor in hypertensive disease. Kidney Int Suppl. 1979:S107–114. [PubMed] [Google Scholar]
- Cashin-Hemphill L, Holmvang G, Chan RC, et al. Angiotensin-converting enzyme inhibition as antiatherosclerotic therapy: no answer yet. QUIET Investigators. QUinapril Ischemic Event Trial. Am J Cardiol. 1999;83:43–7. doi: 10.1016/s0002-9149(98)00780-2. [DOI] [PubMed] [Google Scholar]
- Castro CH, Santos RA, Ferreira AJ, et al. Evidence for a functional interaction of the angiotensin-(1–7) receptor Mas with AT1 and AT-receptors in the mouse heart. Hypertension. 2005;46:937–42. doi: 10.1161/01.HYP.0000175813.04375.8a. [DOI] [PubMed] [Google Scholar]
- Chansel D, Vandermeersch S, Oko A, et al. Effects of angiotensin IV and angiotensin-(1–7) on basal and angiotensin II-stimulated cytosolic Ca2+ in mesangial cells. Eur J Pharmacol. 2001;414:165–75. doi: 10.1016/s0014-2999(01)00791-9. [DOI] [PubMed] [Google Scholar]
- Chappell M, Tallant E, Brosnihan K, et al. Conversion of angiotensin I to angiotensin-(1–7) by thimet oligopeptidase (EC 3.4.24.15) in vascular smooth muscle cells. J Vasc Med Biol. 1994;5:129–37. [Google Scholar]
- Chappell MC, Pirro NT, Sykes A, et al. Metabolism of angiotensin-(1–7) by angiotensin-converting enzyme. Hypertension. 1998;31:362–7. doi: 10.1161/01.hyp.31.1.362. [DOI] [PubMed] [Google Scholar]
- Chobanian AV, Bakris GL, Black HR, et al. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension. 2003;42:1206–52. doi: 10.1161/01.HYP.0000107251.49515.c2. [DOI] [PubMed] [Google Scholar]
- Cipollone F, Fazia M, Iezzi A, et al. Blockade of the angiotensin II type 1 receptor stabilizes atherosclerotic plaques in humans by inhibiting prostaglandin E2-dependent matrix metalloproteinase activity. Circulation. 2004;109:1482–8. doi: 10.1161/01.CIR.0000121735.52471.AC. [DOI] [PubMed] [Google Scholar]
- Crackower MA, Sarao R, Oudit GY, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–8. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- Cuspidi C, Ciulla M, Zanchetti A. Hypertensive myocardial fibrosis. Nephrol Dial Transplant. 2006;21:20–3. doi: 10.1093/ndt/gfi237. [DOI] [PubMed] [Google Scholar]
- Davie AP, Mcmurray JJ. Effect of angiotensin-(1–7) and bradykinin in patients with heart failure treated with an ACE inhibitor. Hypertension. 1999;34:457–60. doi: 10.1161/01.hyp.34.3.457. [DOI] [PubMed] [Google Scholar]
- Deddish PA, Marcic B, Jackman HL, et al. N-domain-specific substrate and C-domain inhibitors of angiotensin-converting enzyme: angiotensin-(1–7) and keto-ACE. Hypertension. 1998;31:912–17. doi: 10.1161/01.hyp.31.4.912. [DOI] [PubMed] [Google Scholar]
- Dellipizzi AM, Hilchey SD, Bell-Quilley CP. Natriuretic action of angiotensin(1–7) Br J Pharmacol. 1994;111:1–3. doi: 10.1111/j.1476-5381.1994.tb14014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dendorfer A, Dominiak P, Schunkert H. ACE inhibitors and angiotensin II receptor antagonists. Handb Exp Pharmacol. 2005:407–42. doi: 10.1007/3-540-27661-0_15. [DOI] [PubMed] [Google Scholar]
- Diz DI, Pirro NT. Differential actions of angiotensin II and angioten-sin-(1–7) on transmitter release. Hypertension. 1992;19:II41–8. doi: 10.1161/01.hyp.19.2_suppl.ii41. [DOI] [PubMed] [Google Scholar]
- Donoghue M, Hsieh F, Baronas E, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res. 2000;87:E1–9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- Faxon DP. Effect of high dose angiotensin-converting enzyme inhibition on restenosis: final results of the MARCATOR Study, a multicenter, double-blind, placebo-controlled trial of cilazapril. The Multicenter American Research Trial With Cilazapril After Angioplasty to Prevent Transluminal Coronary Obstruction and Restenosis (MARCATOR) Study Group. J Am Coll Cardiol. 1995;25:362–9. doi: 10.1016/0735-1097(94)00368-z. [DOI] [PubMed] [Google Scholar]
- Fernandes L, Fortes ZB, Nigro D, et al. Potentiation of bradykinin by angiotensin-(1–7) on arterioles of spontaneously hypertensive rats studied in vivo. Hypertension. 2001;37:703–9. doi: 10.1161/01.hyp.37.2.703. [DOI] [PubMed] [Google Scholar]
- Ferrario CM, Averill DB, Brosnihan KB, et al. Vasopeptidase inhibition and Ang-(1–7) in the spontaneously hypertensive rat. Kidney Int. 2002a;62:1349–57. doi: 10.1111/j.1523-1755.2002.kid559.x. [DOI] [PubMed] [Google Scholar]
- Ferrario CM, Brosnihan KB, Diz DI, et al. Angiotensin-(1–7): a new hormone of the angiotensin system. Hypertension. 1991;18:126–33. doi: 10.1161/01.hyp.18.5_suppl.iii126. [DOI] [PubMed] [Google Scholar]
- Ferrario CM, Chappell MC, Tallant EA, et al. Counterregulatory actions of angiotensin-(1–7) Hypertension. 1997;30:535–41. doi: 10.1161/01.hyp.30.3.535. [DOI] [PubMed] [Google Scholar]
- Ferrario CM, Jessup J, Chappell MC, et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. 2005a;111:2605–10. doi: 10.1161/CIRCULATIONAHA.104.510461. [DOI] [PubMed] [Google Scholar]
- Ferrario CM, Jessup J, Gallagher PE, et al. Effects of renin-angiotensin system blockade on renal angiotensin-(1–7) forming enzymes and receptors. Kidney Int. 2005b;68:2189–96. doi: 10.1111/j.1523-1755.2005.00675.x. [DOI] [PubMed] [Google Scholar]
- Ferrario CM, Martell N, Yunis C, et al. Characterization of angiotensin-(1–7) in the urine of normal and essential hypertensive subjects. Am J Hypertens. 1998;11:137–46. doi: 10.1016/s0895-7061(97)00400-7. [DOI] [PubMed] [Google Scholar]
- Ferrario CM, Smith RD, Brosnihan B, et al. Effects of omapatrilat on the renin-angiotensin system in salt-sensitive hypertension. Am J Hypertens. 2002b;15:557–64. doi: 10.1016/s0895-7061(02)02268-9. [DOI] [PubMed] [Google Scholar]
- Feterik K, Smith L, Katusic ZS. Angiotensin-(1–7) causes endo-thelium-dependent relaxation in canine middle cerebral artery. Brain Res. 2000;873:75–82. doi: 10.1016/s0006-8993(00)02482-3. [DOI] [PubMed] [Google Scholar]
- Freeman EJ, Chisolm GM, Ferrario CM, et al. Angiotensin-(1–7) inhibits vascular smooth muscle cell growth. Hypertension. 1996;28:104–8. doi: 10.1161/01.hyp.28.1.104. [DOI] [PubMed] [Google Scholar]
- Gironacci MM, Valera MS, Yujnovsky I, et al. Angiotensin-(1–7) inhibitory mechanism of norepinephrine release in hypertensive rats. Hypertension. 2004;44:783–7. doi: 10.1161/01.HYP.0000143850.73831.9d. [DOI] [PubMed] [Google Scholar]
- Hall JE. The renin-angiotensin system: renal actions and blood pressure regulation. Compr Ther. 1991;17:8–17. [PubMed] [Google Scholar]
- Handa RK, Ferrario CM, Strandhoy JW. Angiotensin-(1–7) inhibits oxygen consumption in rat proximal tubules [abstract] J Am Soc Nephrol. 1994;5:659. [Google Scholar]
- Handa RK, Ferrario CM, Strandhoy JW. Renal actions of angiotensin-(1–7): in vivo and in vitro studies. Am J Physiol. 1996;270:F141–7. doi: 10.1152/ajprenal.1996.270.1.F141. [DOI] [PubMed] [Google Scholar]
- Harmer D, Gilbert M, Borman R, et al. Quantitative mRNA expression profiling of ACE2, a novel homologue of angiotensin converting enzyme. FEBS Lett. 2002;532:107–10. doi: 10.1016/s0014-5793(02)03640-2. [DOI] [PubMed] [Google Scholar]
- Hayoz D. AT1 receptor antagonists: vascular protection beyond blood pressure reduction? J Hypertens. 2002;20:2345–6. doi: 10.1097/00004872-200212000-00008. [DOI] [PubMed] [Google Scholar]
- Hecker M, Blaukat A, Bara AT, et al. ACE inhibitor potentiation of bradykinin-induced venoconstriction. Br J Pharmacol. 1997;121:1475–81. doi: 10.1038/sj.bjp.0701281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitsch H, Brovkovych S, Malinski T, et al. Angiotensin-(1–7)-stimulated nitric oxide and superoxide release from endothelial cells. Hypertension. 2001;37:72–76. doi: 10.1161/01.hyp.37.1.72. [DOI] [PubMed] [Google Scholar]
- Hilchey SD, Bell-Quilley CP. Association between the natriuretic action of angiotensin-(1–7) and selective stimulation of renal prostaglandin I2 release. Hypertension. 1995;25:1238–44. doi: 10.1161/01.hyp.25.6.1238. [DOI] [PubMed] [Google Scholar]
- Igase M, Strawn WB, Gallagher PE, et al. Angiotensin II AT1 receptors regulate ACE2 and angiotensin-(1–7) expression in the aorta of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2005;289:H1013–9. doi: 10.1152/ajpheart.00068.2005. [DOI] [PubMed] [Google Scholar]
- Ishiyama Y, Gallagher PE, Averill DB, et al. Upregulation of angio-tensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension. 2004;43:970–6. doi: 10.1161/01.HYP.0000124667.34652.1a. [DOI] [PubMed] [Google Scholar]
- Iyer SN, Averill DB, Chappell MC, et al. Contribution of angiotensin-(1–7) to blood pressure regulation in salt-depleted hypertensive rats. Hypertension. 2000;36:417–22. doi: 10.1161/01.hyp.36.3.417. [DOI] [PubMed] [Google Scholar]
- Iyer SN, Chappell MC, Averill DB, et al. Vasodepressor actions of angiotensin-(1–7) unmasked during combined treatment with lisinopril and losartan. Hypertension. 1998a;31:699–705. doi: 10.1161/01.hyp.31.2.699. [DOI] [PubMed] [Google Scholar]
- Iyer SN, Ferrario CM, Chappell MC. Angiotensin-(1–7) contributes to the antihypertensive effects of blockade of the renin-angiotensin system. Hypertension. 1998b;31:356–61. doi: 10.1161/01.hyp.31.1.356. [DOI] [PubMed] [Google Scholar]
- Jaiswal N, Diz DI, Chappell MC, et al. Stimulation of endothelial cell prostaglandin production by angiotensin peptides. Characterization of receptors. Hypertension. 1992;19:II49–55. doi: 10.1161/01.hyp.19.2_suppl.ii49. [DOI] [PubMed] [Google Scholar]
- Jaiswal N, Jaiswal RK, Tallant EA, et al. Alterations in prostaglandin production in spontaneously hypertensive rat smooth muscle cells. Hypertension. 1993a;21:900–5. doi: 10.1161/01.hyp.21.6.900. [DOI] [PubMed] [Google Scholar]
- Jaiswal N, Tallant EA, Jaiswal RK, et al. Differential regulation of prostaglandin synthesis by angiotensin peptides in porcine aortic smooth muscle cells: subtypes of angiotensin receptors involved. J Pharmacol Exp Ther. 1993b;265:664–73. [PubMed] [Google Scholar]
- Jessup J, Gallagher PE, Averill DB, et al. Effect of angiotensin II blockade on a new congenic model of hypertension derived from transgenic ren-2 Rats. Am J Physiol Heart Circ Physiol. 2006;291:42166–72. doi: 10.1152/ajpheart.00061.2006. [DOI] [PubMed] [Google Scholar]
- Johnston CI. Angiotensin receptor antagonists: focus on losartan. Lancet. 1995;346:1403–7. doi: 10.1016/s0140-6736(95)92411-6. [DOI] [PubMed] [Google Scholar]
- Langeveld B, Van Gilst WH, Tio RA, et al. Angiotensin-(1–7) attenuates neointimal formation after stent implantation in the rat. Hypertension. 2005;45:138–41. doi: 10.1161/01.HYP.0000149382.83973.c2. [DOI] [PubMed] [Google Scholar]
- Le Tran Y, Forster C. Angiotensin-(1–7) and the rat aorta: modulation by the endothelium. J Cardiovasc Pharmacol. 1997;30:676–82. doi: 10.1097/00005344-199711000-00019. [DOI] [PubMed] [Google Scholar]
- Li P, Chappell MC, Ferrario CM, et al. Angiotensin-(1–7) augments bradykinin-induced vasodilation by competing with ACE and releasing nitric oxide. Hypertension. 1997;29:394–400. doi: 10.1161/01.hyp.29.1.394. [DOI] [PubMed] [Google Scholar]
- Lima CV, Paula RD, Resende FL, et al. Potentiation of the hypotensive effect of bradykinin by short-term infusion of angiotensin-(1–7) in normotensive and hypertensive rats. Hypertension. 1997;30:542–8. doi: 10.1161/01.hyp.30.3.542. [DOI] [PubMed] [Google Scholar]
- Machado RD, Ferreira MA, Belo AV, et al. Vasodilator effect of angiotensin-(1–7) in mature and sponge-induced neovasculature. Regul Pept. 2002;107:05–13. doi: 10.1016/s0167-0115(02)00070-8. [DOI] [PubMed] [Google Scholar]
- MacMahon S, Sharpe N, Gamble G, et al. Randomized, placebo-controlled trial of the angiotensin-converting enzyme inhibitor, ramipril, in patients with coronary or other occlusive arterial disease. PART-2 Collaborative Research Group. Prevention of Atherosclerosis with Ramipril. J Am Coll Cardiol. 2000;36:438–43. doi: 10.1016/s0735-1097(00)00736-1. [DOI] [PubMed] [Google Scholar]
- Mahon JM, Carr RD, Nicol AK, et al. Angiotensin(1–7) is an antagonist at the type 1 angiotensin II receptor. J Hypertens. 1994;12:1377–81. [PubMed] [Google Scholar]
- Maia LG, Ramos MC, Fernandes L, et al. Angiotensin-(1–7) antagonist A-779 attenuates the potentiation of bradykinin by captopril in rats. J Cardiovasc Pharmacol. 2004;43:685–91. doi: 10.1097/00005344-200405000-00011. [DOI] [PubMed] [Google Scholar]
- Meng W, Busija DW. Comparative effects of angiotensin-(1–7) and angiotensin II on piglet pial arterioles. Stroke. 1993;24:2041–4; discussion 2045. doi: 10.1161/01.str.24.12.2041. [DOI] [PubMed] [Google Scholar]
- Metzger R, Bader M, Ludwig T, et al. Expression of the mouse and rat mas proto-oncogene in the brain and peripheral tissues. FEBS Lett. 1995;357:27–32. doi: 10.1016/0014-5793(94)01292-9. [DOI] [PubMed] [Google Scholar]
- Moan A, Hoieggen A, Seljeflot I, et al. The effect of angiotensin II receptor antagonism with losartan on glucose metabolism and insulin sensitivity. J Hypertens. 1996;14:1093–7. doi: 10.1097/00004872-199609000-00008. [DOI] [PubMed] [Google Scholar]
- Moritz KM, Campbell DJ, Wintour EM. Angiotensin-(1–7) in the ovine fetus. Am J Physiol Regul Integr Comp Physiol. 2001;280:R404–9. doi: 10.1152/ajpregu.2001.280.2.R404. [DOI] [PubMed] [Google Scholar]
- Nakao N, Yoshimura A, Morita H, et al. Combination treatment of angiotensin-II receptor blocker and angiotensin-converting-enzyme inhibitor in non-diabetic renal disease (COOPERATE): a randomised controlled trial. Lancet. 2003;361:117–24. doi: 10.1016/S0140-6736(03)12229-5. [DOI] [PubMed] [Google Scholar]
- Oliveira MA, Fortes ZB, Santos RA, et al. Synergistic effect of angiotensin-(1–7) on bradykinin arteriolar dilation in vivo. Peptides. 1999;20:1195–201. doi: 10.1016/s0196-9781(99)00123-0. [DOI] [PubMed] [Google Scholar]
- Osei SY, Ahima RS, Minkes RK, et al. Differential responses to angiotensin-(1–7) in the feline mesenteric and hindquarters vascular beds. Eur J Pharmacol. 1993;234:35–42. doi: 10.1016/0014-2999(93)90703-k. [DOI] [PubMed] [Google Scholar]
- Pals DT, Masucci FD, Denning GS, Jr, et al. Role of the pressor action of angiotensin II in experimental hypertension. Circ Res. 1971;29:673–81. doi: 10.1161/01.res.29.6.673. [DOI] [PubMed] [Google Scholar]
- Paula RD, Lima CV, Khosla MC, et al. Angiotensin-(1–7) potentiates the hypotensive effect of bradykinin in conscious rats. Hypertension. 1995;26:1154–9. doi: 10.1161/01.hyp.26.6.1154. [DOI] [PubMed] [Google Scholar]
- Peters S, Gotting B, Trummel M, et al. Valsartan for prevention of restenosis after stenting of type B2/C lesions: the VAL-PREST trial. J Invasive Cardiol. 2001;13:93–7. [PubMed] [Google Scholar]
- Pfeffer MA, Swedberg K, Granger CB, et al. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet. 2003;362:759–66. doi: 10.1016/s0140-6736(03)14282-1. [DOI] [PubMed] [Google Scholar]
- Pinheiro SV, Simoes E, Silva AC, Sampaio WO, et al. Nonpeptide AVE 0991 is an angiotensin-(1–7) receptor Mas agonist in the mouse kidney. Hypertension. 2004;44:490–6. doi: 10.1161/01.HYP.0000141438.64887.42. [DOI] [PubMed] [Google Scholar]
- Pitt B, Segal R, Martinez FA, et al. Randomised trial of losartan versus captopril in patients over 65 with heart failure (Evaluation of Losartan in the Elderly Study, ELITE) Lancet. 1997;349:747–52. doi: 10.1016/s0140-6736(97)01187-2. [DOI] [PubMed] [Google Scholar]
- Porsti I, Bara AT, Busse R, et al. Release of nitric oxide by angioten-sin-(1–7) from porcine coronary endothelium: implications for a novel angiotensin receptor. Br J Pharmacol. 1994;111:652–4. doi: 10.1111/j.1476-5381.1994.tb14787.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Y, Garvin JL, Carretero OA. Vasodilator action of angiotensin-(1–7) on isolated rabbit afferent arterioles. Hypertension. 2002;39:799–802. doi: 10.1161/hy0302.104673. [DOI] [PubMed] [Google Scholar]
- Reudelhuber TL. A place in our hearts for the lowly angiotensin 1–7 peptide? Hypertension. 2006;47:811–15. doi: 10.1161/01.HYP.0000209020.69734.73. [DOI] [PubMed] [Google Scholar]
- Reyes-Engel A, Morcillo L, Aranda FJ, et al. Influence of gender and genetic variability on plasma Angiotensin peptides. J Renin Angiotensin Aldosterone Syst. 2006;7:92–7. doi: 10.3317/jraas.2006.015. [DOI] [PubMed] [Google Scholar]
- Roks AJ, Van Geel PP, Pinto YM, et al. Angiotensin-(1–7) is a modulator of the human renin-angiotensin system. Hypertension. 1999;34:296–301. doi: 10.1161/01.hyp.34.2.296. [DOI] [PubMed] [Google Scholar]
- Sampaio WO, Nascimento AA, Santos RA. Systemic and regional hemodynamic effects of angiotensin-(1–7) in rats. Am J Physiol Heart Circ Physiol. 2003;284:H1985–94. doi: 10.1152/ajpheart.01145.2002. [DOI] [PubMed] [Google Scholar]
- Santos RA, Brosnihan KB, Chappell MC, et al. Converting enzyme activity and angiotensin metabolism in the dog brainstem. Hypertension. 1988;11:I153–7. doi: 10.1161/01.hyp.11.2_pt_2.i153. [DOI] [PubMed] [Google Scholar]
- Santos RA, Brosnihan KB, Jacobsen DW, et al. Production of angiotensin-(1–7) by human vascular endothelium. Hypertension. 1992;19:II56–61. doi: 10.1161/01.hyp.19.2_suppl.ii56. [DOI] [PubMed] [Google Scholar]
- Santos RA, Campagnole-Santos MJ, Andrade SP. Angiotensin-(1–7): an update. Regul Pept. 2000;91:45–62. doi: 10.1016/s0167-0115(00)00138-5. [DOI] [PubMed] [Google Scholar]
- Santos RA, Campagnole-Santos MJ, Baracho NC, et al. Characterization of a new angiotensin antagonist selective for angiotensin-(1–7): evidence that the actions of angiotensin-(1–7) are mediated by specific angiotensin receptors. Brain Res Bull. 1994;35:293–8. doi: 10.1016/0361-9230(94)90104-x. [DOI] [PubMed] [Google Scholar]
- Santos RA, Simoes E, Silva AC, Magaldi AJ, et al. Evidence for a physiological role of angiotensin-(1–7) in the control of hydroelectrolyte balance. Hypertension. 1996;27:875–84. doi: 10.1161/01.hyp.27.4.875. [DOI] [PubMed] [Google Scholar]
- Santos RA, Simoes E, Silva AC, Maric C, et al. Angiotensin-(1–7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc Natl Acad Sci USA. 2003;100:8258–63. doi: 10.1073/pnas.1432869100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki S, Higashi Y, Nakagawa K, et al. Effects of angiotensin-(1–7) on forearm circulation in normotensive subjects and patients with essential hypertension. Hypertension. 2001;38:90–4. doi: 10.1161/01.hyp.38.1.90. [DOI] [PubMed] [Google Scholar]
- Schiavone MT, Santos RA, Brosnihan KB, et al. Release of vasopressin from the rat hypothalamo-neurohypophysial system by angiotensin-(1–7) heptapeptide. Proc Natl Acad Sci USA. 1988;85:4095–8. doi: 10.1073/pnas.85.11.4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler C, Brosnihan KB, Ferrario CM, et al. Comparison of inhibitory effects of irbesartan and atorvastatin treatment on the renin angiotensin system (RAS) in veins: a randomized double-blind cross-over trial in healthy subjects. J Clin Pharmacol. 2007;47:112–20. doi: 10.1177/0091270006294280. [DOI] [PubMed] [Google Scholar]
- Schindler C, Schweizer J, Mueller A, et al. Candesartan treatment for peripheral occlusive arterial disease after stent angioplasty: a randomised, placebo-controlled trial. Clin Drug Invest. 2005;25:89–97. doi: 10.2165/00044011-200525020-00001. [DOI] [PubMed] [Google Scholar]
- Silva LC, Fontes MA, Campagnole-Santos MJ, et al. Cardiovascular effects produced by micro-injection of angiotensin-(1–7) on vasopressor and vasodepressor sites of the ventrolateral medulla. Brain Res. 1993;613:321–5. doi: 10.1016/0006-8993(93)90920-i. [DOI] [PubMed] [Google Scholar]
- Stergiou GS, Efstathiou SP, Skeva Ii, et al. Assessment of drug effects on blood pressure and pulse pressure using clinic, home and ambulatory measurements. J Hum Hypertens. 2002;16:729–35. doi: 10.1038/sj.jhh.1001477. [DOI] [PubMed] [Google Scholar]
- Stergiou GS, Skeva Ii. Renin-angiotensin system blockade at the level of the angiotensin converting enzyme or the angiotensin type-1 receptor: similarities and differences. Curr Top Med Chem. 2004;4:473–81. doi: 10.2174/1568026043451320. [DOI] [PubMed] [Google Scholar]
- Tallant EA, Ferrario CM, Gallagher PE. Angiotensin-(1–7) inhibits growth of cardiac myocytes through activation of the mas receptor. Am J Physiol Heart Circ Physiol. 2005;289:H1560–6. doi: 10.1152/ajpheart.00941.2004. [DOI] [PubMed] [Google Scholar]
- Tallant EA, Jaiswal N, Diz DI, et al. Human astrocytes contain two distinct angiotensin receptor subtypes. Hypertension. 1991;18:32–9. doi: 10.1161/01.hyp.18.1.32. [DOI] [PubMed] [Google Scholar]
- Tipnis SR, Hooper NM, Hyde R, et al. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–43. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- Tom B, De Vries R, Saxena PR, et al. Bradykinin potentiation by angiotensin-(1–7) and ACE inhibitors correlates with ACE C-and N-domain blockade. Hypertension. 2001;38:95–9. doi: 10.1161/01.hyp.38.1.95. [DOI] [PubMed] [Google Scholar]
- Trachte GJ, Ferrario CM, Khosla MC. Selective blockade of angiotensin responses in the rabbit isolated vas deferens by angiotensin receptor antagonists. J Pharmacol Exp Ther. 1990;255:929–34. [PubMed] [Google Scholar]
- Ueda S, Masumori-Maemoto S, Ashino K, et al. Angiotensin-(1–7) attenuates vasoconstriction evoked by angiotensin II but not by noradrenaline in man. Hypertension. 2000;35:998–1001. doi: 10.1161/01.hyp.35.4.998. [DOI] [PubMed] [Google Scholar]
- Ueda S, Masumori-Maemoto S, Wada A, et al. Angiotensin(1–7) potentiates bradykinin-induced vasodilatation in man. J Hypertens. 2001;19:2001–9. doi: 10.1097/00004872-200111000-00010. [DOI] [PubMed] [Google Scholar]
- Unger T. The role of the renin-angiotensin system in the development of cardiovascular disease. Am J Cardiol. 2002;89:3A–9A; discussion 10A. doi: 10.1016/s0002-9149(01)02321-9. [DOI] [PubMed] [Google Scholar]
- Walther T, Balschun D, Voigt JP, et al. Sustained long term potentiation and anxiety in mice lacking the Mas protooncogene. J Biol Chem. 1998;273:11867–73. doi: 10.1074/jbc.273.19.11867. [DOI] [PubMed] [Google Scholar]
- Welches WR, Brosnihan KB, Ferrario CM. A comparison of the properties and enzymatic activities of three angiotensin processing enzymes: angiotensin converting enzyme, prolyl endopeptidase and neutral endopeptidase 24.11. Life Sci. 1993;52:1461–80. doi: 10.1016/0024-3205(93)90108-f. [DOI] [PubMed] [Google Scholar]
- Wiemer G, Dobrucki LW, Louka FR, et al. AVE 0991, a nonpeptide mimic of the effects of angiotensin-(1–7) on the endothelium. Hypertension. 2002;40:847–52. doi: 10.1161/01.hyp.0000037979.53963.8f. [DOI] [PubMed] [Google Scholar]
- Wilsdorf T, Gainer JV, Murphey LJ, et al. Angiotensin-(1–7) does not affect vasodilator or TPA responses to bradykinin in human forearm. Hypertension. 2001;37:1136–40. doi: 10.1161/01.hyp.37.4.1136. [DOI] [PubMed] [Google Scholar]
- Yoshida O, Hirayama H, Nanasato M, et al. The angiotensin II receptor blocker candesartan cilexetil reduces neointima proliferation after coronary stent implantation: a prospective randomized study under intravascular ultrasound guidance. Am Heart J. 2005;149:e2. doi: 10.1016/j.ahj.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Yusuf S, Sleight P, Pogue J, et al. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145–53. doi: 10.1056/NEJM200001203420301. [DOI] [PubMed] [Google Scholar]