

Abstract
Pulmonary arterial hypertension (PAH) is a rare fatal disease. Current disease-specific therapeutic interventions in PAH target 1 of 3 established pathways in disease pathobiology: prostacyclin, nitric oxide, and endothelin-1. Endothelin receptor antagonists (ERAs) act on the endothelin pathway by blocking binding of endothelin-1 to its receptors (endothelin type-A [ETA] and/or type-B [ETB]) on the surface of endothelial and smooth muscle cells. Ambrisentan is an oral, once-daily, ETA-selective ERA in development for the treatment of PAH. In Phase 3 clinical trials in patients with PAH, ambrisentan (2.5–10 mg orally once-daily) improved exercise capacity, Borg dyspnea index, time to clinical worsening, WHO functional class, and quality of life compared with placebo. Ambrisentan provided durable (at least 2 years) improvement in exercise capacity in a Phase 2 long-term extension study. Ambrisentan was well tolerated with a lower incidence and severity of liver function test abnormalities compared with the ETA/ETB ERA, bosentan, and the ETA-selective ERA, sitaxsentan. Ambrisentan does not induce or inhibit P450 enzymes; therefore, ambrisentan is unlikely to affect the pharmacokinetics of P450-metabolized drugs. The demonstration of clinical efficacy, low incidence of acute hepatic toxicity, and low risk of drug–drug interactions support the role of ambrisentan for the treatment of PAH.
Keywords: endothelin receptor antagonist, pulmonary arterial hypertension, endothelin-1, time to clinical worsening, Borg dyspnea index
Pulmonary arterial hypertension
Pulmonary arterial hypertension (PAH) is a chronic, progressive disease characterized by increased pulmonary vascular resistance of the lung microvasculature, intimal hyperplasia and smooth muscle cell hypertrophy, and in situ thrombosis (Rubin 2006). PAH disease progression leads to right heart failure and death (Vlahakes et al 1981; D’Alonzo et al 1991; Rich 2001).
PAH is defined by mean pulmonary arterial pressure that exceeds 25 mm Hg at rest or 30 mm Hg during exercise, with mean pulmonary-capillary wedge pressure or left ventricular end diastolic pressure ≤15 mm Hg and pulmonary vascular resistance greater than 3 Wood units (Hatano et al 1975; Barst et al 2004b). Unfortunately and despite significant efforts to diagnose patients earlier in the disease process, the disease is most often diagnosed months or years after symptoms first appear. As a consequence, the majority of patients present with advanced disease and marked functional impairment (Hoeper 2005).
Clinical classification
According to the Venice 2003 World Health Organization (WHO) symposium on PAH classification, the broader category of pulmonary hypertension (PH) is subdivided into 5 categories based on association with heart disease, lung disease, thromboembolic disease or miscellaneous conditions (Table 1) (Simonneau et al 2004). PAH can occur in the absence of an associated disorder as either idiopathic PAH (IPAH) or familial PAH (FPAH) (Rubin et al 2005a). Additionally, PAH can occur as a complication of systemic conditions, such as connective tissue disease, congenital heart disease, portal hypertension, HIV infection, or from the use of anorexigens, amphetamines, or cocaine (Rubin et al 2005a).
Table 1.
Clinical classification of pulmonary hypertension (Venice 2003). Reprinted from Simonneau G, Galie N, Rubin LJ, et al. 2004. Clinical classification of pulmonary hypertension. J Am Coll Cardiol, 43:5S–12S. Copyright © 2004 with permission from American College of Cardiology Foundation
| 1. Pulmonary arterial hypertension (PAH) |
| 1.1 Idiopathic (IPAH) |
| 1.2 Familial (FPAH) |
| 1.3 Associated with (APAH): |
| 1.3.1 Collagen vascular disease |
| 1.3.2 Congenital systemic-to-pulmonary shunts |
| 1.3.3 Portal hypertension |
| 1.3.4 HIV infection |
| 1.3.5 Drugs and toxins |
| 1.3.6 Other (thyroid disorders, glycogen storage disease, Gaucher disease, hereditary hemorrhagic telangiectasia, hemoglobinopathies, myeloproliferative disorders, splenectomy) |
| 1.4 Associated with significant venous or capillary involvement |
| 1.4.1 Pulmonary veno-occlusive disease (PVOD) |
| 1.4.2 Pulmonary capillary hemangiomatosis (PCH) |
| 1.5 Persistent pulmonary hypertension of the newborn |
| 2. Pulmonary hypertension with left heart disease |
| 2.1 Left-sided atrial or ventricular heart disease |
| 2.2 Left-sided valvular heart disease |
| 3. Pulmonary hypertension associated with lung diseases and/or hypoxemia |
| 3.1 Chronic obstructive pulmonary disease |
| 3.2 Interstitial lung disease |
| 3.3 Sleep-disordered breathing |
| 3.4 Alveolar hypoventilation disorders |
| 3.5 Chronic exposure to high altitude |
| 3.6 Developmental abnormalities |
| 4. Pulmonary hypertension due to chronic thrombotic and/or embolic disease |
| 4.1 Thromboembolic obstruction of proximal pulmonary arteries |
| 4.2 Thromboembolic obstruction of distal pulmonary arteries |
| 4.3 Non-thromboembolic pulmonary embolism (tumor, parasites, foreign material) |
| 5. Miscellaneous |
| Sarcoidosis, histiocytosis X, lymphangiomatosis, compression of pulmonary vessels (adenopathy, tumor, fibrosing mediastinitis) |
Abbreviations: HIV, human immunodeficiency virus.
One to 2 persons per million per year are diagnosed with either IPAH or FPAH (Abenhaim et al 1996), with at least 6% of these patients having FPAH (Rich et al 1987). However, IPAH comprises the minority of PAH cases, and the incidence of PAH associated with other conditions is generally higher than that for IPAH/FPAH. Histologic features consistent with PAH and clinically evident pulmonary hypertension have been observed in connective tissue diseases including scleroderma, systemic lupus erythematosus, mixed connec-tive-tissue disease, polymyositis, dermatomyositis, and rheumatoid arthritis (Rich 2001; Farber et al 2004). Estimates for PAH in scleroderma patients vary widely from 11% to 35%, representing an incidence of 50 to 230 cases per million (Rich 2001). In patients infected with HIV, the incidence of HIV associated PAH is estimated at 0.1% (Humbert et al 2001). Unlike PAH associated with noninfectious diseases, PAH associated with HIV demonstrates a geographic distribution with sub-Saharan Africa, South Asia, and Southeast Asia representing >80% of total HIV-associated PAH (UNAIDS 2006). The incidence of PAH associated with anorexigens is cyclical in nature and varies depending on the availability of specific appetite suppressants. An association was first identified in the 1960s when an epidemic of PAH occurred in Switzerland, Austria, and Germany in persons taking the anorexigen aminorex fumarate (Rich et al 2000). Use of anorexigens including fenfluramine and dexfenfluramine, previously available in the United States, have also been causally associated with an increased risk for PAH (Abenhaim et al 1996; Simonneau et al 1998).
Prior to the development of disease-specific targeted PAH therapies, the median survival for subjects diagnosed with IPAH was approximately 2.8 years (D’Alonzo et al 1991). However, 2.8 years likely underestimates current survival as the course of the disease has been favorably altered by therapeutic medical advances since that early report. Prognosis is also dependent on the underlying etiology of the disease. The prognosis for patients with PAH associated with connective tissue disease appears to be worse than those with IPAH. Estimates for 2-year survival of scleroderma patients with associated PAH are 40% compared with 48% for 3-year survival for patients with IPAH (Stupi et al 1986; D’Alonzo et al 1991). Survival in patients with HIV-associated PAH is similar to patients with IPAH (Opravil et al 1997; McLaughlin et al 2004). With current HIV therapies, most of the deaths in patients with HIV and associated PAH are now attributed to PAH.
Although the exact cause(s) of PAH is unknown, current dogma suggests that PAH likely represents the final outcome of multiple biologic abnormalities within the pulmonary circulation. Of significance to this review, many studies implicate pulmonary endothelial cell dysfunction in PAH pathobiology. Vasoactive agents produced from endothelial cells modulate pulmonary vascular smooth muscle cell tone in healthy subjects and maintain the normal pulmonary vascular smooth muscle in a state of relaxation (Vane et al 1990). The increased pulmonary vascular reactivity and vasoconstriction that characterize patients with PAH may be a result of loss of endothelial cell integrity (Higenbottam et al 1998). Indeed, dysfunctional endothelial cells are associated with increased production of vasoconstrictor mediators such as thromboxane A2, decreased synthesis of prostacyclin and nitric oxide, and uncontrolled proliferation (Rich 2001). In some patients with PAH, intimal proliferation progresses to almost complete occlusion of pulmonary arterioles (Edwards et al 1977; Pietra et al 1989). In addition, vasoactive mediators produced by endothelial cells may contribute to vascular remodeling (Botney 1999). Progression of PAH is accompanied by impaired endothelial cell functioning that is characterized by an increase in vasoconstrictor and proliferative mediators and a decrease in vasodilator and antiproliferative mediators (Rich 2001). Drugs or drug classes that restore balance to modulation of pulmonary microvasculature tone could be expected to provide therapeutic benefits.
Current treatment options for PAH
Only since 1996, with the introduction of intravenous epoprostenol, have disease-specific targeted medical therapies for PAH become available (Humbert et al 2004). Current treatments primarily target 3 established pathways in PAH pathobiology: the prostacyclin pathway, the nitric oxide pathway, and the endothelin-1 pathway (Fig. 1) (Humbert et al 2004).
Figure 1.
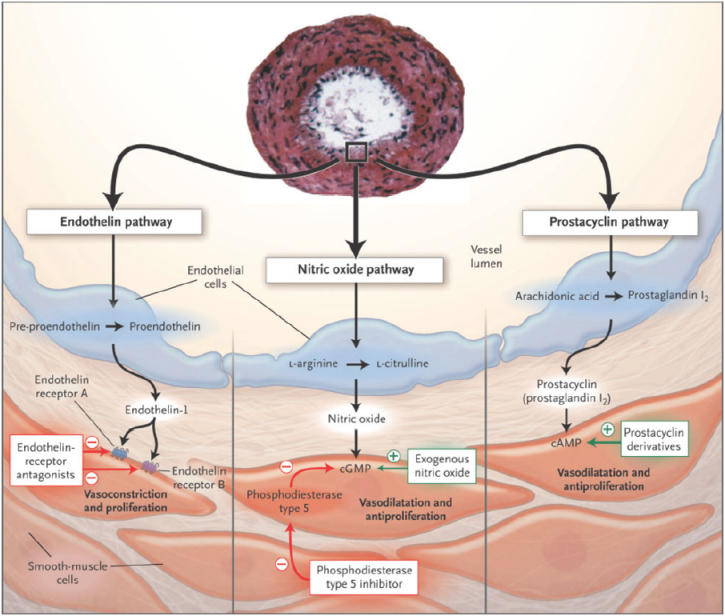
Postulated pathways in the pathobiology of pulmonary arterial hypertension (PAH) and drug classes targeting these pathways. Reprinted from Humbert M, Sitbon O, Simonneau G. 2004. Treatment of pulmonary arterial hypertension. N Engl J Med, 351:1425-36. Copyright © 2004 with permission from Massachusetts Medical Society.
Prostacyclin pathway
Prostacyclin and thromboxane A2 are metabolites of arachidonic acid that are produced by the vascular endothelium. Prostacyclin is a potent vasodilator in both the pulmonary and systemic circulations and has antiproliferative properties (Badesch et al 2004b; Rubin et al 2005a). Thromboxane A2 works in opposition to prostacyclin, inducing vasoconstriction and platelet aggregation (Gerber et al 1980; Farber et al 2004). In PAH, the balance between these two vasoactive mediators is shifted towards thromboxane A2, with decreases in the production of prostacyclin synthase leading to prostacyclin deficiency and thromboxane A2 excess (Christman et al 1992; Tuder et al 1999; Badesch et al 2004b). Exogenously administered prostacyclin or prostanoid analogues may help overcome the adverse effects of prostacyclin depletion (Badesch et al 2004b).
Continuous intravenous (IV) prostacyclin, that is, epoprostenol, has been shown in prospective randomized controlled trials to improve exercise capacity, quality of life (as measured by the Chronic Heart Failure Questionnaire, the Nottingham Health Profile, and the Dyspnea-Fatigue Rating), symptoms, and cardiopulmonary hemodynamics in PAH (Barst et al 1994, 1996, 1999; Shapiro et al 1997; McLaughlin et al 1998). In addition, epoprostenol is the only available therapy shown to improve survival in IPAH in a placebo-controlled clinical trial (Barst et al 1994, 1996). The beneficial effects of prostacyclin therapy are likely due to its vasodilating effects on pulmonary arterioles, inhibition of platelet aggregation, and inhibition of smooth muscle cell proliferation. In addition, epoprostenol appears to have positive inotropic properties and may have anti-inflammatory actions (Baumhakel et al 2005). Two other prostacyclin analogues, subcutaneous or IV treprostinil, and inhaled iloprost, are also approved in the United States. Currently, prostacyclin treatments are most often reserved for more severe cases based on overall risk-benefit considerations. Members of this drug class are limited by short half-life, which requires continuous IV administration (epoprostenol or treprostinil), subcutaneous administration (treprostinil), or frequent inhalation therapy (iloprost) (Rubin et al 2005a). Additional limitations of these treatments include risk of infections with IV epoprostenol or IV treprostinil from central venous catheters, and site pain with subcutaneous treprostinil, not infrequently leading to treatment discontinuation with subcutaneous treprostinil. Epoprostenol therapy remains the treatment of choice for severely ill patients.
Nitric oxide pathway
Nitric oxide (NO) is a potent pulmonary vasodilator and inhibitor of platelet activation and vascular smooth muscle cell proliferation. Synthesis of NO is catalyzed by the family of NO synthase enzymes; decreased levels of the endothelial isoform of NO synthase have been reported in pulmonary vascular tissue in patients with PAH (Giaid et al 1995). The effects of NO are mediated via cyclic guanosine monophosphate (cGMP) in vascular smooth muscle cells. The intracellular concentration of cGMP is regulated by phosphodiesterases, which rapidly degrade cGMP in vivo (Beavo et al 1990; Ahn et al 1991). Phosphodiesterase-5 (PDE-5) is highly expressed in the lung, and its expression is increased in PAH (Braner et al 1993). Therefore, drugs that selectively inhibit PDE-5 may prolong endogenous NO signaling and prove efficacious in PAH. Sildenafil is a PDE-5 inhibitor that was originally approved for erectile dysfunction and was recently approved for the treatment of PAH (Weimann et al 2000; Rubin et al 2005a). In the SUPER-1 trial, three times daily sildenafil improved exercise capacity (assessed by the 6-minute walk test), WHO functional class, and decreased pulmonary arterial pressure and pulmonary vascular resistance (Galie et al 2005b). Tadalafil, another PDE-5 inhibitor, is currently being evaluated for the treatment of PAH.
Endothelin pathway
Endothelin-1 (ET-1) is an endogenous peptide produced by vascular endothelial cells; it is one of the most potent vasoconstrictors and smooth-muscle cell mitogens (Hassoun et al 1992; Stelzner et al 1992). Endothelin-1 expression is elevated in plasma (Rubens et al 2001) and lung tissue (Giaid et al 1993) of patients with PAH. The magnitude of overexpression in ET-1 has been shown to correlate with disease severity (Galie et al 2004) and cardiopulmonary hemodynamics (Cacoub et al 1993; Nootens et al 1995; Cacoub et al 1997). Furthermore, the expression of ET-1 was inversely proportional to survival in a study of PAH patients treated with conventional therapy (Galie et al 2004).
Endothelin-1 exerts its effects through 2 endothelin receptor isoforms: endothelin type-A (ETA) and type-B (ETB). The ETA receptors are primarily expressed on vascular smooth muscle cells and cardiac myocytes; in contrast, ETB receptors are localized predominantly on endothelial cells, and to a lesser extent, on smooth muscle cells and on fibroblasts (Fig. 2) (Dupuis 2001). Activation of the ETA isoform (and the ETB isoform on smooth muscle cells) induces vasoconstriction and proliferation of vascular smooth muscle cells through the activation of phospholipase C and subsequent increase of inositol triphosphate, diacylglycerol, and intracellular calcium (Pollock et al 1995). The mitogenic activity of ET-1 is mediated by the activation of protein kinase C secondary to increases in diacylglycerol and calcium (Ohlstein et al 1992). The ETB receptors are principally involved in the clearance of ET-1, particularly in the vascular beds of the lungs and kidney, and may induces vasodilation via release of NO and prostacyclin from the endothelial cells (Hirata et al 1993; Rubin et al 2005a).
Figure 2.
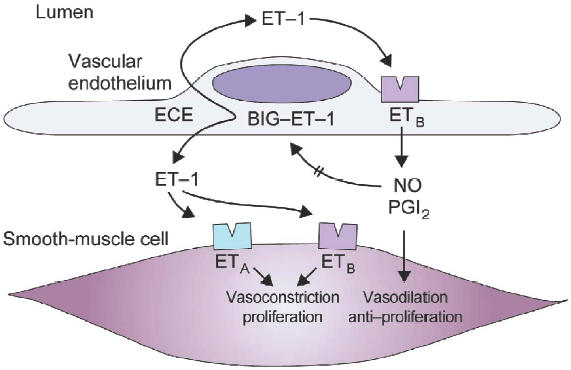
Schematic representation of the endothelin system in vascular tissue. Interactions of endothelin-1 with ETA on endothelial cells and ETA and ETB receptors on smooth muscle cells are shown. Reprinted from Dupuis J. 2001. Endothelin-receptor antagonists in pulmonary hypertension. Lancet, 358:1113-4. Copyright © 2001 with permission from Elsevier.
Abbreviations: ET-1, endothelin-1; BIG-ET-1, proendothelin-1; ECE, endothelin-converting enzyme; NO, nitric oxide; PGI2, prostacyclin.
Endothelin receptor antagonism is a well-established approach to blocking the ET-1 system in PAH (Galie et al 2004). Endothelin receptor antagonists (ERAs) are either ETA selective, such as sitaxsentan and ambrisentan, or non-selective for the ETA and ETB receptors, such as bosentan. Selective inhibition of ETA receptors may be preferential to non-selective receptor antagonism by permitting maintenance of vasodilator and clearance functions specific to ETB receptors on the endothelial cells, while preventing the vasoconstriction and cellular proliferation mediated by ETA. Bosentan, an ETA/ETB non-selective, sulfonamide-class ERA, was the first approved ERA for the treatment of PAH. More recently, the ETA-selective, sulfonamide-class ERA sitaxsentan was approved in the EU. Ambrisentan, a non-sulfonamide, propanoic-acid class, ETA-selective ERA, is in late-stage clinical development.
Ambrisentan in PAH
Chemistry
Ambrisentan, (+)-(2S)-2-[(4,6-dimethylpyrimidin-2-yl)oxy]-3-methoxy-3,3-diphenylpropanoic acid, is an ETA-selective ERA. In vitro studies with ambrisentan have demonstrated that it is a potent and selective inhibitor of the ETA receptor. Studies using human ventricular myocyte-derived ETA and ETB receptors have shown that ambrisentan has a high binding affinity for the ETA receptor, with a Ki (dissociation constant for the inhibitor) of approximately 0.011 nM and a selectivity for the ETA receptor over the ETB receptor of >4000:1, with no relevant binding to other receptors (Greene et al 2006). At therapeutically relevant plasma concentrations, ambrisentan has high receptor occupancy with ETA (>90%), and low affinity for ETB (<10%). Ambrisentan is a unique ERA. It differs from bosentan and sitaxsentan in that it is a propanoic acid-class molecule rather than a sulfonamide-class agent. The chemical composition of ambrisentan, also known as LU 208075 or BSF 208075, is C22 H22N2O4, and its molecular weight is 378.4 g/mol (Rubin et al 2005b). The structural formula of ambrisentan is shown in Figure 3.
Figure 3.
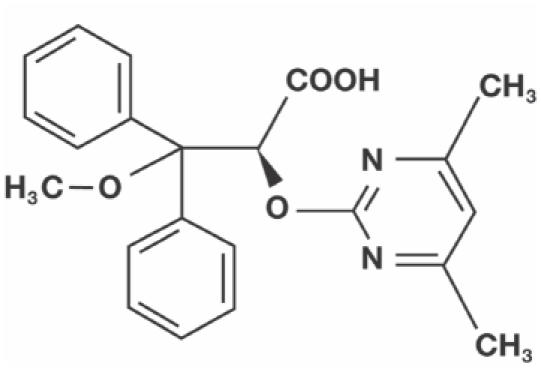
The chemical structure of ambrisentan.
Pharmacokinetics
Ambrisentan is rapidly absorbed into the systemic circulation following oral administration, with high oral bioavailability (Rubin et al 2005b). The pharmacokinetics of ambrisentan are dose-linear over a range of 1–100 mg and are not affected by food intake. Steady-state is achieved after 3–4 days of once-daily oral dosing with ambrisentan, and the pharmacokinetics of multiple doses (ie, steady-state ambrisentan) are consistent with observations after a single dose (Rubin et al 2005b). The steady-state elimination half-life of ambrisentan in PAH patients is approximately 15 hours for the 5 mg dose, providing the rationale for once-daily dosing (Oudiz 2006; Rubin et al 2005b).
The main metabolic pathways of ambrisentan appears to be hepatic phase II glucuronidation of the parent compound, and to a lesser extent phase I hydroxylation. Moreover, in vitro and in vivo animal model studies with ambrisentan concentrations that exceeded therapeutic levels demonstrated that ambrisentan had little effect on hepatic enzyme (eg, CYP2C9, CYP3A4, or CYP1A2) induction or inhibition. Following oral administration of a radiolabeled dose in preclinical studies, ambrisentan was primarily detected in the liver and plasma 2–4 hours after administration. The majority of the radiolabeled dose was recovered in the feces as unchanged or glucuronide-conjugated drug.
Drug–drug interactions
Current sulfonamide-class ERAs developed for the treatment of PAH are associated with potentially significant drug-drug interactions. Bosentan induces the cytochrome P450 isoenzymes CYP2C9 and CYP3A4 and may decrease the systemic exposure of other drugs that share this metabolic pathway (Kenyon et al 2003). Similarly, sitaxsentan inhibits the activity of CYP2C9, thereby increasing systemic exposure to drugs metabolized by this cytochrome P450 isozyme (Barst et al 2004a). Both bosentan and sitaxsentan alter the pharmacokinetics of warfarin (Kenyon et al 2003; Badesch et al 2004a; Dingemanse et al 2004; Barst et al 2006). While the effects of concomitant bosentan may or may not require adjustment of warfarin dose, an 80% reduction in the dose of warfarin was used in the clinical studies of sitaxsentan to prevent over-anticoagulation and the potential for bleeding (Barst et al 2006). Additional drugs commonly prescribed to patients with PAH that are metabolized by the cytochrome P450 system include sildenafil, hormonal contraceptives, glyburide, cyclosporin A, and statins.
Potential drug–drug interactions between ambrisentan and sildenafil, and between ambrisentan and warfarin were evaluated in healthy subjects co-administered ambrisentan plus sildenafil or ambrisentan plus warfarin, respectively. Pharmacokinetic parameters of ambrisentan and sildenafil were similar when administered as monotherapy or in combination (Dufton et al 2006). The pharmacokinetics of n-desmethyl-sildenafil, the active metabolite of sildenafil, were also unaffected by multiple doses of ambrisentan (Dufton et al 2006). Similarly, the pharmacokinetics of warfarin enantiomers and ambrisentan were not appreciably influenced by concomitant administration (Gerber et al 2006). Moreover, co-administration of multiple ambrisentan doses had no clinically relevant effect on the prothrombin time or international normalized ratio following a single dose of warfarin (Gerber et al 2006). In both studies, ambrisentan was well tolerated and no safety concerns arose with the combination therapies.
The lack of pharmacodynamic effect of ambrisentan on warfarin has been confirmed in ambrisentan clinical trials to date, which allowed the use of warfarin or warfarin-like anticoagulants as concomitant medications. In a Phase 2, dose-ranging study, prothrombin time, international normalized ratio, and anticoagulant dose were unaffected by ambrisentan treatment (Galie et al 2005a). Similar results were reported for warfarin-type anticoagulant dosing in the Phase 3 ambrisentan trials (Olschewski et al 2006; Oudiz 2006). These data suggest that dosage adjustment of warfarin is not needed during concomitant ambrisentan therapy.
Clinical efficacy
To date, a total of 7 Phase 2 or 3 trials of ambrisentan have been completed or are ongoing. In the Phase 2, double-blind, dose-ranging study, 64 patients with IPAH or PAH associated with connective tissue disease, anorexigen use, or HIV were randomized to receive oral ambrisentan 1, 2.5, 5, or 10 mg once-daily for 12 weeks, followed by a 12-week open-label treatment period (Galie et al 2005a). The primary endpoint was the change from baseline in 6-minute walk distance (6MWD). Secondary endpoints included change from baseline in WHO functional class, Borg dyspnea index (BDI), subject global assessment, and cardiopulmonary hemodynamics. By week 12, 6MWD had significantly improved for all dose groups (p < 0.02), with a mean increase of +36 m for the combined analysis (p < 0.001) (Fig. 4a) and the magnitude of increase in 6MWD was comparable regardless of baseline WHO functional class. Exercise capacity continued to increase throughout the study, reaching a maximum improvement from baseline of +54 m at week 24 (Fig. 4b).
Figure 4A.
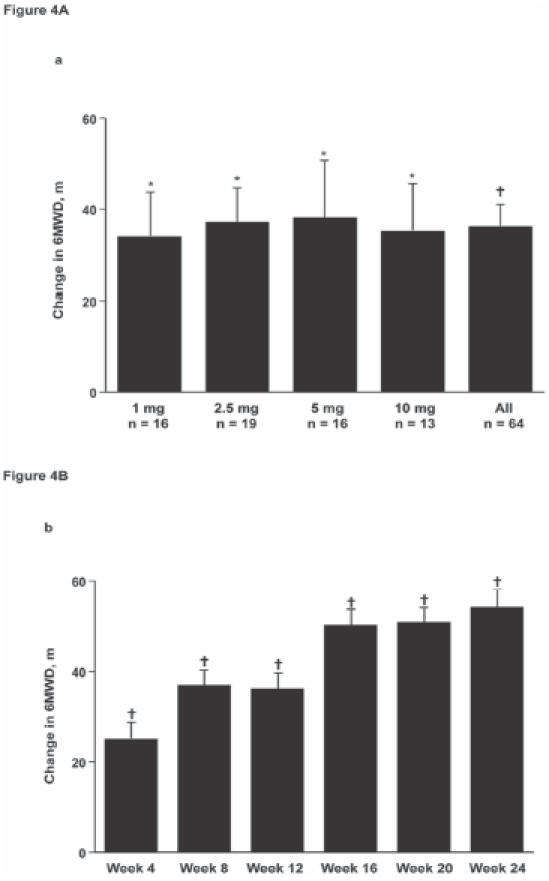
Ambrisentan at all dose levels significantly increased exercise capacity as assessed by the 6-minute walk test from baseline to week 12 in patients with pulmonary arterial hypertension. (4B) Improvements in 6-minute walk distance (6MWD) were maintained over 24 weeks; Data are mean ± standard error. *p < 0.02, †p < 0.001 versus baseline. Reprinted from Galie N, Badesch D, Oudiz R, et al. 2005a. Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol, 46:529–35. Copyright © 2005 with permission from American College of Cardiology Foundation.
Measurable improvements with ambrisentan treatment were also observed in WHO functional class (Fig. 5), BDI, subject global assessment, and cardiopulmonary hemodynamics (Galie et al 2005a). Eighteen (36%) patients experienced clinical improvements of sufficient magnitude to improve WHO functional class by 1 or more classes, whereas only 2 (3.4%) patients deteriorated in WHO functional class. Decreases (ie, improvement) in BDI ranged from −0.6 ± 0.5 in the 1 mg dose group to −1.0 ± 0.6 in the 5 and 10 mg dose groups at week 12, and the average reduction in BDI across dose groups was −1.3 ± 0.3 at week 24 (p < 0.0001). Significant improvements were also observed in quality of life, as measured by a subject global assessment (11.3 ± 2.4 and 12.1 ± 2.7 mm at weeks 12 and 24, respectively; p < 0.0001). In the combined analysis of all dose groups, cardiac index increased, and mean pulmonary arterial pressure and pulmonary vascular resistance decreased compared with baseline (p < 0.05).
Figure 5.
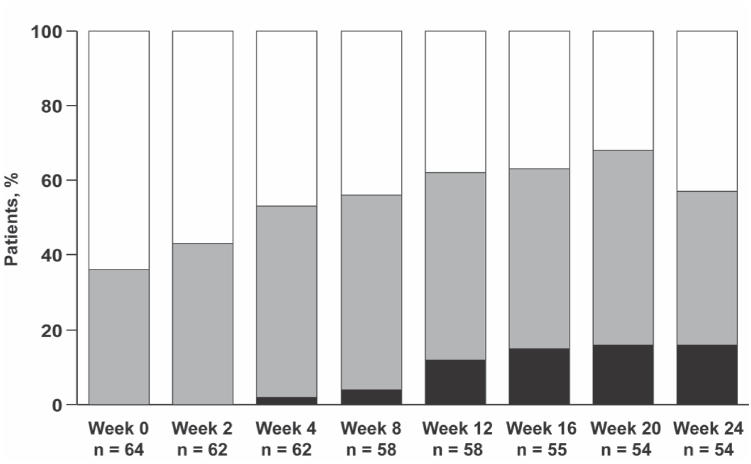
Ambrisentan improved World Health Organization functional class in patients with pulmonary arterial hypertension. Black bars = Class I, grey bars = Class II, white bars = Class III. Reprinted from Galie N, Badesch D, Oudiz R, et al. 2005a. Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol, 46:529–35. Copyright © 2005 with permission from American College of Cardiology Foundation.
Patients who completed the 24-week study were eligible to participate in a long-term, open-label extension study. Fifty-four patients continued treatment and 50 of these patients were receiving ambrisentan monotherapy after 1 year (48 weeks) of treatment (Galie et al 2005c). A combined analysis of the overall patient population demonstrated that the clinical benefits of ambrisentan were sustained over the 1-year study period, including improvements in 6MWD (mean increase 55 ± 55 m, p < 0.0001 for all dose groups combined), BDI, and WHO functional class.
Ambrisentan was also evaluated in two Phase 3 randomized, double-blind, placebo-controlled, multicenter studies (ARIES-1 and ARIES-2: Ambrisentan in PAH-A Phase III, Randomized, Double-blind, Placebo-controlled, Multicenter Efficacy Study of Ambrisentan in Subjects with Pulmonary Arterial Hypertension) in patients with IPAH or PAH associated with connective tissue disease, anorexigen use, or HIV infection. Both Phase 3 studies have been completed, and a combined long-term extension study is ongoing.
The ARIES-1 study assessed the efficacy and safety of once-daily ambrisentan 5 or 10 mg versus placebo. A total of 202 patients were randomized to 1 of these 3 treatment groups for the 12-week study duration. Significant improvements in the placebo-corrected 6MWD change from baseline to week 12 versus placebo were seen with both dose groups (+31 m for 5 mg, p = 0.008; +51 m for 10 mg; p < 0.001). Improvements in WHO functional class and Borg dyspnea index were also observed (Oudiz et al 2006). Although 6 patients in the placebo group developed clinical worsening, compared with 3 patients in each of the ambrisentan groups, this difference was not statistically significant. Clinical worsening of PAH was predefined as: death, lung transplantation, hospitalization for PAH, atrial septostomy, or study withdrawal due to initiation of other PAH therapies or 2 or more early escape criteria.
In the ARIES-2 study, 192 patients with PAH were randomized to receive ambrisentan 2.5 or 5 mg or placebo once-daily for 12 weeks (Olschewski et al 2006). Ambrisentan significantly improved the placebo-corrected 6MWD at week 12 for patients treated with ambrisentan 2.5 mg (+32 m, p = 0.022) or 5 mg (+59 m, p < 0.001). In addition, ambrisentan delayed time to clinical worsening in each dose group versus placebo (2.5 mg: p = 0.005; 5 mg: p = 0.008). Improvements in BDI and the SF-36® Health Survey were also reported to be better in the ambrisentan-treated patients compared with the placebo-treated patients. Six patients died: 4 patients in the placebo group, 2 patients treated with ambrisentan 2.5 mg, and no patients who received ambrisentan 5 mg.
An integrated analysis of ARIES-1 and ARIES-2 confirmed a significant and dose-dependent increase in 6MWD with ambrisentan treatment. All secondary efficacy endpoints demonstrated significant improvements for the combined ambrisentan group and in the individual 5 and 10 mg dose groups (p < 0.05). In addition, significant improvements in time to clinical worsening were seen in the combined ambrisentan group (p = 0.0003) and in each of the individual dose groups (p <0.03).
Safety
Ambrisentan was generally safe and well tolerated in all the PAH clinical studies. The most frequently reported adverse events during the Phase 2, dose-ranging study were peripheral edema, nasal congestion, upper respiratory tract infection, headache, flushing, and nausea (Galie et al 2005a) and did not appear to be dose related. Ambrisentan was well tolerated throughout the 1-year extension study, with no emergent safety signals apparent during long-term therapy (Galie et al 2005c).
Hepatic safety
Liver abnormalities have been associated with the sulfonamide-class ERAs and to date necessitate monthly liver function testing (LFT). In the placebo-controlled PAH studies of bosentan (Bosentan Randomized Trial of Endothelin Antagonist Therapy, BREATHE-1 and Study 351), 12% and 14% of patients, respectively, developed hepatic aminotransferase concentrations >3 × the upper limit of normal (ULN) and 3% and 7% of patients developed hepatic aminotransferase concentrations >8 × ULN respectively following the oral twice-daily administration of 125 and 250 mg bosentan. Three (2%) patients (all in the 250 mg group) withdrew from treatment because of LFT abnormalities (Rubin et al 2002; Channick et al 2001). In the 18-week Sitaxsentan To Relieve ImpaireD Exercise-2 (STRIDE-2) study, the incidence of hepatic aminotransferases > 3 × ULN was 5% for sitaxsentan 50 mg, 3% for sitaxsentan 100 mg, and 11% for bosentan (Barst et al 2006).
In contrast to bosentan, ambrisentan has demonstrated a lower incidence of acute hepatotoxicity in the Phase 2 and 3 clinical trials. During the Phase 2, 24-week study, 2 (3%) patients experienced elevations in hepatic aminotransferases >3 × ULN that required dose reduction or drug discontinuation; an additional 2 patients had isolated elevations that were unconfirmed upon retest and required no change in treatment (Galie et al 2005a). At the start of the long-term extension study (ie. after 24 weeks of ambrisentan treatment), 48% of patients were receiving the maximal dose of ambrisentan (10 mg); no additional elevations of ALT and/or AST >3 × ULN were observed after one year follow-up (Galie et al 2005c). In the pivotal 12-week ARIES-2 study, no patients treated with ambrisentan developed hepatic aminotransferases >3 × ULN, compared with 1 patient in the placebo group (Olschewski et al 2006). Similar results were reported for the pivotal 12-week ARIES-1 study, that is, no patients treated with ambrisentan developed hepatic aminotransferases >3 × ULN, compared with 2 patients in the placebo group (Oudiz et al 2006).
An open-label Phase 2 study of ambrisentan was performed to evaluate the risk of LFT abnormalities in patients with PAH who had previously discontinued bosentan and/or sitaxsentan because of liver toxicity. Thirty-six patients were evaluated, 86% of whom had discontinued bosentan, 6% had discontinued sitaxsentan, and 8% had discontinued both. The median duration of treatment with ERA prior to discontinuation was 9 weeks (McGoon et al 2006). None of the 36 patients enrolled in the study had a recurrence of LFT abnormalities that resulted in discontinuation of ambrisentan during the initial 12-week evaluation period. One patient experienced a transient increase in hepatic aminotransferase >3 × ULN that resulted in dose reduction. No further elevations >3 × ULN have been observed with ambrisentan exposure of more than 1 year.
Decreases in hemoglobin concentration have also been recognized as a class effect associated with ERAs (Galie et al 2005a). In the Phase 2 ambrisentan trial, decreases in hemoglobin concentrations were observed as early as week 2 and remained stable throughout the 12 week study (mean change from baseline to week 12 was −0.8 g/dL); however no further decreases were observed during the subsequent 12-week, open-label treatment period. Furthermore, although both sitaxsentan and bosentan alter the pharmacokinetics of warfarin (Kenyon et al 2003; Badesch et al 2004a; Dingemanse et al 2004; Barst et al 2006), no clinically relevant changes in warfarin-like anticoagulant therapy have been observed in any of the Phase 2 or 3 ambrisentan clinical trials to date (Galie et al 2005a, 2005c; McGoon et al 2006; Olschewski et al 2006; Oudiz et al 2006).
Conclusions
Ambrisentan is a selective ETA receptor antagonist that appears to provide significant clinical benefit in the treatment of patients with PAH. In Phase 2 and 3 clinical trials, ambrisentan improved exercise capacity, dyspnea, time to clinical worsening, WHO functional class, quality of life, and cardiopulmonary hemodynamic parameters. Ambrisentan appears to be safe and well tolerated, with a low incidence of acute hepatotoxicity. Ambrisentan has an improved safety profile compared with sulfonamide-class ERAs with respect to the potential for hepatic toxicity and the potential for drug-drug interactions with agents metabolized by P450 enzymes such as warfarin and sildenafil. These data support the role of ambrisentan for the treatment of PAH.
Expert opinion
Given the significant role that ET-1 seems to play in the pathobiology of PAH, there is a strong rationale for the use of ERAs in the treatment of PAH. A selective ETA receptor antagonist may afford us the opportunity of blocking the actions of ET-1 at the predominant vasoconstrictor receptor subtype, that is, ETA receptor, while permitting ongoing stimulation of the vasodilatory ETB receptor on endothelial cells, in addition to preserving ET-1 pulmonary clearance. However, as the functions of both the ETA and ETB receptors may differ in various disease states as compared with the normal pulmonary vasculature, it remains unclear whether a selective ETA receptor antagonist or an ETA/ETB receptor antagonist will prove to be more efficacious in the treatment of PAH. In addition, whether various PAH subgroups such as PAH associated with connective tissue disease or associated with congenital heart disease may respond more or less favorably with a selective ETA receptor antagonist than with an ETA/ETB receptor antagonist is also currently unknown.
The ETA-selective ERA ambrisentan holds promise in advancing both the safety and convenience of oral PAH treatment. Ambrisentan, a once-daily oral treatment, does not induce or inhibit cytochrome P450 enzymes; therefore, ambrisentan may be preferable to other currently available ERAs when used in conjunction with other medications prescribed for patients with PAH. In clinical trials with ambrisentan, there appears to be a low risk for acute hepatotoxicity. Current evidence suggests that the hepatic aminotransferase increases seen with bosentan may result from intermittent uncoupling of lipid and bile salt excretion that results in intracellular accumulation of bile salts (Fattinger et al 2001; Fouassier et al 2002). Therefore, an ERA metabolized by a different pathway that does not inhibit bile salt secretion may be less likely to lead to acute hepatotoxicity. This is an intriguing hypothesis that is currently under investigation.
It has been an exciting time for the treatment of patients with PAH, with new drugs becoming available, such as prostacyclin analogues, ERAs, and PDE-5 inhibitors. Whether there is an additive or synergistic effect of an ERA with a prostanoid (eg, epoprostenol, iloprost, or treprostinil) and/or a PDE-5 inhibitor (eg, sildenafil or tadalafil) is yet to be determined. As observed in many chronic diseases, for example, diabetes, congestive heart failure and asthma, combination therapy may improve the overall efficacy for treating patients with PAH. An oral once-daily agent with a low risk for liver function abnormalities and drug–drug interactions could confer advantages to the agent’s use in combination regimens, a current area of active investigation and optimism.
The treatment algorithm continues to evolve as newer agents become available. Future developments in vascular biology will improve our understanding of the pathobiology of PAH, as well as provide rationale for more disease-specific targeted therapies. Preclinical and clinical investigations are currently being explored with PDGF inhibitors, vasoactive intestinal peptide, selective 5HT2B antagonists, statins, kinase inhibitors and/or gene therapy. With the advent of genomic technologies and methods, the necessary tools are now becoming available to begin pinpointing the genes that contribute to disease susceptibility and progression. Candidate gene discovery, that is, gene analysis using microarrays, can identify genes that may provide valuable insight into disease biology and may represent an initial step towards the identification of genetic polymorphisms that may help predict efficacy, or lack thereof, with various disease-specific targeted PAH therapeutic modalities. By identifying the genes and gene variants that determine individual disease susceptibility, we might be able to identify patients in preclinical stages of disease as well as allow for individualized therapies that are most efficacious and least likely to cause side effects. Ultimately, as these novel therapeutic options are developed, individualized treatment regimens will evolve. We hope that by further increasing our understanding of the pathobiology of PAH, we will one day be able to prevent and cure this disease.
Further development of ambrisentan for the treatment of PAH is anticipated, as are long-term survival data from the ARIES-1 and ARIES-2 trials.
Acknowledgments
Editorial assistance was provided by Melissa Callahan, PhD, and Crystal Murcia, PhD.
References
- Abenhaim L, Moride Y, Brenot F, et al. Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996;335:609–16. doi: 10.1056/NEJM199608293350901. [DOI] [PubMed] [Google Scholar]
- Ahn HS, Foster M, Cable M, et al. Ca/CaM-stimulated and cGMP-specific phosphodiesterases in vascular and non-vascular tissues. Adv Exp Med Biol. 1991;308:191–7. doi: 10.1007/978-1-4684-6015-5_15. [DOI] [PubMed] [Google Scholar]
- Badesch DB, Abman SH, Ahearn GS, et al. Medical therapy for pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004a;126:35S–62S. doi: 10.1378/chest.126.1_suppl.35S. [DOI] [PubMed] [Google Scholar]
- Barst RJ, Langleben D, Badesch D, et al. Treatment of pulmonary arterial hypertension with the selective endothelin-A receptor antagonist sitaxsentan. J Am Coll Cardiol. 2006;47:2049–56. doi: 10.1016/j.jacc.2006.01.057. [DOI] [PubMed] [Google Scholar]
- Barst RJ, Langleben D, Frost A, et al. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004a;169:441–7. doi: 10.1164/rccm.200307-957OC. [DOI] [PubMed] [Google Scholar]
- Badesch DB, McLaughlin VV, Delcroix M, et al. Prostanoid therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2004b;43:56S–61S. doi: 10.1016/j.jacc.2004.02.036. [DOI] [PubMed] [Google Scholar]
- Barst RJ, McGoon M, Torbicki A, et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol. 2004b;43:40S–7S. doi: 10.1016/j.jacc.2004.02.032. [DOI] [PubMed] [Google Scholar]
- Barst RJ, Maislin G, Fishman AP. Vasodilator therapy for primary pulmonary hypertension in children. Circulation. 1999;99:1197–208. doi: 10.1161/01.cir.99.9.1197. [DOI] [PubMed] [Google Scholar]
- Barst RJ, Rubin LJ, Long WA, et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study Group. N Engl J Med. 1996;334:296–302. doi: 10.1056/NEJM199602013340504. [DOI] [PubMed] [Google Scholar]
- Barst RJ, Rubin LJ, McGoon MD, et al. Survival in primary pulmonary hypertension with long-term continuous intravenous prostacyclin. Ann Intern Med. 1994;121:409–15. doi: 10.7326/0003-4819-121-6-199409150-00003. [DOI] [PubMed] [Google Scholar]
- Baumhakel M, Cremers B, Bohm M. Current Therapy of Pulmonary Hypertension. Herz. 2005;30:303–10. doi: 10.1007/s00059-005-2693-6. [DOI] [PubMed] [Google Scholar]
- Beavo JA, Reifsnyder DH. Primary sequence of cyclic nucleotide phosphodiesterase isozymes and the design of selective inhibitors. Trends Pharmacol Sci. 1990;11:150–5. doi: 10.1016/0165-6147(90)90066-H. [DOI] [PubMed] [Google Scholar]
- Billman GE. Ambrisentan (Myogen) Curr Opin Investig Drugs. 2002;3:1483–6. [PubMed] [Google Scholar]
- Botney MD. Role of hemodynamics in pulmonary vascular remodeling: implications for primary pulmonary hypertension. Am J Respir Crit Care Med. 1999;159:361–4. doi: 10.1164/ajrccm.159.2.9805075. [DOI] [PubMed] [Google Scholar]
- Braner DA, Fineman JR, Chang R, et al. M&B 22948, a cGMP phosphodiesterase inhibitor, is a pulmonary vasodilator in lambs. Am J Physiol. 1993;264:H252–8. doi: 10.1152/ajpheart.1993.264.1.H252. [DOI] [PubMed] [Google Scholar]
- Cacoub P, Dorent R, Maistre G, et al. Endothelin-1 in primary pulmonary hypertension and the Eisenmenger syndrome. Am J Cardiol. 1993;71:448–50. doi: 10.1016/0002-9149(93)90452-i. [DOI] [PubMed] [Google Scholar]
- Cacoub P, Dorent R, Nataf P, et al. Endothelin-1 in the lungs of patients with pulmonary hypertension. Cardiovasc Res. 1997;33:196–200. doi: 10.1016/s0008-6363(96)00189-7. [DOI] [PubMed] [Google Scholar]
- Channick RN, Simonneau G, Sitbon O, et al. Effect of the dual endothelin-receptor antagonist bosentan in patients with pulmonary hypertension: a randomised placebo-controlled study. Lancet. 2001;Oct 6; 358(9288):1119–23. doi: 10.1016/S0140-6736(01)06250-X. [DOI] [PubMed] [Google Scholar]
- Christman BW, McPherson CD, Newman JH, et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med. 1992;327:70–5. doi: 10.1056/NEJM199207093270202. [DOI] [PubMed] [Google Scholar]
- D’Alonzo GE, Barst RJ, Ayres SM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med. 1991;115:343–9. doi: 10.7326/0003-4819-115-5-343. [DOI] [PubMed] [Google Scholar]
- Dingemanse J, van Giersbergen PL. Clinical pharmacology of bosentan, a dual endothelin receptor antagonist. Clin Pharmacokinet. 2004;43:1089–115. doi: 10.2165/00003088-200443150-00003. [DOI] [PubMed] [Google Scholar]
- Dupuis J. Endothelin-receptor antagonists in pulmonary hypertension. Lancet. 2001;358:1113–4. doi: 10.1016/S0140-6736(01)06298-5. [DOI] [PubMed] [Google Scholar]
- Dufton C, Gerber MJ, Yin O, et al. No clinically relevant pharmacokinetic interaction between ambrisentan and sildenafil. Chest. 2006;130:254S. [Google Scholar]
- Edwards WD, Edwards JE. Clinical primary pulmonary hypertension: three pathologic types. Circulation. 1977;56:884–8. doi: 10.1161/01.cir.56.5.884. [DOI] [PubMed] [Google Scholar]
- Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med. 2004;351:1655–65. doi: 10.1056/NEJMra035488. [DOI] [PubMed] [Google Scholar]
- Fattinger K, Funk C, Pantze M, et al. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther. 2001;69:223–31. doi: 10.1067/mcp.2001.114667. [DOI] [PubMed] [Google Scholar]
- Fouassier L, Kinnman N, Lefevre G, et al. Contribution of mrp2 in alterations of canalicular bile formation by the endothelin antagonist bosentan. J Hepatol. 2002;37:184–91. doi: 10.1016/s0168-8278(02)00107-1. [DOI] [PubMed] [Google Scholar]
- Galie N, Badesch D, Oudiz R, et al. Ambrisentan therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2005a;46:529–35. doi: 10.1016/j.jacc.2005.04.050. [DOI] [PubMed] [Google Scholar]
- Galie N, Ghofrani HA, Torbicki A, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. 2005b;353:2148–57. doi: 10.1056/NEJMoa050010. [DOI] [PubMed] [Google Scholar]
- Galie N, Keogh AM, Frost A, et al. Ambrisentan long-term safety and efficacy in pulmonary arterial hypertension-one year follow up. Proc Am Thorac Soc. 2005c;2:A299. [Google Scholar]
- Galie N, Manes A, Branzi A. The endothelin system in pulmonary arterial hypertension. Cardiovasc Res. 2004;61:227–37. doi: 10.1016/j.cardiores.2003.11.026. [DOI] [PubMed] [Google Scholar]
- Gerber MJ, Dufton C, Pentikis H, et al. Ambrisentan has no clinically relevant effect on the pharmacokinetics or pharmacodynamics of warfarin. Chest. 2006;130:256S. [Google Scholar]
- Gerber JG, Voelkel N, Nies AS, et al. Moderation of hypoxic vasoconstriction by infused arachidonic acid: role of PGI2. J Appl Physiol. 1980;49:107–12. doi: 10.1152/jappl.1980.49.1.107. [DOI] [PubMed] [Google Scholar]
- Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333:214–21. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- Giaid A, Yanagisawa M, Langleben D, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328:1732–9. doi: 10.1056/NEJM199306173282402. [DOI] [PubMed] [Google Scholar]
- Greene S, Nunley K, Weber S, et al. ETA vs. ETB receptor selectivity of endothelin-1 receptor antagonists in human myocardial membranes. J Am Coll Cardiol. 2006;47:307A. [Google Scholar]
- Hassoun PM, Thappa V, Landman MJ, et al. Endothelin 1: mitogenic activity on pulmonary artery smooth muscle cells and release from hypoxic endothelial cells. Proc Soc Exp Biol Med. 1992;199:165–70. doi: 10.3181/00379727-199-43342. [DOI] [PubMed] [Google Scholar]
- Hatano S, Strasser T, editors. Report on a WHO Meeting. Geneva: World Health Organization; 1975. Primary Pulmonary Hypertension; pp. 7–45. [Google Scholar]
- Higenbottam TW, Laude EA. Endothelial dysfunction providing the basis for the treatment of pulmonary hypertension: Giles F. Filley lecture. Chest. 1998;114:72S–9S. doi: 10.1378/chest.114.1_supplement.72s. [DOI] [PubMed] [Google Scholar]
- Hirata Y, Emori T, Eguchi S, et al. Endothelin receptor subtype B mediates synthesis of nitric oxide by cultured bovine endothelial cells. J Clin Invest. 1993;91:1367–73. doi: 10.1172/JCI116338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeper MM. Drug treatment of pulmonary arterial hypertension: current and future agents. Drugs. 2005;65:1337–54. doi: 10.2165/00003495-200565100-00003. [DOI] [PubMed] [Google Scholar]
- Humbert M, Nunes H, Sitbon O, et al. Risk factors for pulmonary arterial hypertension. Clin Chest Med. 2001;22:459–75. doi: 10.1016/s0272-5231(05)70284-7. [DOI] [PubMed] [Google Scholar]
- Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425–36. doi: 10.1056/NEJMra040291. [DOI] [PubMed] [Google Scholar]
- Kenyon KW, Nappi JM. Bosentan for the treatment of pulmonary arterial hypertension. Ann Pharmacother. 2003;37:1055–62. doi: 10.1345/aph.1C256. [DOI] [PubMed] [Google Scholar]
- McGoon M, Frost A, Oudiz R, et al. Ambrisentan rescue therapy in patients with pulmonary arterial hypertension who discontinued bosentan or sitaxsentan due to liver function abnormalities. Chest. 2006;130:254S. doi: 10.1378/chest.08-1028. [DOI] [PubMed] [Google Scholar]
- McLaughlin VV, Genthner DE, Panella MM, et al. Reduction in pulmonary vascular resistance with long-term epoprostenol (prostacyclin) therapy in primary pulmonary hypertension. N Engl J Med. 1998;338:273–7. doi: 10.1056/NEJM199801293380501. [DOI] [PubMed] [Google Scholar]
- McLaughlin VV, Presberg KW, Doyle RL, et al. Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest. 2004;126:78S–92S. doi: 10.1378/chest.126.1_suppl.78S. [DOI] [PubMed] [Google Scholar]
- Nootens M, Kaufmann E, Rector T, et al. Neurohormonal activation in patients with right ventricular failure from pulmonary hypertension: relation to hemodynamic variables and endothelin levels. J Am Coll Cardiol. 1995;26:1581–5. doi: 10.1016/0735-1097(95)00399-1. [DOI] [PubMed] [Google Scholar]
- Ohlstein EH, Arleth A, Bryan H, et al. The selective endothelin ETA receptor antagonist BQ123 antagonizes endothelin-1-mediated mitogenesis. Eur J Pharmacol. 1992;225:347–50. doi: 10.1016/0922-4106(92)90109-9. [DOI] [PubMed] [Google Scholar]
- Olschewski H, Galie N, Ghofrani HA, et al. Ambrisentan improves exercise capacity and time to clinical worsening in patients with pulmonary arterial hypertension: Results of the ARIES-2 study. Proc Am Thorac Soc. 2006;3:A728. [Google Scholar]
- Opravil M, Pechere M, Speich R, et al. HIV-associated primary pulmonary hypertension. A case control study. Swiss HIV Cohort Study. Am J Respir Crit Care Med. 1997;155:990–5. doi: 10.1164/ajrccm.155.3.9117037. [DOI] [PubMed] [Google Scholar]
- Oudiz R, Torres F, Frost A, et al. ARIES-1: A placebo-controlled efficacy and safety study of ambrisentan in patients with pulmonary arterial hypertension. Chest. 2006;130:121S. [Google Scholar]
- Pietra GG, Edwards WD, Kay JM, et al. Histopathology of primary pulmonary hypertension. A qualitative and quantitative study of pulmonary blood vessels from 58 patients in the National Heart, Lung, and Blood Institute, Primary Pulmonary Hypertension Registry. Circulation. 1989;80:1198–206. doi: 10.1161/01.cir.80.5.1198. [DOI] [PubMed] [Google Scholar]
- Pollock DM, Keith TL, Highsmith RF. Endothelin receptors and calcium signaling. Faseb J. 1995;9:1196–204. doi: 10.1096/fasebj.9.12.7672512. [DOI] [PubMed] [Google Scholar]
- Rich S, Dantzker DR, Ayres SM, et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med. 1987;107:216–23. doi: 10.7326/0003-4819-107-2-216. [DOI] [PubMed] [Google Scholar]
- Rich S, Rubin L, Walker AM, et al. Anorexigens and pulmonary hypertension in the United States: results from the surveillance of North American pulmonary hypertension. Chest. 2000;117:870–4. doi: 10.1378/chest.117.3.870. [DOI] [PubMed] [Google Scholar]
- Rich S. Pulmonary Hypertension. In: Braunwald E, Zipes DP, Libby P, editors. Heart Disease: A Textbook of Cardiovascular Medicine. Philadelphia, PA: WB Saunders; 2001. pp. 1912–29. [Google Scholar]
- Rubens C, Ewert R, Halank M, et al. Big endothelin-1 and endothelin-1 plasma levels are correlated with the severity of primary pulmonary hypertension. Chest. 2001;120:1562–9. doi: 10.1378/chest.120.5.1562. [DOI] [PubMed] [Google Scholar]
- Rubin LJ, Badesch DB, Barst RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med. 2002;346:896–903. doi: 10.1056/NEJMoa012212. [DOI] [PubMed] [Google Scholar]
- Rubin LJ. Pulmonary arterial hypertension. Proc Am Thorac Soc. 2006;3:111–5. doi: 10.1513/pats.200510-112JH. [DOI] [PubMed] [Google Scholar]
- Rubin LJ, Badesch DB. Evaluation and management of the patient with pulmonary arterial hypertension. Ann Intern Med. 2005a;143:282–92. doi: 10.7326/0003-4819-143-4-200508160-00009. [DOI] [PubMed] [Google Scholar]
- Rubin LJ, Dufton C, Gerber MJ. Ambrisentan for pulmonary arterial hypertension. Future Cardiol. 2005b;1:1–8. doi: 10.2217/14796678.1.4.425. [DOI] [PubMed] [Google Scholar]
- Shapiro SM, Oudiz RJ, Cao T, et al. Primary pulmonary hypertension: improved long-term effects and survival with continuous intravenous epoprostenol infusion. J Am Coll Cardiol. 1997;30:343–9. doi: 10.1016/s0735-1097(97)00187-3. [DOI] [PubMed] [Google Scholar]
- Simonneau G, Fartoukh M, Sitbon O, et al. Primary pulmonary hypertension associated with the use of fenfluramine derivatives. Chest. 1998;114:195S–9S. doi: 10.1378/chest.114.3_supplement.195s. [DOI] [PubMed] [Google Scholar]
- Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43:5S–12S. doi: 10.1016/j.jacc.2004.02.037. [DOI] [PubMed] [Google Scholar]
- Stelzner TJ, O’Brien RF, Yanagisawa M, et al. Increased lung endothelin-1 production in rats with idiopathic pulmonary hypertension. Am J Physiol. 1992;262:L614–20. doi: 10.1152/ajplung.1992.262.5.L614. [DOI] [PubMed] [Google Scholar]
- Stupi AM, Steen VD, Owens GR, et al. Pulmonary hypertension in the CREST syndrome variant of systemic sclerosis. Arthritis Rheum. 1986;29:515–24. doi: 10.1002/art.1780290409. [DOI] [PubMed] [Google Scholar]
- Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159:1925–32. doi: 10.1164/ajrccm.159.6.9804054. [DOI] [PubMed] [Google Scholar]
- UNAIDS. 2006 Report on the global AIDS epidemic. 10th ed. Geneva: Joint United Nations Programme on HIV/AIDS (UNAIDS); 2006. pp. 8–50. [Google Scholar]
- Vane JR, Anggard EE, Botting RM. Regulatory functions of the vascular endothelium. N Engl J Med. 1990;323:27–36. doi: 10.1056/NEJM199007053230106. [DOI] [PubMed] [Google Scholar]
- Vlahakes GJ, Turley K, Hoffman JI. The pathophysiology of failure in acute right ventricular hypertension: hemodynamic and biochemical correlations. Circulation. 1981;63:87–95. doi: 10.1161/01.cir.63.1.87. [DOI] [PubMed] [Google Scholar]
- Weimann J, Ullrich R, Hromi J, et al. Sildenafil is a pulmonary vasodilator in awake lambs with acute pulmonary hypertension. Anesthesiology. 2000;92:1702–12. doi: 10.1097/00000542-200006000-00030. [DOI] [PubMed] [Google Scholar]