

Abstract
The interferon (IFN) system is a well-controlled network of signaling, transcriptional, and post-transcriptional processes that orchestrate host defense against microbes. The IFN response comprises a multi-array of IFN-stimulated gene products that mediate a variety of biological processes designed to control infection and regulate specific immune responses. In this review, we focus on post-transcriptional mechanisms of gene regulation that occur during the course of IFN induction and during the response of cells to IFN. Post-transcriptional mechanisms involve different levels of regulation such as mRNA stability, alternative splicing, and translation. Such controls offer a fine tuning mechanism for efficient and rapid response and as a negative feedback control in IFN biosynthesis and response.
Introduction
Since the discovery of IFN fifty years ago, research on the regulation of the IFN system began several years later and the description of the “super-induction” phenomenon [1, 2] triggered an appreciation of the role of post-transcriptional regulation not only in the IFN system but in general cellular gene expression. In this review, the focus is on the post-transcriptional control of the IFN system during biosynthesis of IFN or during response of cells to IFN. The IFN system consists of latent intracellular enzymes such as 2′-5′ oligoadenylate synthetase (OAS)/ribonuclease L (RNase L), double-stranded RNA dependent protein kinase (PKR), and others acting as the first line of defense against viruses, as evident from studies with specific gene knockout mice [3-6]. Both RNAse L and PKR have post-transcriptional roles that will be discussed here. IFN comes into play as a second line of defense for viruses and amplifies the IFN response with the induction of additional gene products that participate in the antiviral action. Post-transcriptional regulation is one of the several types of regulatory mechanisms that also include receptor-mediated signaling and transcriptional activation. The post-transcriptional control mechanisms confer a stringent mechanism in host defense to viruses and other microbes and contribute to the characteristics of the transient nature of IFN biosynthesis and response. This review is also complementary to other reviews in this special “Biochimie” issue of on the occasion of 50th anniversary of the discovery of IFN.
Post-transcriptional regulation of IFN-beta: the “Super-Induction” phenomenon and involvement of AU-rich elements
Virus infection of fibroblast and epithelial cells leads to the transient production of type-I IFN, e.g., IFN-α/β, to induce an antiviral state in neighboring non-infected cells. The transient nature of IFN-β expression is due to repression of transcriptional activity and rapid turnover of IFN-β mRNA. Earlier work revealed the important role of post-transcriptional control of IFN production through the observation of a “super-induction” phenomenon. This phenomenon occurs when an inhibitor of RNA or protein synthesis is used in cultured cells during early treatment with an IFN inducer, resulting in increased mRNA levels of IFN [1, 2, 7, 8]. For example, when confluent fibroblast cultures are treated with the double stranded RNA polyI-polyC in the presence of the protein synthesis inhibitor cycloheximide, prolongation of IFN mRNA accumulation occurs [2, 7, 9]. The super-induction is thought to be largely due to a pos-transcriptional mechanism that leads to stabilization of IFN mRNA [2, 7]. It was postulated that a labile repressor, probably encoded by an unstable mRNA, when blocked by the inhibitors of RNA or protein synthesis, may account for the increased stability of IFN mRNA [7, 10].
A role of the 3′untranslated region (3′UTR) has been demonstrated by showing that the IFN-β 3′UTR has an inhibitory effect on the translation of IFN-β or Xenopus beta globin [11]; likewise, the murine IFN-α 3′UTR also has an inhibitory effect on translation of beta globin mRNA [12]. The translation inhibitory action of the IFN-β 3′UTR is thought to be mediated by interactions of adenylate uridylate (AU)-rich elements (AREs) and the polyA tail, since the removal of either region improves translation efficiency [13]. The role of IFN-β mRNA stability has been investigated by analyzing mRNA accumulation of a reporter gene fused with various portions of the IFN-β 3′UTR [14, 15]. The destabilization region was minimally mapped to a 28 nucleotide AU-rich region in the IFN-β 3′UTR; however an additional destabilization region in the 5′UTR has been also identified [15]. Deadenylation has been shown to be a pre-requisite for IFN-β mRNA degradation and mediated by the 3′UTR ARE and an additional sequence in the coding region [16]. Figure 1A shows the mRNA sequence of IFN-β and the putative destabilization regions.
Figure 1. ARE variations in Type-I IFN 3′UTRs.

A The mRNA sequence of the human IFN-β is shown. Sequence in bold refers to 3′UTR. Underlined are putative ARE patterns such as pentamers and hexamers. Sequence in italics is destabilization region in the coding region as defined in Ref. [16]. B Sequences of putative ARE regions in the 3′UTRs of type-I IFN including several sub-types of IFN-α. Underlined are AU-rich patterns such as pentamers or hexamers while sequences in Bold are ARE that matches possibly functional 13 nucleotide consensus [18, 46, 87].
The ARE of IFN-β is a Cluster-III ARE, a Class 2 ARE found in many of labile cytokine geness and other genes involved in diverse biological functions [17, 18]. Clusters are based on the number of overlapping pentamers in a continuous stretch [17]. Cluster I to IV are similar to Class 2 AREs while Cluster-V is Class 1 AREs; Classes 1 and 2 are based on Chen and Shyu classification [19]. Members of IFN-α which has more than 20 subtypes, contain heterogeneous AREs as they also differ in the ARE patterns in their 3′UTR. Most of the IFN-α AREs belong to Cluster-III ARE such as IFNA5 and IFNA14, but several members also have Cluster-IV (two overlapping pentamers). Many of the new members of the type-I family, such IFN-lambda, have Cluster V (Class 2 ARE). Figure 1B shows examples of the ARE clusters in IFN-α 3′UTRs.
The 3′UTR and particularly AREs are a target for regulation by a number of RNA binding proteins. These RNA binding proteins either participate in mRNA stabilization (e.g. HuR which is a mammalian homolog of embryonic lethal abnormal vision (ELAV) protein) or in mRNA destabilization (e.g. the zinc finger binding protein, tristetraprolin (TTP), and K homology splicing-regulatory protein (KSRP)). For reviews on RNA binding proteins, see references [20-22]. There has not been any details about the identities of RNA binding proteins that bind type-I IFN mRNAs, but, a 65 kDa protein was identified to bind to a 28 nucleotide AU-rich element in the IFN-β 3′UTR and it is inducible by polyI- polyC [23].
Post-transcriptional control of IFN-β occurs as a result of different stimuli, such as viruses and cytokines. Viruses may be able to induce type-I IFN via post-transcriptional mechanisms in addition to transcriptional induction. HSV-1 infection of fibroblast cell lines leads to stabilization of IFN-β mRNA, as demonstrated by experiments utilizing reporter constructs containing the IFN-β 3′UTR [24]. In particular, the HSV-1 protein ICP27, binds to the AU-rich region and stabilizes IFN-β mRNA [25].
Post-transcriptional control of IFN-γ production
IFN-γ, first reported on in 1965 [27], has been cloned from more than 60 species and based on sequence analysis, portions of its genomic nucleotide structure have been conserved through evolution. This genetic conservation has led a number of laboratories to investigate how expression of this gene is controlled at the transcriptional and post-transcriptional levels. One aspect of the regulation of IFN-γ gene expression that has been investigated by a few laboratories is that of changes in mRNA stability. As described earlier in this review, the presence of ARE motifs in the 3′ UTR region of cytokine contributes to mRNA instability. IFN-γ is no exception to this finding as it has a number of these elements in the 3′ UTR region (Figure 2A). In fact, the portion of the 3′ UTR region that contains these motifs has been highly conserved through evolution, as shown in Figure 2B.
Figure 2.

A The human IFN-γ mRNA sequence. Underlined are putative ARE regions with the 3′ UTR region shown in bold. B Alignment of IFN-γ 3′ UTR regions containing AU-rich motifs.
Over the last few years, there have been a number of reports demonstrating that treatment of both NK cells and T cells with various stimuli results in the stabilization of the IFN-γ mRNA. In almost all cases, this increased stability was shown to be dependent upon p38 mitogen activated protein kinase (p38 MAPK) activity, as the stability was not observed when inhibitors of this pathway were included in the assay. These various stimulations include: treatment of T and NK cells with IL-2 + IL-12 [28-32]; IL-12 + IL-18 treatment of NK cells [32]; anti-CD3 + anti-CD28 or anti-CD2 treatment of T cells [33] with a similar study comparing cells obtained from old and young mice [34]; CD4+ memory T cells activated by OX40L [35]; treatment of both CD4+ and CD8+ T cells with IL-2 and bryostatin [36]; and stimulation of Jurkat T cells with anti-LFA1 and anti-CD3 [37]. In addition, our laboratory (H.A.Y.) has derived a mouse where a 100 bp region containing the ARE elements of the 3′ UTR region of the murine IFN-γ mRNA has been deleted. In preliminary experiments, significantly more IFN-γ was expressed in vitro and in vivo in response to IL-12 or IL-18 treatment (unpublished data), supporting a role for this region of the mRNA in promoting mRNA instability.
Thus far, there have not been any reports directly demonstrating that a specific protein binds to the IFN-γ 3′ UTR region and alters IFN-γ expression. However, in two of the papers mentioned above, there is evidence that the 3′ UTR region is involved in this process. Mavropoulos and co-workers [38] linked the 3′ UTR region of the IFN-γ mRNA to the beta globin gene, transiently transfected the appropriate constructs into HeLa cells and showed that mRNA half-life decreased when activation of the p38 MAPK was inhibited. In another report, Wang and colleagues demonstrated that siRNA to HuR, a protein known to interact with the 3′ UTR regions of cytokine mRNAs, resulted in loss of mRNA stabilization seen in response to treatment of Jurkat cells with anti-LFA1 and anti-CD3 [37]. This paper is the first report of a unique RNA binding protein altering IFN-γ mRNA levels but experiments demonstrating a direct binding of HuR to the IFN-γ mRNA were not shown.
Thus, a common theme from the limited number of reports on IFN-γ mRNA stabilization is that IFN-γ mRNA stability is mediated by a p38 MAPK, a kinase involved in signal transduction in response to many different extracellular signals. Whether or not this kinase targets proteins that may directly interact with the IFN-γ mRNA will likely be elucidated in future studies.
Recently, microRNAs (10-22 nucleotides) have been found to play an important role in regulating gene expression (for reviews see [39, 40]. While a direct interaction of a specific miRNA with the IFN-γ mRNA has not been demonstrated, deletion of Dicer, a gene essential for microRNA processing and maturation, in the T cell compartment resulted in a higher percentage of IFN-γ producing CD4+ T cells that made more IFN-γ in response to stimulation than cells obtained from wild-type mice [41]. Furthermore, computational analysis has revealed that IFN-γ may be a good target for known miRNAs (H.A.Y., unpublished observations). Thus an area of significant future interest will be the possible involvement of miRNAs in the post-transcriptional regulation of IFN expression.
Post-transcriptional control of IFN-γ expression is another possible mechanism by which the host may control levels of IFN-γ protein. While this topic has not been studied in great depth, Kaempfer group [42-44] has reported that the secondary structure of the very 5′UTR portion of the IFN-γ gene has been conserved through evolution, even though the primary nucleotide sequence has not been conserved. The consequence of this evolutionary conservation is that the 5′UTR 14 nucleotides forms a pseudoknot. This pseudoknot strongly activates PKR, an IFN inducible kinase that is activated by double-stranded RNA [42, 44]. This active PKR then phosphorylates EIF2α, resulting in the inhibition of further IFN-γ mRNA translation. This translational control has not been observed with type-I IFN since the 5′UTR pseudoknot is not found [42]. The Kaempfer lab also demonstrated that mutations that decrease the stability of this pseudoknot increase IFN-γ protein expression. Thus IFN-γ RNA secondary structure adds an additional level of complexity to our understanding of the mechanisms by which the host regulates expression of this critical immunoregulatory protein.
In summary, mRNA stability, miRNAs and RNA secondary structure all seem to play a role in regulating host IFN-γ gene expression. It is becoming quite evident that numerous mechanisms have evolved to limit prolonged expression of this gene and that RNA structure is an important component in this process.
Role of IFN in post-transcriptional control of cellular gene expression
In addition to suppression of viral mRNA and viral protein synthesis, IFNs possess a multi-array of functions that are reflected in the large number of IFN-stimulated genes [45, 46]. IFN possesses both stimulatory and inhibitory functions on the immune system which include post-transcriptional regulation of gene expression, including mRNA stability and translation. Type-I IFNs may increase major histocompatibility antigens (MHC) Class I and II expression, partially by stabilization of the MHC mRNA [47]. The major histocompatibility antigens (MHC class I and II) are increased by IFN treatment of target cells leading to macrophage activation, augmentation of antigen presentation, increased natural killer activity, and cytotoxic T cell activity. Thus, IFNs augment the immune response through MHC I and II mediated processes and this process involves post-transcriptional effects on specific gene expression.
Type-I IFNs have several suppressive effects, including anti-proliferative and down-modulatory effects which include both transcriptional and post-transcriptional regulation of gene expression. For example, IFN-α can reduce c-myc mRNA stability in Daudi cells [48, 49] but increases c-myc mRNA in murine fibroblasts [50]; IFN-β serine 17 can also inhibit c-myc mRNA stability in a colon carcinoma cell line [51]. While it is known that c-myc gene expression is decreased by IFN-γ in a Stat1-dependent manner in embryonic mouse fibroblasts[52], IFN-γ increases c-myc mRNA stability in a human breast cancer cell line [53]. Thus, IFNs have contra-indicatory roles on stability of c-myc mRNA, a transcriptional factor that regulates cell proliferation, and the response seems to be dependent on both of the type of IFN and the target cell.
IFN, particularly Type-I, may exert suppressive action on certain immune response components similar to that reported for IL-4 and IL-10,. For example, IFN-α/β can suppress the pro-inflammatory cytokines TNF-α and IL-8 by both transcriptional and post-transcriptional mechanisms [54, 55]. IFN-β induces destabilization of TNF-α and IL-1β mRNA but increase the stability of IL-1ra mRNA leading to a negative feedback control of the monokine network [55]. A recent report demonstrates a role for the RNA binding protein, TTP, in the cellular response to IFN-β or -γ action on LPS-induced macrophages; TTP downregulates several pro-inflammatory cytokines and chemokines including TNF-α, IL-6, CCCl2 (MCP-1), and CCL3 or MIP-1α [56]. This IFN effect is due to induction of the TTP promoter and it is dependent upon both Stat-1 phosphorylation induced by IFN signaling and by p38 MAPK kinase activation that is triggered by LPS [56]. TTP is a zinc finger protein that promotes the decay of a number of mRNAs that contain Class 2 AREs, such as TNF-α, GM-CSF, IL-3, and COX-2 [57, 58]. The p38 MAPK pathway is important in TTP-mediated activity by the fact that its downstream target, MK2, phosphorylates TTP, resulting in the loss of mRNA destabilization activity [59, 60].
The suppressive effects of type-I IFN can occur post-transcriptionally via mechanisms different from the regulation of mRNA stability. IFN-α inhibits IgM mu-chain synthesis in IFN-treated sensitive Daudi lymphoma cells as a result of decreased steady-state levels of mu-chain mRNA [49]. This effect has been attributed to a post-transcriptional mechanism, that is not thought to involve increased mRNA decay [49]. The α-chemokine, interleukin-8 (CXCL8) inhibits the antiviral action of IFN-α, presumably by a further downstream post-transcriptional mechanism that affects the OAS/RNase L pathway, though the exact mechanism is not known [61, 62].
The role of IFN-regulated RNAse L and PKR in the post-transcriptional control of gene expression
IFN induces gene expression of OAS which, upon binding to viral double-stranded RNA intermediates, becomes activated and synthesizes short 2′-5′oligoadenylates (2-5A). The latter, in turn, activates RNAse L, an enzyme that potently degrades viral mRNAs. This pathway is reviewed by Silverman and Bisbal in this same issue. In recent years, it has been accepted that RNase L participates in the degradation of selected cellular mRNAs [63-67]. Specifically, RNase L has been shown to downregulate PKR mRNA [64]. During the IFN antiviral response in normal cells, PKR mRNA expression is transient, but in RNase L-null cells, extended kinetics of PKR mRNA expression is observed, due to increased mRNA stability [64]. The effect results in prolongation of the PKR-dependent phosphorylation of the subunit of eukaryotic translation initiation factor 2, eIF2α; a process that leads to inhibition of viral protein synthesis [64]. Thus, RNAse L contributes to the transient nature of the IFN response in order to ensure a brief translational arrest imparted by PKR. A similar role of RNAse L negative regulation of the IFN response has also been suggested by the report that a novel IFN-stimulated gene encoding a 43-kDa ubiquitin-specific protease, designated ISG43, is downregulated by RNase L [67]. This regulation occurs at the mRNA stability level since the ISG43 mRNA half life increases in RNase L-null cells [67].
RNAse L can downregulate another functionally important cellular mRNA, myoD, an important transcription factor essential for muscle differentiation. RNase L and its inhibitor RLI are sequentially induced during C2 cell line myoblast differentiation to myotubes [63]. Inhibition or over-expression of RNAse L prolongs or decreases MyoD mRNA half life, respectively[63]. Activated by the localization of a fraction of RNase L in mitochondria that increases with IFN-α treatment, a role of RNAse L in down-regulating miotchondria mRNAs such as those of CYTB), ATPase 6 (ATP6), and cytochrome oxidase II (CO) has been established and proposed as a mechanism of the anti-proliferative action of IFN [66]. This was demonstrated by reducing RNAse L activity through the introduction of an antisense construct or by directly activating RNase L activity by 2-5A [66]. The effects of RNase L on cellular mRNAs appear to be highly restricted to specific mRNAs since no global effects on cellular mRNAs are observed in the studies that dealt with this topic. None of the previous studies however, demonstrate a direct binding of RNase L or association with the target mRNA, and the mechanism of RNAse L activity remains to be explored.
RNase L has a post-transcriptional regulatory role that is different from mRNA turnover; it can modulate translation termination by interacting with human translation termination factor eRF3/GSPT1 [68]. It is proposed that this interaction brings RNase L into close contact with its target mRNA. Moreover, activated RNase L, may compete with eRF3/GSPT1-PABP, a complex that is essential for termination of mRNA translation and release for subsequent rounds of translation [68].
PKR, an important effecter arm of the antiviral action against many viruses including EMCV, VSV, and HSV-1 [69-71], is a serine/threonine kinase and its mechanism of action is mediated through inhibition of translation initiation due to phosphorylation of EIF 2α. A detailed description of the PKR pathway is given in this issue by Meurs and Esteban. It is expected that PKR can mediate several pathways as a result of translational control. Specific expression of selected proteins (Fas, p53, Bax and others) that trigger apoptosis by engagement of the caspase pathway, can be mediated by PKR activation [72]. PKR also has novel protein substrates and cellular functions in calcium signaling, nuclear factor-k B (NF-kB) signaling, IgE class switching, and cellular growth [5, 73]. Though translation arrest is transient and global, particularly for housekeeping proteins, translation of stress-induced transcripts encoding heat shock proteins and selected transcription factors (e.g, GCN4 and ATF4) are not inhibited and may actually increase as a result of stress [74]. PKR-mediated stabilization of mRNA, which is dependent on the presence of 3′UTR AU-rich sequences, has been demonstrated during heat shock, with heat shock 70 (HSP70) mRNA [75].
Other types of post-transcriptional control in the IFN system
An interesting observation regarding PKR and an example of alternative splicing in the IFN system, is the finding of a spliced variant with deletion of exon 7 of the PKR gene [76] - in transformed Jurkat T cells, but not normal T cells. This PKR variant has a dominant negative function that inhibits PKR autophosphorylation and eIF2α subunit phosphorylation [77]. Whether this is a general phenomenon in transformed cells is not known. Another example of alternative splicing operating in the IFN system is IFN regulatory factor (IRF) 3 (IRF-3) which, along with IRF-7, is indispensable for IFN-β promoter activation. There is an alternatively spliced form, called IRF-3a, that lacks the DNA binding domain and is unable to bind its target sites in the IFN promoter. Thus this variant acts as a negative modulator that inhibits virus-induced activation of the IFN-β promoter [78]. Alternative splicing can also be induced by IFN-β resulting in the production of soluble isoforms of CD28 and CTLA-4 and these altered forms of the proteins can in turn affect T cell activation [79].
RNA editing by the IFN-inducible form of adenosine deaminase, ADAR1, may also be considered a post-transcriptional mechanism of gene regulation. This enzyme recognizes RNA with a double-stranded structure and causes Adenosine (A) deamination to Inosine (I). Inosine is recognized as guanosine by the cell machinery [80, 81]. Reviews citing work from the Samuel laboratory focused on this topic is found elsewhere [80-82].
The role of post-translational effects of IFN-α has been demonstrated by examples such as the reduction of eIF5A activity in a tumor cell line [72]. The eIF5A activity is modulated by a series of post-translational modifications that culminates in the formation of the unusual amino acid hypusine. that the synthesis of this amino acid has been correlated with cell proliferation and apoptosis [83, 84]. Another post-translational effect of IFN is ubiquitin conjugation, a process that targets proteins for degradation in the 26 S proteasome. ISG15 is an IFN-inducible ubiquitin-like protein that has been experimentally linked to the proteasome [85]. A possible ramification of this pathway has been recently demonstrated in which virus- and IFN-induced ISG15 conjugation leads to inhibition of IRF-3 degradation and therefore increased IRF-3 activity and an enhanced antiviral innate response [86].
Conclusions
IFNs are key gene products that play diverse roles in host defense and regulation of the immune system. Regulation of the IFN system, whether during IFN biosynthesis or during the cellular response to IFN, occurs at many different levels. This complex regulation requires immediate signaling events, rapid transcriptional activation, and post-transcriptional control. The post-transcriptional mechanism itself occurs at multiple levels, including mRNA stability, alternative splicing, translation, and post-translational effects. IFNs themselves stimulate a large number of IFN-stimulated genes, notably RNAse L, PKR, and ADAR1 and these genes participate in the post-transcriptional regulation of the host immune response. Stringent control of the IFN system allows swift elimination of microbes and beneficial immunomodulatory functions prior to the onset of deleterious consequence for the host (Figure 3). Understanding these pathways can lead to new approaches to harnessing the IFN system for further therapeutic applications.
Figure 3. Post-transcriptional control of gene expression during IFN response.
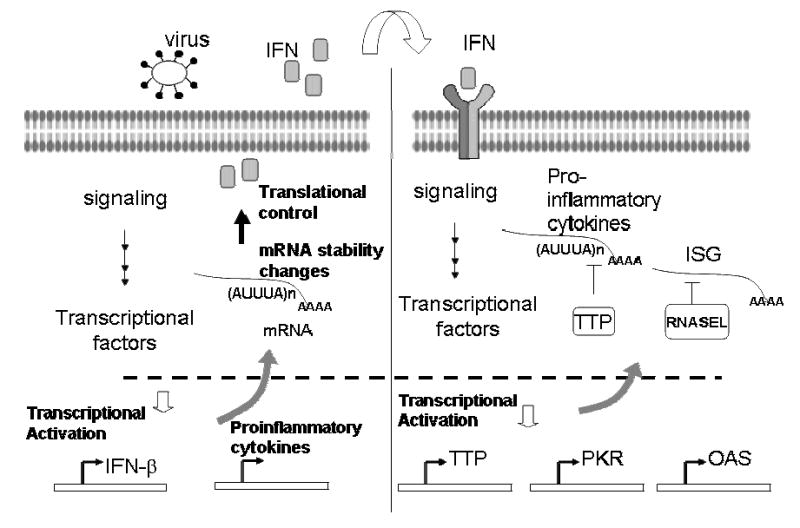
Viruses induce the transcription of type-I IFN in several cell types. The biosynthesis of IFN-β is well-regulated and involves post-transcriptional mechanisms of regulation such as mRNA stability and translation control. The IFN-β mRNA contains ARE(AUUUA)n in the Figure, which is subject to modulation by RNA binding proteins. IFN produced as a result of virus infection binds to IFN receptors in nearby healthy cells to protect them from virus infection. IFN initiates a cascade of events that leads to transcription of many genes (IFN-stimulated genes, ISG) and also pro-inflammatory cytokines. Some of these gene products act as negative feedback inhibitors that attenuate IFN and cytokine response. The RNA binding protein, tristetraprolin (TTP), may destabilize mRNAs that belong to the pro-inflammatory cytokines, particularly in the presence of inflammatory stimulus. RNAse L which requires the activation of the IFN-inducible oligoadenylate synthetase (OAS) act as a negative feedback inhibitor resulting in downmodulation of a selected class of ISG mRNAs including the PKR and ISG43. Additional details are found in Text. Also, details of the individual pathways are found elsewhere in this Review Issue.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sehgal PB, Tamm I, Vilcek J. Human interferon production: superinduction by 5,6-dichloro-1-beta-D-ribofuranosylbenzimidazole. Science. 1975;190:282–284. doi: 10.1126/science.1179208. [DOI] [PubMed] [Google Scholar]
- 2.Cavalieri RL, Havell EA, Vilcek J, Pestka S. Induction and decay of human fibroblast interferon mRNA. Proc Natl Acad Sci U S A. 1977;74:4415–4419. doi: 10.1073/pnas.74.10.4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khabar KS, Dhalla M, Siddiqui Y, Zhou A, Al-Ahdal MN, Der SD, Silverman RH, Williams BR. Effect of deficiency of the double-stranded RNA-dependent protein kinase, PKR, on antiviral resistance in the presence or absence of ribonuclease L: HSV-1 replication is particularly sensitive to deficiency of the major IFN-mediated enzymes. J Interferon Cytokine Res. 2000;20:653–659. doi: 10.1089/107999000414835. [DOI] [PubMed] [Google Scholar]
- 4.Zhou A, Paranjape J, Brown TL, Nie H, Naik S, Dong B, Chang A, Trapp B, Fairchild R, Colmenares C, Silverman RH. Interferon action and apoptosis are defective in mice devoid of 2′,5′- oligoadenylate-dependent RNase L. Embo J. 1997;16:6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou A, Paranjape JM, Der SD, Williams BR, Silverman RH. Interferon action in triply deficient mice reveals the existence of alternative antiviral pathways. Virology. 1999;258:435–440. doi: 10.1006/viro.1999.9738. [DOI] [PubMed] [Google Scholar]
- 6.Samuel MA, Whitby K, Keller BC, Marri A, Barchet W, Williams BR, Silverman RH, Gale M, Jr, Diamond MS. PKR and RNase L contribute to protection against lethal West Nile Virus infection by controlling early viral spread in the periphery and replication in neurons. J Virol. 2006;80:7009–7019. doi: 10.1128/JVI.00489-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sehgal PB, Tamm I. Two mechanisms contribute to the superinduction of poly(I).poly(C)-induced human fibroblast interferon production. Virology. 1979;92:240–244. doi: 10.1016/0042-6822(79)90230-7. [DOI] [PubMed] [Google Scholar]
- 8.Lebendiker MA, Tal C, Sayar D, Pilo S, Eilon A, Banai Y, Kaempfer R. Superinduction of the human gene encoding immune interferon. Embo J. 1987;6:585–589. doi: 10.1002/j.1460-2075.1987.tb04794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tan YH, Berthold W. A mechanism for the induction and regulation of human fibroblastoid interferon genetic expression. J Gen Virol. 1977;34:401–411. doi: 10.1099/0022-1317-34-3-401. [DOI] [PubMed] [Google Scholar]
- 10.Whittemore LA, Maniatis T. Postinduction repression of the beta-interferon gene is mediated through two positive regulatory domains. Proc Natl Acad Sci U S A. 1990;87:7799–7803. doi: 10.1073/pnas.87.20.7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kruys V, Wathelet M, Poupart P, Contreras R, Fiers W, Content J, Huez G. The 3′ UTR region of the human interferon-beta mRNA has an inhibitory effect on translation. Proc Natl Acad Sci U S A. 1987;84:6030–6034. doi: 10.1073/pnas.84.17.6030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Heuvel M, Bosveld IJ, Luyten W, Trapman J, Zwarthoff EC. Transient expression of murine interferon-alpha genes in mouse and monkey cells. Gene. 1986;45:159–165. doi: 10.1016/0378-1119(86)90250-7. [DOI] [PubMed] [Google Scholar]
- 13.Grafi G, Sela I, Galili G. Translational regulation of human beta interferon mRNA: association of the 3′ AU-rich sequence with the poly(A) tail reduces translation efficiency in vitro. Mol Cell Biol. 1993;13:3487–3493. doi: 10.1128/mcb.13.6.3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mosca JD, Pitha PM. Transcriptional and posttranscriptional regulation of exogenous human beta interferon gene in simian cells defective in interferon synthesis. Mol Cell Biol. 1986;6:2279–2283. doi: 10.1128/mcb.6.6.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whittemore LA, Maniatis T. Postinduction turnoff of beta-interferon gene expression. Mol Cell Biol. 1990;10:1329–1337. doi: 10.1128/mcb.10.4.1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paste M, Huez G, Kruys V. Deadenylation of interferon-beta mRNA is mediated by both the AU-rich element in the 3′-UTR region and an instability sequence in the coding region. Eur J Biochem. 2003;270:1590–1597. doi: 10.1046/j.1432-1033.2003.03530.x. [DOI] [PubMed] [Google Scholar]
- 17.Bakheet T, Frevel M, Williams BR, Greer W, Khabar KS. ARED: human AU-rich element-containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res. 2001;29:246–254. doi: 10.1093/nar/29.1.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bakheet T, Williams BR, Khabar KS. ARED 3.0: the large and diverse AU-rich transcriptome. Nucleic Acids Res. 2006;34:D111–114. doi: 10.1093/nar/gkj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen CY, Shyu AB. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- 20.Barreau C, Paillard L, Osborne HB. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2005;33:7138–7150. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khabar KS. The AU-rich transcriptome: more than interferons and cytokines and its role in disease. J Interferon Cytokine Res. 2005;25:1–10. doi: 10.1089/jir.2005.25.1. [DOI] [PubMed] [Google Scholar]
- 22.Fan J, Heller NM, Gorospe M, Atasoy U, Stellato C. The role of post-transcriptional regulation in chemokine gene expression in inflammation and allergy. Eur Respir J. 2005;26:933–947. doi: 10.1183/09031936.05.00120204. [DOI] [PubMed] [Google Scholar]
- 23.Raj NB, Pitha PM. 65-kDa protein binds to destabilizing sequences in the IFN-beta mRNA coding and 3′ UTR. Faseb J. 1993;7:702–710. doi: 10.1096/fasebj.7.8.8500695. [DOI] [PubMed] [Google Scholar]
- 24.Mosca JD, Pitha PM, Hayward GS. Herpes simplex virus infection selectively stimulates accumulation of beta interferon reporter gene mRNA by a posttranscriptional mechanism. J Virol. 1992;66:3811–3822. doi: 10.1128/jvi.66.6.3811-3822.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown CR, Nakamura MS, Mosca JD, Hayward GS, Straus SE, Perera LP. Herpes simplex virus trans-regulatory protein ICP27 stabilizes and binds to 3′ ends of labile mRNA. J Virol. 1995;69:7187–7195. doi: 10.1128/jvi.69.11.7187-7195.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peppel K, Vinci JM, Baglioni C. The AU-rich sequences in the 3′ UTR region mediate the increased turnover of interferon mRNA induced by glucocorticoids. J Exp Med. 1991;173:349–355. doi: 10.1084/jem.173.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wheelock EF, Sibley WA. Circulating Virus, Interferon and Antibody after Vaccination with the 17-D Strain of Yellow-Fever Virus. N Engl J Med. 1965;273:194–198. doi: 10.1056/NEJM196507222730404. [DOI] [PubMed] [Google Scholar]
- 28.Chan SH, Kobayashi M, Santoli D, Perussia B, Trinchieri G. Mechanisms of IFN-gamma induction by natural killer cell stimulatory factor (NKSF/IL-12). Role of transcription and mRNA stability in the synergistic interaction between NKSF and IL-2. J Immunol. 1992;148:92–98. [PubMed] [Google Scholar]
- 29.Gollob JA, Schnipper CP, Murphy EA, Ritz J, Frank DA. The functional synergy between IL-12 and IL-2 involves p38 mitogen-activated protein kinase and is associated with the augmentation of STAT serine phosphorylation. J Immunol. 1999;162:4472–4481. [PubMed] [Google Scholar]
- 30.Hodge DL, Martinez A, Julias JG, Taylor LS, Young HA. Regulation of nuclear gamma interferon gene expression by interleukin 12 (IL-12) and IL-2 represents a novel form of posttranscriptional control. Mol Cell Biol. 2002;22:1742–1753. doi: 10.1128/MCB.22.6.1742-1753.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagy E, Buhlmann JE, Henics T, Waugh M, Rigby WF. Selective modulation of IFN-gamma mRNA stability by IL-12/NKSF. Cell Immunol. 1994;159:140–151. doi: 10.1006/cimm.1994.1303. [DOI] [PubMed] [Google Scholar]
- 32.Matikainen S, Paananen A, Miettinen M, Kurimoto M, Timonen T, Julkunen I, Sareneva T. IFN-alpha and IL-18 synergistically enhance IFN-gamma production in human NK cells: differential regulation of Stat4 activation and IFN-gamma gene expression by IFN-alpha and IL-12. Eur J Immunol. 2001;31:2236–2245. [PubMed] [Google Scholar]
- 33.Musgrave BL, Watson CL, Haeryfar SM, Barnes CA, Hoskin DW. CD2-CD48 interactions promote interleukin-2 and interferon-gamma synthesis by stabilizing cytokine mRNA. Cell Immunol. 2004;229:1–12. doi: 10.1016/j.cellimm.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 34.Pioli C, Pucci S, Barile S, Frasca D, Doria G. Role of mRNA stability in the different patterns of cytokine production by CD4+ cells from young and old mice. Immunology. 1998;94:380–387. doi: 10.1046/j.1365-2567.1998.00523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mestas J, Crampton SP, Hori T, Hughes CC. Endothelial cell co-stimulation through OX40 augments and prolongs T cell cytokine synthesis by stabilization of cytokine mRNA. Int Immunol. 2005;17:737–747. doi: 10.1093/intimm/dxh255. [DOI] [PubMed] [Google Scholar]
- 36.Curiel RE, Garcia CS, Farooq L, Aguero MF, Espinoza-Delgado I. Bryostatin-1 and IL-2 synergize to induce IFN-gamma expression in human peripheral blood T cells: implications for cancer immunotherapy. J Immunol. 2001;167:4828–4837. doi: 10.4049/jimmunol.167.9.4828. [DOI] [PubMed] [Google Scholar]
- 37.Wang JG, Collinge M, Ramgolam V, Ayalon O, Fan XC, Pardi R, Bender JR. LFA-1-dependent HuR nuclear export and cytokine mRNA stabilization in T cell activation. J Immunol. 2006;176:2105–2113. doi: 10.4049/jimmunol.176.4.2105. [DOI] [PubMed] [Google Scholar]
- 38.Mavropoulos A, Sully G, Cope AP, Clark AR. Stabilization of IFN-gamma mRNA by MAPK p38 in IL-12- and IL-18-stimulated human NK cells. Blood. 2005;105:282–288. doi: 10.1182/blood-2004-07-2782. [DOI] [PubMed] [Google Scholar]
- 39.Mattick JS, Makunin IV. Non-coding RNA. Hum Mol Genet. 2006;15(Spec No 1):R17–29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- 40.Massirer KB, Pasquinelli AE. The evolving role of microRNAs in animal gene expression. Bioessays. 2006;28:449–452. doi: 10.1002/bies.20406. [DOI] [PubMed] [Google Scholar]
- 41.Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K. Aberrant T cell differentiation in the absence of Dicer. J Exp Med. 2005;202:261–269. doi: 10.1084/jem.20050678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ben-Asouli Y, Banai Y, Pel-Or Y, Shir A, Kaempfer R. Human interferon-gamma mRNA autoregulates its translation through a pseudoknot that activates the interferon-inducible protein kinase PKR. Cell. 2002;108:221–232. doi: 10.1016/s0092-8674(02)00616-5. [DOI] [PubMed] [Google Scholar]
- 43.Kaempfer R. RNA sensors: novel regulators of gene expression. EMBO Rep. 2003;4:1043–1047. doi: 10.1038/sj.embor.7400005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaempfer R. Interferon-gamma mRNA attenuates its own translation by activating PKR: a molecular basis for the therapeutic effect of interferon-beta in multiple sclerosis. Cell Res. 2006;16:148–153. doi: 10.1038/sj.cr.7310020. [DOI] [PubMed] [Google Scholar]
- 45.de Veer MJ, Holko M, Frevel M, Walker E, Der S, Paranjape JM, Silverman RH, Williams BR. Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol. 2001;69:912–920. [PubMed] [Google Scholar]
- 46.Khabar KS, Al-Haj L, Al-Zoghaibi F, Marie M, Dhalla M, Polyak SJ, Williams BR. Expressed gene clusters associated with cellular sensitivity and resistance towards anti-viral and anti-proliferative actions of interferon. J Mol Biol. 2004;342:833–846. doi: 10.1016/j.jmb.2004.07.065. [DOI] [PubMed] [Google Scholar]
- 47.Kuchtey J, Chefalo PJ, Gray RC, Ramachandra L, Harding CV. Enhancement of dendritic cell antigen cross-presentation by CpG DNA involves type I IFN and stabilization of class I MHC mRNA. J Immunol. 2005;175:2244–2251. doi: 10.4049/jimmunol.175.4.2244. [DOI] [PubMed] [Google Scholar]
- 48.Knight E, Jr, Anton ED, Fahey D, Friedland BK, Jonak GJ. Interferon regulates c-myc gene expression in Daudi cells at the post-transcriptional level. Proc Natl Acad Sci U S A. 1985;82:1151–1154. doi: 10.1073/pnas.82.4.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meurs E, Hovanessian AG. Alpha-interferon inhibits the expression of heavy chain mu messenger RNA in Daudi cells. Embo J. 1988;7:1689–1696. doi: 10.1002/j.1460-2075.1988.tb02997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Levine RA, Seshadri T, Hann SR, Campisi J. Posttranscriptional changes in growth factor-inducible gene regulation caused by antiproliferative interferons. Cell Regul. 1990;1:215–226. doi: 10.1091/mbc.1.2.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chatterjee D, Savarese TM. Posttranscriptional regulation of c-myc proto-oncogene expression and growth inhibition by recombinant human interferon-beta ser17 in a human colon carcinoma cell line. Cancer Chemother Pharmacol. 1992;30:12–20. doi: 10.1007/BF00686479. [DOI] [PubMed] [Google Scholar]
- 52.Ramana CV, Grammatikakis N, Chernov M, Nguyen H, Goh KC, Williams BR, Stark GR. Regulation of c-myc expression by IFN-gamma through Stat1-dependent and -independent pathways. Embo J. 2000;19:263–272. doi: 10.1093/emboj/19.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hamburger AW, Pinnamaneni G. Interferon induced increases in c-myc expression in a human breast carcinoma cell line. Anticancer Res. 1991;11:1891–1894. [PubMed] [Google Scholar]
- 54.Abu-Khabar KS, Armstrong JA, Ho M. Type I interferons (IFN-alpha and -beta) suppress cytotoxin (tumor necrosis factor-alpha and lymphotoxin) production by mitogen-stimulated human peripheral blood mononuclear cell. J Leukoc Biol. 1992;52:165–172. doi: 10.1002/jlb.52.2.165. [DOI] [PubMed] [Google Scholar]
- 55.Jungo F, Dayer JM, Modoux C, Hyka N, Burger D. IFN-beta inhibits the ability of T lymphocytes to induce TNF-alpha and IL-1beta production in monocytes upon direct cell-cell contact. Cytokine. 2001;14:272–282. doi: 10.1006/cyto.2001.0884. [DOI] [PubMed] [Google Scholar]
- 56.Sauer I, Schaljo B, Vogl C, Gattermeier I, Kolbe T, Muller M, Blackshear PJ, Kovarik P. Interferons limit inflammatory responses by induction of tristetraprolin. Blood. 2006;107:4790–4797. doi: 10.1182/blood-2005-07-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carballo E, Lai WS, Blackshear PJ. Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood. 2000;95:1891–1899. [PubMed] [Google Scholar]
- 58.Carrick DM, Lai WS, Blackshear PJ. The tandem CCCH zinc finger protein tristetraprolin and its relevance to cytokine mRNA turnover and arthritis. Arthritis Res Ther. 2004;6:248–264. doi: 10.1186/ar1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hitti E, Iakovleva T, Brook M, Deppenmeier S, Gruber AD, Radzioch D, Clark AR, Blackshear PJ, Kotlyarov A, Gaestel M. Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol Cell Biol. 2006;26:2399–2407. doi: 10.1128/MCB.26.6.2399-2407.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carballo E, Cao H, Lai WS, Kennington EA, Campbell D, Blackshear PJ. Decreased sensitivity of tristetraprolin-deficient cells to p38 inhibitors suggests the involvement of tristetraprolin in the p38 signaling pathway. J Biol Chem. 2001;276:42580–42587. doi: 10.1074/jbc.M104953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khabar KS, Al-Zoghaibi F, Al-Ahdal MN, Murayama T, Dhalla M, Mukaida N, Taha M, Al-Sedairy ST, Siddiqui Y, Kessie G, Matsushima K. The alpha chemokine interleukin 8, inhibits the antiviral action of interferon alpha. J Exp Med. 1997;186:1077–1085. doi: 10.1084/jem.186.7.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Polyak SJ, Khabar KS, Paschal DM, Ezelle HJ, Duverlie G, Barber GN, Levy DE, Mukaida N, Gretch DR. Hepatitis C virus nonstructural 5A protein induces interleukin-8, leading to partial inhibition of the interferon-induced antiviral response. J Virol. 2001;75:6095–6106. doi: 10.1128/JVI.75.13.6095-6106.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bisbal C, Silhol M, Laubenthal H, Kaluza T, Carnac G, Milligan L, Le Roy F, Salehzada T. The 2′-5′ oligoadenylate/RNase L/RNase L inhibitor pathway regulates both MyoD mRNA stability and muscle cell differentiation. Mol Cell Biol. 2000;20:4959–4969. doi: 10.1128/mcb.20.14.4959-4969.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Khabar KS, Siddiqui YM, al-Zoghaibi F, al-Haj L, Dhalla M, Zhou A, Dong B, Whitmore M, Paranjape J, Al-Ahdal MN, Al-Mohanna F, Williams BR, Silverman RH. RNase L mediates transient control of the interferon response through modulation of the double-stranded RNA-dependent protein kinase PKR. J Biol Chem. 2003;278:20124–20132. doi: 10.1074/jbc.M208766200. [DOI] [PubMed] [Google Scholar]
- 65.Chandrasekaran K, Mehrabian Z, Li XL, Hassel B. RNase-L regulates the stability of mitochondrial DNA-encoded mRNAs in mouse embryo fibroblasts. Biochem Biophys Res Commun. 2004;325:18–23. doi: 10.1016/j.bbrc.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 66.Le Roy F, Bisbal C, Silhol M, Martinand C, Lebleu B, Salehzada T. The 2-5A/RNase L/RNase L inhibitor (RLI) [correction of (RNI)] pathway regulates mitochondrial mRNAs stability in interferon alpha-treated H9 cells. J Biol Chem. 2001;276:48473–48482. doi: 10.1074/jbc.M107482200. [DOI] [PubMed] [Google Scholar]
- 67.Li XL, Blackford JA, Judge CS, Liu M, Xiao W, Kalvakolanu DV, Hassel BA. RNase-L-dependent destabilization of interferon-induced mRNAs. A role for the 2-5A system in attenuation of the interferon response. J Biol Chem. 2000;275:8880–8888. doi: 10.1074/jbc.275.12.8880. [DOI] [PubMed] [Google Scholar]
- 68.Le Roy F, Salehzada T, Bisbal C, Dougherty JP, Peltz SW. A newly discovered function for RNase L in regulating translation termination. Nat Struct Mol Biol. 2005;12:505–512. doi: 10.1038/nsmb944. [DOI] [PubMed] [Google Scholar]
- 69.Meurs EF, Watanabe Y, Kadereit S, Barber GN, Katze MG, Chong K, Williams BR, Hovanessian AG. Constitutive expression of human double-stranded RNA-activated p68 kinase in murine cells mediates phosphorylation of eukaryotic initiation factor 2 and partial resistance to encephalomyocarditis virus growth. J Virol. 1992;66:5804–5814. doi: 10.1128/jvi.66.10.5805-5814.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Balachandran S, Roberts PC, Brown LE, Truong H, Pattnaik AK, Archer DR, Barber GN. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity. 2000;13:129–141. doi: 10.1016/s1074-7613(00)00014-5. [DOI] [PubMed] [Google Scholar]
- 71.Khabar KS, Dhalla M, Siddiqui Y, Zhou A, Al-Ahdal MN, Der SD, Silverman RH, Williams BR. Effect of deficiency of the double-stranded RNA-dependent protein kinase, PKR, on antiviral resistance in the presence or absence of ribonuclease L: HSV-1 replication is particularly sensitive to deficiency of the major IFN-mediated enzymes. J Interferon Cytokine Res. 2000;20:653–659. doi: 10.1089/107999000414835. [DOI] [PubMed] [Google Scholar]
- 72.Caraglia M, Vitale G, Marra M, Del Prete S, Lentini A, Budillon A, Beninati S, Abbruzzese A. Translational and post-translational modifications of proteins as a new mechanism of action of alpha-interferon: review article. Amino Acids. 2004;26:409–417. doi: 10.1007/s00726-004-0085-5. [DOI] [PubMed] [Google Scholar]
- 73.Chawla-Sarkar M, Lindner DJ, Liu YF, Williams BR, Sen GC, Silverman RH, Borden EC. Apoptosis and interferons: role of interferon-stimulated genes as mediators of apoptosis. Apoptosis. 2003;8:237–249. doi: 10.1023/a:1023668705040. [DOI] [PubMed] [Google Scholar]
- 74.Anderson P, Kedersha N. Visibly stressed: the role of eIF2, TIA-1, and stress granules in protein translation. Cell Stress Chaperones. 2002;7:213–221. doi: 10.1379/1466-1268(2002)007<0213:vstroe>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhao M, Tang D, Lechpammer S, Hoffman A, Asea A, Stevenson MA, Calderwood SK. Double-stranded RNA-dependent protein kinase (pkr) is essential for thermotolerance, accumulation of HSP70, and stabilization of ARE-containing HSP70 mRNA during stress. J Biol Chem. 2002;277:44539–44547. doi: 10.1074/jbc.M208408200. [DOI] [PubMed] [Google Scholar]
- 76.Xu Z, Williams BR. Genomic features of human PKR: alternative splicing and a polymorphic CGG repeat in the 5′-UTR region. J Interferon Cytokine Res. 1998;18:609–616. doi: 10.1089/jir.1998.18.609. [DOI] [PubMed] [Google Scholar]
- 77.Li S, Koromilas AE. Dominant negative function by an alternatively spliced form of the interferon-inducible protein kinase PKR. J Biol Chem. 2001;276:13881–13890. doi: 10.1074/jbc.M008140200. [DOI] [PubMed] [Google Scholar]
- 78.Karpova AY, Ronco LV, Howley PM. Functional characterization of interferon regulatory factor 3a (IRF-3a), an alternative splice isoform of IRF-3. Mol Cell Biol. 2001;21:4169–4176. doi: 10.1128/MCB.21.13.4169-4176.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Giorelli M, Livrea P, Defazio G, Ricchiuti F, Pagano E, Trojano M. IFN-beta1a modulates the expression of CTLA-4 and CD28 splice variants in human mononuclear cells: induction of soluble isoforms. J Interferon Cytokine Res. 2001;21:809–812. doi: 10.1089/107999001753238042. [DOI] [PubMed] [Google Scholar]
- 80.Samuel CE. RNA editing minireview series. J Biol Chem. 2003;278:1389–1390. doi: 10.1074/jbc.R200032200. [DOI] [PubMed] [Google Scholar]
- 81.Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14:778–809. doi: 10.1128/CMR.14.4.778-809.2001. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Toth AM, Zhang P, Das S, George CX, Samuel CE. Interferon action and the double-stranded RNA-dependent enzymes ADAR1 adenosine deaminase and PKR protein kinase. Prog Nucleic Acid Res Mol Biol. 2006;81:369–434. doi: 10.1016/S0079-6603(06)81010-X. [DOI] [PubMed] [Google Scholar]
- 83.Caraglia M, Marra M, Giuberti G, D’Alessandro AM, Baldi A, Tassone P, Venuta S, Tagliaferri P, Abbruzzese A. The eukaryotic initiation factor 5A is involved in the regulation of proliferation and apoptosis induced by interferon-alpha and EGF in human cancer cells. J Biochem (Tokyo) 2003;133:757–765. doi: 10.1093/jb/mvg097. [DOI] [PubMed] [Google Scholar]
- 84.Caraglia M, Budillon A, Vitale G, Lupoli G, Tagliaferri P, Abbruzzese A. Modulation of molecular mechanisms involved in protein synthesis machinery as a new tool for the control of cell proliferation. Eur J Biochem. 2000;267:3919–3936. doi: 10.1046/j.1432-1327.2000.01465.x. [DOI] [PubMed] [Google Scholar]
- 85.Liu M, Li XL, Hassel BA. Proteasomes modulate conjugation to the ubiquitin-like protein, ISG15. J Biol Chem. 2003;278:1594–1602. doi: 10.1074/jbc.M208123200. [DOI] [PubMed] [Google Scholar]
- 86.Lu G, Reinert JT, Pitha-Rowe I, Okumura A, Kellum M, Knobeloch KP, Hassel B, Pitha PM. ISG15 enhances the innate antiviral response by inhibition of IRF-3 degradation. Cell Mol Biol (Noisy-le-grand) 2006;52:29–41. [PubMed] [Google Scholar]
- 87.Raghavan A, Dhalla M, Bakheet T, Ogilvie RL, Vlasova IA, Khabar KS, Williams BR, Bohjanen PR. Patterns of coordinate down-regulation of ARE-containing transcripts following immune cell activation. Genomics. 2004;84:1002–1013. doi: 10.1016/j.ygeno.2004.08.007. [DOI] [PubMed] [Google Scholar]