

Abstract
In this review we outline clinical features, presentation and pathogenesis of polycystic ovarian syndrome (PCOS), treatment objectives and therapeutic options. We focus on and outline the changing role of the clinical laboratory in diagnosis and treatment of this condition. We also review recent information on the involvement of insulin resistance in the syndrome. We provide some explanation for confusion over the selection of the best hormone measurements for diagnosis. Finally, we outline the best current and future laboratory support for this common condition in young women.
Introduction
PCOS is a common disorder of chronically abnormal ovarian function and hyperandrogenism affecting 5–10% of the female population of reproductive age.1 Although first described in 1935 by Stein and Leventhal,2 the primary aetiology remains unclear and historically there has been no consensus on absolute defining features of the phenotype. At the National Institutes of Health Conference in 1990, three key features of PCOS were generally agreed as oligomenorrhoea, hyperandrogenism (clinical or laboratory evidence), and the absence of other endocrine disorders [congenital adrenal hyperplasia (CAH), hyperprolactinaemia, thyroid dysfunction and androgen secreting tumours].3 The presence of polycystic ovaries, determined by ultrasound evaluation, was not included at that stage as a definitive requirement. A more recent joint consensus statement between the European Society for Human Reproduction and Embryology and the American Society for Reproductive Medicine (ESHRE/ASRM) has revised the criteria for diagnosis of PCOS to include two from three of the following criteria: i) oligomenorrhoea /anovulation; ii) clinical or biochemical evidence of hyperandrogenism; iii) polycystic ovaries, with the exclusion of other aetiologies.4 Thus, there is now some acceptance that ultrasound is not necessarily essential for the diagnosis of this condition. However, the inclusion of polycystic ovaries as one possible diagnostic feature has been promoted by improvements in ultrasound technology and more robust criteria for diagnosis. It is noteworthy that it has been accepted for some years now that abnormal insulin metabolism is a major underlying feature of PCOS, but no aspect of it is included in the most recent definition.4
Patients with PCOS tend to present at clinics complaining of infertility, menstrual disturbance or hirsutism, with or without acne. The clinical biochemist should therefore be aware that laboratory diagnostic services may be used by a wide range of clinicians beyond gynaecologists, and will include requests from primary care physicians, endocrinologists and dermatologists, and potentially specialists in other fields.
Clinical Features
The hallmark clinical features of PCOS are menstrual irregularities (amenorrhoea, oligomenorrhoea, or other signs of irregular uterine bleeding), signs of androgen excess, and obesity.
Hirsutism is the best clinical marker of hyperandrogenism, although different degrees of hirsutism should be expected based on ethnicity. Acne is a more variable marker of hyperandrogenism. Hirsutism is defined as the presence of unwanted terminal (coarse) hairs in females in a pattern more typically seen in adult males. This is in contrast to hypertrichosis, which is independent of androgen influence and is manifested by the superfluous and uniform growth of non-terminal (vellus) hair over the body, particularly in non-sexual areas.5
Visual assessment of the degree of excess terminal hair can be made using the Ferriman-Gallwey scale.6 The original scale scored hair density on a scale 1–4 at 11 androgen-sensitive sites (upper lip, chin, chest, upper back, lower back, upper abdomen, lower abdomen, arm, forearm, thigh, and lower leg), however this has been modified to include nine body sites because arms and legs are currently not considered to be androgen sensitive sites.
PCOS affects women of any age. Originally thought only to impact women of child-bearing age because of the presence of infertility, it is becoming clear that the disease, or early stages of the disease, can occur during adolescence.7 PCOS often manifests around the time of menarche as irregular and often lengthened menstrual cycles, unfortunately this is often not recognised or diagnosed at this age. These girls are often prescribed oral contraception to regulate menstrual cycles and may not receive a diagnosis until much later when seeking treatment for infertility.8
Pathophysiology
The aetiology of PCOS is complex, multifactorial and still poorly understood. In 1958, McArthur et al. documented high urinary levels of both luteinising hormone (LH) and follicle stimulating hormone (FSH) in women with amenorrhoea, hirsutism, obesity, and a characteristic polycystic appearance of the ovaries9 as first described by Stein and Leventhal.2 Later, when an elevation in the serum concentration of LH relative to that of FSH was demonstrated by Yen and colleagues in 1970, a gonadal dysfunction secondary to a regulatory breakdown in the hypothalamic-pituitary axis was hypothesised.10
There is, however, now a consensus that the key features include insulin resistance, androgen excess and abnormal gonadotrophin dynamics but it is unlikely that any one cause in isolation is responsible for this heterogeneous defect. Whatever the cause in addition to ovarian dysregulation, the adrenal, pituitary, adipose tissue and liver are implicated leading to a wide range of associated hormonal imbalances (Figure 1) resulting in a disturbance in the selection of the dominant follicle and hence anovulation.
Figure 1.
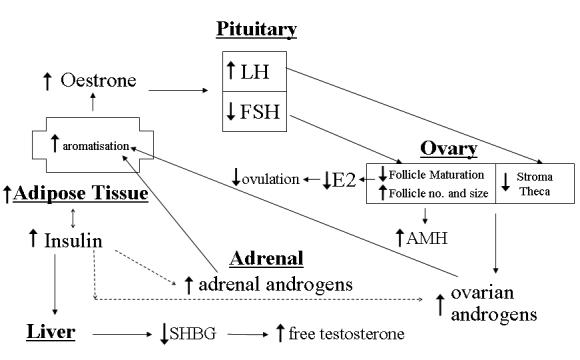
Hormonal imbalances associated with polycystic ovarian syndrome. The key features of PCOS include insulin resistance, androgen excess and abnormal gonadotrophin dynamics. The adrenal, pituitary, adipose tissue and liver are implicated in addition to ovarian dysregulation in PCOS, leading to a wide range of associated hormonal imbalances resulting in a disturbance in the selection of the dominant follicle, hence anovulation.
Laboratory Investigation – Past, Present and Future
Laboratory investigation of patients with suspected PCOS plays an important part in both excluding other aetiologies (Table 1) and diagnosis (Table 2). Measurement of circulating concentrations of LH, FSH, oestradiol, androgens (both of ovarian and adrenal origin), sex hormone binding globulin (SHBG), insulin and anti-Müllerian hormone (AMH), could all, in theory, be of diagnostic value. In practice, however, there are many pitfalls in the selection of a biochemical test or tests with adequate diagnostic specificity when dealing with such a heterogeneous group of patients. In addition, laboratory investigation of PCOS spans fifty years, over which time there have been significant changes in the procedures for hormone measurement. This has contributed to some of the conflicting messages on diagnostic accuracy of the various testing procedures. Furthermore, many of the established hormone tests for this condition may be superseded by the improved diagnostic specificity offered by increased availability of newer tests such as AMH. In this review we will outline the strengths and weaknesses of hormone measurements commonly used in support of a diagnosis of PCOS.
Table 1.
Laboratory tests used to exclude other causes of amenorrhoea or hyperandrogenism.
| Test | Exclusion | When to Perform |
|---|---|---|
| Free thyroxine (fT4)
Thyroid stimulating hormone (TSH) |
Thyroid disease | All Cases |
| Human Chorionic Gonadotrophin (hCG) | Pregnancy | If clinical suspicion or unexplained low LH/FSH |
| Prolactin | Hyperprolactinaemia | All cases |
| 17- Hydroxyprogesterone
Urinary steroid profile |
Congenital adrenal hyperplasia | If total testosterone >4 nmol/L – see text and Table 4 |
| Urinary free cortisol
Evening serum or salivary cortisol |
Cushing’s Syndrome | If clinical suspicion |
Table 2.
Laboratory tests used to investigate PCOS.
| Test | Process assessed |
|---|---|
| LH and FSH | extent of associated pituitary dysregulation; premature ovarian failure if FSH >25 |
| Dehydroepiandrosterone Sulphate (DHEAS) | adrenal androgen production |
| Androstenedione | both adrenal and ovarian androgen production |
| Testosterone | both adrenal and ovarian androgen production |
| Free testosterone | both adrenal and ovarian androgen production |
| Sex Hormone Binding Globulin (SHBG) | hepatic production which can be decreased by insulin resistance and excess androgen |
| Free testosterone (direct or calculated) | biologically active proportion of testosterone which is controlled by both total testosterone and SHBG |
| Anti Müllerian Hormone | follicular number and size – emerging test – needs more study |
| Fasting insulin | insulin resistance (HOMA index) – not routinely recommended associated type 2 diabetes mellitus – restrict to those at elevated |
| Glucose, HbA1c and oral glucose tolerance test | risk e.g. if BMI >30 and family history – see text |
| Lipids (total cholesterol, HDL-cholesterol, LDL-cholesterol) | associated lipid abnormalities and cardiovascular risk – not routinely indicated unless significant other risk factors or over 40 years |
Exclusions
A diagnosis of PCOS should only be made after prolactin and thyroid function tests are performed to exclude hyperprolactinaemia and thyroid disorders. The non-classical 21-hydroxylase deficiency (NC21OHD) variant of CAH and androgen-secreting tumours should be considered if testosterone is significantly elevated (see later). Also remember that the most common cause of amenorrhoea is pregnancy and, in our experience, it is not uncommon for a patient in early pregnancy to be investigated for amenorrhoea. Pregnancy should be suspected if gonadotrophins are low despite adequate or elevated oestradiol and should be confirmed by measuring human chorionic gonadotrophin. The relationship between endocrine disorders and polycystic ovaries was investigated prospectively in 350 patients presenting with hirsutism or androgenic alopecia by O’Driscoll et al.11 Eight patients were identified with relevant additional endocrine disorders. One was acromegalic and one had a microprolactinoma, probably both being unrelated to PCOS. Three had NC21OHD, one a rare hepatic deficiency of 11β-reductase, one a virilising adrenal carcinoma and one a Leydig cell tumour. Interestingly in the latter six cases circulating testosterone was >5 nmol/L and the authors recommended that this cut-off be used to identify patients who required further detailed investigation. Our own experience supports this recommendation but given the considerable concern related to the performance of testosterone immunoassays (see later) we suggest it best to err on the side of caution and suggest that in practice a cutoff of 4 nmol/L be adopted. Hence, 17-hydroxyprogesterone (17-OHP, a marker for NC21OHD) and more complex tests related to androgen/steroid metabolism could be withheld in most women with suspected PCOS unless total testosterone is >4 nmol/L.
In relation to hyperprolactinaemia, early studies did suggest a link with PCOS. In 1954, Forbes et al. indirectly suggested an association between multicystic ovaries in amenorrhoeic patients with galactorrhoea.12 Additional supportive evidence was presented during the 1960s and 1970s and the relationship became generally accepted at that time. More recent studies in which transient elevations of prolactin have been excluded have shown a less frequent association,13 and in 2001 Bracero and Zacur reviewed all the evidence and concluded that PCOS and hyperprolactinaemia have independent origins.14 They suggest that the earlier evidence is based on a limited number of patients and that given the high incidence of PCOS it is probably not surprising that occasionally patients present with both pathologies.
There is, however, a well-described association between PCOS and NC21OHD and it is most important to exclude NC21OHD before a diagnosis of PCOS is made. NC21OHD is a recessive genetic disorder affecting 1–6% of all hyperandrogenic women.15,16 It is easily treatable with glucocorticoid replacement but must first be diagnosed correctly. Unfortunately the elevated androgens associated with this condition may also induce PCOS and there is little clinically to differentiate patients with NC21OHD from those with PCOS. Because the latter condition is extremely common the correct diagnosis may be delayed or tragically, given that we are dealing with an easily treatable disease, missed completely. Even initial biochemical investigation can be misleading. The most commonly used biochemical marker for the 21-hydroxylase deficiency variant of CAH is serum 17-OHP. In NC21OHD, whilst 17-OHP is elevated early in the day (before 10 a.m.) it is often within reference limits later, a time when most patients attend clinic.17 Accordingly, it is recommended that further detailed investigation if the serum 17-OHP concentration is >6 nmol/L (reference range <13 nmol/L in adults).18
Fortunately there are two definitive biochemical tests. The most commonly used is an adrenocorticotropic hormone (ACTH) stimulation test, in which the serum concentration of 17-OHP is measured at 0 and 60 minutes after ACTH (Synacthen 0.25 mg) administration, an exaggerated response being diagnostic.16 Measurement of adrenal steroid metabolites in a 24h urine collection is also diagnostic if 17-hydroxypregnenolone, pregnanetriol and 11-oxo pregnanetriol are elevated19 and has the additional advantage of specifically defining which adrenal steroid biosynthetic enzyme is responsible.
The pattern of urinary steroid metabolites might also be indicative of an androgen-secreting tumour. If an androgen-secreting tumour is suspected a 48h low dose (2 mg/day) dexamethasone should be administered. Lack of response of an elevated circulating testosterone concentration has been shown to clearly differentiate tumourous from non-tumourous hyperandrogenism.20
Gonadotrophins
As discussed earlier, abnormal gonadotrophin secretion, especially increased LH concentrations, is one of the most common findings in PCOS. Both increased LH pulse frequency and amplitude occur.21 FSH may be normal or low, leading to an elevated LH/FSH ratio compared to normally cycling early follicular phase young control women. In the early 1970s it was first demonstrated that PCOS was characterised by an increase in the ratio of LH to FSH as measured by radioimmunoassay (RIA).10 These and other studies around this time favoured an LH to FSH ratio >3 as diagnostic in the absence of the mid-cycle preovulatory LH surge or the menopausal transition. Unfortunately this picture changed with the replacement of RIA by immunometric assays for measurement of LH and FSH in the early 1990s. RIAs were based on the use of polyclonal antibodies and although results obtained between methods were not fully identical they were acceptable.22 After the introduction of monoclonal antibodies, an essential component for most immunometric assays, agreement between methods deteriorated since each monoclonal antibody targeted specific but often different epitopes. Even more worrying, when compared with results obtained by RIA, values tended to be lower probably because a ‘unique’ determinant of antigen structure is being measured rather than a range of isoforms.23
In 2003, Milsom et al. performed a detailed study of the effect that the then newer monoclonal assays had on the LH/FSH ratio in PCOS.24 They found that a ratio of one or greater provided the most reliable separation of women with PCOS from controls. Despite this, not all centres re-evaluated the diagnostic power of the LH/FSH ratio, and altered the PCOS diagnostic cut-off level when monoclonal assays became firmly established. In addition to these methodological considerations, it has been known for some years that LH tends to more elevated in lean compared to obese PCOS patients.25 One may speculate that this pattern signifies a greater contribution to excess androgen synthesis from LH in lean women, whereas in the obese women the significantly elevated insulin levels provide a more major stimulus, and thus there is less need for LH to be high. Body mass index (BMI) should, therefore, also be considered when assessing whether the LH or the LH/FSH ratio are abnormally high. Additional valuable information on the stage of the menstrual cycle or the presence of other abnormalities such as premature ovarian failure may be obtained by measuring circulating concentrations of oestradiol and progesterone.
In summary, both methodological and physiological considerations are important to correctly interpret circulating concentrations of LH and the LH/FSH ratio in the diagnosis of PCOS. Lack of adherence to these considerations has led to much confusion in this area and it is, perhaps, not surprising that measurement of gonadotrophins was not recommended in the recent ESHRE/ASRM consensus on diagnostic criteria for PCOS. Notwithstanding the group did indicate that LH concentrations could be useful as a secondary parameter especially in lean women with amenorrhoea, or in research. They suggested that additional research is needed to clarify further the clinical relevance of LH in PCOS.3
It must not be forgotten, however, that measurements of gonadotrophins can be valuable to detect other causes of anovulation such as primary ovarian failure, which results in significant gonadotrophin elevation, and in this situation FSH concentrations are higher than LH.
Androgens
Excessive androgen production with or without clinical signs is a consistent, but not universal, feature of PCOS. To satisfy current PCOS diagnostic criteria the patient should either have clinical and/or biochemical signs of hyperandrogenism. If hirsutism is unequivocally present then the criteria are matched but other signs of hyperandrogenism such as acne, hair loss or mild hirsutism may be more difficult to determine. In this situation androgen measurement is often chosen as the preferred option, but there remains considerable debate about which androgen or androgens to measure and what constitutes an elevated concentration.
The major circulating androgens are testosterone, androstenedione, dehydroepiandrosterone (DHEA) and DHEA sulphate (DHEAS). In women they arise from the adrenal cortex, the ovarian theca (and to a lesser extent, ovarian stromal cells), and by peripheral conversion of circulating androgenic prohormones (Figure 2). In the normal premenopausal woman DHEAS is produced almost exclusively by the adrenal cortex, circulates unbound to protein and has virtually no androgenic action. DHEA is produced by the adrenal cortex (50%) in addition to peripheral conversion of DHEAS (30%) with a small ovarian contribution (20%). Androstenedione is produced in both the adrenal (30%), ovary (30%) with about 40% from peripheral conversion of DHEA.26 The ovary produces androstenedione and testosterone under trophic LH stimulation with negative feedback by oestradiol. The circulating concentration of androstenedione is subject to significant diurnal variation, caused by the adrenal contribution as well as the variation in ovarian contribution over the menstrual cycle, but is invariably raised in PCOS.
Figure 2.
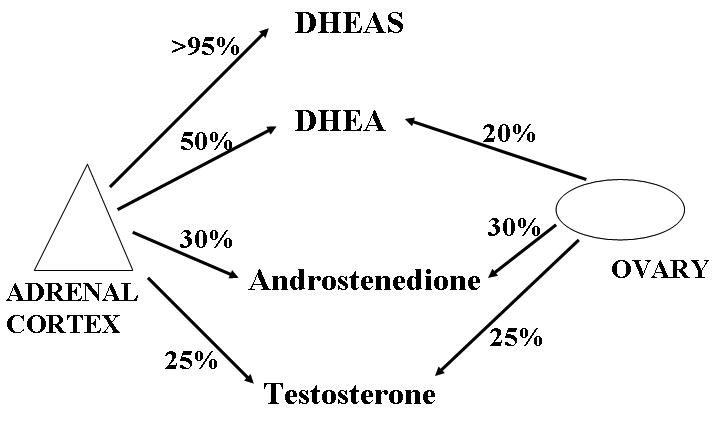
Source of circulating androgens in premenopausal women. In women the major circulating androgens testosterone, androstenedione, DHEAS and DHEA arise from the adrenal cortex, the ovarian theca (and to a lesser extent, ovarian stromal cells), and by peripheral conversion of circulating androgenic pro-hormones.
In adult females testosterone is the most clinically relevant circulating androgen, and has both an adrenal (25%) and ovarian (25%) contribution, but is mainly produced by peripheral conversion from circulating androstenedione. Although testosterone is converted to a more active androgen (dihydrotestosterone) by 5α-reductase, this conversion occurs almost exclusively within target tissues and is not reflected by the circulating concentration of dihydrotestosterone.27
The range of androgens most commonly associated with PCOS is included in Table 2. There is currently no consensus on what is the best androgen to measure or the upper cutoff consistent with PCOS but it is generally accepted that testosterone is the measurement of choice for the investigation of female hyperandrogenism. Total testosterone is not, however, invariably elevated in PCOS. In a study to determine which hormone test was best for the diagnosis of PCOS, it was concluded that an increase in serum total testosterone was found in 70% of patients with PCOS.28 In contrast, a study involving over 1700 women with PCOS showed only a third had an elevated serum testosterone,29 a finding consistent with our own experience.
Surprisingly, as with immunoassays for LH, technological advances have hindered rather than improved the quality of testosterone measurement. This is due to the move away from cumbersome, but specific, solvent extraction assays to more easily automated but less specific direct (non-solvent extraction) procedures. In these newer direct immunoassays testosterone is displaced from carrier proteins by chemical agents competing for protein binding e.g. danazol,30 rather than physically removing binding proteins (and conjugated steroids) during the solvent extraction. The exact nature of the kit components is, however, usually only known to the manufacturers and is protected by property rights. Although many non-extraction methods perform satisfactorily for male serum measurements, measurement in female serum is fraught with problems. Interferences related to the presence of incompletely blocked binding proteins and conjugated interfering steroids can cause falsely elevated results. Recently, Taieb et al. reported on the measurement of female testosterone by using ten direct commercially available immunoassays compared to an isotope-dilution gas chromatography – mass spectrometry reference method.31 They concluded that most non-extraction immunoassays showed a large positive bias. Such was the extent of the problem that it prompted a hard hitting editorial in Clinical Chemistry entitled ‘Immunoassays for testosterone in women: better than a guess?’.32 Although the exact nature of the interference is unknown, some recent evidence does implicate DHEAS.33,34 It is, however, probable that other conjugated steroids and also binding protein related interferences play a part and that different direct immunoassays are affected to different degrees.
The measurement of free testosterone or non-specifically bound testosterone may represent, more closely, the biologically active component and therefore may be of more value than total testosterone in the assessment of hyperandrogenism. The apparent free testosterone concentration obtained by equilibrium dialysis as well as the fraction of serum testosterone not precipitated by 50% ammonium sulphate concentration (non-SHBG-testosterone), often referred to as bioavailable testosterone,35 appear to represent reliable indexes of biologically readily available testosterone, but are not well suited for clinical routine use, being labour intensive, time consuming and expensive. Several other parameters have been used without complete validation: direct immunoassay of free testosterone with a labelled testosterone analogue, calculation of free testosterone from total testosterone and immunoassayed SHBG concentrations, and the free androgen index (FAI = the ratio 100*testosterone /SHBG).36 The formula, and some examples of how to calculate free testosterone starting from total testosterone and SHBG concentrations, is available online.37 The calculated FAI is by far the most widely used parameter for the assessment of female hyperandrogenism.
Information that incorporates SHBG measurements is especially valuable in investigation of PCOS. SHBG is the main serum transporter for testosterone, which binds with strong affinity. Both androgens and insulin have a direct effect on lowering SHBG in hepatocytes,38 and it has been suggested by some39 and contested by others40 that SHBG could be used as a surrogate marker of insulin resistance in PCOS. It has, however, been clearly demonstrated that the increased circulating insulin concentration in PCOS can result in elevated FAI by decreasing circulating SHBG.41 The involvement of insulin and insulin resistance in PCOS will be discussed in more detail later in this review. Although an elevated FAI due to reduced SHBG is more common in patients with PCOS this is not a unique marker for the disorder; it can often be found in non-PCOS women especially if obese, and results should be interpreted with care and together with the wider clinical presentation. In patients who have already a clear diagnosis of PCOS, reduced SHBG may be of use to identify insulin-resistant individuals for targeted treatment with insulin-sensitising agents.39
PCOS undoubtedly leads to overproduction of ovarian androgens but there has been considerable debate over the years relating to the extent of adrenal involvement.42,43 In a recent study Kumar et al. suggested that the prevalence of supranormal DHEAS, an androgen produced exclusively in the adrenal, is approximately 20% among white and 30% among black PCOS patients, when using age- and race-adjusted normative values.44 These prevalences are lower than the 40–65% reported in earlier studies.45 Given that an isolated increase in DHEAS is a marker of other adrenal disorders and that this weak androgen is usually associated with increased testosterone, FAI and androstenedione in PCOS, we do not recommend that it is routinely measured in PCOS patients. Rather, it is useful in differentiation of an adrenal from an ovarian disorder in situations of diagnostic uncertainty and should therefore be considered as a secondary test in a small subset of women.
Anti Müllerian Hormone
The internal genitalia derive from the differentiation of two pairs of ducts, the Wolffian ducts and the Müllerian ducts. In mammals at an early stage of development, foetuses of both sexes have both pairs of ducts. In the male, regression of the Müllerian ducts occurs under the influence of a Sertoli cell factor, AMH, also known as Müllerian inhibiting substance (MIS). A recent excellent review by Rey et al. describes the fascinating detective work by Alfred Jost and other pioneering French scientists around the middle of last century, which led to the discovery of AMH.46
AMH is a homodimeric disulfide-linked glycoprotein with a molecular weight of 140 kDa. The gene is located on the short arm of chromosome 19 in humans and is strongly expressed in Sertoli cells from the time of testicular differentiation up to puberty and to a much lesser degree in granulosa cells from birth up to menopause.46,47 It has recently been shown that AMH is produced by the growing antral follicles in the human ovary up to the selection stage (4–6 mm).48 Peripheral concentrations of the hormone therefore provide a signal of the growing follicle pool and this marker is being increasingly used as part of in vitro fertilisation programmes to give an indication of follicle reserve.49 In addition, serum AMH can serve as a marker for PCOS since, as outlined earlier, the antral follicle pool is enlarged in this condition. AMH concentrations also correlate with other clinical features of PCOS such as cycle duration, mean ovarian volume, testosterone and androstenedione concentrations, and FAI.50,51 A rapidly increasing volume of research confirms that circulating concentrations of AMH are elevated in PCOS and that AMH measurement is a specific (92%) and sensitive (67%) marker for the disease.51–54 These initial findings are extremely encouraging and the outcome of more detailed prospective studies is awaited with interest. If confirmed, measurement of the circulating concentration of AMH could provide the most reliable biochemical marker for PCOS. Such work could lead to increasing demand for AMH measurements with a potential decrease in the use of other biochemical markers. However, studies relating AMH not simply to diagnosis of PCOS but also to clinical benefit in terms of leading to improved outcomes are needed to help support the case for AMH measurements. It is important to emphasise that although AMH measurements are valuable for research they are not yet established for the routine investigation of PCOS.
Insulin resistance and metabolic consequences
Studies over two decades have convincingly shown insulin resistance to be an integral pathogenic feature of PCOS, particularly in obese individuals.55,56 Women with PCOS, however, appear to have greater insulin resistance even after adjusting for BMI, in line with their greater visceral fat accumulation and higher waist to hip ratios compared to control women without PCOS. The associated hyperinsulinaemia appears to directly promote ovarian androgen secretion (i.e. gonadotrophic effect) in susceptible women, and abnormal follicular development, which ultimately leads to dysfunctional ovarian and menstrual activity (Figure 3).57
Figure 3.
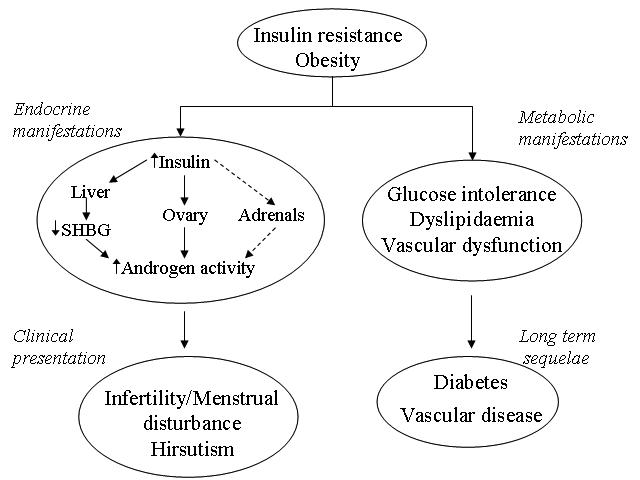
Endocrine and metabolic manifestations of insulin resistance. Hyperinsulinaemia in PCOS promotes ovarian androgen secretion in susceptible women, abnormal follicular development and, ultimately, dysfunctional ovarian and menstrual activity. Metabolic abnormalities include high triglycerides and tissue plasminogen activator and low-grade chronic inflammation. Patients with PCOS have an increased risk of diabetes and cardiovascular disease.
As mentioned earlier, conditions of increased androgen concentrations and hyperinsulinaemia are associated with reduced circulating SHBG, which results in high concentrations of free testosterone. Other metabolic abnormalities commonly linked to insulin resistance are also evident in patients with PCOS. These include high concentrations of triglyceride and tissue plasminogen activator58 and low-grade chronic inflammation.59 In line with these metabolic features is preliminary evidence that patients with a history of PCOS have an increased risk of diabetes (around 2–4 fold higher compared to weight-matched women) and cardiovascular disease (about 50% higher relative risk) in later life.57,60 Should the clinical biochemist, therefore, provide a service for or advise our clinical colleagues to investigate insulin resistance in this group of patients? Some information in this respect is already readily available, indirectly, by measuring SHBG, which not only helps provide an index of free testosterone but also gives some indication of degree of insulin resistance in the patient. To measure insulin resistance directly, however, requires complex dynamic techniques such as glucose clamps but these procedures remain within the research domain. Some investigators have taken to the measurement of fasting insulin in some of their women with possible PCOS to determine the degree of insulin resistance. The premise here is the higher the fasting insulin, the more insulin resistant is the woman. We suggest, however, that this is not commonly needed since, not only does the woman’s appearance (i.e. BMI or waist circumference) give some indication of the likely degree of insulin resistance, but SHBG also adds more information. Moreover, measurement of fasting insulin requires that the woman is fasted and that the samples taken be rapidly processed, centrifuged, and plasma frozen within an hour. Finally, as fasting insulin assays are not universally standardised and the results can vary many-fold in an individual, its value is questionable.
Since women with PCOS are insulin resistant, they have approximately a 2–4 fold higher risk of developing diabetes compared to BMI similar women without PCOS.61 It does not follow, however, that all women should be necessarily screened for type 2 diabetes; this would represent a significant work load and as yet is not evidence-based. Rather, screening for diabetes should be restricted to those at significantly elevated risk, namely those who are obese (BMI>30 kg/m2) or have a family history of type 2 diabetes. The vast majority of women who had either impaired glucose tolerance or frank type 2 diabetes had a BMI >30 kg/m2 in a series of PCOS women screened by Legro.62
Furthermore, the exact mode of screening for diabetes is not clear or universally accepted; some advocate an oral glucose tolerance test (OGTT) on all, others a fasting glucose followed by an OGTT in those with higher fasting glucose results. A practical solution may be to perform an initial non-fasting glycated haemoglobin (HbA1c) in the clinic combined with a non-fasting glucose. Whilst this measure is not accepted for screening, nor is it currently Medicare rebatable in Australia for this purpose, there is some evidence that an HbA1c of >6.1% has a good sensitivity in picking up individuals likely to have diabetes, although specificity is not ideal. The point we are trying to make is that to screen every woman with PCOS for diabetes is not sensible or useful. Women at high risk should be screened and the mode of screening remains contentious. A two stage process could be used with an initial non-fasting test to be followed up with a more formal test (OGTT) in those with high non-fasting glucose or HbA1c. It is important to point out that this is very much a pragmatic approach and clearly, further research is needed to provide better guidance in this complex area.
Should we also conduct a cardiovascular risk assessment? In reality most women with PCOS, who are generally young and attending to seek treatment for irregular periods, infertility or hirsutism, do not have lipids or indeed blood pressure measured in the context of a gynaecological clinic or even in a general practice arena. The best evidence estimating coronary heart disease risk in such women suggests around a 50% higher relative risk compared to women of similar age and weight. This level of risk does not justify wide screening since absolute risk is generally low in young women. Therefore, a 50% higher relative risk will still not put most women with PCOS at high absolute risk sufficient to warrant any intervention. We do not recommend that assessment of lipids and blood pressure be included in the majority and only done so if there is a strong family history of premature heart disease or stroke or if the woman has several other risk factors e.g. obesity, smoker, diabetes or known hypertension. Of course, recent recommendations suggest all adults from 40 years onwards, who have no history of cardiovascular disease or diabetes, and who are not already on treatment for blood pressure or lipids, should be considered for an opportunistic comprehensive cardiovascular disease risk assessment in primary care. Thus, women above 40 with PCOS should have lipids and blood pressure measured.63
Current and Potential Treatments
Women with PCOS are currently treated according to their presenting features: irregular menses, hirsutism, or infertility; current and potential treatments are summarised in Table 3 in relation to the problems addressed.
Table 3.
Current treatments for PCOS.
| Treatment | Problems Addressed |
|---|---|
| Current Treatments | |
| Oral contraceptives | Menstrual disturbance |
| Clomiphene Ovarian diathermy or laser treatment Assisted conception techniques | Anovulatory infertility |
| Cyproterone acetate + ethinyloestradiol Spironolactone | Hirsutism and acne |
| Weight loss | Menstrual disturbance and anovulatory infertility, but also improvement of metabolic perturbances and thus risk of coronary heart disease |
| Potential Treatments | |
| Insulin sensitising agents (such as metformin) |
|
Irregular menses
The combined oral contraceptive pill is commonly used to regulate menses. By increasing concentrations of SHBG while decreasing androgen secretion, it reduces the circulating free testosterone activity. The combined pill can exacerbate insulin resistance, and, since many patients are overweight and obesity is a relative contraindication, this treatment may be unsuitable.64
Hirsutism
Hirsutism may be addressed by using combination therapy, to possibly include i) hormonal suppression (oral contraceptives, long-acting gonadotrophin-releasing hormone analogues, and insulin sensitisers), ii) peripheral androgen blockade (spirinolactone, flutamide, cyproterone acetate, or finasteride), and iii) mechanical/cosmetic amelioration and destruction of the unwanted hairs (electrology and potentially, laser hair removal).65 With anti-androgen treatment beneficial effects can be seen after three months, but excessive hair growth returns soon after cessation of treatment. Cyproterone acetate may exacerbate irregularity of the menstrual cycle, and both cyproterone and spirinolactone are unsuitable for use in those trying to conceive.
Infertility
For patients wishing to become pregnant, clomiphene citrate, an orally active antioestrogen, may be successful in stimulating ovulation but carries an increased risk of multiple pregnancy.66 By inhibiting the oestrogen mediated negative feedback loop at the hypothalamus, it enhances secretion of FSH which stimulates the development of the ovarian follicle, inducing ovulation. Guidelines suggest that the duration of clomiphene treatment should not exceed six months because of the potential increased risk of ovarian cancer.67 Those failing to conceive after clomiphene treatment usually respond to exogenous gonadotrophins, but this requires intensive monitoring to reduce the risk of multiple conceptions.
Weight Reduction
Weight reduction has multiple benefits for obese women with PCOS.68 The resultant reduction in insulin resistance corrects the hormonal imbalance, promotes ovulation and regular menses, and improves the metabolic consequences of the disorder. Weight loss should therefore be encouraged, but it seems to be hard to achieve for this group of patients.
Insulin Sensitising Agents
Metformin, a biguanide often used in noninsulin dependent diabetes, has been the most commonly used insulin sensitising agent for treating PCOS. A meta-analysis of thirteen randomised trials comparing metformin with placebo, or metformin plus clomiphene with clomiphenalone, in women with PCOS concluded that metformin increased the ovulation rate by around 20%, and thus metformin does not represent a cure and in many cases, it may have limited benefit.69 Metformin’s effects may also be dependent on BMI, with very obese women (e.g. BMI >35 kg/m2) receiving less benefit than those with lesser degrees of obesity. Whether insulin sensitising agents can modify the vascular risk factors associated with the syndrome remains to be seen, but although data are relatively sparse, metformin seems to achieve modest improvements in LDL-cholesterol, HDL-cholesterol, tissue plasminogen activator and C-reactive protein, at least in some circumstances.61 Additionally, some studies have reported that treated subjects have shown some weight loss despite continuation of their normal diet and lifestyle70 and others have demonstrated a reduction in central obesity.71 It is therefore probable that longer term treatment with metformin in PCOS will attenuate the progress of diabetes and perhaps vascular risk.
An important report on a large multicentre, randomised trial which compared the effects of clomiphene, extended-release metformin, and combination therapy with both agents in 626 infertile women with PCOS (mean BMI 35) has recently been published.72 At six months, the live-birth rate (the primary end point) in the clomiphene group was three times that in the metformin group, and there was no significant benefit of the combination of metformin and clomiphene as compared to clomiphene alone. Although the ovulation rate in the combination therapy group was significantly higher than that in other groups, the increase did not translate into a higher live-birth rate. It does therefore now appear that although metformin may improve the metabolic consequences of the disease, it may not offer any additional benefit in relation to increasing the live-birth rate, at least in a group with a very high BMI. The results from Legro’s study72 underscore the limitations of the use of ovulation as a surrogate marker of live birth in infertility trials, thus explaining the possible false hopes induced by some of the earlier metformin treatment trials. Other large randomised trials are ongoing and should provide better guidance as regards use of metformin in women with PCOS.
Alternatives
Alternatives to medical treatment include laser or electro-cautery of the ovary. This is often used as a last resort, is not available in all centres, and is difficult to perform on obese patients. Although effective in aiding ovulation and regulating menses, its beneficial effects are usually short term.
Conclusion
Given that PCOS is a common disease that has received intensive study over the last 50 years we still know remarkably little about its complex aetiology. We have, however, learned much about the consequences and how to diagnosis the syndrome. The recent diagnostic criteria outlined in the EHSHRE/ARM consensus statement4 is a move in the right direction and does indicate that the laboratory has an important role to play. Methods for the reliable biochemical identification of women with hyperandrogenism are an important requirement but, as outlined in this review, measurement of the androgen of choice, testosterone, is fraught with problems. Those of us who run such services should be aware of and circumvent these problems. In addition, as outlined, important additional information can be obtained by measuring SHBG and reporting a calculated FAI. Measurement of androstenedione can, in some instance, produce important supportive information but the calculation of an LH/FSH ratio is no longer routinely recommended. Exclusion of other androgen-related abnormalities should be considered if the circulating testosterone concentration is significantly elevated and we suggest that a safe cut-off of >4 nmol/L be adopted. Our current recommended protocol for laboratory investigation of PCOS is detailed in Table 4.
Table 4.
Recommended protocol for laboratory investigation of PCOS.
| First line tests |
| To confirm hyperandrogenaemia and exclude significant hyperprolactinaemia, thyroid disease or premature ovarian failure |
| Testosterone, SHBG, prolactin, thyroid function tests, FSH |
| Follow up tests (in order) |
| Only instigate if testosterone is >4 nmol/L (after solvent extraction if methodology suspect – see text) |
| 1) To investigate extent of hyperandrogenaemia and provide supportive evidence for late onset congenital adrenal hyperplasia (CAH) or Cushing’s syndrome. |
| Androstenedione, DHEAS, 17-OHP, urinary free cortisol. |
| 2) To diagnose CAH |
| Only instigate if 17-OHP is >6 nmol/L |
| Serum 17-OHP before and 60 mins after Synacthen (250 ug i.v.) |
| Urinary steroid profile |
| 3) To diagnose androgen secreting tumour |
| These tumours are not common; only perform test if no other explanation for significantly elevated testosterone |
| Serum testosterone before and after 48h low dose (2 mg/day) dexamethasone |
| Urinary steroid profile |
Recent research has indicated that insulin resistance plays an important role in the syndrome but the significance of this in relation to diagnosis and treatment is not yet clear. Screening for diabetes should be restricted to those who are obese (BMI >30) or with strong family history, and we suggest that, in the first instance, the measurement of choice could simply be HbA1c with values of 6.0% or less not requiring further investigation. In relation to cardiovascular risk we do not recommend routine assessment of lipids and blood pressure be included for the majority but should be restricted to those woman with several other risk factors or women older than 40 years.
New treatment regimes based on insulin sensitising agents should await the outcome of more trials but recent trial data information cast some doubts on the initial enthusiasm. In relation to biochemical diagnosis it is envisaged that measurement of AMH may become a significant player in the years ahead but more research is needed.
Footnotes
Competing Interests: None declared.
References
- 1.Franks S. Polycystic ovary syndrome. N Engl J Med. 1995;333:853–61. doi: 10.1056/NEJM199509283331307. [DOI] [PubMed] [Google Scholar]
- 2.Stein IF, Leventhal ML. Amenorrhea associated with bilateral polycystic ovaries. Am J Obstet Gynecol. 1935;29:181–91. [Google Scholar]
- 3.Dunaif A, Thomas A. Current concepts in the polycystic ovary syndrome. Annu Rev Med. 2001;52:401–19. doi: 10.1146/annurev.med.52.1.401. [DOI] [PubMed] [Google Scholar]
- 4.Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004;81:19–25. doi: 10.1016/j.fertnstert.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Dawber RP. Guidance for the management of hirsutism. Curr Med Res Opin. 2005;21:1227–34. doi: 10.1185/030079905X56475. [DOI] [PubMed] [Google Scholar]
- 6.Ferriman D, Gallwey JD. Clinical assessment of body hair growth in women. J Clin Endocrinol Metab. 1961;21:1440–7. doi: 10.1210/jcem-21-11-1440. [DOI] [PubMed] [Google Scholar]
- 7.Lewy VD, Danadian K, Witchel SF, Arslanian S. Early metabolic abnormalities in adolescent girls with polycystic ovarian syndrome. J Pediatr. 2001;138:38–44. doi: 10.1067/mpd.2001.109603. [DOI] [PubMed] [Google Scholar]
- 8.Richardson MR. Current perspectives in polycystic ovary syndrome. Am Fam Physician. 2003;68:697–704. [PubMed] [Google Scholar]
- 9.McArthur JW, Ingersoll FM, Worcester J. The urinary excretion of interstitial-cell and follicle-stimulating hormone activity by women with diseases of the reproductive system. J Clin Endocrinol Metab. 1958;18:1202–15. doi: 10.1210/jcem-18-11-1202. [DOI] [PubMed] [Google Scholar]
- 10.Yen SS, Vela P, Rankin J. Inappropriate secretion of follicle-stimulating hormone and luteinizing hormone in polycystic ovarian disease. J Clin Endocrinol Metab. 1970;30:435–42. doi: 10.1210/jcem-30-4-435. [DOI] [PubMed] [Google Scholar]
- 11.O’Driscoll JB, Mamtora H, Higginson J, Pollock A, Kane J, Anderson DC. A prospective study of the prevalence of clear-cut endocrine disorders and polycystic ovaries in 350 patients presenting with hirsutism or androgenic alopecia. Clinical Endocrinol (Oxf) 1994;41:231–6. doi: 10.1111/j.1365-2265.1994.tb02535.x. [DOI] [PubMed] [Google Scholar]
- 12.Forbes AP, Henneman PH, Griswold GC, Albright F. Syndrome characterized by galactorrhea, amenorrhea and low urinary FSH: comparison with acromegaly and normal lactation. J Clin Endocrinol Metab. 1954;14:265–71. doi: 10.1210/jcem-14-3-265. [DOI] [PubMed] [Google Scholar]
- 13.Murdoch AP, Dunlop W, Kendall-Taylor P. Studies of prolactin secretion in polycystic ovary syndrome. Clin Endocrinol (Oxf) 1986;24:165–75. doi: 10.1111/j.1365-2265.1986.tb00759.x. [DOI] [PubMed] [Google Scholar]
- 14.Bracero N, Zacur HA. Polycystic ovary syndrome and hyperprolactinemia. Obstet Gynecol Clin North Am. 2001;28:77–84. doi: 10.1016/s0889-8545(05)70186-8. [DOI] [PubMed] [Google Scholar]
- 15.Azziz R, Woods KS, Reyna R, Key TJ, Knochenhauer ES, Yildiz BO. The prevalence and features of the polycystic ovary syndrome in an unselected population. J Clin Endocrinol Metab. 2004;89:2745–9. doi: 10.1210/jc.2003-032046. [DOI] [PubMed] [Google Scholar]
- 16.New MI. Extensive clinical experience: nonclassical 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2006;91:4205–14. doi: 10.1210/jc.2006-1645. [DOI] [PubMed] [Google Scholar]
- 17.Zerah M, Ueshiba H, Wood E, Speiser PW, Crawford C, McDonald T, et al. Prevalence of nonclassical steroid 21-hydroxylase deficiency based on a morning salivary 17-hydroxyprogesterone screening test: a small sample study. J Clin Endocrinol Metab. 1990;70:1662–7. doi: 10.1210/jcem-70-6-1662. [DOI] [PubMed] [Google Scholar]
- 18.Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, Fox L, Boots LR. Screening for 21-hydroxylase-deficient nonclassic adrenal hyperplasia among hyperandrogenic women: a prospective study. Fertil Steril. 1999;72:915–25. doi: 10.1016/s0015-0282(99)00383-0. [DOI] [PubMed] [Google Scholar]
- 19.Wallace AM. Analytical support for the detection and treatment of congenital adrenal hyperplasia. Ann Clin Biochem. 1995;32:9–27. doi: 10.1177/000456329503200102. [DOI] [PubMed] [Google Scholar]
- 20.Kaltsas GA, Isidori AM, Kola BP, Skelly RH, Chew SL, Jenkins PJ, et al. The value of the low-dose dexamethasone suppression test in the differential diagnosis of hyperandrogenism in women. J Clin Endocrinol Metab. 2003;88:2634–43. doi: 10.1210/jc.2002-020922. [DOI] [PubMed] [Google Scholar]
- 21.Waldstreicher J, Santoro NF, Hall JE, Filicori M, Crowley WF., Jr Hyperfunction of the hypothalamic-pituitary axis in women with polycystic ovarian disease: indirect evidence for partial gonadotroph desensitization. J Clin Endocrinol Metab. 1988;66:165–72. doi: 10.1210/jcem-66-1-165. [DOI] [PubMed] [Google Scholar]
- 22.Seth J, Hanning I, Bacon RR, Hunter WM. Progress and problems in immunoassays for serum pituitary gonadotrophins: evidence from the UK external quality assessment schemes, (EQAS) 1980–1988. Clin Chim Acta. 1989;186:67–82. doi: 10.1016/0009-8981(89)90205-2. [DOI] [PubMed] [Google Scholar]
- 23.Vetterlein D. Monoclonal antibodies: production, purification, and technology. Adv Clin Chem. 1989;27:303–54. doi: 10.1016/s0065-2423(08)60186-9. [DOI] [PubMed] [Google Scholar]
- 24.Milsom SR, Sowter MC, Carter MA, Knox BS, Gunn AJ. LH levels in women with polycystic ovarian syndrome: have modern assays made them irrelevant? Br J Obstet Gynaecol. 2003;110:760–4. [PubMed] [Google Scholar]
- 25.Taylor AE, McCourt B, Martin KA, Anderson EJ, Adams JM, Schoenfeld D, et al. Determinants of abnormal gonadotropin secretion in clinically defined women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1997;82:2248–56. doi: 10.1210/jcem.82.7.4105. [DOI] [PubMed] [Google Scholar]
- 26.Burger HG. Androgen production in women. Fertil Steril. 2002;77(Suppl 4):S3–5. doi: 10.1016/s0015-0282(02)02985-0. [DOI] [PubMed] [Google Scholar]
- 27.Wilson JD. The role of 5alpha-reduction in steroid hormone physiology. Reprod Fertil Dev. 2001;13:673–8. doi: 10.1071/rd01074. [DOI] [PubMed] [Google Scholar]
- 28.Robinson S, Rodin DA, Deacon A, Wheeler MJ, Clayton RN. Which hormone tests for the diagnosis of polycystic ovary syndrome? Br J Obstet Gynaecol. 1992;99:232–8. doi: 10.1111/j.1471-0528.1992.tb14505.x. [DOI] [PubMed] [Google Scholar]
- 29.Balen AH, Conway GS, Kaltsas G, Techatrasak K, Manning PJ, West C, et al. Polycystic ovary syndrome: the spectrum of the disorder in 1741 patients. Hum Reprod. 1995;10:2107–11. doi: 10.1093/oxfordjournals.humrep.a136243. [DOI] [PubMed] [Google Scholar]
- 30.Pugeat MM, Dunn JF, Nisula BC. Transport of steroid hormones: interaction of 70 drugs with testosterone-binding globulin and corticosteroid-binding globulin in human plasma. J Clin Endocrinol Metab. 1981;53:69–75. doi: 10.1210/jcem-53-1-69. [DOI] [PubMed] [Google Scholar]
- 31.Taieb J, Mathian B, Millot F, Patricot MC, Mathieu E, Queyrel N, et al. Testosterone measured by 10 immunoassays and by isotope-dilution gas chromatography-mass spectrometry in sera from 116 men, women, and children. Clin Chem. 2003;49:1381–95. doi: 10.1373/49.8.1381. [DOI] [PubMed] [Google Scholar]
- 32.Herold DA, Fitzgerald RL. Immunoassays for testosterone in women: better than a guess? Clin Chem. 2003;49:1250–1. doi: 10.1373/49.8.1250. [DOI] [PubMed] [Google Scholar]
- 33.Heald AH, Butterworth A, Kane JW, Borzomato J, Taylor NF, Layton T, et al. Investigation into possible causes of interference in serum testosterone measurement in women. Ann Clin Biochem. 2006;43:189–95. doi: 10.1258/000456306776865106. [DOI] [PubMed] [Google Scholar]
- 34.Warner MH, Kane JW, Atkin SL, Kilpatrick ES. Dehydroepiandrosterone sulphate interferes with the Abbott Architect direct immunoassay for testosterone. Ann Clin Biochem. 2006;43:196–9. doi: 10.1258/000456306776865034. [DOI] [PubMed] [Google Scholar]
- 35.Manni A, Pardridge WM, Cefalu W, Nisula BC, Bardin CW, Santner SJ, et al. Bioavailability of albumin-bound testosterone. J Clin Endocrinol Metab. 1985;61:705–10. doi: 10.1210/jcem-61-4-705. [DOI] [PubMed] [Google Scholar]
- 36.Rosner W. An extraordinarily inaccurate assay for free testosterone is still with us. J Clin Endocrinol Metab. 2001;86:2903. doi: 10.1210/jcem.86.6.7643. [DOI] [PubMed] [Google Scholar]
- 37.Free and Bioavailable Testosterone Calculator. [Accessed 26 June 2007]; http://www.issam.ch/freetesto.htm.
- 38.Plymate SR, Matej LA, Jones RE, Friedl KE. Inhibition of sex hormone-binding globulin production in the human hepatoma (Hep G2) cell line by insulin and prolactin. J Clin Endocrinol Metab. 1988;67:460–4. doi: 10.1210/jcem-67-3-460. [DOI] [PubMed] [Google Scholar]
- 39.Jayagopal V, Kilpatrick ES, Jennings PE, Hepburn DA, Atkin SL. The biological variation of testosterone and sex hormone-binding globulin (SHBG) in polycystic ovarian syndrome: implications for SHBG as a surrogate marker of insulin resistance. J Clin Endocrinol Metab. 2003;88:1528–33. doi: 10.1210/jc.2002-020557. [DOI] [PubMed] [Google Scholar]
- 40.Dahan MH, Goldstein J. Serum sex hormone-binding globulin levels show too much variability to be used effectively as a screening marker for insulin resistance in women with polycystic ovary syndrome. Fertil Steril. 2006;86:934–41. doi: 10.1016/j.fertnstert.2006.02.108. [DOI] [PubMed] [Google Scholar]
- 41.Nestler JE, Powers LP, Matt DW, Steingold KA, Plymate SR, Rittmaster RS, et al. A direct effect of hyperinsulinemia on serum sex hormone-binding globulin levels in obese women with the polycystic ovary syndrome. J Clin Endocrinol Metab. 1991;72:83–9. doi: 10.1210/jcem-72-1-83. [DOI] [PubMed] [Google Scholar]
- 42.Loughlin T, Cunningham S, Moore A, Culliton M, Smyth PP, McKenna TJ. Adrenal abnormalities in polycystic ovary syndrome. J Clin Endocrinol Metab. 1986;62:142–7. doi: 10.1210/jcem-62-1-142. [DOI] [PubMed] [Google Scholar]
- 43.Doi SA, Al-Zaid M, Towers PA, Scott CJ, Al-Shoumer KA. Steroidogenic alterations and adrenal androgen excess in PCOS. Steroids. 2006;71:751–9. doi: 10.1016/j.steroids.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 44.Kumar A, Woods KS, Bartolucci AA, Azziz R. Prevalence of adrenal androgen excess in patients with the polycystic ovary syndrome (PCOS) Clin Endocrinol (Oxf) 2005;62:644–9. doi: 10.1111/j.1365-2265.2005.02256.x. [DOI] [PubMed] [Google Scholar]
- 45.Carmina E, Koyama T, Chang L, Stanczyk FZ, Lobo RA. Does ethnicity influence the prevalence of adrenal hyperandrogenism and insulin resistance in polycystic ovary syndrome? Am J Obstet Gynecol. 1992;167:1807–12. doi: 10.1016/0002-9378(92)91779-a. [DOI] [PubMed] [Google Scholar]
- 46.Rey R, Lukas-Croisier C, Lasala C, Bedecarras P. AMH/MIS: what we know already about the gene, the protein and its regulation. Mol Cell Endocrinol. 2003;211:21–31. doi: 10.1016/j.mce.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 47.La Marca A, Volpe A. Anti-Mullerian hormone (AMH) in female reproduction: is measurement of circulating AMH a useful tool? Clin Endocrinol (Oxf) 2006;64:603–10. doi: 10.1111/j.1365-2265.2006.02533.x. [DOI] [PubMed] [Google Scholar]
- 48.Weenen C, Laven JS, Von Bergh AR, Cranfield M, Groome NP, Visser JA, et al. Anti-Mullerian hormone expression pattern in the human ovary: potential implications for initial and cyclic follicle recruitment. Mol Hum Reprod. 2004;10:77–83. doi: 10.1093/molehr/gah015. [DOI] [PubMed] [Google Scholar]
- 49.Visser JA, de Jong FH, Laven JS, Themmen AP. Anti-Mullerian hormone: a new marker for ovarian function. Reproduction. 2006;131:1–9. doi: 10.1530/rep.1.00529. [DOI] [PubMed] [Google Scholar]
- 50.Pigny P, Merlen E, Robert Y, Cortet-Rudelli C, Decanter C, Jonard S, et al. Elevated serum level of anti-mullerian hormone in patients with polycystic ovary syndrome: relationship to the ovarian follicle excess and to the follicular arrest. J Clin Endocrinol Metab. 2003;88:5957–62. doi: 10.1210/jc.2003-030727. [DOI] [PubMed] [Google Scholar]
- 51.Laven JS, Mulders AG, Visser JA, Themmen AP, De Jong FH, Fauser BC. Anti-Mullerian hormone serum concentrations in normoovulatory and anovulatory women of reproductive age. J Clin Endocrinol Metab. 2004;89:318–23. doi: 10.1210/jc.2003-030932. [DOI] [PubMed] [Google Scholar]
- 52.Fallat ME, Siow Y, Marra M, Cook C, Carrillo A. Mullerian-inhibiting substance in follicular fluid and serum: a comparison of patients with tubal factor infertility, polycystic ovary syndrome, and endometriosis. Fertil Steril. 1997;67:962–5. doi: 10.1016/s0015-0282(97)81417-3. [DOI] [PubMed] [Google Scholar]
- 53.Mulders AG, Laven JS, Eijkemans MJ, de Jong FH, Themmen AP, Fauser BC. Changes in anti-Mullerian hormone serum concentrations over time suggest delayed ovarian ageing in normogonadotrophic anovulatory infertility. Hum Reprod. 2004;19:2036–42. doi: 10.1093/humrep/deh373. [DOI] [PubMed] [Google Scholar]
- 54.Cook CL, Siow Y, Brenner AG, Fallat ME. Relationship between serum mullerian-inhibiting substance and other reproductive hormones in untreated women with polycystic ovary syndrome and normal women. Fertil Steril. 2002;77:141–6. doi: 10.1016/s0015-0282(01)02944-2. [DOI] [PubMed] [Google Scholar]
- 55.Chang JR, Nakamura RM, Howard LJ, Kaplan SA. Insulin resistance in nonobese patients with polycystic ovarian disease. J Clin Endocrinol Metab. 1983;57:356–9. doi: 10.1210/jcem-57-2-356. [DOI] [PubMed] [Google Scholar]
- 56.Nestler JE, Jakubowicz DJ. Lean women with polycystic ovary syndrome respond to insulin reduction with decreases in ovarian P450c17 activity and serum androgens. J Clin Endocrinol Metab. 1997;82:4075–9. doi: 10.1210/jcem.82.12.4431. [DOI] [PubMed] [Google Scholar]
- 57.Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev. 1997;18:774–800. doi: 10.1210/edrv.18.6.0318. [DOI] [PubMed] [Google Scholar]
- 58.Kelly CJ, Lyall H, Petrie JR, Gould GW, Connell JM, Rumley A, et al. A specific elevation in tissue plasminogen activator antigen in women with polycystic ovarian syndrome. J Clin Endocrinol Metab. 2002;87:3287–90. doi: 10.1210/jcem.87.7.8634. [DOI] [PubMed] [Google Scholar]
- 59.Kelly CC, Lyall H, Petrie JR, Gould GW, Connell JM, Sattar N. Low grade chronic inflammation in women with polycystic ovarian syndrome. J Clin Endocrinol Metab. 2001;86:2453–5. doi: 10.1210/jcem.86.6.7580. [DOI] [PubMed] [Google Scholar]
- 60.Wild RA. Polycystic ovary syndrome: a risk for coronary artery disease? Am J Obstet Gynecol. 2002;186:35–43. doi: 10.1067/mob.2002.119180. [DOI] [PubMed] [Google Scholar]
- 61.Sattar N. Polycystic ovarian syndrome: vascular and metabolic issues. In: Greer IA, Ginsberg J, Forbes CD, editors. Women’s Vascular Health. London: Hodder Arnold; 2007. pp. 265–79. [Google Scholar]
- 62.Legro RS, Kunselman AR, Dodson WC, Dunaif A. Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab. 1999;84:165–9. doi: 10.1210/jcem.84.1.5393. [DOI] [PubMed] [Google Scholar]
- 63.British Cardiac Society; British Hypertension Society; Diabetes UK; HEART UK; Primary Care Cardiovascular Society; Stroke Association. JBS 2: Joint British Societies’ guidelines on prevention of cardiovascular disease in clinical practice. Heart. 2005;91(Suppl 5):v 1–52. doi: 10.1136/hrt.2005.079988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sheu WH, Hsu CH, Chen YS, Jeng CY, Fuh MM. Prospective evaluation of insulin resistance and lipid metabolism in women receiving the oral contraceptives. Clin Endocrinol (Oxf) 1994;40:249–55. doi: 10.1111/j.1365-2265.1994.tb02476.x. [DOI] [PubMed] [Google Scholar]
- 65.Azziz R. The evaluation and management of hirsutism. Obstet Gynecol. 2003;101:995–1007. doi: 10.1016/s0029-7844(02)02725-4. [DOI] [PubMed] [Google Scholar]
- 66.Kettel LM, Hummel WP. Ovulation induction in the oestrogenised anovulatory patient. Semin Reprod Endocrinol. 1996;14:309–15. doi: 10.1055/s-2008-1067976. [DOI] [PubMed] [Google Scholar]
- 67.Rossing MA, Daling JR, Weiss NS, Moore DE, Self SG. Ovarian tumours in a cohort of infertile women. N Engl J Med. 1994;331:771–6. doi: 10.1056/NEJM199409223311204. [DOI] [PubMed] [Google Scholar]
- 68.Crave JC, Fimbel S, Lejeune H, Cugnardey N, Dechaud H, Pugeat M. Effects of diet and metformin administration on sex hormone inding globulin, androgens and insulin in hirsute and obese women. J Clin Endocrinol Metab. 1995;80:2057–62. doi: 10.1210/jcem.80.7.7608255. [DOI] [PubMed] [Google Scholar]
- 69.Lord JM, Flight IH, Norman RJ. Metformin in polycystic ovary syndrome: systematic review and meta-analysis. BMJ. 2003;327:951–3. doi: 10.1136/bmj.327.7421.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Velazquez E, Acosta A, Mendoza SG. Menstrual cyclicity after metformin therapy in polycystic ovary syndrome. Obstet Gynecol. 1997;90:392–5. doi: 10.1016/s0029-7844(97)00296-2. [DOI] [PubMed] [Google Scholar]
- 71.Nestler JE, Jakubowicz D. Decrease in ovarian cytochrome P450c17a activity and serum free testosterone after reduction of insulin secretion in polycystic ovary syndrome. N Engl J Med. 1996;335:617–23. doi: 10.1056/NEJM199608293350902. [DOI] [PubMed] [Google Scholar]
- 72.Legro RS, Barnhart HX, Schlaff WD, Carr BR, Diamond MP, Carson SA, et al. Clomiphene, metformin, or both for infertility in the polycystic ovary syndrome. N Engl J Med. 2007;356:551–66. doi: 10.1056/NEJMoa063971. [DOI] [PubMed] [Google Scholar]