

Abstract
Introduction
Sepsis is associated with growth hormone (GH) insensitivity and in the intact animal the major surface component of the bacterial cell wall, lipopolysaccharide (LPS), inhibits GH receptor (GHR) gene expression. The prevailing explanation for LPS-induced effects on the GHR promoter is that this effect is indirect via generation of cytokines. Our recent studies demonstrate that saturated free fatty acids (FFAs) inhibit the activity of the murine GHR promoter. Saturated FFAs are an essential component of the lipid A moiety of LPS required for biological activity of LPS.
Hypothesis
LPS directly modulates the activity of the dominant GHR promoter via interaction with Toll-like receptor(s) (TLR)/MD2 complex and activation of cognate signaling pathway(s).
Results
In transient transfection experiments with RAW 264.7 cells which express endogenous TLR4 and MD2, LPS treatment inhibited GHR promoter activity. Co-transfection of dominant negative TLR4 abrogated this effect on GHR promoter activity. In HEK 293T cells, which are devoid of endogenous TLR4 or MD2, ectopic expression of TLR4 and MD2 resulted in LPS-induced inhibition of the GHR promoter activity. The inhibition of GHR promoter activity was demonstrable by 5-6 h after exposure to LPS and persisted at 24 h. Fatty-acid free LPS failed to elicit a similar effect on the GHR promoter and the effect of LPS was abrogated by Polymyxin B. The essential role of the cofactor MD2 on the effect of LPS on the GHR promoter was established in experiments using ectopic expression of wild type and mutant MD2. Cotransfection of CD14 in these cells failed to alter the effect of LPS on the activity of the GHR promoter. Analysis of cell culture supernatant excluded the possibility that the effect of LPS was secondary to release of cytokines from the transfected cells. The effect of LPS on the endogenous GHR promoter activity and protein expression was confirmed in F442A preadipocyte cells. In HEK 293T cells, ectopic expression of mutant MyD88 or mutant TRIF abrogated the effect of LPS on the GHR promoter, suggesting that the effect of LPS on the GHR promoter was via both MyD88-dependent and -independent pathways.
Conclusions
LPS acts through both MyD88-dependent and -independent TLR4 signaling pathways to directly inhibit GHR gene expression. Our results establish a novel cytokine-independent mechanism for decrease in GHR expression in bacterial sepsis.
Introduction
Pituitary Growth Hormone (GH) is essential for postnatal growth in mammals. In addition to growth, GH affects the metabolism of fat, protein, and carbohydrate. GH exerts these actions both by its direct effect on target organs and by stimulating the production of insulin-like growth factor-I (IGF-I). At the tissue level, these pleiotropic actions of GH result from the interaction of GH with a specific cell surface receptor, i.e., GH receptor (GHR). GHRs are present in all the tissues towards which GH actions are directed. Thus, the ability of GH to exert biological effects is intimately linked to the number, function, and regulation of GHRs in these tissues. A feature common to GHR transcripts from different species is the heterogeneity in the 5′-untranslated region (Edens and Talamantes, 1998). In the mouse, three 5′-UTRs (termed L1, L2, and L5) have been identified (Menon et al., 1997; Menon et al., 1995; Moffat et al., 1999; Schwartzbauer et al., 1998; Yu et al., 1999). The L2 transcript is the dominant transcript expressed in postnatal life, constituting 50 to 80% of the hepatic GHR transcripts in the nonpregnant adult animal (Southard et al., 1995).
Sepsis is characterized by a state of GH insensitivity contributing to enhanced rate of protein catabolism, ensuing cachexia and wasting, and associated increase in mortality rate. GH insensitivity in sepsis is characterized by decreased expression of GHR, inhibition of GHR signaling pathways, and consequent decrease in circulating levels of IGF-I (Yumet et al., 2006). However the pathogenesis of sepsis-induced alterations in the GHR expression and signaling pathways are incompletely understood. LPS (lipopolysaccharide) is a major component of the outer membranes of gram-negative bacteria and plays a dominant role in host response to gram-negative bacterial infection (Trent et al., 2006). Previous studies have established that GH insensitivity induced by LPS is characterized by down-regulation of GHR mRNA expression and up-regulation of expression of SOCS-3 mRNA, a canonical inhibitor of GHR signaling pathways (Yumet et al., 2006). The prevailing explanation for LPS-induced effects on the GHR mRNA expression is that these effects are secondary to LPS-induced generation of cytokines such as IL-1, IL-6 and TNF-α (Denson et al., 2003; Denson et al., 2001). Thus Denson et al., reported on the molecular mechanisms involved in the pathogenesis of TNF-α-induced suppression of murine hepatic GHR (Denson et al., 2001). These investigators identified a specific role for the transcription factors Sp1 and Sp3 in the transduction of the effect of TNF-α on GHR expression.
The cellular effects of LPS are transduced through interaction with Toll-like receptor-4 (TLR4) (Imler and Zheng, 2004). LPS is composed of lipid A, core antigen, and O antigen. Saturated free fatty acids (FFAs) are an essential component of the lipid A moiety which is obligatory for biological activity of LPS (Trent et al., 2006). Previous studies have demonstrated that saturated FFAs interact with TLR and stimulate post-receptor signaling pathways (Lee et al., 2003). Based on our recent studies demonstrating that saturated free fatty acids (FFAs) decrease GHR expression via inhibition of GHR promoter activity (Thimmarayappa et al., 2006), we hypothesized that one of the mechanisms for LPS-induced inhibition of GHR gene expression is direct effect(s) of LPS-dependent TLR pathways on GHR promoter activity. Thus the present study was undertaken to investigate the direct effect of LPS on the GHR promoter and the role of TLR signaling pathways in LPS-induced inhibition of GHR expression. Our results demonstrate that LPS directly modulates the activity of the GHR promoter in a cytokine-independent manner via interaction with Toll-like receptor-4/MD2 complex and activation of the cognate signaling pathway(s).
Materials and Methods
Plasmids
The design and construction of the promoter-luciferase reporter gene construct (pGL3-L2-2.0 kb) containing 2.0 kb of the 5-flanking region of the L2 promoter of the murine GHR gene has been described previously (Yu et al., 1999). pUNO-mTLR4 expressing wild type murine TLR4, pUNO-mMD2 expressing wild type murine MD2, pUNO-mCD14 expressing wild type murine CD14, and pDeNy-hTRIF expressing dominant negative human TRIF were purchased from Invivogen. TLR4P712H (TLRDN) was obtained from Dr. Daniel Hwang (University of California, Davis, CA)(Rhee and Hwang, 2000) and MyD88-ΔDD (MyD88[Δ110-296]) was obtained from Dr. Jürg Tschopp (University of Lausanne, Switzerland)(Burns et al., 1998). The mouse MD2C95Y was constructed by site-directed mutagenesis (QuikChange, Stratagene) using the following primers: forward primer, 5′-GCG TAA GGA AGT TCT GTA CCA TGG ACA TGA TGA TG-3′; reverse primer, CAT CAT CAT GTC CAT GGT ACA AAA CTT CCT TAC GC-3′. Plasmid DNA was prepared using the EndoFree plasmid maxi-kit (Qiagen Inc.) and all constructs were verified by DNA sequencing.
Reagents
Purified LPS from E.Coli 011:B4 strain and fatty acid free LPS were purchased from Invivogen. Polymyxin B, tumor necrosis factor-alpha (TNF-α), and anti-TNF-α antibody were purchased from Sigma. Routine laboratory chemicals and reagents were purchased from Sigma unless otherwise specified.
Cell culture
RAW 264.7 cells (murine macrophage cells, TIB-71, ATCC) and HEK 293T cells (human embryonic kidney cells, ATCC) were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (endotoxin-free grade), penicillin G (100 unit/ml) and streptomycin (100 μg/ml) at 37 °C in an atmosphere of 5% CO2/95% air. BNL.CL2 cells (mouse embryonic hepatocyte-like cells, ATCC:) stably expressing the L2 promoter-luciferase construct (pGL3-L2-2.0)(Denson et al., 2001) were cultured in DMEM supplemented with G418 (1.6 mg/ml) in an atmosphere of 5% CO2/95% air at 37 °C. 3T3-F442A cells (mouse preadipocyte cells, ATCC) were maintained in DMEM supplemented with 10% fetal calf serum, penicillin and streptomycin in an atmosphere of 10% CO2/90% air at 37 °C.
Transient transfection and luciferase assay
RAW 264.7 cells (5×105 cells/well in 6-well plates) or HEK 293T cells (1.5×105 cells/well in 24-well plates) were co-transfected with luciferase reporter plasmids containing GHR promoter (pGL3B-L2-2.0; 2 and 0.125μg/well for 6- and 24-well plates respectively) and internal control, pRL-TK (Promega; 0.2 and 0.05μg/well for 6- and 24-well plates respectively) expressing the Renilla luciferase. On occasions plasmids expressing various proteins used to interrogate cognate signaling pathways were also co-transfected. Transfection was routinely achieved using Superfect (Qiagen Inc.) for RAW 264.7 cells or the calcium phosphate protocol (Invitrogen) for HEK 293T cells. The day following transfection, the cells were exposed for 5-6 h to varying concentration of LPS followed by lysis of the cells in passive lysis buffer (Promega). For estimation of luciferase activity the plates were rinsed twice with phosphate-buffered saline and the cells harvested by the addition of 200 μl lysis buffer (Dual Luciferase Assay System; Promega). Following a brief freeze-thaw cycle the insoluble debris was removed by centrifugation at 4 °C for 2-3 min at 14000xg. 20 μl aliquots of the supernatant were then immediately processed for sequential quantitation of both firefly and Renilla luciferase activity (Dual Luciferase Assay System; Promega) using a Monolight TD 20/20 Luminometer (Turner Designs). The activity of the co-transfected Renilla control reporter plasmid was used to normalize the L2 promoter activity for transfection efficiency. All transfections were performed in triplicates.
Real-time PCR analysis of L2 promoter expression
3T3-F442A preadipocyte cells were stimulated with LPS (1 μg/ml) for 5-6 h. Total RNA was extracted using Trizol-reagent as specified by the manufacturer (Molecular Research Center)(Chomczynski and Sacchi, 1987) and quantitated by the absorbance at 260 nm. 5 ng of total RNA was used for RT-PCR. Real-time quantitative reverse-transcription PCR (QT-PCR) was performed using the ABI Prism 7700 Sequence Detection System (PE Biosystems) and analyzed following protocols described previously (Denson et al., 2001). The primers used for detecting mouse L2 transcript have been described previously (Denson et al., 2001). Normalization of expression of GHR transcripts was carried out by concomitant measurement of the steady state abundance levels of the housekeeping gene GAPDH.
Measurement of cytokines production
Cytokine levels in conditioned media were measured using the Human Cytokine Twenty-Five-Plex Antibody Bead Kit (Catalog #LHC0009) from Biosource International, Inc. (Camarillo, CA) on the Luminex100® platform from Bio-Rad Laboratories, Inc. (Hercules, CA). The assay characteristics for the relevant cytokines were: TNF-α (sensitivity: 10 pg/ml, inter-assay variation: 8.3%), IL-6 (sensitivity: 3 pg/ml, inter-assay variation: 7%), and IL-1 beta (sensitivity: 15 pg/ml, inter-assay variation: 8.8%).
Antibodies
Anti-GHR antibody AL-47 obtained from Dr. Stuart Frank (University of Alabama at Birmingham) was used in 1 in 1000 dilution for Western blot analysis; the anti-rabbit secondary antibody was used in a 1:5000 dilution.
Western blot analysis
3T3-F442A preadipocyte cells stimulated with LPS for 5-6 h were harvested in 400 μl of lysis buffer containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 2 mM EGTA, 0.1% Triton X-100, 1 mmol/L sodium pyrophosphate, 10 mmol/L sodium fluoride, and 1 mmol/L sodium orthovanadate. The protein concentration was quantitated by colorimetric protein assay (Biorad) and equal amounts of protein resolved on gradient SDS-polyacrylamide gels, and blotted onto nitrocellulose membrane for Western blot analysis. The blot was probed with suitable primary and secondary antibodies and was developed by the ECL-chemiluminescent method (Amersham Biosciences) according to the manufacturer's instructions.
Statistical analysis
Data are presented as mean ± S.E. unless otherwise indicated. Mann-Whitney U and Kruskal-Wallis nonparametric tests were performed to analyze statistical significance of the difference between the distributions of two or multiple independent samples respectively using SPSS software, version 11.5 for windows (SPSS, Inc.). p values equal to or less than 0.05 were considered significant.
Results
LPS inhibits expression of the dominant (L2) GHR transcript
The acquired state of GH insensitivity in bacterial sepsis is in part due to decreased expression of GHR. Prevailing dogma implicates inhibition of GHR gene promoter activity by sepsis-induced cytokines as the mechanism for decreased GHR expression in bacterial sepsis. We hypothesized that an additional mechanism resulting in sepsis-induced inhibition of GHR gene expression is a direct cytokine-independent effect of LPS on the GHR promoter. To investigate this hypothesis we analyzed the effect of LPS on promoter activity of the dominant GHR transcript, the L2 transcript. RAW 264.7 cells that express endogenous TLR4 and MD2 were transiently transfected with L2-promoter-luciferase plasmid and exposed to LPS and luciferase activity measured. These results indicated that exposure of LPS for 5-6 h resulted in a significant decrease in activity of the GHR promoter (Fig.1A). This effect was dose dependent and was evident at a LPS concentration of 0.05 μg/ml. Concomitant studies evaluating cell viability established that the decrease in promoter activity was not secondary to cell death (data not shown).
Fig.1. LPS, but not fatty acid free LPS, inhibits expression of the dominant GHR (L2-GHR) transcript.

(A) RAW 264.7 cells, which express endogenous TLR4 and MD2, were co-transfected with L2-promoter-luciferase reporter (pGL3-L2-2.0 kb) and pRL-TK (internal control) plasmids. 24 h after transfection, cells were stimulated with either vehicle or the indicated concentration of LPS for 5-6 h and cells harvested and luciferase activity measured. Results are expressed as mean ± SEM; n = 3-4, p < 0.05 (Kruskal Wallis test) compared to the luciferase activity in the absence of LPS (vehicle). (B) BNL.CL2 cells, stably expressing the L2 promoter-luciferase construct were exposed to 1μg/ml of either LPS or fatty acid free LPS. 48 h later, the cells were harvested and the luciferase activity measured. Luciferase activity, normalized for protein content, is expressed relative to activity observed with exposure of the cells to vehicle (PBS) alone. Results are expressed as mean ±SEM; n=4. p < 0.01 (Mann-Whitney U test) compared to the luciferase activity in the absence of LPS (vehicle).
Saturated FA is an essential component of the Lipid A moiety of LPS. Our hypothesis posits that the saturated FFA component of LPS plays an essential role in LPS' effects on GHR expression. To investigate the role of fatty acids on the effect of LPS on GHR promoter, BNL.CL2 cells stably expressing the L2 promoter-luciferase construct were exposed to either LPS or fatty-acid free LPS. Whereas, LPS resulted in a significant decrease in L2 promoter activity, fatty acid-free LPS did not alter the activity of the L2 promoter (Fig.1B). Furthermore, the specificity of the observed effect of LPS on GHR promoter activity was established by demonstrating that Polymyxin B, a known inhibitor of LPS action (Lynn and Golenbock, 1992), abrogated the effect of LPS on GHR promoter activity (data not shown). These results demonstrate that fatty acid is essential for the effect of LPS on GHR-L2 promoter activity and verify the specificity of the observed effect of LPS on the GHR promoter.
The effect of LPS on the GHR promoter is not secondary to release of cytokines into the cell culture medium
Previous studies had reported that cytokines such as TNF-α can suppress GHR gene expression (Denson et al., 2001). To exclude the possibility that the observed effect of LPS on GHR was due to the release into the cell culture medium of cytokines such as TNF-α, we measured cytokine levels in cell culture supernatants of HEK 293T cells transfected with wild type TLR4 and MD2 and then stimulated with LPS for 5-6 h. These results revealed minimal elevations of IL-6, IL-β and TNF-α concentrations in the cell culture medium following LPS stimulation (Fig. 2A). TNF-α was the relevant cytokine with the most consistent elevation with concentrations averaging 40 pg/ml. It is noteworthy that previous reports demonstrating effect of TNF-α on the GHR promoter had used significantly higher (10 ng/ml) concentrations of TNF-α. We next investigated the effect of graded doses (40 pg/ml and 10 ng/ml) of TNF-α on GHR promoter activity in HEK 293T cells transfected with wild type TLR4, MD2, and L2-luciferase reporter (Fig. 2B). These result demonstrated that 40 pg/ml of TNF-α had no measurable effect on L2 promoter activity. On the contrary, and consistent with previous reports, 10 ng/ml of TNF-α significantly decreased L2 promoter activity. Concomitant exposure to LPS and 10 ng/ml of TNF-α did not result in an additive effect on GHR promoter activity, suggesting overlap in intracellular signaling pathways transducing the effects of LPS and TNF-α on the L2 promoter. These results established that the observed effect of LPS on GHR promoter activity was not secondary to generation of cytokines.
Fig.2. Effect of LPS on GHR promoter is not secondary to release of cytokines into the cell culture medium.

(A) HEK 293T cells were transfected with L2-promoter-luciferase reporter (pGL3-L2-2.0 kb), pRL-TK (internal control), wild type TLR-4 (0.1 μg/well), and wild type MD2 (0.1 μg/well) plasmids. 48 h after transfection, cells were stimulated with LPS (1 μg/ml) for 5-6 h. After stimulation, the cells were harvested and the supernatant analyzed for cytokine levels. Results are expressed as mean ±SEM; n= 3. (B) HEK 293T cells were transfected with a L2-promoter-luciferase reporter (pGL3-L2-2.0 kb), pRL-TK (internal control), wild type TLR-4 (0.1 ng/well), and wild type MD2 (0.1 ng/well). 48 h after transfection, cells were stimulated for 5-6 h with TNF-α (10 or 40 pg/ml), LPS (1 μg/ml), or a combination of LPS (1 μg/ml) and TNF-α (10 ng/ml). After stimulation, the cells were harvested and the luciferase activity measured. Results are expressed as mean ±SEM; n= 3. p < 0.05 (Mann-Whitney U test) compared to the luciferase activity in the absence of LPS (vehicle).
LPS inhibits the expression of the endogenous GHR
To verify the biological significance of the observed effect of LPS on the GHR promoter, we next investigated the effect of LPS on endogenous GHR gene expression in 3T3-F442A preadipocytes which express endogenous GHR. 3T3-F442A preadipocytes were exposed to LPS for 5-6 h and the steady state abundance of the L2 GHR transcript was quantitated by real time RT-PCR assay (Fig. 3A). These results demonstrated that exposure of the cells to LPS resulted in a significant decrease in steady state abundance of the L2 transcript. The magnitude of the decrease (40-50%) in L2-GHR mRNA levels was similar to that observed for the activity for the L2-GHR promoter. We next analyzed the effect of LPS on endogenous GHR protein expression in these cells using Western blot analysis with an anti-GHR antibody (Fig. 3B). These experiments revealed that exposure of 3T3-F442A cells to LPS resulted in a significant decrease in GHR protein expression. These results thus establish that LPS inhibits the expression of the endogenous GHR gene and protein.
Fig.3. LPS inhibits expression of endogenous GHR.
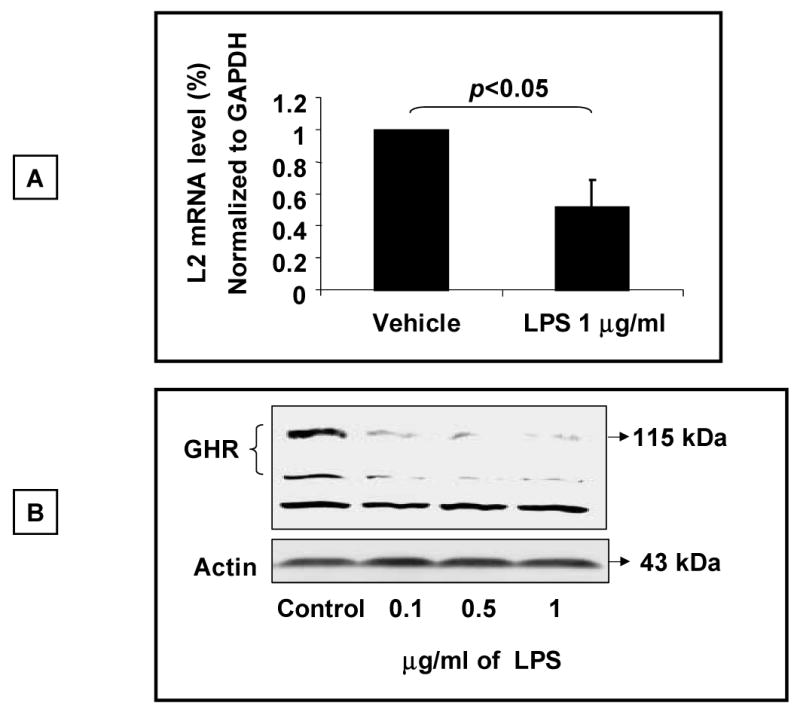
(A) 3T3-F442A preadipocytes were exposed to LPS (1 μg/ml) for 5-6 h and cells harvested for extraction of total RNA. The steady state abundance of the L2 mRNA transcript of the GHR was measured by Real Time RT-PCR analysis. Expression of the housekeeping gene GAPDH was used as internal control to normalize the results. Results are expressed as mean ± SEM; n=3. p < 0.05 (Mann-Whitney U test) compared to absence of LPS (vehicle). (B) Analysis of GHR protein expression. 3T3-F442A preadipocyte cells were stimulated with LPS (1 μg/ml) for 5-6 h and whole cell lysates prepared. Aliquots of equal amounts of protein were size-fractionated by electrophoresis, transferred on to nitrocellulose membrane by Western blotting, and the membrane probed with antibody specific for the GHR (AL-47). Following detection of the GHR using the chemiluminescence system as described under “Experimental Procedures, the blot was stripped and reprobed for actin. The positions of the molecular weight markers, the GHR, non-specific band (NS), and actin are indicated. Results depicted are representative of two such experiments.
The effect of LPS on the growth hormone receptor promoter is mediated via interaction of LPS with TLR4/MD2 complex
LPS is recognized by the receptor complex composed of TLR4 and MD2 (Akira and Takeda, 2004). We further investigated the hypothesis that the putative effect of LPS on GHR promoter activity was mediated via interaction of LPS with TLR4 by exploiting a TLR4 construct that possesses a P712H mutation in the cytoplasmic domain and functions in a dominant negative manner (Rhee and Hwang, 2000). The effect of ectopic expression of the TLR4P712H mutant on LPS' effect on GHR promoter activity in RAW 264.7 cells was analyzed. These results demonstrate that, whereas LPS significantly decreased L2 promoter activity in these cells, ectopic expression of the dominant-negative TL4 protein abrogated the effect of LPS on the L2-GHR promoter (Fig. 4A). These results support the model wherein the effect of LPS on the GHR promoter is mediated via interaction of LPS with the TLR4 receptor.
Fig. 4. The effect of LPS on GHR-L2 promoter activity is mediated through TLR4-MD2 complex.

(A) Dominant negative TLR4 construct abrogates the effect of LPS on GHR-L2 promoter activity. RAW 264.7 cells were transfected with L2-promoter-luciferase reporter (pGL3-L2-2.0 kb) and pRL-TK (internal control) plasmids with or without dominant negative TLR4 (0.2 ng/well). 24 h after transfection, cells were stimulated with LPS (0.5 μg/ml) for 5-6 h and cells harvested and luciferase activity measured. Results are expressed as mean ± SEM; n = 5. p < 0.05 (Mann-Whitney U test) compared to the luciferase activity in the absence of LPS (vehicle). (B) MD2 is required for TLR-4 mediated inhibition of L2 promoter activity by LPS. HEK 293T cells were transfected with L2-promoter-luciferase reporter (pGL3-L2-2.0 kb) and pRL-TK (internal control) plasmids with or without wild type TLR4 (0.1 ng/well) and either wild type MD2 (0.1 ng/well) or mutant MD2 (MD-2C95Y; 0.1 ng/well). 48 h after transfection, cells were stimulated with LPS (1 μg/ml) for 5-6 h and cells harvested and the luciferase activity measured. Results are expressed as mean ± SEM; n = 5. p < 0.05 (Mann-Whitney U test) compared to the luciferase activity in the absence of LPS (vehicle).
The MD2 protein is essential for interaction of LPS with TLR4 receptor (Fitzgerald et al., 2004) and cells which are devoid of MD2 or express MD2 mutant protein are hyporesponsive to LPS (Schromm et al., 2001). To determine the role of MD2 in LPS-induced inhibition of GHR promoter activity, HEK 293T cells which are devoid of endogenous TLR4 or MD2, were co-transfected with wild type TLR4 in the presence or absence of MD2. Analysis of luciferase activity of these cells established that cotransfection of TLR4 alone was not sufficient to elicit LPS-induced inhibition of L2 promoter activity (Fig. 4B). In contrast, cotransfection of both TLR4 and MD2 resulted in a significant LPS-dependent inhibition of L2 promoter activity. The specificity of the effect of MD2 was verified by demonstrating that ectopic expression of MD2C95Y mutant, which has been proven to lack LPS-binding ability (Schromm et al., 2001), did not enable LPS-dependent inhibition of L2-GHR promoter activity. These results establish the essential role of MD2 for LPS-induced inhibition of GHR promoter activity.
CD14 is not essential for LPS-induced GHR promoter suppression and fails to potentiate the effect of LPS on GHR promoter activity
CD14 is a cofactor that binds LPS and plays a role in the presentation of LPS to the MD-2 / TLR4 complex (Miyake, 2006). CD14 is not essential for LPS-stimulated signaling through the TLR but serves to potentiate LPS's interaction with TLR4. To investigate the role of CD14 in LPS induced inhibition of GHR promoter activity, HEK 293T cells were transfected with wild type TLR4 and MD2 in the presence or absence of CD14. Whereas LPS inhibited L2-GHR promoter activity in cells cotransfected with wild type TLR4 and MD2, cotransfection of CD14 did not potentiate this effect of LPS on the L2-GHR promoter activity (Fig. 5). These result suggested that CD14 is not essential for and does not modify LPS-induced inhibition of GHR promoter activity.
Fig.5. CD14 does not potentiate LPS's effect on the GHR promoter.

HEK 293T cells were transfected with L2-promoter-luciferase reporter (pGL3-L2-2.0 kb) and pRL-TK (internal control) plasmids with or without wild type TLR-4 (0.1 ng/well), wild type MD2 (0.1 ng/well), and CD14 (0.1 ng/well). 48 h after transfection, cells were stimulated with LPS (1 μg/ml) for 5-6 h. and cells harvested and luciferase activity measured. Results are expressed as mean ± SEM; n = 5, p < 0.05 (Mann-Whitney U test) compared to the luciferase activity in the absence of LPS (vehicle).
Suppression of the GHR promoter by LPS is transduced via both MyD88-dependent and -independent pathways
Canonical TLR signaling cascades consist of MyD88-dependent and -independent pathways (Akira and Takeda, 2004). MyD88 is an adaptor molecule recruited by the activation of TLR4 and stimulates downstream signaling pathways. TRIF is an adaptor protein and has been established as a key adapter protein in the MyD88-independent signaling pathways stimulated by TLR4. We next study the canonical TLR signaling cascades in LPS-induced inhibition of GHR promoter activity using the dominant negative mutants of MyD88 or TRIF (Fig. 6). Whereas LPS induced a significant decrease in L2 promoter activity in cells transfected with TLR4 and MD2, ectopic expression of either the dominant negative MyD88 construct or the dominant negative TRIF constructed resulted in abrogation of the effect of LPS on the L2 promoter. These results indicate that the effect of LPS on the GHR promoter is mediated via both MyD88-dependent and -independent pathways.
Fig.6. Effect of LPS on the GHR promoter is mediated via both MyD88-dependent and -independent pathways.

HEK 293T cells were transfected with L2-promoter-luciferase reporter (pGL3-L2-2.0 kb), pRL-TK (internal control), wild type TLR4 (0.1 ng/well), and wild type MD2 (0.1 ng/well) plasmids with or without dominant negative MyD88 or dominant negative TRIF plasmids. 48 h after transfection, cells were stimulated with LPS (1 μg/ml) for 5-6 h and cells harvested and luciferase activity measured. Results are expressed as mean ±SEM; n= 4 p < 0.05 (Mann-Whitney U test) compared to the luciferase activity in the absence of LPS (vehicle).
Discussion
The major novel findings of this study are that LPS directly suppresses GHR expression in a cytokine-independent manner and that this direct effect of LPS is transduced via MyD88-dependent and -independent Toll-like receptor-4/MD2 complex signaling pathways.
It is well established that bacterial sepsis results in a state of growth hormone insensitivity. The accepted hypothesis is that the growth hormone insensitivity in bacterial sepsis is mediated via the actions of cytokines on the GHR /IGF-1 axis. Hence TNF-α had been demonstrated to downregulate hepatic GHR expression via inhibition of the ability of Sp1/Sp3 to transactivate the GHR gene (Denson et al., 2001). Similarly, IL-1 suppresses the promoter activity of the major GHR transcript via a Sp response element (Denson et al., 2001). In contrast IL-6 suppresses GHR expression by upregulation of Cis and Socs-3, signaling molecules that inhibit GHR-STAT5 signaling pathways (Denson et al., 2003; Wang et al., 2002). The results of the current study demonstrate that LPS can directly inhibit GHR promoter activity. The possibility that the observed effect of LPS on the GHR promoter could be a non-specific effect was excluded by demonstrating that FA-free LPS was devoid of effect on the GHR promoter and that Polymyxin B, a known inhibitor of LPS action (Bhor et al., 2005), also abrogated the effect of LPS on the GHR promoter. The possibility that the observed effect of LPS is secondary to LPS-induced generation of cytokines acting in an autocrine/paracrine manner was excluded by establishing that the concentrations of cytokines in the cell culture supernatant was significantly below the threshold required to obtain a direct effect of cytokines on GHR promoter activity. Furthermore, addition of anti-TNF-α antibody failed to abrogate the effect of LPS on the L2 promoter (data not shown). However since we only profiled a limited number of cytokines in the cell culture medium of the LPS-stimulated cells, our results do leave open the possibility that a disparate cytokine could contribute to the observed effect of LPS on GHR expression. This caveat notwithstanding, our results support a model wherein in a cytokine-independent manner LPS directly inhibits GHR gene expression. These results thus represent a paradigm shift in the molecular pathogenesis of LPS-induced effects on the GHR-IGF1 axis from the current model which posits that LPS-induced inhibition of the GH/GHR axis is secondary to cytokines generated by LPS action (Fig. 7).
Fig.7. Proposed model of LPS' regulation of GHR gene expression.

LPS indirectly suppresses GHR promoter by stimulation of release of inflammatory cytokine such as IL-1, IL-6 and TNF-α from monocytes. LPS also directly inhibits GHR gene expression through MyD88-dependent and independent Toll-like receptor-4/MD2 signaling pathways. LPS, Lipopolysaccharide; IL-1, interleukin-1; IL-6, interleukin-6; TNF-α, tumor necrosis factor-α; TLR4, Toll-like receptor-4.
Our results support a model in which the direct effects of LPS on GHR expression are dependent on LPS' interaction with TLR4. Previous studies have determined that MD2 is a cofactor necessary for activation of canonical TLR4 signaling pathways (Schromm et al., 2001). Our studies also establish that LPS-dependent inhibition of GHR requires MD2. These conclusions are supported by the findings that LPS suppressed GHR expression in disparate cell lines that expressed TLR4 and MD2 either endogenously or ectopically. Moreover, abrogation of LPS' effect in cells transfected with either nonfunctional TLR4 (pro712his TLR4 mutant) or nonfunctional MD2 (cys95tyr MD2 mutant) further supports the conclusion that the effect of LPS on GHR is mediated through TLR-MD2 complex. In contrast, our studies indicate that the cofactor CD14, which has been demonstrated to amplify the effect of LPS in certain systems, was not essential for LPS' effect on the GHR promoter and did not serve to amplify the effect of LPS on the GHR promoter. However CD14 is present in two forms, a membrane-bound form and a soluble form (Palsson-McDermott and O'Neill, 2004), and our studies do not exclude the possibility that the soluble form of CD14 present in serum containing cell culture medium could play a role in modifying LPS' actions on the GHR promoter.
Our results indicate that LPS acts through MyD88-dependent and independent Toll-like receptor-4/MD2 signaling pathways to directly inhibit GHR gene expression. LPS-induced TLR signaling pathways are bipartite with MyD88-dependent and MyD88-independent pathways (Palsson-McDermott and O'Neill, 2004). The MyD88-dependent pathway is common to all TLRs; however, the MyD88-independent pathway is unique to TLR3 and TLR4. The MyD88 adaptor, which is the crucial adaptor for MyD88 dependent pathway, associates with the cytoplasmic domain of TLRs and enhances the activity of the inhibitor of nuclear factor-κB (NF-κB)–kinase complex (IKK complex). The activated IKK complex enables NF-κB to translocate to the nucleus and induce expression of cognate inflammatory cytokines. The MyD88 independent pathway utilizes the adaptor TRIF to mediate activation of IRF-3 and delayed activation of NF-κB and production of type 1 interferon (IFN-β). Our results indicate that LPS suppresses GHR expression through both MyD88- dependent and MyD88-indendent pathways. These results predict that the GHR gene would be a target for NF-κB action. Preliminary results (data not shown) using NF-κB inhibitors suggest that the NF-κB pathway plays a role in the modulating the activity of the GHR gene. Our results also suggest that LPS and TNF-α inhibit GHR gene expression via common intracellular signaling pathways. It is noteworthy that previous studies have indicated that NF-κB can be activated by TNF-α through the ubiquitin-mediated degradation of IKK complex (Chen, 2005). Hence our results would be compatible with a model wherein both LPS and TNF-α inhibit GHR gene expression via NF-κB-dependent mechanisms.
Whereas the current study establishes a cytokine-independent effect of LPS on GHR expression, the significance of this mechanism vis-a-vis the traditional cytokine-dependent actions of LPS on the GHR expression and GH action remain to be elucidated. Since the expression profiles of TLRs vary in different tissues in the body, our results predict that the importance of the cytokine-independent mechanism of LPS' effects on GH action will be tissue/organ-specific. In this regard it is noteworthy that our previous studies had demonstrated that LPS' action on hepatic GHR expression was abrogated in the TNFR null mice, thereby suggesting dominance of cytokine-dependent mechanisms in the liver in the intact animal (Denson et al., 2001). Further studies will be needed to ascertain the relative importance of cytokine-dependent versus cytokine-independent mechanisms for LPS induced GH insensitivity in other tissues/organs. Previous trials of GH designed to ameliorate the anabolic status of critically ill patients with sepsis resulted in increased mortality (in particular unexplained cardiac related deaths) in these patients, warranting termination of such trials (Carroll and Van den Berghe, 2001; Takala et al., 1999). The explanation for this unexpected adverse outcome remains unclear. We hypothesize that differential sensitivity of various tissues to LPS induced decrease in GHR expression results in some tissues (e.g. macrophages or heart) being relatively more sensitive than other tissues/organs (e.g. liver) to exogenously administered GH with resultant adverse side effects. The findings of the current study predict that this differential sensitivity of tissues to LPS' effect on GHR expression is not only determined by LPS-induced cytokine production, but also by the profile of expression of TLR in the concerned tissues/organs.
In summary, the current report establishes the direct effect of LPS in the regulation of the GHR gene. Our results indicate that LPS acts through MyD88-dependent and independent Toll-like receptor-4/MD2 signaling pathways to directly inhibit GHR gene expression. Our studies establish a novel cytokine-independent mechanism for resistance to growth hormone action in bacterial sepsis.
Acknowledgments
The authors acknowledge the assistance and the generous provision of reagents by Drs. Antonello Punturieri (University of Michigan), Stuart Frank (University of Alabama), Daniel Hwang (University of California) and Jürg Tschopp (University of Lausanne). This work was supported in part by grants from the NIH (DK49845 [RKM], and P60DK-20572 [Michigan Diabetes Research and Training Center]).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Bhor VM, Thomas CJ, Surolia N, Surolia A. Polymyxin B: an ode to an old antidote for endotoxic shock. Mol Biosyst. 2005;1:213–22. doi: 10.1039/b500756a. [DOI] [PubMed] [Google Scholar]
- Burns K, Martinon F, Esslinger C, Pahll H, Schneider P, Bodmer JL, Di Marco F, French L, Tschopp J. MyD88, an adapter protein Involved in Interleukin-1 signaling. J Biol Chem. 1998;273:12203–12209. doi: 10.1074/jbc.273.20.12203. [DOI] [PubMed] [Google Scholar]
- Carroll PV, Van den Berghe G. Safety aspects of pharmacological GH therapy in adults. Growth Horm IGF Res. 2001;11:166–72. doi: 10.1054/ghir.2001.0242. [DOI] [PubMed] [Google Scholar]
- Chen ZJ. Ubiquitin signalling in the NF-[kappa]B pathway. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Denson LA, Held MA, Menon RK, Frank SJ, Parlow AF, Arnold DL. Interleukin-6 inhibits hepatic growth hormone signaling via upregulation of Cis and Socs-3. Am J Physiol Gastrointest Liver Physiol. 2003;284:G646–54. doi: 10.1152/ajpgi.00178.2002. [DOI] [PubMed] [Google Scholar]
- Denson LA, Menon RK, Shaufl A, Bajwa HS, Williams CR, Karpen SJ. TNF-alpha downregulates murine hepatic growth hormone receptor expression by inhibiting Sp1 and Sp3 binding. J Clin Invest. 2001;107:1451–8. doi: 10.1172/JCI10994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denson LA, Shaufl A, Karpen SJ, Menon RK. Tumor necrosis factor alpha down regulates murine hepatic growth hormone receptor gene expression via inhibition of Sp1 and Sp3 binding. J Clin Invest. 2001;107:1451–58. doi: 10.1172/JCI10994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edens A, Talamantes F. Alternative processing of growth hormone receptor transcripts. Endocr Rev. 1998;19:559–82. doi: 10.1210/edrv.19.5.0347. [DOI] [PubMed] [Google Scholar]
- Fitzgerald KA, Rowe DC, Golenbock DT. Endotoxin recognition and signal transduction by the TLR4/MD2-complex. Microbes and Infection. 2004;6:1361–1367. doi: 10.1016/j.micinf.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Imler JL, Zheng L. Biology of Toll receptors: lessons from insects and mammals. J Leukoc Biol. 2004;75:18–26. doi: 10.1189/jlb.0403160. [DOI] [PubMed] [Google Scholar]
- Lee JY, Ye J, Gao Z, Youn HS, Lee WH, Zhao L, Sizemore N, Hwang DH. Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem. 2003;278:37041–51. doi: 10.1074/jbc.M305213200. [DOI] [PubMed] [Google Scholar]
- Lynn WA, Golenbock DT. Lipopolysaccharide antagonists. Immunology Today. 1992;13:271–276. doi: 10.1016/0167-5699(92)90009-V. [DOI] [PubMed] [Google Scholar]
- Menon R, Cheng H, Singh M. Identification and characterization of single strand DNA-binding protein that represses growth hormone receptor gene expression. Mol Endocrinol. 1997;11:1291–1304. doi: 10.1210/mend.11.9.9967. [DOI] [PubMed] [Google Scholar]
- Menon RK, Stephan DA, Singh M, Morris SM, Jr, Zou L. Cloning of the promoter-regulatory region of the murine growth hormone receptor gene. Identification of a developmentally regulated enhancer element. J Biol Chem. 1995;270:8851–9. doi: 10.1074/jbc.270.15.8851. [DOI] [PubMed] [Google Scholar]
- Miyake K. Roles for accessory molecules in microbial recognition by Toll-like receptors. Journal of Endotoxin Res. 2006;12:195–204. doi: 10.1179/096805106X118807. [DOI] [PubMed] [Google Scholar]
- Moffat JG, Edens A, Talamantes F. Structure and expression of the mouse growth hormone receptor/growth hormone binding-protein gene. J Mol Endocrinol. 1999;23:33–44. doi: 10.1677/jme.0.0230033. [DOI] [PubMed] [Google Scholar]
- Palsson-McDermott EM, O'Neill LAJ. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology. 2004;113:153–162. doi: 10.1111/j.1365-2567.2004.01976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SH, Hwang D. Murine TOLL-like receptor 4 confers lipopolysaccharide responsiveness as determined by activation of NFkappa B and expression of the Inducible cyclooxygenase. J Biol Chem. 2000;275:34035–34040. doi: 10.1074/jbc.M007386200. [DOI] [PubMed] [Google Scholar]
- Schromm AB, Lien E, Henneke P, Chow JC, Yoshimura A, Heine H, Latz E, Monks BG, Schwartz DA, Miyake K, Golenbock DT. Molecular genetic analysis of an endotoxin nonresponder mutant cell line: a point mutation in a conserved region of MD-2 abolishes endotoxin-induced signaling. J Exp Med. 2001;194:79–88. doi: 10.1084/jem.194.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzbauer G, Yu JH, Cheng H, Menon RK. Transcription factor MSY-1 regulates expression of the murine growth hormone receptor gene. J Biol Chem. 1998;273:24760–9. doi: 10.1074/jbc.273.38.24760. [DOI] [PubMed] [Google Scholar]
- Southard JN, Barrett BA, Bikbulatova L, Ilkbahar Y, Wu K, Talamantes F. Growth hormone (GH) receptor and GH-binding protein messenger ribonucleic acids with alternative 5′-untranslated regions are differentially expressed in mouse liver and placenta. Endocrinology. 1995;136:2913–2921. doi: 10.1210/endo.136.7.7789316. [DOI] [PubMed] [Google Scholar]
- Takala J, Ruokonen E, Webster NR, Nielsen MS, Zandstra DF, Vundelinckx G, Hinds CJ. Increased mortality associated with growth hormone treatment in critically ill adults. N Engl J Med. 1999;341:785–792. doi: 10.1056/NEJM199909093411102. [DOI] [PubMed] [Google Scholar]
- Thimmarayappa J, Sun J, Schultz LE, Dejkhamron P, Lu C, Giallongo A, Merchant JL, Menon RK. Inhibition of growth hormone receptor gene expression by saturated fatty acids: role of Kruppel-like zinc finger factor, ZBP-89. Mol Endocrinol. 2006;20:2747–60. doi: 10.1210/me.2006-0128. [DOI] [PubMed] [Google Scholar]
- Trent M, Stead C, Tran A, Hankins J. Diversity of endotoxin and its impact on pathogenesis. Journal of Endotoxin Res. 2006;12:205–223. doi: 10.1179/096805106X118825. [DOI] [PubMed] [Google Scholar]
- Wang P, Li N, Li JS, Li WQ. The role of endotoxin, TNF-alpha, and IL-6 in inducing the state of growth hormone insensitivity. World J Gastroenterol. 2002;8:531–6. doi: 10.3748/wjg.v8.i3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JH, Schwartzbauer G, Kazlman A, Menon RK. Role of the Sp family of transcription factors in the ontogeny of growth hormone receptor gene expression. J Biol Chem. 1999;274:34327–36. doi: 10.1074/jbc.274.48.34327. [DOI] [PubMed] [Google Scholar]
- Yumet G, Shumate M, Bryant D, Lang C, Cooney R. Hepatic growth hormone resistance during sepsis is associated with increased suppressors of cytokine signaling expression and impaired growth hormone signaling. Crit Care Med. 2006;34:1420–7. doi: 10.1097/01.CCM.0000215113.66070.E0. [DOI] [PubMed] [Google Scholar]