

Abstract
Rapid syntheses of chitotetrose derivatives were achieved in good yields using the newly developed reactivity independent iterative one-pot strategy. The protective groups on donors and acceptors were independently evaluated allowing matching of the two partners in glycosylation. No anomeric reactivity adjustments or intermediate purification were necessary thus significantly improving the overall synthetic efficiency. Only near stoichiometric amounts of building blocks were required for the assembly of target molecules further highlighting the potential of the iterative one-pot method in complex oligosaccharide synthesis.
Keywords: Iterative, One-pot synthesis, Chitotetrose, Reactivity independent
1. Introduction
With the recognition of multifaceted biological properties of oligosaccharides and glyco-conjugates, the development of novel methods to expedite oligosaccharide synthesis has been a major focus of modern synthetic carbohydrate chemistry.1,2 A popular synthetic strategy is the reactivity based chemoselective glycosylation method, where a more reactive armed glycosyl donor is preferentially activated in the presence of a less reactive disarmed acceptor.2,3 Many innovative methods have been developed for tuning anomeric reactivities.3–5 However, the very need to achieve precise anomeric reactivity values necessitates extensive and tedious protecting group and/or aglycon adjustments, which in turn detracts from its broad application and restricts the possibility of matching6 the glycosyl donor with the acceptor.
Recently, we have developed a new iterative one-pot glycosylation method, by which multiple sequential glycosylations of a thioglycosyl donor by a thioglycosyl acceptor can be carried out independent of the anomeric reactivities of donors and acceptors.7 Medium sized oligosaccharides have been assembled in a few hours using this methodology without the need of intermediate purification and aglycon adjustments. Our reactivity independent one-pot approach represents a significant advancement in chemoselective glycosylation as the time-consuming anomeric reactivity adjustment is unnecessary. To explore the scope of this method, we have embarked on application of this strategy in assembly of complex oligosaccharides. Herein, we report our syntheses of chitotetroses, that is, tetrasaccharides containing β-(1→4)-linked glucosamines.
2. Results and discussion
Chitotetroses and their derivatives have diverse biological functions such as immunostimulatory,8 antitumor,9 anticoagulant,10 and anti-HIV activities.11 Chemical synthesis of this class of compounds presents a significant challenge due to the well known low nucleophilicity of 4-hydroxyl group of glucosamine building blocks. To date, there have been a few reports of step-wise assembly of these molecules, which are often characterized by multiple protecting group manipulations and aglycon leaving group adjustments on oligosaccharide intermediates. 12,13 A single one-pot synthesis was accomplished by our group using the reactivity based chemoselective glycosylation approach.4 Even though building blocks with multiple levels of anomeric reactivities were conveniently generated through post-synthetic modification of the aglycon of a common intermediate, the need for reactivity adjustment of our previous one-pot synthesis prompted us to explore the utility of our reactivity independent iterative one-pot method.7
It is known that the low nucleophilicity of 4-hydroxyl group of protected N-acetyl glucosamine is mainly due to the steric hinderance around this secondary hydroxyl group and the intermolecular hydrogen bonding with the 2-acetamido moiety.14 The N-protecting group on a glycosyl acceptor can significantly affect its nucleophilicity, with the following order of activities: N3, NH-Troc > N-Phth > NH-Ac.14,15 Recently, Crich and Vinod have demonstrated that the usage of oxazolidinone protected N-acetyl glucosamine O-glycoside acceptor 1 considerably enhanced the glycosylation yield compared with the corresponding O-glycoside without the oxazolidinone moiety.16,17 We anticipated that oxazolidinone protected thioglycosyl acceptor 2 may lead to higher glycosylation yield and the resulting disaccharide can function as a glycosyl donor.18 To investigate the influence of N-protecting groups on our glycosylation methodology, glycosyl acceptors 2, 3, and 4 were prepared. The azido moiety is unsuitable as an N-protecting group for one-pot synthesis of β-linked oligoglucosamines because it is a well known non-participatory group favoring the formation of α-anomer when used in a donor.19
The synthesis of oxazolidinone protected acceptor 2 started from the benzylidene protected glucosamine derivative 5 (Scheme 1).20 Removal of the phthalimido group followed by oxazolidinone formation and acetylation provided the fully protected glucosamine 6. Interestingly, regioselective reductive opening of the benzylidene ring by treatment of NaCNBH3 and HCl4 yielded a mixture of α-and β-thioglycoside 2, with 2α as the major product (40%). This is in line with the observation by the Crich group where the oxazolidinone bearing O-glycoside 7 was epimerized at the anomeric position under similar reaction conditions.16 Thioglycosyl acceptors 321 and 422 were synthesized in a straightforward manner from building block 5 (Scheme 1b and c).
Scheme 1.
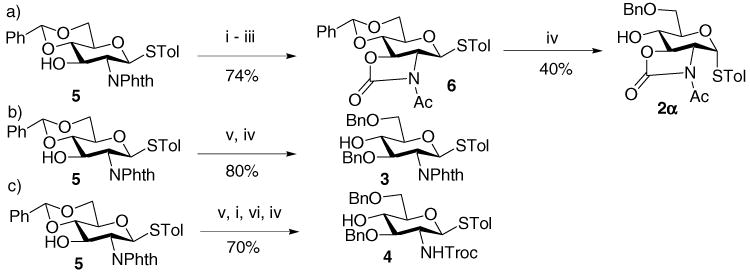
Preparation of acceptors 2–4. Reagents and conditions: (i) ethylene diamine, EtOH, reflux, overnight; (ii) 4-O2NC6H4OCOCl, NaHCO3, CH3CN/H2O, rt, overnight; (iii) AcCl, DIPEA, DCM, rt, overnight; (iv) NaCNBH3, 1 M HCl in Et2O, 0 °C, 1.5 h; (v) NaH, TBAI, DMF, MS; BnBr, 2 h, rt; (vi) Troc-Cl, NaHCO3, H2O, THF.
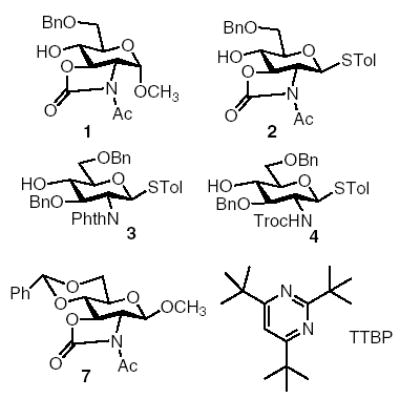
Glycosylations of thioglycosyl donor 820 with acceptors 2 to 4 were carried out following our previously established pre-activation condition.7 The disarmed glycosyl donor 8 was cleanly activated in the absence of any acceptors at −70 °C by the powerful thiophilic promoter p-TolSOTf, formed in situ by reaction of p-TolSCl23 with AgOTf.7,24 Subsequent addition of the glycosyl acceptor (0.9 equiv) to the reaction mixture led to the formation of thioglycosyl disaccharide. The reaction of N-Phth protected acceptor 3 with donor 8 produced the desired disaccharide 9 in 56% yield with trace amount (<5%) of donor elimination product glycal 10 (Table 1, entry 1). It is noteworthy that acceptor 3 has a higher anomeric reactivity than donor 8. This reversal of anomeric reactivity, that is, chemoselective glycosylation of an armed acceptor with a disarmed donor is not possible with the traditional reactivity based glycosylation methods.
Table 1.
Evaluation of N-protective group on glycosylation of various thioglycosyl acceptors by donor 8
| Entry # | Acceptor | Pdt. | Yield (%) |
|---|---|---|---|
| 1 | 3 | 9 | 56 |
| 2 | 4 | 11 | 50 |
| 3 | 12 | 13 | 50 |
| 4 | 2α | 14 | 39 |
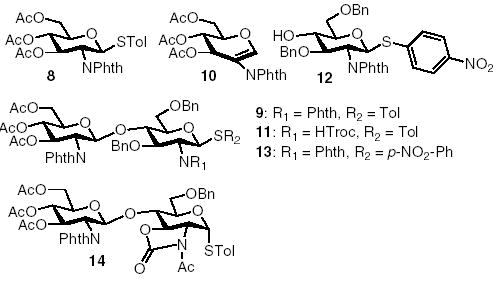
The glycosylation of N-Troc protected acceptor 4 by donor 8 gave a yield of 50% of the disaccharide 11, along with 20% of regenerated donor 8 even after complete consumption of the donor during pre-activation (Table 1, entry 2). This is the first time that a substantial amount of the regenerated donor was observed among all reactions we carried out so far using the pre-activation scheme. The donor regeneration is presumably due to the intermolecular aglycon transfer25 from the acceptor to the activated donor via S-alkylation instead of the desired O-alkylation of the hydroxyl group. This further re-affirms the low nucleophilicity of 4-hydroxyl group of glucosamine acceptors. The usage of p-nitro phenyl thioglycosyl acceptor 124 suppressed the aglycon transfer side reaction, but the disaccharide product 13 was obtained in only 50% yield (Table 1, entry 3). We examined next the glycosylation of oxazolidinone thioglycosyl acceptor 2α by donor 8. Disappointingly, the disaccharide product 14 was produced in only 39% yield with 50% of acceptor 2α recovered after reaction (Table 1, entry 4). The oxazolidinone moiety did not seem to improve the glycosylation and N-Phth was used as the N-protecting group in further studies.
The four-component one-pot assembly of fully protected tetraglucosamine derivative 16 was performed as outlined in Scheme 2. Pre-activation of donor 8 by p-TolSOTf was followed by addition of acceptor 3. The reaction temperature was raised to −10 °C in 20 min. After acceptor 3 was completely consumed as judged by TLC analysis, the reaction mixture was cooled back down to −70 °C, followed by sequential addition of AgOTf, p-TolSCl, acceptor 3, and warming up to −10 °C. Subsequently, the reaction temperature was lowered to −70 °C again, which was followed by addition of acceptor 154 and AgOTf/p-TolSCl. Slight excess of glycosyl donors was employed for the first two glycosylations to ensure complete consumption of the acceptor. The excess activated donor decomposed upon warming up thus not affecting the following reactions. The desired protected chitotetrose 16 was isolated from the one-pot reaction by flash column chromatography in 32% overall yield. It is noteworthy that the same acceptor 3 was used for the formation of the first and the second glycosidic linkages without resorting to anomeric reactivity adjustment.
Scheme 2.
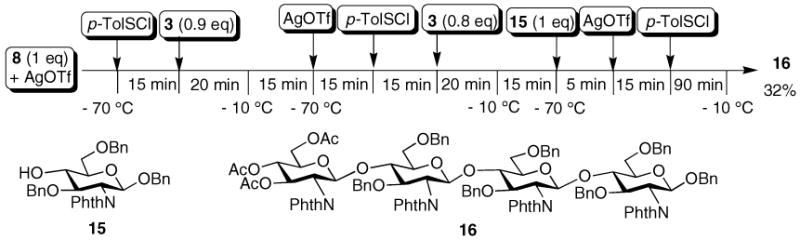
One-pot synthesis of chitotetrose 16.
To enhance the glycosylation yield, we next explored the protecting groups on the glycosyl donor. From the readily available alcohol 5,20 benzylation and silylation gave donor 1721 and 18 in 90% and 85% yields, respectively (Scheme 3a and b). Regioselective opening of the benzylidene group with TMSOTf and borane THF26 followed by benzylation produced the benzyl protected donor 19 in 80% yield for the two steps (Scheme 3c).
Scheme 3.
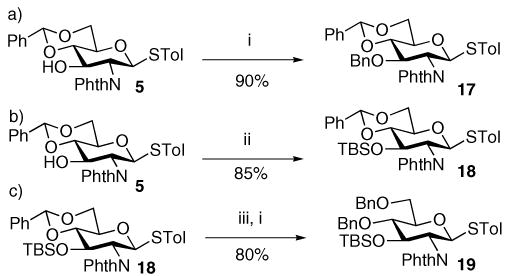
Preparation of donors 17–19. Reagents and conditions: (i) NaH, TBAI, DMF, MS; BnBr, 2 h, rt; (ii) TBSOTf, 2,6-lutidine, DCM, 4 h, 0 °C; (iii) TMSOTf, BH3·THF, 4 h, −20 to 0 °C.
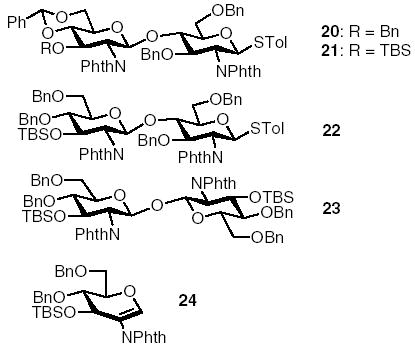
With various donors in hand, we began to evaluate their effects on glycosylation. The benzyl and benzylidene containing donor 17 reacted with acceptor 3 giving 61% of disaccharide 20 (Table 2, entry 1) together with 20% of recovered acceptor 3. Exchange of the benzyl group in 17 with TBS moiety (donor 18) led to a much cleaner reaction with complete consumption of the acceptor, producing disaccharide 21 in 74% yield (Table 2, entry 2). Both the TBS and benzylidene groups are important for high yield, because donor 19, which is devoid of the benzylidene group, glycosylated acceptor 3 in a lower 50% yield (Table 2, entry 3). 1,1′-Linked disaccharide 23 and glycal 24 were isolated as the major side products (~10%) for this reaction. The enhancement effect of TBS group is currently under investigation.
Table 2.
Evaluation of donors on glycosylation of acceptor 3
| Entry # | Donor | Pdt. | Yield (%) |
|---|---|---|---|
| 1 | 17 | 20 | 61 |
| 2 | 18 | 21 | 74 |
| 3 | 19 | 22 | 50 |
One-pot sequential reactions of 18, 3, 25,27 and 2613 promoted by p-TolSOTf led to the formation of tetrasaccharide 27 in 45% yield in less than 6 h (Scheme 4). This corresponds to an average of 85% yield for each step of the five synthetic steps carried out in one pot. The reactivity independent nature of our method allowed us to utilize the PMB containing building block 25 instead of acceptor 3 for the second glycosylation. To prevent the loss of acid labile PMB group, a sterically hindered base TTBP28 was added with each acceptor. In addition to the desired product 27 and final acceptor 26 (50%), we have isolated from the final reaction mixture, glucals 28 (18%), 29 (15%), and hemiacetal 30 (24%) as the major side products, which were formed through elimination or hydrolysis of activated glycosyl donors due to insufficient nucleophilicity of the acceptors. The glucals do not react with the promoter p-Tol- SOTf at low temperature, which do not affect further donor activation. In addition, these side products have sufficiently different polarities from the desired oligosaccharide 27, thus not interfering with product purification by flash chromatography. The PMB and TBS groups in tetrasaccharide 27 can be selectively removed to allow for alkylations of alternating 3-hydroxyl groups in the tetraglucosamine, which are useful intermediates for peptidoglycan synthesis.29
Scheme 4.
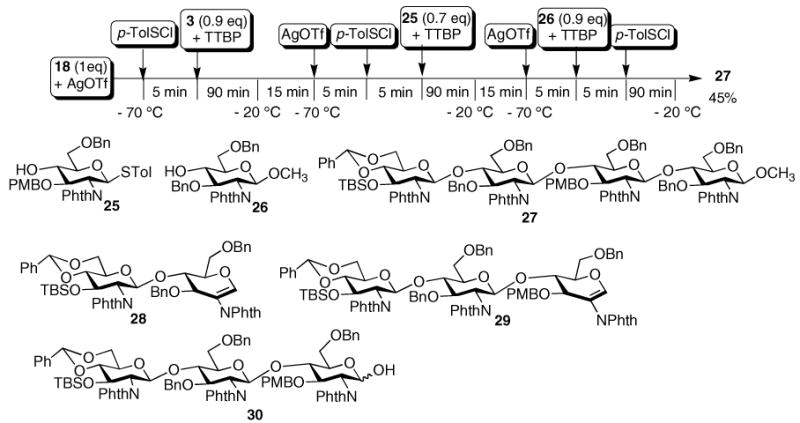
One-pot synthesis of chitotetrose 27.
In conclusion, facile syntheses of chitotetrose derivatives were achieved in good yields using our newly developed reactivity independent iterative one-pot method. Unlike solid phase synthesis,30 only near stoichiometric amounts of building blocks were required. No anomeric reactivity differentiations between donors and acceptors were necessary, thus allowing independent evaluation of protecting groups. As the N-protecting group, the phthalimido moiety gave the most consistent results, while the new oxazolidinone group did not improve the glycosylation. The introduction of benzylidene and TBS groups onto thioglycosyl donor significantly enhanced the yield. Further improvements on this challenging glycosylation reaction will depend upon the augmentation of the nucleophilicity of 4-hydroxyl group of glucosamine acceptors.
3. Experimental
3.1. General procedures
All reactions were carried out under nitrogen with anhydrous solvents in flame-dried glassware, unless otherwise noted. All glycosylation reactions were performed in the presence of molecular sieves, which were flame-dried right before the reaction under high vacuum. Glycosylation solvents were dried using an MBraun solvent purification system. Chemicals used were reagent grade as supplied except where noted. Analytical thin-layer chromatography was performed using silica gel 60 F254 glass plates (EM Science); compound spots were visualized by UV light (254 nm) and by staining with a yellow solution containing Ce(NH4)2(NO3)6 (0.5 g) and (NH4)6Mo7O244H2O (24.0 g) in 6% H2SO4 (500 mL). Flash column chromatography was performed on silica gel 60 (230–400 Mesh, EM Science). 1H NMR, 13C NMR, 1H–1H COSY, and 1H–13C HMQC spectra were recorded on a Varian VXRS-400 or Inova-600 instrument and were referenced using Me4Si (0 ppm), residual CHCl3 (δ 1H NMR 7.26 ppm, 13C NMR 77.0 ppm). Optical rotations were measured at 25 °C. ESI mass spectra were recorded on ESQUIRE LC–MS operated in positive ion mode. High-resolution mass spectra were recorded on a Micromass electrospray Tof™ II (Micromass, Wythenshawe, UK) mass spectrometer equipped with an orthogonal electrospray source (Z-spray) operated in positive ion mode, which is located at the Mass Spectrometry and Proteomics Facility, the Ohio State University.
3.2. p-Tolyl 2-acetamido-4,6-O-benzylidene-2-N-3-O-carbonyl-2-deoxy-1-thio-β-D-glucopyranoside (6)
p-Tolyl 4,6-O-benzylidene-2-deoxy-2-phthalimido-1-thio-β-D-glucopyranoside 5 (1.5 g, 2.98 mmol, 1 equiv) and ethylenediamine (9 g, 150 mmol, 50 equiv) were dissolved in dry ethanol (15 mL). The mixture was heated at reflux for 16 h. The reaction mixture was cooled and neutralized with 2 N HCl until slightly basic. After concentration, the crude product was dissolved in EtOAc (10 mL) and washed with H2O, brine, then dried, and concentrated. Column chromatography (15:1, CH2Cl2–MeOH) on silica gel afforded the free amine as a yellow solid (0.98 g, 88%). The obtained amine (0.98 g, 2.62 mmol, 1 equiv) and NaHCO3 (1.1 g, 13.09 mmol, 5 equiv) were dissolved in H2O/CH3CN mixture (15 mL/15 mL) and cooled to 0 °C. p-Nitrophenyl chloroformate (1.59 g, 7.89 mmol, 3 equiv) in CH3CN (5 mL) was added slowly and the mixture was stirred at 0 °C for 3 h. The resulting aqueous mixture was extracted with EtOAc and the combined extracts were washed with water, brine, then dried, and concentrated. Silica gel column chromatography (2.5:1, hexanes–EtOAc) afforded the oxazolidinone protected glucosamine as a white solid (0.88 g, 84%). The oxazolidinone protected glucosamine (0.42 g, 1.05 mmol, 1 equiv) and Hünig’s base (0.92 mL, 5.29 mmol, 5 equiv) were dissolved in CH2Cl2 (6 mL). Acetyl chloride (0.38 mL, 5.34 mmol, 5.1 equiv) was added under N2. The mixture was stirred for 6 h and then poured into saturated aqueous NaHCO3. The mixture was diluted with CH2Cl2 (5 mL) and the organic layer was separated. The aqueous layer was extracted twice with CH2Cl2 and the combined extracts were washed with H2O, brine, then dried, and concentrated. Column chromatography (1:1, hexanes–EtOAc) on silica gel afforded the product 11 as a white solid (0.39 g, 84%). 1H NMR (600 MHz, CDCl3): δ 2.33 (s, 3H), 2.58 (s, 3H), 3.50–3.54 (m, 1H), 3.92 (t, J = 10.2 Hz, 1H), 4.07 (t, J = 9.3 Hz, 1H), 4.13 (dd, J = 9.0, 10.8 Hz, 1H), 4.27 (dd, J = 4.8, 10.5 Hz, 1H), 4.33 (t, J = 0.5 Hz, 1H), 4.91 (d, J = 9.0 Hz, 1H), 5.59 (s, 1H), 7.11–7.12 (m, 2H), 7.38–7.35 (m, 5H), 7.43–7.44 (m, 2H); 13C NMR (150 MHz, CDCl3): δ 21.42, 25.17, 61.01, 68.46, 73.31, 78.60, 78.97, 88.95, 101.77, 126.35, 128.60, 129.65, 129.87, 130.02, 133.20, 136.45, 138.79, 153.81, 173.54; HRMS: [M+Na]+ C23H23NNaO6S calcd 464.1144, obsd 464.1181.
3.3. p-Tolyl 2-acetamido-6-O-benzyl-2-N,3-O-carbonyl-2-deoxy-1-thio-α-D-glucopyranoside (2α)
The mixture of compound 6 (0.11 g, 0.25 mmol, 1 equiv), NaCNBH3 (0.19 g, 3.0 mmol, 12 equiv) in THF (5 mL) was cooled to 0 °C. One molar of HCl in Et2O (~3 mL) was added under N2 until the solution was slightly acidic. The reaction mixture was stirred at 0 °C for 1.5 h and diluted with 10 mL of CH2Cl2 and then poured into aqueous NaHCO3. The organic layer was separated and washed with H2O, brine, then dried, and concentrated. Column chromatography (1:1, hexanes–EtOAc) on silica gel afforded the product 2α as a white solid (44 mg, 40%). [α]D +176.4 (c, 0.0056 g/mL, CH2Cl2); 1H NMR (600 MHz, CDCl3): δ 2.31 (s, 3H), 2.53 (s, 3H), 3.11 (br, 1H), 3.72–3.74 (m, 1H), 3.83–3.86 (m, 1H), 4.01–4.04 (m, 1H), 4.08–4.11 (m, 1H), 4.16–4.19 (m, 1H), 4.37 (dd, J = 12, 9.6 Hz, 1H), 4.53 (d, J = 12.0 Hz, 1H), 4.61 (d, J = 12 Hz, 1H), 6.07 (d, J = 4.2 Hz, 1H), 7.08–7.09 (m, 2H), 7.29–7.36 (m, 7H); 13C NMR (150 MHz, CDCl3): δ 21.37, 24.06, 59.91, 69.72, 70.70, 72.51, 74.07, 78.41, 86.88, 128.06, 128.31, 128.82, 128.92, 130.21, 133.27, 137.55, 138.70, 153.32, 171.45; HRMS: [M+Na]+ C23H25NNaO6S calcd 466.1300, obsd 466.1271.
3.4. p-Tolyl 3,6-di-O-benzyl-2-deoxy-2-N-phthalimido-1-thio-β-D-glucopyranoside (3)
The mixture of compound 17 (1 g, 1.68 mmol), NaCNBH3 (1.06 g, 17 mmol), and molecular sieves MS AW300 in THF (20 mL) was cooled to 0 °C. Two molar of HCl in Et2O (~9 mL) was added under N2 until no more gas evolved from the reaction. The reaction mixture was stirred at 0 °C for 30 min. The reaction was quenched with Et3N and filtered. The filtrate was diluted with CH2Cl2 (30 mL) and extracted with saturated solutions of aqueous NH4Cl (20 mL) and NaHCO3 (20 mL). The organic layer was separated and dried with Na2SO4. Column chromatography (2:1, hexanes–EtOAc) on silica gel afforded the product 3 as a white solid in 90% yield (80% yield for the two steps from compound 5). Comparison of the NMR data with those reported in the literature21 confirmed the identity of 3.
3.5. p-Tolyl 3,6-di-O-benzyl-2-deoxy-1-thio-2-N-trichloroethoxycarbonyl-β-D-glucopyranoside (4)
Compound 17 (1.9 g, mmol) and ethylenediamine (11.8 mL, 175 mmol, 55 equiv) were dissolved in dry ethanol (30 mL). The mixture was heated at reflux for 16 h. The reaction mixture was evaporated and dissolved in EtOAc (30 mL), which was neutralized with 1 N H2SO4 until slightly basic. The resulting suspension was kept in ice-water for 1 h, and then filtered to afford the glucosamine sulfate salt (1.53 g, 85%) as a white solid. The solid (1.05 g, 1.86 mmol) was dissolved in a mixture of THF and saturated aqueous NaHCO3 solution (30 mL) followed by addition of TrocCl (340 μL, 2.51 mmol). The mixture was stirred for 1 h, and then extracted with EtOAc (60 mL) and saturated aqueous NaHCO3 (20 mL). The organic layer was dried over Na2SO4 and purified by silica gel column to give 2-deoxy-2-N-Troc glucopyranoside (1.19 g, 100%). The desired compound 4 (660 mg, 90%) was then obtained as a white solid from the 2-deoxy-2-N-Troc glucopyranoside (730 mg, 1.14 mmol) following the same procedure as in the synthesis of compound 3. The overall yield for synthesis of compound 4 was 70% for the four steps starting from compound 5. Comparison of the NMR data with those reported in the literature22 confirmed the identity of 4.
3.6. General procedure for single step glycosylation
A solution of donor (0.067 mmol) and freshly activated molecular sieve MS—4 Å (300 mg) in a mixture of CH2Cl2 and Et2O (1:1, 4 mL) (100% Et2O was used for donors 18 and 19) was stirred for 30 min, and cooled to −70 °C, which was followed by sequential additions of AgOTf (52 mg, 0.201 mmol) dissolved in Et2O (1 mL) and p-TolSCl (10.5 μL, 0.067 mmol) through syringes. After the donor was completely consumed according to TLC analysis (about 15 min at −70 °C), a solution of acceptor (0.060 mmol) in CH2Cl2 (0.5 mL) was injected via a syringe. The reaction mixture was warmed to −10 °C under stirring in 2 h. Then the mixture was diluted with CH2Cl2 (20 mL) and filtered. The filtrate was washed twice with saturated aqueous NaHCO3 solution (20 mL) and once with brine (10 mL). The organic layer was collected and dried over MgSO4. After removal of the solvent, the residue was applied to silica gel flash chromatography column using a mixture of hexanes and EtOAc to afford the desired disaccharide.
3.7. p-Tolyl 3,4,6-tri-O-acetyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-D-glucopyranoside (9)
Compound 9 was synthesized from donor 8 and acceptor 3 in 56% yield following the general procedure of single step glycosylation together with trace amount of glycal 10. Compound 9 [α]D +3.5 (c, 0.058 g/mL, CH2Cl2); 1H NMR (600 MHz, CDCl3): δ 1.82 (s, 3H), 1.93 (s, 3H), 1.98 (s, 3H), 2.20 (s, 3H), 3.35 (dd, J = 3.0, 9.6Hz, 1H), 3.44–3.48 (m, 2H), 3.58 (d, J = 10.8 Hz, 1H), 3.89 (dd, J = 2.4, 12.0 Hz, 1H), 4.08–4.22 (m, 4H), 4.30 (dd, J = 8.4, 10.2 Hz, 1H), 4.44 (d, J = 12.6 Hz, 1H), 4.48 (d, J = 11.4 Hz, 1H), 4.54 (d, J = 11.4 Hz, 1H), 4.79 (d, J = 12.6 Hz, 1H), 5.11 (t, J = 9.6 Hz, 1H), 5.26 (d, J = 9.6 Hz, 1H), 5.49 (d, J = 8.4 Hz, 1H), 5.78 (dd, J = 9.0, 10.2 Hz, 1H), 6.79–7.88 (m, 22 H); 13C NMR (150 MHz, CDCl3): δ 20.68, 20.86, 20.86, 21.31, 54.93, 55.46, 60.64, 68.38, 68.96, 70.82, 71.70, 72.94, 74.63, 76.15, 77.89, 78.81, 83.50, 97.03, 127.27–138.64 (aromatic carbons), 169.75, 170.36, 170.93 (CH3CO); HRMS: [M+Na]+ C55H52N2NaO15S calcd 1035.2986, obsd 1035.2985. Compound 10: [α]D −22.5 (c, 0.0067 g/mL, CH2Cl2); 1H NMR (600 MHz, CDCl3): δ 1.92 (s, 3H), 2.12 (s, 3H), 2.14 (s, 3H), 4.36 (dd, J = 4.2, 12.0 Hz, 1H), 4.47–4.58 (m, 2H), 5.31 (t, J = 4.2 Hz, 1H), 5.59 (d, J = 4.2 Hz, 1H), 6.76 (s, 1H), 7.72–7.76 (m, 2H), 7.83–7.88 (m, 2H); HRMS [M+Na]+ C20H19NNaO9 calcd 440.0958, obsd 440.0961.
3.8. p-Tolyl 3,4,6-tri-O-acetyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-1-thio-2-N-trichloroethoxycarbonyl-β-D-glucopyranoside (11)
Compound 11 was synthesized from donor 8 and acceptor 4 in 50% yield following the general procedure of single step glycosylation together with recovered acceptor 4 (20%). [α]D −10.7 (c, 0.012 g/mL, CH2Cl2); 1H NMR (600 MHz, CDCl3): δ 1.82 (s, 3H), 1.94 (s, 3H), 1.98 (s, 3H), 2.25 (s, 3H), 3.26 (m, 2H), 3.40 (d, J = 10.2 Hz, 1H), 3.45 (dd, J = 3.6, 10.8Hz, 1H), 3.56 (d, J = 10.2 Hz, 1H), 3.75–3.83 (m, 2H), 4.08 (t, J = 9.0 Hz, 1H), 4.11 (dd, J = 4.2, 12.6 Hz, 1H), 4.27 (dd, J = 9.0, 10.8 Hz, 1H), 4.42 (d, J = 12 Hz, 1H), 4.48 (d, J = 12 Hz, 1H), 4.62–4.65 (m, 2H), 4.70 (d, J = 12 Hz, 1H), 4.76 (d, J = 4.2 Hz, 1H), 4.91 (d, J = 11.4 Hz, 1H), 5.03 (d, J = 7.8 Hz, 1H), 5.10 (t, J = 9.6 Hz, 1H), 5.50 (d, J = 8.4 Hz, 1H), 5.74 (t, J = 10.2 Hz, 1H), 6.95–7.84 (18, 2H); 13C NMR (150 MHz, CDCl3): δ 20.66, 20.84, 20.89, 21.33, 55.43, 56.30, 61.66, 68.44, 68.86, 70.79, 71.88, 73.00, 74.40, 74.59, 75.33, 78.80, 79.76, 85.62, 97.10, 127.63–138.42 (aromatic carbons), 153.71(carbonyl of Troc), 169.70, 170.35, 170.88 (CH3CO); HRMS: [M+Na]+ C50H51Cl3N2NaO15S calcd 1079.1973, obsd 1079.2067.
3.9. p-Nitrophenyl 3,4,6-tri-O-acetyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-D-glucopyranoside (13)
Compound 13 was synthesized from donor 8 and acceptor 12 in 50% yield following the general procedure of single step glycosylation. [α]D +21.8 (c, 0.010 g/mL, CH2Cl2); 1H NMR (600 MHz, CDCl3): δ 1.84 (s, 3H), 1.94 (s, 3H), 1.99 (s, 3H), 3.49–3.55 (m, 3H), 3.61 (d, J = 9.6 Hz, 1H), 3.96 (dd, J = 2.4, 12.0 Hz, 1H), 4.19–4.23 (m, 3H), 4.25 (dd, J = 3.6, 12.0 Hz, 1H), 4.33 (dd, J = 8.4, 10.8 Hz, 1H), 4.44 (d, J = 12 Hz, 1H), 4.45 (d, J = 12.6 Hz, 1H), 4.53 (d, J = 11.4 Hz, 1H), 4.82 (d, J = 12.6 Hz, 1H), 5.13 (t, J = 10.2 Hz, 1H), 5.47 (d, 1H, J = 10.2 Hz, 1H), 5.50 (d, J = 8.4 Hz, 1H), 5.79 (dd, J = 9.0, 10.8 Hz, 1H), 6.79–7.94 (m, 22H); 13C NMR (150 MHz, CDCl3): δ 20.68, 20.86, 20.87, 54.52, 55.45, 61.69, 68.43, 68.90, 70.77, 71.81, 73.06, 74.89, 76.42, 77.70, 78.79, 82.03, 97.25, 123.94–146.65 (aromatic carbons), 169.74, 170.36, 170.88 (CH3CO); HRMS: [M+Na]+ C54H49N3NaO17S calcd 1066.2680, obsd 1066.2742.
3.10. p-Tolyl 3,4,6-tri-O-acetyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→4)-2-acetamido-6-O-benzyl-2-N-3-O-carbonyl-2-deoxy-1-thio-α-D-glucopyranoside (14)
Compound 14 was synthesized from donor 8 and acceptor 2α in 39% yield following the general procedure of single step glycosylation together with recovered acceptor 2α (50%). [α]D +98.8 (c, 0.0093 g/mL, CH2Cl2); 1H NMR (600 MHz, CDCl3): δ 1.86 (s, 3H), 2.04 (s, 3H), 2.14 (s, 3H), 2.29 (s, 3H), 2.52 (s, 3H), 3.39 (m, 1H), 3.49 (dd, J = 4.8 Hz, 13.8 Hz, 1H), 3.88–4.25 (m, 8H), 4.33–4.37 (m, 2H), 4.48 (dd, J = 14.4 Hz, 18.6 Hz, 1H) 5.21 (t, J = 15.0 Hz, 1H), 5.56 (d, J = 13.2 Hz, 1H), 5.78 (dd, J = 13.2 Hz, 15.6 Hz, 1H), 6.05 (d, J = 6.6 Hz, 1H), 6.98–7.88 (m, 13H); 13C NMR (150 MHz, CDCl3): δ 20.69, 20.89, 21.04, 21.35, 24.12, 54.94, 60.05, 61.93, 67.82, 68.80, 71.03, 72.26, 72.46, 73.20, 75.40, 77.45, 86.45, 97.75, 123.86–138.61 (aromatic carbons), 152.98, 169.75, 170.41, 171.12, 171.41; HRMS: [M+Na]+ C43H44N2NaO15S calcd 883.2360, obsd 883.2333.
3.11. Benzyl 3,4,6-tri-O-acetyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranoside (16)
After the donor 8 (60 mg, 0.11 mmol) and activated molecular sieve MS—4 Å (500 mg) were stirred for 30 min at room temperature in the mixed solvents of CH2Cl2 and Et2O (1:1) (6 mL), the solution was cooled to −70 °C followed by sequential additions of AgOTf (86 mg, 0.335 mmol) in Et2O (1.5 mL) and p-TolSCl (17.3 μL, 0.11 mmol). The mixture was vigorously stirred for 15 min and a solution of acceptor 3 (59 mg, 0.099 mmol) in CH2Cl2 (0.5 mL) was added. The mixture was stirred for 5 min, then the flask was warmed to −10 °C in 15 min. Then the mixture was cooled down to −70 °C again followed by sequential additions of AgOTf (76 mg, 0.297 mmol) in Et2O (1.5 mL) and p-TolSCl (15.5 μL, 0.099 mmol). After 15 min, a solution of acceptor 3 (53 mg, 0.089 mmol) in CH2Cl2 (0.5 mL) was injected though a syringe. The mixture was stirred for 5 min and warmed to −10 °C in 15 min by switching the cold bath. A solution of acceptor 15 (64 mg, 0.11 mmol) in CH2Cl2 (0.5 mL) was added to the reaction mixture, and the resulting mixture was cooled down to −70 °C followed by sequential additions of AgOTf (68 mg, 0.089 mmol) in Et2O (1.5 mL) and p-TolSCl (14 μL, 0.089 mmol). The reaction mixture was stirred for 1.5 h from −70 to 10 °C and then was diluted with CH2Cl2 (30 mL) followed by filtration and extraction. The collected organic phase was dried over Na2SO4, and the residue was applied to flash column chromatography with a mixed solvent system of CH2Cl2, hexanes, and EtOAc to give the desired tetrasaccharide 16 (55 mg) in 32% yield as colorless foam. Its structure was confirmed by 1H NMR, 13C NMR, 1H–1H COSY experiments, and HRMS. [α]D −20.0 (c, 0.038 g/mL, CH2Cl2); 1H NMR (600 MHz, CDCl3): (a, b, c, and d denote the four monosaccharide units in the sequence of its anomeric proton appearance in the 1H NMR spectrum from the most downfield to the most upfield shifted. Monosaccharide unit a is the one at the non-reducing end) δ 1.82 (s, 3H, CH3CO), 1.88 (s, 3H, CH3CO), 1.96 (s, 3H, CH3CO), 2.76 (broad d, J = 10.2 Hz, 1H, c/d-H5), 2.89 (broad d, J = 10.2 Hz, 1H, b-H5), 3.02 (dd, J = 3.0, 10.2 Hz, 1H, c/d-H6), 3.15 (dd, J = 2.4, 10.2 Hz, 1H, b-H6), 3.18 (dd, J = 3.0, 10.2 Hz, 1H, c/d-H5), 3.27 (dd, J = 3.6, 10.2 Hz, 1H, c/d-H6), 3.29 (d, J = 10.2 Hz, 1H, c/d-H6), 3.37–3.42 (m, 2H, a-H5, b-H6), 3.44 (broad d, J = 10.2 Hz, 1H, c/d-H6), 3.88 (dd, J = 1.8, 12.0 Hz, 1H, a-H6), 3.97–4.10 (m, 7H, b-H2, c-H2, c-H3, c-H4, d-H2, d-H3, d-H4), 4.14 (dd, J = 9.0, 10.8 Hz, 1H, a-H3), 4.16 (dd, J = 8.4, 12.0 Hz, 1H, a-H6), 4.24 (d, J = 9.0 Hz, 1H, a-H4), 4.27–4.44 (m, 10H, 9PhCH2, a-H2), 4.52 (d, J = 11.4 Hz, 1H, PhCH2), 4.62(d, J = 12.6 Hz, 1H, PhCH2), 4.67 (d, J = 12.6 Hz, 1H, PhCH2), 4.81 (d, J = 12.6 Hz, 1H, PhCH2), 4.84 (d, 1H, J = 12.6 Hz, PhCH2), 4.85 (d, J = 8.4 Hz, 1H, d-H1), 5.01 (m, 1H, c-H1), 5.07 (d, 1H, J = 9.0 Hz, 1H, b-H1), 5.10 (t, J = 9.0 Hz, a-H4), 5.46 (d, J = 8.4 Hz, 1H, a-H1), 5.75 (dd, J = 9.0, 10.8 Hz, 1H, a-H3), 6.63–7.91 (m, 51 H); 13C NMR (150 MHz, CDCl3): δ 20.68, 20.82, 20.85, 55.49, 55.86, 56.79, 56.82, 61.57, 67.19, 67.27, 68.28, 68.87, 70.64, 70.91, 71.53, 72.35, 72.50, 72.72, 74.18, 74.36, 74.42, 74.60, 74.63, 74.73, 75.47, 75.57, 76.08, 76.54, 96.81, 96.81, 96.91, 97.21, 123.26–139.00 (aromatic carbons), 167.78–168.41 (phthalimido carbonyl carbons), 169.74, 170.36, 170.91 (CH3CO); HRMS: [M+Na]+ C111H102N4NaO28 calcd 1961.6578, obsd 1961.6393.
3.12. p-Tolyl 3-O-benzyl-4,6-O-benzylidene-2-deoxy-2-phthalimido-1-thio-β-D-glucopyranoside (17)
To a mixture of compound 5 (1 g, 2 mmol), tetrabutylammonium iodide (0.15 g, 0.4 mmol), and freshly activated molecular sieve MS—4 Å (300 mg) in anhydrous DMF (10 mL) was added 95% NaH (80 mg, 3.2 mmol) at 0 °C. The mixture was stirred for 30 min and benzyl bromide (290 μL, 0.42 g, 2.4 mmol) was added via a syringe. After 90 min, the reaction was quenched with acetic acid and filtered. Et2O (40 mL) was added to the filtrate, which was extracted with saturated aqueous solutions of NH4Cl (20 mL) and NaHCO3 (20 mL). The organic layer was separated and dried over Na2SO4. Column chromatography on silica gel (3:1, hexanes–EtOAc) afforded the product 17 as white foam (1.07 g, 90%). Comparison of the NMR data with those reported in the literature21 confirmed the identity of 17.
3.13. p-Tolyl 4,6-O-benzylidene-3-O-tert-butyldimethylsilyl-2-deoxy-2-phthalimido-1-thio-β-D-glucopyranoside (18)
To a mixture of compound 5 (1 g, 2 mmol) and molecular sieve MS—4 Å (300 mg) in anhydrous CH2Cl2 (10 mL) were added 2,6-lutidine (460 μL, 4 mmol) and tert-butyldimethylsilyl triflate (550 μL, 2.4 mmol) at 0 °C. The mixture was stirred for 4 h. The reaction mixture was filtered and the filtrate was extracted with a satd aqueous solution of NH4Cl (20 mL). The organic layer was separated and dried over Na2SO4. Column chromatography on silica gel (15% EtOAc in hexanes) afforded the product 18 as white foam (1.04 g, 85%). 1H NMR (600 MHz, CDCl3): δ −0.32 (s, 3H), −0.16 (s, 3H), 0.56 (s, 9H), 2.28 (s, 3H), 3.55 (t, J = 9.6 Hz, 1H), 3.67 (dt, J = 4.8, 9.6 Hz, 1H), 3.80 (t, J = 10.2 Hz, 1H), 4.29 (t, J = 10.2 Hz, 1H), 4.37 (dd, J = 4.8, 10.2 Hz, 1H), 4.61 (t, J = 9.6 Hz, 1H), 5.51 (s, 1H), 5.57 (d, J = 10.2 Hz, 1H), 7.01–7.04 (m, 2H), 7.20–7.35 (m, 7H), 7.75–7.95 (m, 4H); 13C NMR (150 MHz, CDCl3): δ −5.11, −3.93, 17.96, 21.39, 25.63, 57.02, 68.92, 70.79, 82.70, 84.86, 102.24, 123.47–138.47 (aromatic carbons), 167.64, 168.61 (carbonyl groups of phthalimido); ESI-MS [M+Na]+ C34H39NNaO6SSi calcd 640.8, obsd 640.3.
3.14. p-Tolyl 4,6-di-O-benzyl-3-O-tert-butyldimethylsilyl-2-deoxy-2-phthalimido-1-thio-β-D-glucopyranoside (19)
To a solution of compound 18 (0.43 g, 0.7 mmol) and 1 M borane THF (7.2 mL) in anhydrous CH2Cl2 (10 mL) was added trimethylsilyl triflate (100 μL, 0.5 mmol) at −20 °C. After 2 h, trimethylsilyl triflate (200 μL, 1 mmol) was added again. The mixture was stirred for 2 h more. The reaction was quenched by the addition of Et3N, and MeOH was added until no gas was formed. All solvents were evaporated and the desired primary alcohol (0.39 g) was purified through column chromatography on silica gel (25% EtOAc in hexanes). To a mixture of the primary alcohol (0.3 g, 0.5 mmol), tetrabutyl-ammonium iodide (40 mg, 0.1 mmol) and freshly activated molecular sieve MS—4 Å (100 mg) in anhydrous DMF (5 mL) was added 95% NaH (20 mg, 0.8 mmol) at 0 °C. The mixture was stirred for 30 min and benzyl bromide (180 μL, 0.26 g, 1.5 mmol) was added via a syringe. After 90 min, the reaction was quenched by the addition of acetic acid and filtered. Et2O (10 mL) was added to the filtrate, which was extracted with saturated aqueous solutions of NH4Cl (10 mL) and NaHCO3 (10 mL). The organic layer was separated and dried over Na2SO4. Column chromatography on silica gel (4:1, hexanes–EtOAc) afforded the product 19 (0.32 g) as white foam in overall 80% yield for the two steps. 1H NMR (600 MHz, CDCl3): δ −0.44 (s, 3H), −0.08 (s, 3H), 0.72 (s, 9H), 2.27 (s, 3H), 3.58 (t, J = 9.6 Hz, 1H), 3.64–3.69 (m, 1H), 3.72–3.80 (m, 2H), 4.27 (t, J = 10.2 Hz, 1H), 4.47 (t, J = 9.0 Hz, 1H), 4.53 (d, J = 12.0 Hz, 1H), 4.59 (d, J = 12.0 Hz, 1H), 4.65 (d, J = 12.0 Hz, 1H), 4.78 (d, J = 12.0 Hz, 1H), 5.52 (d, J = 10.2 Hz, 1H), 7.01–7.04 (m, 2H), 7.20–7.35 (m, 12H), 7.75–7.95 (m, 4H); 13C NMR (150 MHz, CDCl3): δ −4.46, −3.94, 17.84, 21.35, 25.87, 56.99, 69.18, 73.64, 73.71, 74.70, 79.67, 80.01, 83.69, 123.39, 123.78, 127.25, 127.59, 127.74, 127.87, 127.90, 128.45–128.53, 129.75, 133.34, 134.38, 138.51, 138.52; ESI-MS [M+Na]+, C41H47NNaO6SSi calcd 732.9, obsd 732.5.
3.15. p-Tolyl 3-O-benzyl-4,6-benzylidene-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-D-glucopyranoside (20)
Compound 20 was synthesized from donor 17 and acceptor 3 in 61% yield following the general procedure of single step glycosylation. [α]D +17.0 (c, 0.024 g/mL, CH2Cl2); 1H NMR (600 MHz, CDCl3): δ 2.21 (s, 3H), 3.33–3.42 (m, 3H), 3.48–3.52 (m, 2H), 3.70 (t, J = 9.0 Hz, 1H), 4.10–4.22 (m, 5H), 4.37 (d, J = 20 Hz, 1H), 4.42 (dd, 1H, J = 9.0, 10.2 Hz, 1H), 4.43–4.48 (m, 3H), 4.77 (dd, J = 4.8, 12.6 Hz, 2H), 5.27 (d, J = 10.2 Hz, 1H), 5.35 (d, J = 8.4 Hz, 1H), 5.50 (s, 1H), 6.86–7.88 (m, 32 H); 13C NMR (150 M Hz, CDCl3): δ 21.32, 54.91, 56.72, 65.95, 68,35, 68.96, 72.92, 74.32, 74.67, 74.80, 76.32, 78.00, 78.93, 83.35, 83.39, 97.95, 101.41, 126.29–138.58 (aromatic carbons); HRMS: [M+Na]+ C63H56N2NaO12S calcd 1087.3452, obsd 1087.3494.
3.16. p-Tolyl 4,6-O-benzylidene-3-O-tert-butyldimethylsilyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-D-glucopyranoside (21)
Compound 21 was synthesized from donor 18 and acceptor 3 in 74% yield following the general procedure of single step glycosylation. [α]D −4.4 (c, 0.011 g/mL, CH2Cl2); 1H NMR (600 MHz, CDCl3): δ −0.28 (s, 3H), −0.14 (s, 3H), 0.56 (s, 9H), 2.21 (s, 3H), 3.32–3.38 (m, 2H), 3.42 (dd, J = 7.2, 10.8 Hz, 1H), 3.46 (t, J = 9.0 Hz, 1H), 3.52 (t, J = 10.2 Hz, 1H), 3.54 (t, J = 10.2 Hz, 1H), 4.08–4.24 (m, 5H), 4.46 (d, J = 12.0 Hz, 1H), 4.49 (d, J = 12.0 Hz, 1H), 4.53 (d, J = 12.0 Hz, 1H), 4.61 (t, J = 9.6 Hz, 1H), 4.77 (d, J = 12.0 Hz, 1H), 5.27 (d, J = 10.2 Hz, 1H), 5.34 (d, J = 8.4 Hz, 1H), 5.41 (s, 1H), 6.84–6.92 (m, 5H), 7.00–7.03 (m, 2H), 7.16–7.20 (m, 2H), 7.28–7.39 (m, 8H), 7.42–7.46 (m, 2H), 7.54–7.58 (m, 1H), 7.62–7.69 (m, 2H), 7.71–7.79 (m, 3H), 7.84–7.88 (m, 1H), 7.92–7.94 (m, 1H); 13C NMR (100 MHz, CDCl3): −5.46, −4.12, 17.67, 21.07, 25.36, 54.67, 58.29, 65.92, 68.17, 68.75, 69.30, 72.71, 74.56, 76.03, 77.55, 78.74, 82.78, 83.10, 97.60, 101.85, 123.07, 123.19, 123.41, 123.78, 126.34, 127.06, 127.33, 127.50, 127.74, 127.91, 128.05, 128.11, 128.26, 129.03, 129.41, 131.59, 131.65, 131.66, 133.48, 133.61, 133.78, 134.28, 134.33, 137.15, 138.04, 138.33, 138.37, 167.21, 167.63, 167.86, 168.64; HRMS: [M+Na]+ C62H64N2NaO12SSi calcd 1111.3925, obsd 1111.3877.
3.17. p-Tolyl 4,6-di-O-benzyl-3-O-tert-butyldimethylsilyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-1-thio-β-D-glucopyranoside (22)
Compound 22 was synthesized from donor 19 and acceptor 3 in 50% yield following the general procedure of single step glycosylation together with disaccharide 23 and glycal 24 (10%). [α]D +20.1 (c, 0.035 g/mL, CH2Cl2); 1H NMR (600 MHz, CDCl3): δ −0.40 (s, 3H), −0.07 (s, 3H), 0.70 (s, 9H), 2.20 (s, 3H), 3.32–3.36 (m, 2H), 3.48 (dd, J = 3.6 Hz, 10.8 Hz, 1H), 3.58–3.70 (m, 4H), 4.10–4.24 (m, 5H), 4.47–4.56 (m, 6H), 4.69 (d, J = 12.0 Hz, 1H), 4.75 (d, J = 11.4Hz, 1H), 4.88 (d, J = 12.6 Hz, 1H), 5.26 (d, J = 10.3 Hz, 1H), 4.30 (d, J = 10.2 Hz, 1H), 6.71–7.89 (m, 32 H); 13C NMR (150 MHz, CDCl3): δ −4.82, −3.88, 17.89, 21.32, 25.87, 55.05, 58.59, 68.10, 68.62, 72.21, 72.92, 73.33, 74.37, 74.86, 75.31, 75.46, 77.78, 79.09, 80.11, 83.58, 96.77, 123.24–138.79 (aromatic carbons), 167.51, 167.92, 168.01, 169.25 (carbonyl groups of phthalimido); HRMS: [M+Na]+ C69H72N2NaO12SSi calcd 1203.4473, obsd 1203.4505.
3.18. 4,6-di-O-benzyl-3-O-tert-butyldimethylsilyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→1)-4,6-di-O-benzyl-3-O-tert-butyldimethylsilyl-2-deoxy-2-phthalimido-D-glucopyranoside (23)
[α]D = 0 (c, 0.013 g/mL, CH2Cl2); 1H NMR (600 MHz, CDCl3): δ −0.48 (s, 6H), −0.13 (s, 6H), 0.66 (s, 18H), 3.37 (m, 2H), 3.47 (m, 4H), 3.65 (m, 2H), 4.05 (d, J = 18 Hz, 2H), 4.07 (dd, J = 13.2 Hz, 15.6 Hz, 4H), 4.20 (d, J = 18 Hz, 2H), 4.43 (m, 2H), 4.54 (d, J = 17.4 Hz, 2H), 4.65 (d, J = 7.4 Hz, 2H), 5.41 (d, J = 13.2 Hz, 2H), 7.13–7.55 (m, 28H); 13C NMR (150 MHz, CDCl3): δ −4.55, −4.04, 17.78, 25.82, 57.48, 69.12, 72.20, 73.72, 74.41, 75.47, 79.62, 96.88, 127.17–138.69 (aromatic carbons); HRMS: [M+Na]+ C68H80N2NaO13Si2 calcd 1211.5096, obsd 1211.5143. Compound 24 was isolated as a mixture with compound 23, which was characterized by ESI-MS. ESI-MS: [M+Na]+ C34H39NNaO6Si calcd 608.2, obsd 608.3.
3.19. Methyl 4,6-O-benzylidene-3-O-tert-butyldimethylsilyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranosyl-(1→4)-6-O-benzyl-2-deoxy-3-O-p-methoxybenzyl-2-phthalimido-β-D-glucopyranosyl-(1→4)-3,6-di-O-benzyl-2-deoxy-2-phthalimido-β-D-glucopyranoside (27)
Compound 27 was synthesized in 45% overall yield using building blocks 18, 3, 25, and 26 in a similar manner as in the assembly of tetrasaccharide 16 with the exception that pure Et2O was used to dissolve donor 18, and TTBP (1 equiv) was added together with each acceptor. For reaction time, see Scheme 4. The desired product was purified by silica gel flash chromatography using the solvent systems of hexanes–EtOAc (1:1) and hexanes–CH2Cl2–EtOAc (5:5:3). Its structure was confirmed by 1H NMR, 13C NMR, 1H–1H COSY, 1H–13C HMQC experiments, and HRMS. [α]D −31.4 (c, 0.013 g/mL, CH2Cl2); 1H NMR (600 MHz, CDCl3): (a, b, c, and d denote the four monosaccharide units in the sequence of its anomeric proton appearance in the 1H NMR spectrum from the most downfield to the most upfield shifted) δ −0.31 (s, 3H, SiCH3), −0.17 (s, 3H, SiCH3), 0.54 (s, 9H, SiC(CH3)3), 2.76 (broad d, J = 9.0 Hz, 1H, c-H5), 2.90 (broad d, J = 9.0 Hz, 1H, b-H5), 3.01 (dd, J = 3.0, 10.8 Hz, 1H, c-H6), 3.12 (dd, J = 2.4, 10.8 Hz, 1H, b-H6), 3.18–3.24 (m, 1H, d-H4), 3.21 (s, 3H, OCH3), 3.24–3.32 (m, 3H, c-H6, d-H5, d-H6), 3.37–3.47 (m, 4H, a-H4, a-H6, b-H6, d-H6), 3.54 (s, 3H, ArOCH3), 3.96–4.03 (m, 4H, c-H2, c-H3, d-H2, d-H3), 4.04–4.12 (m, 3H, a-H6, b-H2, c-H4), 4.13–4.21 (m, 3H, a-H2, a-H5, b-H3), 4.23 (t, J = 8.4 Hz, 1H, b-H4), 4.29 (d, J = 12.0 Hz, 1H, ArCH2), 4.32–4.42 (m, 6H, 6ArCH2), 4.47 (d, J = 12.0 Hz, 1H, ArCH2), 4.50 (d, J = 12.0 Hz, 1H, ArCH2), 4.59 (t, J = 9.0 Hz, 1H, a-H3), 4.70 (d, J = 12.6 Hz, 1H, ArCH2), 4.71 (d, J = 12.6 Hz, 1H, ArCH2), 4.77 (d, J = 7.8 Hz, 1H, d-H1), 4.82 (d, J = 12.0 Hz, 1H, ArCH2), 5.01–5.04 (m, 1H, c-H1), 5.07 (d, J = 7.8 Hz, 1H, b-H1), 5.33 (d, J = 8.4 Hz, 1H, a-H1), 5.38 (s, 1H, PhCHbenzylidene), 6.18–6.21 (m, 2H), 6.62–7.96 (m, 48H); 13C NMR (100 MHz, CDCl3): −5.18 (SiCH3), −3.85 (SiCH3), 17.94 (SiC(CH3)3), 25.65 (SiC(CH3)3), 54.88 (ArOCH3), 55.76, 56.60 (OCH3), 56.85 (b-C2), 56.90, 58.58 (a-C2), 66.05, 67.33, 68.30, 68.97, 69.61, 72.45, 72.61, 72.74, 74.36, 74.46, 74.52, 74.72, 75.51, 75.80, 76.26, 76.58, 83.04 (a-C4), 96.75 (b-C1), 96.95 (c-C1), 97.76 (a-C1), 99.18 (d-C1), 102.09 (PhCHbenzylidene), 113.25, 123.05, 123.35, 123.52, 123.79, 123.95, 126.59, 126.88, 127.16, 127.23, 127.48, 127.53, 127.56, 127.59, 127.87, 127.99, 128.11, 128.14, 128.22, 128.27, 128.33, 128.52, 129.25, 129.88, 131.33, 131.77, 131.99, 133.71, 133.73, 133.82, 134.02, 134.19, 134.53, 137.42, 138.53, 138.60, 138.63, 138.88, 138.97, 158.50, 167.82, 167.84, 168.27, 168.38, 168.75 (carbonyl groups of phthalimido); ESI-MS C113H112N4NaO26Si [M+Na]+ calcd 1991.7, found 1991.6; HRMS: [M+Na]+ C113H112N4NaO26Si calcd 1991.7231, obsd 1991.7139. Compounds 28, 29, and 30 were characterized by ESI-MS. Compound 28, C55H56N2NaO12Si [M+Na]+calcd 987.4, obsd 987.6. Compound 29, C84H83N3NaO19Si [M+Na]+calcd 1488.5, obsd 1488.7. Compound 30, C84H85N3NaO20Si [M+Na]+calcd 1506.5, obsd 1506.7.
Acknowledgments
We are grateful for the financial support from the University of Toledo, the National Institutes of Health (R01 GM 72667), and the Pardee Foundation.
Footnotes
Dedicated to Professor Koji Nakanishi on the occasion of his 80th birthday
References
- 1.Codee JDC, Litjens REJN, Van den Bos LJ, Overkleeft HS, Van der Marel GA. Chem Soc Rev. 2005;34:769–782. doi: 10.1039/b417138c. (a) [DOI] [PubMed] [Google Scholar]; Carbohydrates in Chemistry and Biology. 1–4 Wiley-VCH; Weinheim: 2000. (b) [Google Scholar]; Davis BG. J Chem Soc, Perkin Trans 1. 2000:2137–2160. (c) [Google Scholar]
- 2.Yu B, Yang Z, Cao H. Curr Org Chem. 2005;9:179–194. [Google Scholar]
- 3.Koeller KM, Wong CH. Chem Rev. 2000;100:4465–4493. doi: 10.1021/cr990297n. [DOI] [PubMed] [Google Scholar]
- 4.Huang L, Wang Z, Huang X. Chem Commun. 2004:1960–1961. doi: 10.1039/b405886k. [DOI] [PubMed] [Google Scholar]
- 5.Pornsuriyasak P, Gangadharmath UB, Rath NP, Demchenko AV. Org Lett. 2004;6:4515–4518. doi: 10.1021/ol048043y. (a) [DOI] [PubMed] [Google Scholar]; Lahmann M, Oscarson S. Org Lett. 2000;2:3881–3882. doi: 10.1021/ol006621e. (b) [DOI] [PubMed] [Google Scholar]; Geurtsen R, Holmes DS, Boons GJ. J Org Chem. 1997;62:8145–8154. doi: 10.1021/jo971233k. (c) [DOI] [PubMed] [Google Scholar]
- 6.Lopez JC, Agocs A, Uriel C, Gomez AM, Fraser-Reid B. Chem Commun. 2005:5088–5090. doi: 10.1039/b507468a. (a) [DOI] [PubMed] [Google Scholar]; Fraser-Reid B, Lopez JC, Gomez AM, Uriel C. Eur J Org Chem. 2004:1387–1395. (b) [Google Scholar]
- 7.Huang X, Huang L, Wang H, Ye XS. Angew Chem, Int Ed. 2004;43:5221–5224. doi: 10.1002/anie.200460176. [DOI] [PubMed] [Google Scholar]
- 8.Semenuk T, Krist P, Pavlicek J, Bezouska K, Kuzma M, Novak P, Kren V. Glycoconjugate J. 2002;18:817–826. doi: 10.1023/a:1021111703443. (a) [DOI] [PubMed] [Google Scholar]; Shibata Y. Rec Res Dev Immun. 1999;1:551–558. (b) [Google Scholar]; Bezouska K, Sklenar J, Dvorakova J, Havlicek V, Pospisil M, Thiem J, Kren V. Biochem Biophys Res Commun. 1997;238:149–153. doi: 10.1006/bbrc.1997.7260. (c) [DOI] [PubMed] [Google Scholar]
- 9.Gorbach VI, Kraiskova IN, Lukyanov PA, Loenko YN, Soloveva TF, Ovodov YS, Deev VV, Pimenov AA. Carbohydr Res. 1994;260:73. doi: 10.1016/0008-6215(94)80023-5. (a) [DOI] [PubMed] [Google Scholar]; Suzuki K, Mikami T, Okawa Y, Tokoro A, Suzuki S, Suzuki M. Carbohydr Res. 1986;151:403–408. doi: 10.1016/s0008-6215(00)90359-8. (b) [DOI] [PubMed] [Google Scholar]
- 10.Vongchan P, Sajomsang W, Subyen D, Kongtawelert P. Adv Chitin Sci. 2002;5:650–655. doi: 10.1016/s0008-6215(02)00098-8. [DOI] [PubMed] [Google Scholar]
- 11.Nishimura S-I, Kai H, Shinada K, Yoshida T, Tokura S, Kurita K, Nakashima H, Yamamoto N, Uryu T. Carbohydr Res. 1998;306:427–433. doi: 10.1016/s0008-6215(97)10081-7. (a) [DOI] [PubMed] [Google Scholar]; Katsuraya K, Ishikawa S, Ichikawa T, Goto K, Uryu T. Seisan Kenkyu. 1997;49:170–173. (b) [Google Scholar]
- 12.Yamago S, Yamada T, Maruyama T, Yoshida JI. Angew Chem, Int Ed. 2004;43:2145–2148. doi: 10.1002/anie.200353552. (a) [DOI] [PubMed] [Google Scholar]; Aly MRE, Ibrahim EI, El Ashry ESH, Schmidt RR. Carbohydr Res. 2001;331:129–142. doi: 10.1016/s0008-6215(01)00024-6. (b) [DOI] [PubMed] [Google Scholar]; Hinou H, Umino A, Matsuoka K, Terunuma D, Takahashi S, Esumi Y, Kuzuhara H. Bull Chem Soc Jpn. 2000;73:163–171. (c) [Google Scholar]; Kanie O, Ito Y, Ogawa T. J Am Chem Soc. 1994;116:12073–12074. (d) [Google Scholar]; Tailler D, Jacquinet JC, Beau JM. J Chem Soc, Chem Commun. 1994:1827–1828. (e) [Google Scholar]; Ikeshita S, Sakamoto A, Nakahara Y, Nakahara Y, Ogawa T. Tetrahedron Lett. 1994;35:3123–3126. (f) [Google Scholar]; Wang LX, Li C, Wang QW, Hui YZ. Tetrahedron Lett. 1993;34:7763–7766. (g) [Google Scholar]; Kuyama H, Nakahara Y, Nukada T, Ito Y, Nakahara Y, Ogawa T. Carbohydr Res. 1993;243:C1–C7. doi: 10.1016/0008-6215(93)84095-n. (h) [DOI] [PubMed] [Google Scholar]; Nicolaou KC, Bockovich NJ, Carcanague DR, Hummel CW, Even LF. J Am Chem Soc. 1992;114:8701–8702. (i) [Google Scholar]
- 13.Schwartz DA, Lee HH, Carver JP, Krepinsky JJ. Can J Chem. 1985;63:1073–1079. [Google Scholar]
- 14.Crich D, Dudkin V. J Am Chem Soc. 2001;123:6819–6825. doi: 10.1021/ja010086b. [DOI] [PubMed] [Google Scholar]
- 15.Xue J, Khaja SD, Locke RD, Matta KL. Synlett. 2004:861–865. [Google Scholar]
- 16.Crich D, Vinod AU. J Org Chem. 2005;70:1291–1296. doi: 10.1021/jo0482559. [DOI] [PubMed] [Google Scholar]
- 17.Crich D, Vinod AU. Org Lett. 2003;5:1297–1300. doi: 10.1021/ol0342305. [DOI] [PubMed] [Google Scholar]
- 18.Boysen M, Gemma E, Lahmann M, Oscarson S. Chem Commun. 2005:3044–3046. doi: 10.1039/b503651h. (a) [DOI] [PubMed] [Google Scholar]; Wei P, Kerns RJ. J Org Chem. 2005;70:4195–4198. doi: 10.1021/jo047812o. (b) [DOI] [PubMed] [Google Scholar]
- 19.Tanaka H, Adachi M, Takahashi S. Tetrahedron Lett. 2004;45:1433–1436. (a) [Google Scholar]; Orgueira HA, Bartolozzi A, Schell P, Litjens REJN, Palmacci ER, Seeberger PH. Chem Eur J. 2003;9:140–169. doi: 10.1002/chem.200390009. (b) [DOI] [PubMed] [Google Scholar]; Paulsen H, Stenzel W. Angew Chem, Int Ed. 1975;14:558–559. (c) [Google Scholar]
- 20.Zhang Z, Ollmann IR, Ye XS, Wischnat R, Baasov T, Wong CH. J Am Chem Soc. 1999;121:734–753. [Google Scholar]
- 21.Wang Y, Huang X, Zhang LH, Ye XS. Org Lett. 2004;6:4415–4417. doi: 10.1021/ol0483246. [DOI] [PubMed] [Google Scholar]
- 22.Mong TKK, Huang CY, Wong CH. J Org Chem. 2003;68:2135–2142. doi: 10.1021/jo0206420. [DOI] [PubMed] [Google Scholar]
- 23.Barrett AGM, Dhanak D, Graboski GG, Taylor SJ. Org Synth Coll. 1993;8:550. [Google Scholar]
- 24.Crich D, Sun S. Tetrahedron. 1998;54:8321–8348. (a) [Google Scholar]; Martichonok V, Whitesides GM. J Org Chem. 1996;61:1702–1706. doi: 10.1021/jo951711w. (b) [DOI] [PubMed] [Google Scholar]
- 25.Zhu T, Boons GJ. Carbohydr Res. 2000;329:709–715. doi: 10.1016/s0008-6215(00)00252-4. (a) [DOI] [PubMed] [Google Scholar]; Belot F, Jacquinet JC. Carbohydr Res. 1996;290:79–86. doi: 10.1016/0008-6215(96)00116-4. (b) [DOI] [PubMed] [Google Scholar]
- 26.Hernandez-Torres JM, Liew ST, Achkar J, Wei A. Synthesis. 2002:487–490. [Google Scholar]
- 27.Kameyama A, Ehara T, Yamada Y, Ishida H, Kiso M, Hasegawa A. J Carbohydr Chem. 1995;14:507–523. doi: 10.1016/0008-6215(95)00353-3. [DOI] [PubMed] [Google Scholar]
- 28.Crich D, Smith M, Yao Q, Picione J. Synthesis. 2001:323–326. [Google Scholar]
- 29.Chowdhury AR, Siriwardena A, Boons GJ. Tetrahedron Lett. 2002;43:7805–7807. [Google Scholar]
- 30.Seeberger PH. Chem Commun. 2003:1115–1121. doi: 10.1039/b210230g. (a) [DOI] [PubMed] [Google Scholar]; Plante OJ, Palmacci ER, Seeberger PH. Science. 2001;291:1523–1527. doi: 10.1126/science.1057324. (b) [DOI] [PubMed] [Google Scholar]; Seeberger PH, Haase WC. Chem Rev. 2000;100:4349–4393. doi: 10.1021/cr9903104. (c) [DOI] [PubMed] [Google Scholar]