

Abstract
Purpose
To determine the presence of calcification markers in the trabecular meshwork tissue from glaucoma donors and in trabecular meshwork cells insulted by dexamethasone (DEX) and transforming growth factor β2 (TGFβ2), factors associated with glaucoma. To investigate as well the effect of silencing the inhibitor of calcification matrix Gla (MGP) in the trabecular meshwork cells.
Methods
Trabecular meshwork tissue was obtained from perfused postmortem anterior segments of glaucomatous and normal eyes. Primary trabecular meshwork cells were obtained from residual corneal rims after surgical corneal transplantation. Calcification marker alkaline phosphatase (ALP) enzyme activity was assayed by fluorescence produced after substrate cleavage. DNA quantification was evaluated by fluorescence produced after binding to the Hoechst dye. Transfection of siRNA to primary cells was accomplished by nucleofector electroporation with trabecular meshwork–optimized conditions. cDNA quantification was performed with the use of TaqMan real-time PCR.
Results
Human trabecular meshworks from glaucoma donors exhibited significantly higher levels of ALP activity than their matched counterparts with normal eyes. The normalized ALP of the control specimens was 7.3 ± 1.6 ng ALP/μg DNA (n = 4), whereas that of the glaucomatous tissue was 37.0 ± 10.7 ng ALP/μg genomic DNA (n = 5; P ≤ 0.04). DEX and TGFβ2 significantly induced the upregulation of ALP activity in two trabecular meshwork primary cell lines. Expression of the gene encoding MGP was reduced in the glaucomatous tissue by −4.4 ± 1.7-fold (n = 9; P ≤ 0.006). Silencing MGP by siRNA resulted in ALP activity that was increased by 197% ± 8.4% (P ≤ 0.0003).
Conclusions
The increased activity of the calcification marker, ALP, in glaucomatous trabecular meshworks might be indicative of an undergoing mineralization process during development of the disease. Inhibition of the calcification mechanism represented by the presence of active MGP appears to be compromised in glaucomatous tissue.
Glaucoma is a complex ocular disease that results in irreversible blindness. It is the second leading cause of blindness worldwide and, in the United States, exhibits a higher prevalence among African Americans and probably also among Hispanic Americans.1,2 It is widely accepted that failure to regulate elevated intraocular pressure (IOP) is the major risk factor for the development of glaucoma.3 Similarly, it is well established that disruption of trabecular meshwork function leads to increased resistance to the aqueous humor outflow and to the generation of elevated IOP.
Aqueous humor flows out of the eye through the trabecular meshwork, a soft spongiform tissue located in the anterior segment at the corner of the iris and cornea. To maintain physiological aqueous humor resistance, the trabecular meshwork uses a variety of cellular functions and mechanisms. Functions such as secretion, phagocytosis, cytoskeletal reorganization, cell-matrix adhesion, ion channel transport, signal transduction, and proteosome involvement have all been shown to have a role in outflow facility. Among them, extra-cellular matrix (ECM) deposition as well as its quality4 has been shown to have a strong correlation with increased resistance and glaucoma.5
Identification of genes expressed in the human trabecular meshwork under different conditions has revealed the gene encoding the inhibitor of calcification Matrix Gla (MGP) as the third most abundant expressed gene in adult trabecular meshwork tissue6,7 and among the 20 highest genes in infant-cultured cells.8 In addition, we have shown that MGP expression is regulated by elevated IOP in a model of perfused human anterior segments from postmortem donors using contralateral eyes as controls for identical genetic backgrounds.9 In the same study, MGP was downregulated in primary human trabecular meshwork (HTM) cultures by transforming growth factor-β1 (TGFβ1), a growth factor found elevated in patients with glaucoma.9
MGP is an 84-amino acid protein that contains five γ-carboxyglutamic acid (Gla) residues. The Gla residues are produced by γ-carboxylation of glutamic acid residues by γ-carboxylase, a vitamin K–dependent enzyme. The only known function of Gla residues is to bind calcium ions.10 Matrix Gla was originally isolated from demineralized bovine bone matrix, but it is also known to be expressed in cartilage, vascular smooth muscle cells (VSMCs), and kidney.10 MGP plays a key role in the formation of atherosclerotic plaques and vascular calcification.11 Protection of soft tissue calcification is thought to be partly attributed to the complexes of calcium to inhibitors of calcification such as MGP.10
A number of studies have drawn parallels between VSMCs and trabecular meshwork cells and between their corresponding pathologic conditions of atherosclerosis and glaucoma. For example, one of the VSMC markers is smooth muscle actin, whose promoter directs specific expression to these cells in the arterial wall.12 Smooth muscle actin is also a well-characterized protein in the trabecular meshwork,13 where it was shown to be responsible for the contractile properties of tissue.14 Similarly, the first reported glaucoma marker, endothelial leukocyte adhesion molecule-1 (ELAM-1),15 happens to be the earliest marker for the atherosclerotic plaque. Other genes that are abundant in the trabecular meshwork, such as cartilage Gp-39,6 are also known to be strongly induced during atherosclerosis.6
Previous studies from our laboratory have shown that, in addition to very high MGP expression, cells of the trabecular meshwork exert very high γ-carboxylase activity, and their MGP protein is indeed γ-carboxylated.16 We also showed that overexpression of MGP reverses bone morphogenetic protein 2 (BMP2)–induced calcification in HTM cells.16 These findings indicate that MGP has the ability to function in the trabecular meshwork as a calcification inhibitor.
In this study, we continued our investigation on the potential calcification of the trabecular meshwork tissue under conditions associated with glaucoma. As a marker of calcification, we used the presence and upregulation of the activity of the enzyme alkaline phosphatase (ALP), a well-established marker of osteogenic differentiation17-23 and vascular calcification.21,23,24 ALP is an enzyme that hydrolyzes a great variety of substrates that lead to increases of Pi and is activated when a pluripotent, undifferentiated mesenchymal cell becomes committed to differentiate into bone-forming cells (osteoblasts).18 ALP further allows inactivation of mineralization inhibitors18 and has been detected in atherosclerotic plaques.23 For the source of tissue, we obtained intact trabecular meshworks from matched donors with and without glaucoma. To revive and stabilize postmortem eyes, we perfused them on arrival by an organ culture system that mimics physiological aqueous humor flow through an intact trabecular meshwork. For the source of conditioned cells, we treated primary HTM cells with two common glaucoma-associated molecules, dexamethasone (DEX) and TGFβ2. Elevated IOP develops in 40% of patients treated with DEX,25 and increased concentrations of TGFβ2 in the aqueous humor have undoubtedly been associated with the pathogenesis of glaucoma.26,27
We observed an increase in ALP activity on all glaucomatous conditions that was accompanied by a decrease in the expression of MGP. Silencing the MGP encoding gene by siRNA in normal HTM cells also led to an increase in ALP activity. We believe these results support the hypothesis that protection against mineralization of the trabecular meshwork by MGP may be a relevant factor in the maintenance of the soft nature of the tissue and thereby may affect resistance to aqueous humor outflow. Most important, our observations revealed that increased mineralization of the trabecular meshwork is concomitant with the presence of glaucomatous conditions and may be a contributing factor to the disease.
Materials and Methods
Glaucoma Tissue and Primary Human Trabecular Meshwork Cells
Glaucomatous and normal human eyes were obtained within 36 to 40 hours of donor death from national eye banks (Lions Eye Bank of Oregon, Portland, OR/National Disease Research Interchange, Philadelphia, PA/North Carolina Eye Bank, Winston-Salem, NC) after signed consent of the donors' families. For organ cultures, whole eye globes were dissected at the equator, cleaned away from the vitreous, iris, and lens, and mounted on the perfusion chambers, as described previously.28,29 Perfusion was conducted at constant flow using serum-free, high-glucose Dulbecco modified Eagle medium (DMEM). All procedures were performed in accordance with the tenets of the Declaration of Helsinki. We collected postmortem eye specimens from glaucoma patients and matched them with those from healthy donors by race, sex, and approximate age. Anterior segments from each person were set in organ culture and perfused between 1 and 6 days. To generate primary human trabecular meshwork cells (HTM), trabecular meshworks were dissected from residual cornea rims after surgical corneal transplantation at the University of North Carolina eye clinic. Specimens were cut in small pieces, carefully attached to the bottom of a 2% gelatin-coated 35-mm dish, and covered with a drop of medium (improved minimal essential medium [IMEM], 20% fetal bovine serum [FBS], 50 μg/mL gentamicin) and a coverslip. Dishes were incubated in a humidified 6% CO2 atmosphere, medium was changed every other day, and cells were allowed to grow from the explant for a period of 4 weeks. Once confluent, cells were passed to a T-25 flask and labeled as passage 1. Subsequently, cells were passed 1:4 to confluence and were maintained in the same medium with 10% FBS. All cells were used at passages 4 to 6. These outflow pathway cultures comprise all cell types involved in maintaining resistance to flow. These include cells from the three distinct regions of the trabecular meshwork plus cells lining the Schlemm canal. Because most of the cells in these cultures come from the trabecular meshwork, they are commonly referred to as trabecular meshwork cells. The cells used in this study were from two women, 29 and 21 years of age (HTM-63 and HTM-70, respectively).
The monobasic phosphate content of the IMEM used in the cell cultures was 150 mg/L, whereas that of the DMEM used in the perfused organ cultures was 130 mg/L. Neither medium was supplemented with β-glycerophosphate, an organic phosphate donor that influences the onset of mineralization in vitro and that is commonly used as a supplement in osteoblast cultures.
Extraction and Measurement of Alkaline Phosphatase
Alkaline phosphatase activity in trabecular meshwork tissue and primary HTM cells was measured with a fluorescence substrate system (AttoPhos AP; Promega, Madison, WI). This system uses 2′-[2-benzothiazoyl]-6′-hydroxybenzothiazole (BBTP) as substrate, which is cleaved by the ALP enzyme to produce inorganic Pi and BBT, a derivative that shifts the fluorescence spectra and results in lower background and higher sensitivity. In contrast to other commercial fluorescence or luminescence ALP assay systems, the fluorescence substrate system (AttoPhos AP; Promega) does not contain an endogenous ALP inhibitor. For the tissue, the trabecular meshworks of perfused anterior segments were dissected and lysed by homogenization in a sterile glass microtissue grinder (Kimble-Kontes, Vineland, NJ) with 200 μL PBS. Tissue homogenates were briefly centrifuged, and aliquots from the supernatants were used to measure ALP and genomic DNA. For the cells, 35-mm culture dishes were washed twice with PBS, scraped with 1 mL PBS, centrifuged, resuspended in 200 μL PBS, and lysed by 3× freeze-thawing. Different aliquots from the same cell extracts were used for ALP and genomic DNA measurements. For the ALP, a given aliquot was adjusted to 100 μL with PBS and incubated with 100 μL substrate (AttoPhos AP; Promega) at room temperature in a black microlon 96-well plate (Greiner Microlon; Greiner Bio-One, Monroe, NC) in the dark. A parallel set of incubations was conducted with different dilutions (5–100 ng) of a purified ALP enzyme (Roche, Indianapolis, IN) to generate a standard curve. Controls containing PBS aliquots without enzyme were run in parallel. Fluorometric values were read usually after 20 minutes of incubation time in a fluorescence plate reader (Fluostar Optima; BMG Labtechnologies, Durham, NC) with 430-nm excitation and 555-nm emission filters. Fluorescence values from the enzyme-free sample were subtracted from each of the samples, and ALP concentrations were calculated by extrapolation on the standard curve and correction for aliquot volume.
Genomic DNA Quantification
DNA was quantified with the use of a DNA fluorometric quantification kit (Invitrogen-Molecular Probes, Carlsbad, CA) according to the manufacturer's instructions. Reactions were performed in a 200-μL final volume, in a black microlon 96-well plate (Greiner Microlon; Greiner Bio-One). Serial dilutions of spectrophotometrically measured calf thymus DNA (Sigma Chemical, St. Louis, MO) were used to generate the standard curve. Aliquots from tissue and cell extracts were adjusted to 100 μL with 0.01 M Tris, pH 7.4, 0.001 M EDTA (TE) buffer and were thoroughly mixed with 100 μL freshly diluted Hoechst dye. After brief centrifugation, plates were directly read in a fluorescence plate reader (Fluostar; BMG Labtechnologies) at 350-nm excitation and 450-nm emission wavelengths. Fluorescence values from a TE control sample were subtracted from each of the extract samples, and DNA concentrations were calculated by extrapolation on the standard curve and correction for aliquot volume.
Alizarin Red Staining
For visualization of calcification, cells were stained with alizarin red. Cells in culture dishes were washed with PBS 3× and were fixed in cold 80% ethanol for at least 1 hour at 4°C. Cells were then washed in distilled water and exposed to fresh 2% alizarin red (pH 4.2; Sigma) for 5 minutes (red/orange = positive staining). After staining, cells were washed with distilled water at least 5 times, followed by one PBS wash for 15 minutes, and were photographed under brightfield with a CK40 inverted fluorescence microscope (Olympus, Tokyo, Japan) equipped with a charge-coupled device (CCD) digital camera (DP70; Olympus) and software.
Drug Treatments
Primary HTM cells were grown to confluence and treated with DEX (Sigma) at a final concentration of 10−7 M in serum-containing medium. DEX was prepared in absolute ethanol at 0.1 mM and was diluted 1000-fold into fresh complete IMEM every other day for the duration of the experiment. Confluent cells were treated with TGFβ2 (R&D Systems, Minneapolis, MN) at final concentrations of 1 ng/mL or 3 ng/mL for 3 days in serum-free medium. The cytokine was reconstituted with 4 mM HCl into a stock solution of 50 μg/mL and kept frozen at −20°C. Cells were washed 3× with PBS before treatment, and the stock solution was diluted to the desired concentration into fresh serum-free IMEM and added to the cells every day. At the end of the experiment, cells were washed with PBS and harvested either for the assay of ALP and genomic DNA or RNA extraction.
RNA Extraction, Reverse Transcription, and Quantification of cDNA
Anterior segments were frozen at −80°C or placed in RNAlater (Ambion, Austin, TX) immediately after perfusion. RNA extraction was performed by homogenizing dissected trabecular meshwork tissues or cellular pellets in 300 μL guanidine thiocyanate buffer and loading the solution onto a column (QIAshredder; Qiagen, Valencia, CA). Extraction continued by the use of the RNeasy Mini kit with RNase-free DNAse digestion on the column provided according to manufacturer's recommendations (Qiagen). Reverse transcription (RT) reactions and subsequent normalized cDNA quantifications were performed by real-time TaqMan PCR (Applied Biosystems [ABI], Foster City, CA). RT was conducted with 1 μg of spectrophotometrically measured total RNA in a 25-μL total volume of proprietary RT buffer containing random primers, dNTP, and 62.5 U murine leukemia virus (MuLV) RT enzyme with RNase inhibitor (High Capacity cDNA kit; ABI) according to the manufacturer's recommendations (25°C for 10 minutes, 37°C for 2 hours). When the starting RNA was 0.5 μg, the volume of reagents was divided by two.
Fluorescence-labeled TaqMan probe/primer sets for MGP, γ-glutamyl carboxylase (γ-GGCX), and control 18S were purchased from Applied Biosystems (TaqMan Gene Expression collection; http://www.allgenes.com). The MGP probe corresponded to sequences from exons 1 and 2 (Hs00179899_m1), the GGCX probe corresponded to sequences from exons 4 and 5 (Hs00168139_m1), and the 18S rRNA probe corresponded to sequences surrounding position nucleotide 609 (Hs99999901_s1). Reactions were performed in 20-μL aliquots using a PCR mix (TaqMan Universal PCR Master Mix, No AmpErase UNG; ABI), run on a PCR system (7500 Real-Time; ABI) and analyzed by software (7500 System Sequence Detection Software; ABI). Relative quantification (RQ) values between treated and untreated samples were calculated by the formula 2−ΔΔCT, where CT is the cycle at threshold (automatic measurement), ΔCT is CT of the assayed gene (MGP, γ-GGCX) minus CT of the endogenous control (18S), and ΔΔCT is the ΔCT of the normalized assayed gene in the treated sample minus the ΔCT of the same gene in the untreated one (calibrator). Because of the high abundance of the 18S rRNA used as endogenous control, and to get a linear amplification, RT reactions from control and experimental samples were diluted 104 times before their hybridization to the 18S TaqMan probe. Relative quantification values greater than 1 corresponded to increased fold changes. Relative quantification values less than 1 were converted to decreased fold changes by the formula 1/2−ΔΔCT. In other words, an RQ of 4 corresponded to an increased expression of fourfold (+4), and an RQ of 0.25 corresponded to a reduction of expression of fourfold (−4).
MGP siRNA Nucleofector Transfection
Predesigned human MGP siRNA (Silencer; ID no. 122163; NM_000900) and nontargeted scramble siRNA 1 (Silencer Negative Control; catalog no. 4611) were purchased from Ambion (Austin, TX), reconstituted to a final concentration of 100 μM in 50 μL RNase-free water, and stored at −20°C. The siRNA sense sequence for the MGP was 5′-GUGAGGGUCAAAGGAGAGUtt-3′, and the antisense strand was 5′-ACUCUCCUUUGACCCUCACtg-3′. siRNAs were transfected to early-passage primary HTM cells using nucleofector technology (Basic Kit for Primary Mammalian Endothelial Cells; Amaxa, Gaithersburg, MD). Pilot experiments with 4 × 105 primary HTM cells and 2 μg pmaxGFP plasmid (Amaxa) were used to determine conditions for maximum transfection efficiency. Of all the built-in programs, one (program T-23; Amaxa) was found to result in an approximate 80% efficiency/80% viability of HTM cells and thus was selected for consecutive studies. For this transfection procedure, cells in 10-cm dishes were split 24 hours before transfection to obtain 70% to 80% confluence. Cells were then trypsinized, counted, and spun at 100g for 10 minutes. Cell pellets were resuspended in proprietary mammalian endothelial solution at a concentration of 4 × 105 cells/100 μL. After the addition of 1.2 μL or 2.4 μL stock 100 μM siRNA solution, the cell-siRNA mix was electroporated on the nucleofector apparatus (program T-23; Amaxa). Immediately after electroporation, the cell-siRNA solution was allowed to recover for 15 minutes at 37°C in 0.5 mL warm IMEM in an Eppendorf tube. After the recovery period, cells were transferred to 3-cm dishes containing warm medium in which the final siRNA concentration was that of 40 and 80 nM, respectively. Cell medium was changed 24 hours after electroporation, and cultured cells were harvested 48 hours later. ALP, genomic DNA, or RNA were then extracted from the cultured cells.
Results
Increased Alkaline Phosphatase Activity in Trabecular Meshwork from Patients with Glaucoma
To follow up on previous findings from our laboratory on the calcification inhibition properties of the human trabecular meshwork,16 we investigated the potential role of calcification in the development of glaucoma by measuring the presence of the marker ALP. Dissected trabecular meshworks from perfused matched donors with glaucomatous or normal eyes were assayed for ALP activity (n = 5 glaucoma; n = 4 normal) and total genomic DNA for normalization. A first experiment comparing ALP activity between eyes perfused during different times indicated that the activity of the enzyme was not affected by the perfusion times used here (not shown).
In every match, normalized active ALP of the glaucoma trabecular meshwork was considerably higher than that of the normal eye (Fig. 1). Average normalized ALP in the control specimens was 7.3 ± 1.6 ng ALP/μg genomic DNA (n = 4), whereas that of the glaucomatous tissue was 37.0 ± 10.7 ng ALP/μg genomic DNA (n = 5; P = 0.04). This 407% increase in the trabecular meshwork from glaucomatous eyes suggested that glaucoma tissue is experiencing a calcification process, which may cause increased resistance to aqueous humor outflow. Attempts to detect von Kossa staining of the trabecular meshwork were negative, perhaps an indication that mineralization in this tissue may occur at levels below those detected by the von Kossa staining. The high ALP levels observed indicate that the trabecular meshwork cells are undergoing differentiation to osteogenic or chondrogenic lineages.
Figure 1.
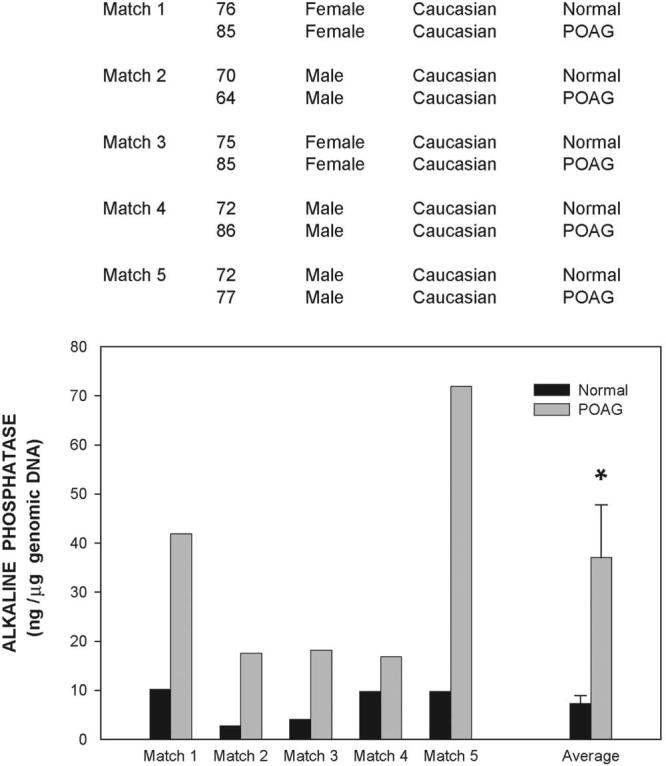
Comparison of alkaline phosphatase activity in the trabecular meshwork of anterior segments of normal and glaucomatous eyes. Top: information on the age, sex, race, and disease state of the nine donors used in this study (normal, n = 4; glaucoma, n = 5). Glaucomatous eyes used in matches 4 and 5 were matched against the normal donor. Bottom: anterior segments were perfused at constant flow between 1 to 6 days. The averaged perfused pressure of the eyes at 24 hours baseline was 19.6 ± 7.1 mm Hg. Extracts of dissected trabecular meshwork tissues were divided into aliquots and evaluated for endogenous ALP and DNA content. ALP values were normalized to total genomic DNA. Normalized ALP of normal and glaucomatous samples were averaged (mean ± SEM) and analyzed by the Student's t-test. *P ≤ 0.04. ALP activity was significantly elevated in glaucomatous trabecular meshworks.
Dexamethasone Induces Calcification in Human Trabecular Meshwork Cells
We next investigated whether glaucoma risk factors had an influence on human trabecular meshwork mineralization. Because prolonged cell exposure to glucocorticoids has been found to be associated with the development of calcification in bone and vascular systems,30-33 as well as with glaucoma,25,34,35 we evaluated whether DEX treatment also had the capacity to induce calcification on HTM cells.
For visualization of calcification, confluent primary cells (HTM-63) at passage 4 were treated in duplicate with 0.1 μM DEX and were stained with alizarin red 7 days after treatment. DEX-treated cells showed intense alizarin red and formed calcification nodules, but the untreated cells did not (Fig. 2, top and middle panels). In addition, cell extracts from 3, 5, and 7 days after treatment showed increased ALP activity (normalized to genomic DNA). Pooled control dishes showed a concentration of the active enzyme of 1.27 ± 0.05 ng/μg genomic DNA. After 3 days of DEX treatment, the concentration of ALP was 1.35 ± 0.07 ng/μg, which was not significantly different from that of the control (P = 0.24). However, at 5 days the normalized ALP value of the treated dishes significantly increased to 2.0 ± 0.02 ng/μg (P = 0.0004) and reached 4.7 ± 0.40 ng/μg after 7 days (P = 0.001), an induction of 3.6 fold (Fig. 2, bottom panel). This high induction of the biochemical calcification marker was observed in all single individual primary HTM cell lines tried, though the extent of induction varied. Thus, experiments using a high responding primary line (HTM-70) showed a 5.0-fold increase at 5 days that reached 29.4-fold over the untreated control at 7 days. These results indicate that a new additional effect of DEX on the trabecular meshwork could be one of inducing mineralization of the tissue. This effect appears to increase considerably with prolonged exposure.
Figure 2.
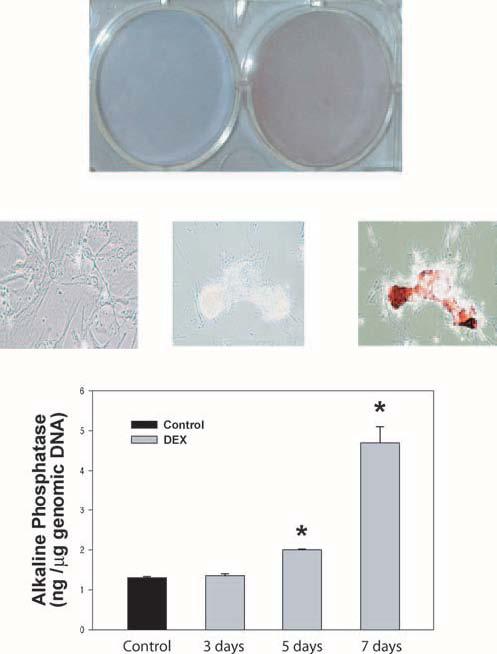
Effect of DEX treatment on calcification of primary HTM cells. Top, middle: HTM-63 cells in passage 4 were grown to confluence, treated with 0.1 μM DEX every other day for 7 days, and stained with 2% alizarin red. DEX-treated cells showed show positive red/orange staining for calcification. Top: general view with standard digital camera. Middle: Brightfield images obtained with a microscope (CK40; Olympus) equipped with a CCD DP70 digital camera (original magnification, ×200). Left: stained-untreated cells. Middle: unstained-treated cells. Right: stained-treated cells showing positive nodules. Bottom: parallel untreated (n = 3) and DEX-treated HTM-63 cells were harvested at 3, 5, and 7 days after treatment (n = 2 each). Cell extracts were divided into aliquots and evaluated for endogenous ALP and DNA content. ALP values were normalized to cell number, represented by total genomic DNA (mean ± SEM). *P ≤ 0.0004 (day 5). *P ≤ 0.001 (day 7). Experiments were repeated in HTM-70 and gave similar results. HTM cells treated with DEX form calcification nodules and show time-dependent increases of ALP activity in the primary HTM cells.
TGFβ2 Induces an ALP Increase in Human Trabecular Meshwork Cells
To next determine the effect on HTM calcification of another well-known associated glaucoma factor, we treated the cells with TGFβ2. Primary HTM-63 cells (passage 5) in triplicate were exposed to TGFβ2, harvested, and aliquots from the same sample were used for ALP assays and DNA quantification. At day 3, ALP was significantly induced in the treated HTM cells. The normalized endogenous ALP was 15.98 ± 0.6 ng/μg DNA (n = 3; P ≤ 0.0002) after treatment with 1 ng/mL TGFβ2 and reached 128.3 ± 2.8 ng/μg DNA (n = 3; P ≤ 0.001) after treatment with 3 ng/mL. These values represent ALP percentage increases from the untreated cells of 138% and 323% (1 ng/mL– and 3 ng/mL–treated cells, respectively) (Fig. 3). Experiments were duplicated in the same and different cell lines (HTM-70) with comparable results. In the second HTM-63 trial, the ALP percentage increases were 66% and 100%, whereas those on the HTM-70 cells were 35% and 57%. As demonstrated in other systems,36-39 these results indicated that TGFβ2 also induces a significant ALP increase in the cells of the trabecular meshwork that could contribute to an initiation of mineralization and a hardening of the outflow tissue.
Figure 3.
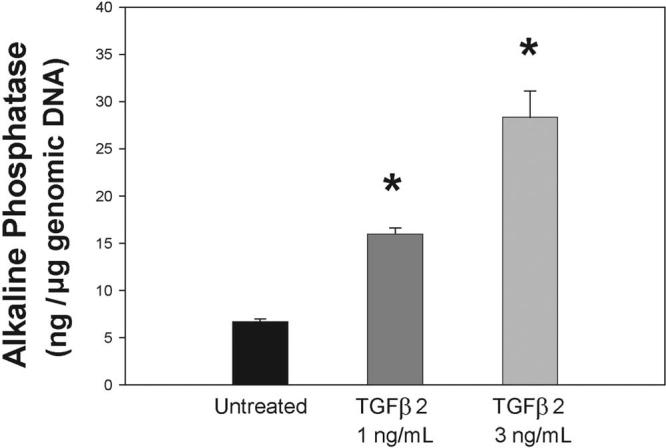
TGFβ2 effect on the alkaline phosphatase activity of primary HTM cells. Confluent HTM-63 cells in passage 5 were treated with 1 ng/mL and 3 ng/mL TGFβ2 for 3 days in serum-free medium. Extracts from harvested cells were divided into aliquots and evaluated for endogenous ALP and genomic DNA content. ALP levels were normalized to cell number, represented by total genomic DNA (mean ± SEM). *P ≤ 0.0002 (1 ng/mL, n = 3). *P ≤ 0.001 (3 ng/mL, n = 3). TGFβ2 significantly induces ALP in a dose-dependent manner in the primary HTM cells.
Expression of the Inhibitor of Calcification MGP and Its Activating Enzyme γ-GGCX in Specimens from Glaucoma Patients
Active MGP is a potent inhibitor of calcification, and it is activated by the vitamin K–dependent γ-carboxylase enzyme (for a review, see Proudfoot and Shanahan10). In our initial studies, we have shown that MGP mRNA is upregulated in the human trabecular meshwork tissue by elevated IOP9 and that HTM cells exhibit high γ-carboxylase activity.16 Because of the higher ALP found in tissues from patients with glaucoma, we wanted to investigate whether expression of MGP and γ-carboxylase genes was affected in the trabecular meshwork of glaucoma patients. Given the limited material obtained from a single trabecular meshwork, we gathered additional data for this study from six other donors (three with glaucomatous eyes, three with normal eyes) and matched them by age, sex, and race (matches 6, 7, and 9; Fig. 4). A fourth match was obtained between the glaucomatous eyes of match 5 (Fig. 1) and new normal eyes (match 8).
Figure 4.
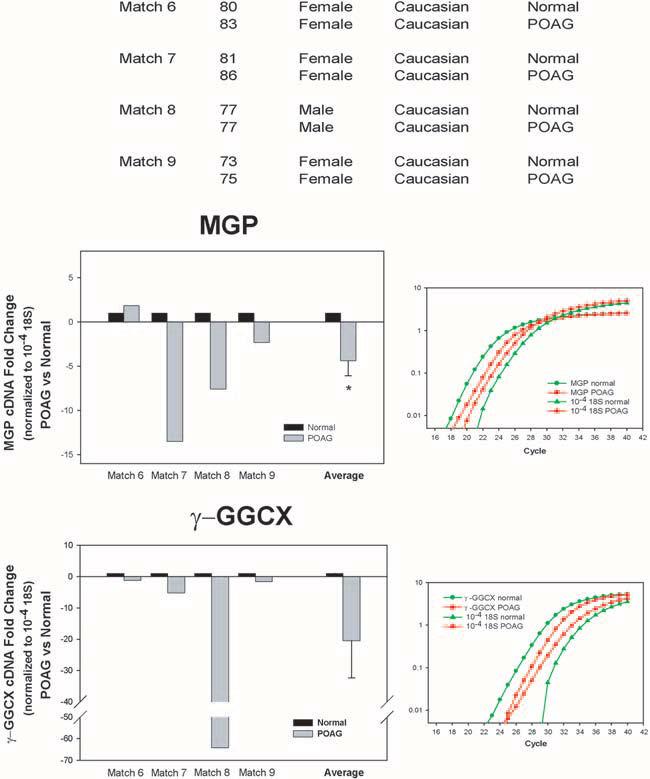
Expression of MGP and γ-carboxylase enzyme (γ-GGCX) in the trabecular meshwork tissue from normal and glaucoma donors. Top: information on the age, sex, race, and disease state of the eight donors used in this study (normal, n = 4; glaucoma, n = 4). Glaucomatous eye used in match 9 was from the same person used in match 5 in Figure 1. Anterior segments were perfused at constant flow between 1 to 5 days. Average perfused pressure of the eyes at 24 hours baseline was 15.9 ± 3.8 mm Hg. Trabecular meshwork tissues were dissected from frozen or RNA-later perfused anterior segments, or both, and were processed for RNA, RT, and exponential amplification with MGP, γ-GGCX, or 18S TaqMan PCR probes, as described in Methods. Normalization was achieved with the 18S TaqMan probe hybridized to 10−4 diluted RTs. Left middle, left lower: individual and average (mean ± SEM) fold changes of MGP and γ-GGCX in POAG versus normal specimens, normalized to 18S cDNA. After normalization, normal samples are given a value of 1.0. Right: representative CT logarithmic curves of the real-time TaqMan hybridizations of MGP, γ-GGCX, and 18S to cDNAs from POAG and normal. *P ≤ 0.006 (n = 9). MGP and its activating enzyme γ-GGCX are downregulated in intact trabecular meshworks obtained after death from donors with glaucoma.
Through real-time TaqMan PCR with the probes described in Methods and normalized by 18S, we found that, in the glaucoma samples, MGP was reduced in three of the four matches whereas γ-carboxylase was reduced in all four (Fig. 4). The average extent of reduction of MGP expression was a fold change of −4.39 ± 1.7 (n = 9; P ≤ 0.006), and that of γ-carboxylase expression was a fold change of −20.5 ± 11.9 (n = 7; P ≤ 0.26). The lack of significance of the activator enzyme expressed in glaucomatous versus normal samples was attributed to the variation introduced by the drastic reduction of this enzyme exhibited in one person (77 years old, white, male [match 8]; Fig. 4), which, if excluded, renders the comparative reduction, albeit much smaller, significant. Interestingly, this same person exhibited the highest ALP activity of the five glaucoma matches used to measure the activity of this enzyme (see match 5, Fig. 1). These results suggest that the tendency of the glaucomatous trabecular meshwork tissue to undergo mineralization observed here by the presence of ALP activity might be mediated in part by the MGP vitamin K–dependant pathway.
Expression of MGP and Its Activating Enzyme γ-GGCX in TGFβ2-Treated Human Trabecular Meshwork Cells
We had previously shown that primary trabecular meshwork cells exposed to TGFβ1 downregulated the expression of MGP.9 Although TGFβ1 has been suggested as a possible contributor to the development of glaucoma,40 TGFβ2 has been shown to be more abundant in the aqueous humor of these patients.26 Because TGFβ2 treatments have been extensively reported as correlated with tissue mineralization,36,38,41,42 in this study we investigated whether treatment with this cytokine altered the expression of MGP and its vitamin K–activating enzyme in HTM cells in culture.
Primary trabecular meshwork cells HTM-63 (passage 5) were treated with TGFβ2 at concentrations of 1 and 3 ng/mL. Total RNA from 3-day TGFβ2 and sham treatments was analyzed for MGP and γ-GGCX expression by real-time TaqMan PCR, as described in Methods. Expression values were normalized to the expression of 18S, used as the endogenous control. The normalized extent of MGP expression was reduced −1.92 ± 0.6-fold (P ≤ 0.006) in the treated samples compared with that of the control at 1 ng/mL (n = 3) and −1.84 ± 0.3-fold (P ≤ 0.0001) at 3 ng/mL (n = 3; Fig. 5 top panel). Experiments were repeated in a different individual-derived cell line (HTM-70), where MGP cDNA showed a reduction of −3.32 ± 0.4-fold (P ≤ 3E-08; n = 3) after similar 1 ng/mL exposure to TGFβ2 (not shown).
Figure 5.

Expression of MGP and γ-carboxylase enzyme (γ-GGCX) in primary trabecular meshwork cells treated with TGFβ2. Primary HTM cells, passage 6, were treated with 1 ng or 3 ng/mL TGFβ2 for 3 days in serum-free medium. Controls cells were left untreated. Wells were processed for RNA, RT, and exponential amplification with MGP, γ-GGCX, or 18S TaqMan PCR probes, as described in Methods. Normalization was achieved with the 18S TaqMan probe hybridized to 10−4 diluted RTs. Untreated samples were normalized to a value of 1.0, and fold change values are expressed as the mean ± SEM. Left: fold changes of MGP (top) and γ-GGCX (bottom) cDNAs in TGFβ2-treated versus untreated samples were normalized to 18S cDNA. Right: representative CT logarithmic curves of the real-time TaqMan hybridizations of MGP, γ-GGCX, and 18S to cDNAs from TGFβ2-treated and untreated cells. *P ≤ 0.006 (MGP; 1 ng/mL; n = 3). *P ≤ 0.0001 (MGP; 3 ng/mL; n = 3). *P ≤ 0.004 (γ-GGCX; 1 ng/mL; n = 3). *P ≤ 0.003 (γ-GGCX; 3 ng/mL; n= 3). TGFβ2 downregulates MGP and its activating enzyme γ-carboxylase in human trabecular meshwork cells.
The effect of TGFβ2 on the expression of the MGP-activating enzyme γ-carboxylase was more pronounced than its effect on MGP. On the HTM-63 cells, normalized γ-GGCX expression was reduced −2.26 ± 0.6-fold (P ≤ 0.004) in the treated samples compared with that of the control at 1 ng/mL (n = 3) and −2.76 ± 0.6-fold (P ≤ 0.003) at 3 ng/mL (n = 3; Fig. 5, bottom panel). In the HTM-70 cell line, reduction of the γ-GGCX enzyme extended to −2.78 ± 0.3-fold (P ≤ 0.0002; n = 3). These results suggest that this factor, significantly more abundant in the aqueous humor of glaucoma patients, might be one of the inducers of reduced expression of MGP and γ-GGCX genes (and consequent mediate increased mineralization) in the trabecular meshworks of these patients.
RNA Interference–Mediated Knockdown of MGP Increases Alkaline Phosphatase Activity
We have previously shown that the MGP gene reverses BMP2-induced calcification in primary HTM cells.16 To further test the involvement of MGP in calcification in this tissue, we silenced the expression of its gene by siRNA and measured the effect of silencing on normalized ALP levels. The extent of silencing was pretested by introducing two siRNA MGP concentrations into the primary HTM cells by nucleofector transfection and comparing it with that of cells transfected with negative control scrambled siRNAs. Forty-eight hours after nucleofector electroporation of the siRNAs, total RNA was extracted, reverse transcribed, and assayed in parallel with MGP and 18S TaqMan probes. CT values of MGP in samples treated with the nontargeting scrambled siRNA or left untreated were in the 18 to 19 value range, confirming the high expression of this gene in the human trabecular meshwork (not shown). After 18S normalization, the percentage of MGP remaining transcripts in the sample transfected with MGP siRNA was 3.8% (26-fold reduction) of those in control cells electroporated with nontargeted scrambled siRNA (1.2 μM in the nucleofector solution during electroporation, 40 nM in the culture dish). At 80 nM (2.4 μM during nucleofector electroporation), the percentage of remaining transcripts in the MGP siRNA sample was reduced to 2.2% (45-fold reduction; Fig. 6, top panel). Two more MGP siRNA-transfected samples resulted in 5% and 4% remaining MGP transcripts compared with non-transfected and scramble-transfected samples, respectively.
Figure 6.
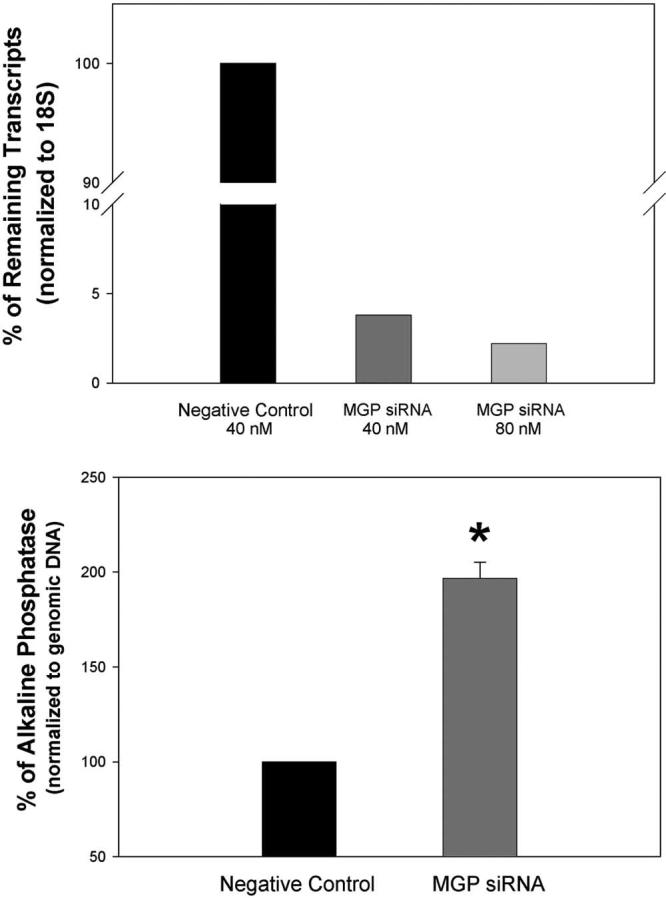
Effect of silencing the MGP gene in human trabecular meshwork cells. Primary human trabecular meshwork cells were transfected by nucleofector electroporation with either MGP siRNA (40 nM and 80 nM final concentrations) or scrambled negative control siRNA (40 nM final). Forty-eight hours after transfection, cells were harvested and homogenized, and aliquots were taken for total RNA, endogenous ALP, and genomic DNA extraction and determination. RNA was reverse transcribed, analyzed, and normalized for MGP expression by real-time TaqMan PCR using probes, as described in Methods. Scrambled negative control siRNA samples are given a value of 100. Top: percentage of remaining transcripts in cells treated with MGP siRNA compared with those treated with the scrambled negative control siRNA. Bottom: normalized endogenous ALP values in MGP and scrambled negative control siRNA-treated samples (n = 3). *P ≤ 0.0003. MGP siRNA silences the MGP gene efficiently in the human trabecular cells. Silencing MGP results in increased endogenous ALP activity.
For the ALP measurement, cells were electroporated at the higher siRNA concentration, harvested, and split into aliquots for assaying of genomic DNA (Hoechst) and ALP using the corresponding standards, as indicated in Methods. After normalization with genomic DNA, cells in which the MGP was silenced showed an increase of 197% ± 8.4% (n = 3) in the calcification marker compared with those transfected with the scrambled siRNA (P ≤ 0.0003; Fig. 6, bottom panel). These results confirm that the high expression of MGP specifically contributed to maintain lower levels of calcification in human trabecular meshwork cells.
Discussion
For many years, ectopic soft tissue calcification, such as that observed in atherosclerosis or in chronic renal failure, has been considered a passive event, a mere physicochemical precipitation of calcium phosphate caused by elevated phosphate and calcium ions. However, recent studies involving arterial calcification in knockout mouse models11,43 and in cell culture vascular calcification models44,45 have provided evidence that such calcification is an actively regulated process. These experiments have also shown that the vascular calcification process shares many molecular and cellular characteristics with bone and cartilage mineralization.10,46
Led by the finding of the high expression of an active inhibitor of calcification, MGP, in the human trabecular meshwork, previous studies from our laboratory showed that aged trabecular meshwork cells undergo calcification in cell culture in much the same way as VSMCs.16 In the present study, we investigated whether signs of calcification were present in glaucoma tissues and known glaucomatous insults. We chose a widely used mineralization marker, ALP,11,17-24,47,48 and measured its—rather than mRNA or protein level—enzymatic activity. We found that trabecular meshwork from donors with glaucoma had higher levels of active ALP than the normal matched counterparts. We interpret this finding as an indication that the trabecular meshwork cell might be undergoing a mineralization process during the course of the disease and that this process could contribute to the development of glaucoma.
Although a calcification process has never before been described for glaucoma, it is interesting to note that genes and mechanisms described during calcifying conditions in other diseases are also present in glaucomatous trabecular meshworks. Such is, for example, the molecular correlation between atherosclerosis and glaucoma.6,15 Endothelial leukocyte adhesion molecule-1 (ELAM-1), the earliest marker for the atherosclerotic plaque, is found only in the trabecular meshwork from glaucoma patients, and it is considered the first diagnostic glaucoma marker.15 Another parallel between the pathology of the two diseases is that calcification of the atherosclerotic plaque is thought to be initiated by its VSMCs and macrophages. Macrophages release TNFα, which has been shown to enhance osteoblastic differentiation, ALP, and mineralization.49 Interestingly, TNFα has been long associated with glaucomatous processes. TNFα is a regulator of trabecular meshwork metalloproteinases,50 induces the glaucoma-linked gene optineurin,51 and is induced by laser trabeculoplasty.52 Whether TNFα also induces calcification in the trabecular meshwork has not yet been tested.
Additional molecular examples associating glaucomatous conditions with mineralization in other systems are connective tissue growth factor (CTGF) and endothelin-1. In the eye, CTGF levels are significantly increased in the aqueous humor of patients with pseudoexfoliation glaucoma.53 Its cDNA is present in the human trabecular meshwork,7 where it is significantly induced by mechanical stretch54 and elevated IOP (T. Borrás, unpublished observations, 2006). At the same time, in primary osteoblast and mesenchymal stem cells, CTGF regulation has been shown to be essential to osteogenic differentiation and bone formation.17,55 The second molecule, endothelin-1, is a potent vasoconstrictor peptide proposed as a potential contributor to the pathogenesis in glaucoma.56,57 It induces contraction in the trabecular meshwork, reduces outflow facility,58 and is elevated in the aqueous humor of patients with glaucoma.59 Endothelin-1 is also associated with vascular wall thickening in renal failure60 and has been reported as a potent regulator of vascular calcification in vivo and in vitro in VSMCs.61 It is tempting to think that these two molecules could also stimulate mineralization of the trabecular meshwork.
In this study, we also report that ALP activity was elevated after treatment with DEX and TGFβ2, two well-established risk factors of glaucoma. The role of steroid treatment in the development of glaucoma is well documented.25,34,35 Concomitantly, glucocorticoids have long been known as potent stimulators of osteogenic differentiation in vitro. This characteristic extends to cells responsible for bone or cement formation62,63 and those responsible for vascular calcification, such as VSMCs and pericytes.31,33 In this study, we show for the first time that DEX induces the formation of calcified multicellular nodules and the upregulation of ALP on postconfluent primary HTM cells. This increased activity was seen at 5 days after treatment and was dramatically increased at 7 days. It appears to be a paradox, though, that treatment with glucocorticoids, which induces osteoporosis in many patients, would simultaneously cause mineralization of the trabecular meshwork in vivo. We do not know whether that paradox occurs in glaucoma because not many studies in patients with osteoporosis and glaucoma are available. However, numerous studies report a similar paradox regarding the simultaneous presence of vascular calcification and osteoporosis.64-66
The association of TGFβ2 with glaucoma is also extensive. It includes elevated levels in patients with primary open-angle glaucoma (POAG)26,67 and increased outflow resistance in human organ cultures.68 In HTM cells, TGFβ2 induces fibronectin and transglutaminase, an enzyme that cross-links fibronectin to complexes that cannot be digested by proteinases.4 Interestingly, transglutaminase is upregulated in the articular cartilage at the time of the pathologic mineralization associated with osteoarthritic conditions and aging.69,70 As for its role in calcification, TGFβ2 is expressed in the VSMCs of calcified arteriosclerotic arteries,36 induces chondrogenic differentiation of mesenchymal stem cells,41,42 and initiates endochondral ossification after injection in the rat femur.38 It does not come as a surprise then that here, TGFβ2 induced calcifying activity in the HTM cells. Extensive evidence also indicates that mesenchyme-originated VSMCs transdifferentiate into chondrocytes and undergo chondrogenic commitment, leading to vascular calcification. Given that trabecular meshwork cells are derived from the mesenchymal cells of the neural crest and exhibit many of the characteristics of VSMCs,71 it is not unconceivable to speculate that trabecular meshwork cells might undergo the same fate, as is represented in Figure 7.
Figure 7.
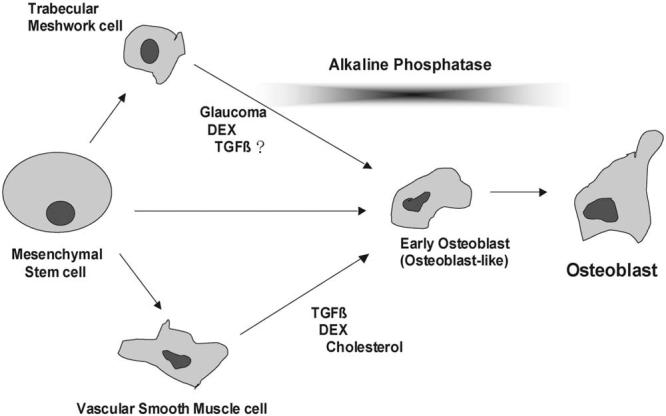
Schematic illustration depicting the transition of vascular precursor cells to osteoblasts, together with the hypothesis presented here of trabecular meshwork cells undergoing similar osteogenic differentiation.
Consequences of potential calcification of the trabecular meshwork tissue could have important implications for the regulation of resistance to aqueous humor outflow. A subtle, ongoing hardening of the tissue could begin as a subclinical effect but contribute with time to the detrimental effects observed on the ECM of glaucomatous trabecular meshworks.72 In atherosclerosis, the cause and effect (mineralization and disease) might be in either direction. Although it seems more rational to speculate that the causality in glaucoma would go mainly in one direction (mineralization of the trabecular meshwork, glaucoma), given the multifactorial nature of this disease, the second direction (glaucoma factors leading to mineralization) could also occur.
To protect themselves against calcification, VSMCs express potent inhibitors such as MGP.10 The trabecular meshwork also highly expresses MGP.6,16 Only active MGP is able to function as a calcification inhibitor, which to date is the only known function for the MGP gene. Therefore, here we investigated the expression not only of MGP but also of its activating enzyme γ-carboxylase (γ-GGCX). Both genes were downregulated in glaucomatous tissues and in primary HTM cells treated with TGFβ2. The extent of downregulation varied, and MGP was, in one of the five POAG/normal matches, slightly upregulated. Except in one instance, the fold change cDNA levels of the γ-GGCX enzyme were small, suggesting that perhaps no great changes in abundance are needed to modulate enzyme activity. The dramatic downregulation of γ-GGCX in the patient exhibiting the highest ALP activity is intriguing and contributes to the notion of the need for active MGP in the trabecular meshwork to protect against calcification. Overall, these findings reflect the fact that in glaucoma, cells of the trabecular meshwork appear to lose their protection against pathologic mineralization.
Studies by Murshed et al.12 have demonstrated that MGP inhibition of vascular calcification is regulated locally, rather than systemically. To investigate whether local inhibition of MGP would affect the mineralization potential of the trabecular meshwork cells, we silenced MPG in culture using RNA interference. Under conditions of greater than 90% MGP transcription reduction, the activity of ALP was significantly induced. These results strengthen our previous findings that at least one of the functions of MGP in the trabecular meshwork is to inhibit ECM mineralization. They also reinforce the notion that calcification may occur independently in different tissues of the body, albeit driven by the same mechanism.
Finally, we are continuing to investigate whether, in addition to MGP, other proteins associated with bone metabolism or vascular calcification73 are expressed in the trabecular meshwork. Analysis of the various microarray data in our laboratory indicated that BMP2, osteoprotegerin (OPG), osteocalcin (bone Gla protein), osteomodulin (osteoadherin), osteopontin (OPN), osteonectin (SPARC), osteoglycin, Cbfa1, ALP, and parathyroid hormone-related peptide (PTHrP) are all expressed either in the meshwork tissue or in its primary cultured cells. Interestingly, in addition to MGP, some of these osteogenic genes were regulated by elevated pressure, and some were among the 20 most upregulated. Thus, osteoblastspecific factor 2, induced after 2- to 4-day IOP insult,9 is the most relevant isoform of Cba1, a key transcription factor in the differentiation of mesenchymal cells to osteoblasts and is also present in calcified arteries.74,75 Osteomodulin, also expressed in odontoblasts and involved in biomineralization, was among the most downregulated genes at 2 to 4 days of elevated IOP.9 Osteoglycin (osteoinductive factor), which induces bone formation in conjunction with TGFβ and is differentially expressed in atherosclerotic plaques,76,77 was induced after 7 days of IOP insult (T. Borrás, unpublished observations, 2005).
In summary, our findings support the presence of higher mineralization characteristics in the trabecular meshwork of glaucoma tissues. Together with our previous findings,16 they support evidence of an inhibition of the calcification mechanism in the human trabecular meshwork. Because of the unique characteristics of the eye, the appearance of this mechanism in the cells of the outflow tissue might have developed in response to a need for strong protection against the potent calcification inducers associated with physiological functions of the eye and with pathologic insults. These would include the physiologically high levels of ascorbic acid (potent inducer of calcification) in the aqueous humor78 and the pathologic agents elevated during conditions of glaucoma, such TGFβ2, DEX, CTGF, and endothelin-1. Compromising the inhibition of the calcification mechanism might contribute to the development of disease while strengthening such inhibition by local administrators could open the way for new treatments of glaucoma.
Acknowledgments
The authors thank W. Craig Fowler and Kenneth L. Cohen for their contributions to the generation of primary cells and Annette Giangiacomo and LaKisha Buie for critical reading of the manuscript.
Supported by National Institutes of Health Grants EY11906 (TB) and EY13126 (TB) and by a Research to Prevent Blindness challenge grant (Department of Ophthalmology, University of North Carolina).
Footnotes
Disclosure: W. Xue, None; N. Comes, None; T. Borrás, None
References
- 1.Sommer A, Tielsch JM, Katz J, et al. Relationship between intraocular pressure and primary open angle glaucoma among white and black Americans: the Baltimore Eye Survey. Arch Ophthalmol. 1991;109:1090–1095. doi: 10.1001/archopht.1991.01080080050026. [DOI] [PubMed] [Google Scholar]
- 2.Wilson MR, Eezzuduemhoi DR. Ophthalmologic disorders in minority populations. Med Clin North Am. 2005;89:795–804. doi: 10.1016/j.mcna.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 3.Kass MA, Heuer DK, Higginbotham EJ, et al. The Ocular Hyper-tension Treatment Study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch Ophthalmol. 2002;120:701–713. doi: 10.1001/archopht.120.6.701. [DOI] [PubMed] [Google Scholar]
- 4.Welge-Lüssen U, May CA, Lütjen-Drecoll E. Induction of tissue transglutaminase in the trabecular meshwork by TGF-β1 and TGF-β2. Invest Ophthalmol Vis Sci. 2000;41:2229–2238. [PubMed] [Google Scholar]
- 5.Lütjen-Drecoll E, Shimizu T, Rohrbach M, Rohen JW. Quantitative analysis of “plaque material” in the inner and outer wall of Schlemm's canal in normal and glaucomatous eyes. Exp Eye Res. 1986;42:443–455. doi: 10.1016/0014-4835(86)90004-7. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez P, Epstein DL, Borrás T. Characterization of gene expression in human trabecular meshwork using single-pass sequencing of 1060 clones. Invest Ophthalmol Vis Sci. 2000;41:3678–3693. [PubMed] [Google Scholar]
- 7.Tomarev SI, Wistow G, Raymond V, Dubois S, Malyukova I. Gene expression profile of the human trabecular meshwork: NEIBank sequence tag analysis. Invest Ophthalmol Vis Sci. 2003;44:2588–2596. doi: 10.1167/iovs.02-1099. [DOI] [PubMed] [Google Scholar]
- 8.Wirtz MK, Samples JR, Xu H, Severson T, Acott TS. Expression profile and genome location of cDNA clones from an infant human trabecular meshwork library. Invest Ophthalmol Vis Sci. 2002;43:3698–3704. [PubMed] [Google Scholar]
- 9.Vittitow J, Borrás T. Genes expressed in the human trabecular meshwork during pressure-induced homeostatic response. J Cell Physiol. 2004;201:126–137. doi: 10.1002/jcp.20030. [DOI] [PubMed] [Google Scholar]
- 10.Proudfoot D, Shanahan CM. Molecular mechanisms mediating vascular calcification: role of matrix Gla protein. Nephrology (Carlton) 2006;11:455–461. doi: 10.1111/j.1440-1797.2006.00660.x. [DOI] [PubMed] [Google Scholar]
- 11.Luo G, Ducy P, McKee MD, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78–81. doi: 10.1038/386078a0. [DOI] [PubMed] [Google Scholar]
- 12.Murshed M, Schinke T, McKee MD, Karsenty G. Extracellular matrix mineralization is regulated locally; different roles of two gla-containing proteins. J Cell Biol. 2004;165:625–630. doi: 10.1083/jcb.200402046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tamm ER, Siegner A, Baur A, Lütjen-Drecoll E. Transforming growth factor-beta 1 induces alpha-smooth muscle-actin expression in cultured human and monkey trabecular meshwork. Exp Eye Res. 1996;62:389–397. doi: 10.1006/exer.1996.0044. [DOI] [PubMed] [Google Scholar]
- 14.Wiederholt M. Direct involvement of trabecular meshwork in the regulation of aqueous humor outflow. Curr Opin Ophthalmol. 1998;9:46–49. doi: 10.1097/00055735-199804000-00009. [DOI] [PubMed] [Google Scholar]
- 15.Wang N, Chintala SK, Fini ME, Schuman JS. Activation of a tissue-specific stress response in the aqueous outflow pathway of the eye defines the glaucoma disease phenotype. Nat Med. 2001;7:304–309. doi: 10.1038/85446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue W, Wallin R, Olmsted-Davis EA, Borrás T. Matrix GLA protein function in human trabecular meshwork cells: inhibition of BMP2-induced calcification process. Invest Ophthalmol Vis Sci. 2006;47:997–1007. doi: 10.1167/iovs.05-1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo Q, Kang Q, Si W, et al. Connective tissue growth factor (CTGF) is regulated by Wnt and bone morphogenetic proteins signaling in osteoblast differentiation of mesenchymal stem cells. J Biol Chem. 2004;279:55958–55968. doi: 10.1074/jbc.M407810200. [DOI] [PubMed] [Google Scholar]
- 18.Magne D, Julien M, Vinatier C, Merhi-Soussi F, Weiss P, Guicheux J. Cartilage formation in growth plate and arteries: from physiology to pathology. Bioessays. 2005;27:708–716. doi: 10.1002/bies.20254. [DOI] [PubMed] [Google Scholar]
- 19.Boskey AL. Mineral-matrix interactions in bone and cartilage. Clin Orthop Relat Res. 1992:244–274. [PubMed] [Google Scholar]
- 20.Hashimoto S, Ochs RL, Rosen F, et al. Chondrocyte-derived apoptotic bodies and calcification of articular cartilage. Proc Natl Acad Sci USA. 1998;95:3094–3099. doi: 10.1073/pnas.95.6.3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jono S, McKee MD, Murry CE, et al. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87:E10–E17. doi: 10.1161/01.res.87.7.e10. [DOI] [PubMed] [Google Scholar]
- 22.Proudfoot D, Davies JD, Skepper JN, Weissberg PL, Shanahan CM. Acetylated low-density lipoprotein stimulates human vascular smooth muscle cell calcification by promoting osteoblastic differentiation and inhibiting phagocytosis. Circulation. 2002;106:3044–3050. doi: 10.1161/01.cir.0000041429.83465.41. [DOI] [PubMed] [Google Scholar]
- 23.Tanimura A, McGregor DH, Anderson HC. Calcification in atherosclerosis, I: human studies. J Exp Pathol. 1986;2:261–273. [PubMed] [Google Scholar]
- 24.Shanahan CM, Proudfoot D, Tyson KL, Cary NR, Edmonds M, Weissberg PL. Expression of mineralisation-regulating proteins in association with human vascular calcification. Z Kardiol. 2000;89(suppl 2):63–68. doi: 10.1007/s003920070101. [DOI] [PubMed] [Google Scholar]
- 25.Weinreb RN, Polansky JR, Kramer SG, Baxter JD. Acute effects of dexamethasone on intraocular pressure in glaucoma. Invest Ophthalmol Vis Sci. 1985;26:170–175. [PubMed] [Google Scholar]
- 26.Tripathi RC, Li J, Chan WF, Tripathi BJ. Aqueous humor in glaucomatous eyes contains an increased level of TGF-beta 2. Exp Eye Res. 1994;59:723–727. doi: 10.1006/exer.1994.1158. [DOI] [PubMed] [Google Scholar]
- 27.Lütjen-Drecoll E. Morphological changes in glaucomatous eyes and the role of TGFβ2 for the pathogenesis of the disease. Exp Eye Res. 2005;81:1–4. doi: 10.1016/j.exer.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 28.Johnson DH, Tschumper RC. Human trabecular meshwork organ culture: a new method. Invest Ophthalmol Vis Sci. 1987;28:945–953. [PubMed] [Google Scholar]
- 29.Borrás T, Rowlette LL, Erzurum SC, Epstein DL. Adenoviral reporter gene transfer to the human trabecular meshwork does not alter aqueous humor outflow: relevance for potential gene therapy of glaucoma. Gene Ther. 1999;6:515–524. doi: 10.1038/sj.gt.3300860. [DOI] [PubMed] [Google Scholar]
- 30.Aubin JE. Osteoprogenitor cell frequency in rat bone marrow stromal populations: role for heterotypic cell-cell interactions in osteoblast differentiation. J Cell Biochem. 1999;72:396–410. [PubMed] [Google Scholar]
- 31.Kirton JP, Wilkinson FL, Canfield AE, Alexander MY. Dexamethasone downregulates calcification-inhibitor molecules and accelerates osteogenic differentiation of vascular pericytes: implications for vascular calcification. Circ Res. 2006;98:1264–1272. doi: 10.1161/01.RES.0000223056.68892.8b. [DOI] [PubMed] [Google Scholar]
- 32.Shalhoub V, Conlon D, Tassinari M, et al. Glucocorticoids promote development of the osteoblast phenotype by selectively modulating expression of cell growth and differentiation associated genes. J Cell Biochem. 1992;50:425–440. doi: 10.1002/jcb.240500411. [DOI] [PubMed] [Google Scholar]
- 33.Mori K, Shioi A, Jono S, Nishizawa Y, Morii H. Dexamethasone enhances in vitro vascular calcification by promoting osteoblastic differentiation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:2112–2118. doi: 10.1161/01.atv.19.9.2112. [DOI] [PubMed] [Google Scholar]
- 34.Johnson DH. Corticosteroid glaucoma. In: Epstein DL, Allingham RR, Schuman JS, editors. Chandler and Grant's Glaucoma. Williams & Wilkins; Baltimore: 1997. pp. 404–411. [Google Scholar]
- 35.Armaly MF. Effect of corticosteroids on intraocular pressure and fluid dynamics, I: the effect of dexamethasone in the normal eye. Arch Ophthalmol. 1963;70:482–491. doi: 10.1001/archopht.1963.00960050484010. [DOI] [PubMed] [Google Scholar]
- 36.Jeziorska M. Transforming growth factor-betas and CD105 expression in calcification and bone formation in human atherosclerotic lesions. Z Kardiol. 2001;90(suppl 3):23–26. doi: 10.1007/pl00022848. [DOI] [PubMed] [Google Scholar]
- 37.Watson KE, Bostrom K, Ravindranath R, Lam T, Norton B, Demer LL. TGF-beta 1 and 25-hydroxycholesterol stimulate osteoblast-like vascular cells to calcify. J Clin Invest. 1994;93:2106–2113. doi: 10.1172/JCI117205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joyce ME, Roberts AB, Sporn MB, Bolander ME. Transforming growth factor-beta and the initiation of chondrogenesis and osteogenesis in the rat femur. J Cell Biol. 1990;110:2195–2207. doi: 10.1083/jcb.110.6.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nurminskaya M, Magee C, Faverman L, Linsenmayer TF. Chondrocyte-derived transglutaminase promotes maturation of preosteoblasts in periosteal bone. Dev Biol. 2003;263:139–152. doi: 10.1016/s0012-1606(03)00445-7. [DOI] [PubMed] [Google Scholar]
- 40.Tripathi RC, Li J, Borisuth NS, Tripathi BJ. Trabecular cells of the eye express messenger RNA for transforming growth factor-beta 1 and secrete this cytokine. Invest Ophthalmol Vis Sci. 1993;34:2562–2569. [PubMed] [Google Scholar]
- 41.Seyedin SM, Thompson AY, Bentz H, et al. Cartilage-inducing factor-A: apparent identity to transforming growth factor-beta. J Biol Chem. 1986;261:5693–5695. [PubMed] [Google Scholar]
- 42.Barry F, Boynton RE, Liu B, Murphy JM. Chondrogenic differentiation of mesenchymal stem cells from bone marrow: differentiation-dependent gene expression of matrix components. Exp Cell Res. 2001;268:189–200. doi: 10.1006/excr.2001.5278. [DOI] [PubMed] [Google Scholar]
- 43.Bucay N, Sarosi I, Dunstan CR, et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Proudfoot D, Skepper JN, Shanahan CM, Weissberg PL. Calcification of human vascular cells in vitro is correlated with high levels of matrix Gla protein and low levels of osteopontin expression. Arterioscler Thromb Vasc Biol. 1998;18:379–388. doi: 10.1161/01.atv.18.3.379. [DOI] [PubMed] [Google Scholar]
- 45.Tintut Y, Demer LL. Recent advances in multifactorial regulation of vascular calcification. Curr Opin Lipidol. 2001;12:555–560. doi: 10.1097/00041433-200110000-00012. [DOI] [PubMed] [Google Scholar]
- 46.Vattikuti R, Towler DA. Osteogenic regulation of vascular calcification: an early perspective. Am J Physiol Endocrinol Metab. 2004;286:E686–E696. doi: 10.1152/ajpendo.00552.2003. [DOI] [PubMed] [Google Scholar]
- 47.Rattazzi M, Bennett BJ, Bea F, et al. Calcification of advanced atherosclerotic lesions in the innominate arteries of ApoE-deficient mice: potential role of chondrocyte-like cells. Arterioscler Thromb Vasc Biol. 2005;25:1420–1425. doi: 10.1161/01.ATV.0000166600.58468.1b. [DOI] [PubMed] [Google Scholar]
- 48.Towler DA, Shao JS, Cheng SL, Pingsterhaus JM, Loewy AP. Osteogenic regulation of vascular calcification. Ann NY Acad Sci. 2006;1068:327–333. doi: 10.1196/annals.1346.036. [DOI] [PubMed] [Google Scholar]
- 49.Tintut Y, Patel J, Parhami F, Demer LL. Tumor necrosis factor-alpha promotes in vitro calcification of vascular cells via the cAMP pathway. Circulation. 2000;102:2636–2642. doi: 10.1161/01.cir.102.21.2636. [DOI] [PubMed] [Google Scholar]
- 50.Alexander JP, Acott TS. Involvement of protein kinase C in TNFα regulation of trabecular matrix metalloproteinases and TIMPs. Invest Ophthalmol Vis Sci. 2001;42:2831–2838. [PubMed] [Google Scholar]
- 51.Vittitow J, Borrás T. Expression of optineurin, a glaucoma-linked gene, is influenced by elevated intraocular pressure. Biochem Biophys Res Commun. 2002;298:67–74. doi: 10.1016/s0006-291x(02)02395-1. [DOI] [PubMed] [Google Scholar]
- 52.Bradley JM, Anderssohn AM, Colvis CM, et al. Mediation of laser trabeculoplasty-induced matrix metalloproteinase expression by IL-1beta and TNFα. Invest Ophthalmol Vis Sci. 2000;41:422–430. [PubMed] [Google Scholar]
- 53.Ho SL, Dogar GF, Wang J, et al. Elevated aqueous humour tissue inhibitor of matrix metalloproteinase-1 and connective tissue growth factor in pseudoexfoliation syndrome. Br J Ophthalmol. 2005;89:169–173. doi: 10.1136/bjo.2004.044685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vittal V, Rose A, Gregory KE, Kelley MJ, Acott TS. Changes in gene expression by trabecular meshwork cells in response to mechanical stretching. Invest Ophthalmol Vis Sci. 2005;46:2857–2868. doi: 10.1167/iovs.05-0075. [DOI] [PubMed] [Google Scholar]
- 55.Safadi FF, Xu J, Smock SL, et al. Expression of connective tissue growth factor in bone: its role in osteoblast proliferation and differentiation in vitro and bone formation in vivo. J Cell Physiol. 2003;196:51–62. doi: 10.1002/jcp.10319. [DOI] [PubMed] [Google Scholar]
- 56.Prasanna G, Narayan S, Krishnamoorthy RR, Yorio T. Eyeing endothelins: a cellular perspective. Mol Cell Biochem. 2003;253:71–88. doi: 10.1023/a:1026005418874. [DOI] [PubMed] [Google Scholar]
- 57.Yorio T, Krishnamoorthy R, Prasanna G. Endothelin: is it a contributor to glaucoma pathophysiology? J Glaucoma. 2002;11:259–270. doi: 10.1097/00061198-200206000-00016. [DOI] [PubMed] [Google Scholar]
- 58.Thieme H, Schimmat C, Munzer G, et al. Endothelin antagonism: effects of FP receptor agonists prostaglandin F2α and fluprostenol on trabecular meshwork contractility. Invest Ophthalmol Vis Sci. 2006;47:938–945. doi: 10.1167/iovs.05-0527. [DOI] [PubMed] [Google Scholar]
- 59.Tezel G, Kass MA, Kolker AE, Becker B, Wax MB. Plasma and aqueous humor endothelin levels in primary open-angle glaucoma. J Glaucoma. 1997;6:83–89. [PubMed] [Google Scholar]
- 60.Amann K, Tyralla K, Gross ML, Eifert T, Adamczak M, Ritz E. Special characteristics of atherosclerosis in chronic renal failure. Clin Nephrol. 2003;60(suppl 1):S13–S21. [PubMed] [Google Scholar]
- 61.Wu SY, Zhang BH, Pan CS, et al. Endothelin-1 is a potent regulator in vivo in vascular calcification and in vitro in calcification of vascular smooth muscle cells. Peptides. 2003;24:1149–1156. doi: 10.1016/j.peptides.2003.07.008. [DOI] [PubMed] [Google Scholar]
- 62.Igarashi M, Kamiya N, Hasegawa M, Kasuya T, Takahashi T, Takagi M. Inductive effects of dexamethasone on the gene expression of Cbfa1, Osterix and bone matrix proteins during differentiation of cultured primary rat osteoblasts. J Mol Histol. 2004;35:3–10. doi: 10.1023/b:hijo.0000020883.33256.fe. [DOI] [PubMed] [Google Scholar]
- 63.Hakki SS, Nohutcu RM, Hakki EE, Berry JE, Akkaya MS, Somerman MJ. Dexamethasone and basic-fibroblast growth factor regulate markers of mineralization in cementoblasts in vitro. J Periodontol. 2005;76:1550–1558. doi: 10.1902/jop.2005.76.9.1550. [DOI] [PubMed] [Google Scholar]
- 64.Parhami F, Morrow AD, Balucan J, et al. Lipid oxidation products have opposite effects on calcifying vascular cell and bone cell differentiation: a possible explanation for the paradox of arterial calcification in osteoporotic patients. Arterioscler Thromb Vasc Biol. 1997;17:680–687. doi: 10.1161/01.atv.17.4.680. [DOI] [PubMed] [Google Scholar]
- 65.Banks LM, Lees B, MacSweeney JE, Stevenson JC. Effect of degenerative spinal and aortic calcification on bone density measurements in post-menopausal women: links between osteoporosis and cardiovascular disease? Eur J Clin Invest. 1994;24:813–817. doi: 10.1111/j.1365-2362.1994.tb02024.x. [DOI] [PubMed] [Google Scholar]
- 66.Yamaguchi T, Sugimoto T. Osteoporosis and vascular calcification (in Japanese) Clin Calcium. 2002;12:1089–1093. [PubMed] [Google Scholar]
- 67.Picht G, Welge-Luessen U, Grehn F, Lutjen-Drecoll E. Transforming growth factor beta 2 levels in the aqueous humor in different types of glaucoma and the relation to filtering bleb development. Graefes Arch Clin Exp Ophthalmol. 2001;239:199–207. doi: 10.1007/s004170000252. [DOI] [PubMed] [Google Scholar]
- 68.Gottanka J, Chan D, Eichhorn M, Lütjen-Drecoll E, Ethier CR. Effects of TGF-β2 in perfused human eyes. Invest Ophthalmol Vis Sci. 2004;45:153–158. doi: 10.1167/iovs.03-0796. [DOI] [PubMed] [Google Scholar]
- 69.Aeschlimann D, Thomazy V. Protein crosslinking in assembly and remodelling of extracellular matrices: the role of transglutaminases. Connect Tissue Res. 2000;41:1–27. doi: 10.3109/03008200009005638. [DOI] [PubMed] [Google Scholar]
- 70.Rosenthal AK, Derfus BA, Henry LA. Transglutaminase activity in aging articular chondrocytes and articular cartilage vesicles. Arthritis Rheum. 1997;40:966–970. doi: 10.1002/art.1780400526. [DOI] [PubMed] [Google Scholar]
- 71.de Kater A, Shahsafaei A, Epstein DL. Localization of smooth muscle and nonmuscle actin isoforms in the human aqueous outflow pathway. Invest Ophthalmol Vis Sci. 1992;33:424–429. [PubMed] [Google Scholar]
- 72.Lütjen-Drecoll E, Rohen JW. Morphology of aqueous outflow pathways in normal and glaucomatous eyes. In: Ritch R, Shields MB, Krupin T, editors. The Glaucomas. CV Mosby; St. Louis: 1996. pp. 89–123. [Google Scholar]
- 73.Wallin R, Wajih N, Greenwood GT, Sane DC. Arterial calcification: a review of mechanisms, animal models, and the prospects for therapy. Med Res Rev. 2001;21:274–301. doi: 10.1002/med.1010. [DOI] [PubMed] [Google Scholar]
- 74.Steitz SA, Speer MY, Curinga G, et al. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–1154. doi: 10.1161/hh2401.101070. [DOI] [PubMed] [Google Scholar]
- 75.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 76.Shanahan CM, Cary NR, Osbourn JK, Weissberg PL. Identification of osteoglycin as a component of the vascular matrix: differential expression by vascular smooth muscle cells during neointima formation and in atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 1997;17:2437–2447. doi: 10.1161/01.atv.17.11.2437. [DOI] [PubMed] [Google Scholar]
- 77.Kukita A, Bonewald L, Rosen D, Seyedin S, Mundy GR, Roodman GD. Osteoinductive factor inhibits formation of human osteoclast-like cells. Proc Natl Acad Sci USA. 1990;87:3023–3026. doi: 10.1073/pnas.87.8.3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reiss GR, Werness PG, Zollman PE, Brubaker RF. Ascorbic acid levels in the aqueous humor of nocturnal and diurnal mammals. Arch Ophthalmol. 1986;104:753–755. doi: 10.1001/archopht.1986.01050170143039. [DOI] [PubMed] [Google Scholar]