

Abstract
The nuclear oxysterol-receptor paralogues LXRα and LXRβ share a high degree of amino acid identity and bind endogenous oxysterol ligands with similar affinities. While LXRα has been established as an important regulator of cholesterol catabolism in cholesterol-fed mice, little is known about the function of LXRβ in vivo. We have generated mouse lines with targeted disruptions of each of these LXR receptors and have compared their responses to dietary cholesterol. Serum and hepatic cholesterol levels and lipoprotein profiles of cholesterol-fed animals revealed no significant differences between LXRβ–/– and wild-type mice. Steady-state mRNA levels of 3-hydroxy-3-methylglutaryl coenzyme A reductase, farnesyl diphosphate synthase, and squalene synthase were increased in LXRβ–/– mice compared with LXRβ+/+ mice, when fed standard chow. The mRNA levels for cholesterol 7α-hydroxylase, oxysterol 7α-hydroxylase, sterol 12α-hydroxylase, and sterol 27-hydroxylase, respectively, were comparable in these strains, both on standard and 2% cholesterol chow. Our results indicate that LXRβ–/– mice — in contrast to LXRα–/– mice — maintain their resistance to dietary cholesterol, despite subtle effects on the expression of genes coding for enzymes involved in lipid metabolism. Thus, our data indicate that LXRβ has no complete overlapping function compared with LXRα in the liver.
Introduction
Cholesterol is a basic component of all cellular membranes and precursor of several hormones, vitamins, and bile acids, and therefore is an essential molecule for life. However, it is well established that elevated levels of cholesterol in plasma represent a risk factor for the development of atherosclerosis (1, 2).
The biosynthesis and catabolism of cholesterol are regulated by a complex network of enzymes, which are tightly controlled by several hormones (3), by cholesterol itself (4), and possibly also by oxidized derivatives of cholesterol — the oxysterols (5). Recent studies have identified several oxysterols, in particular 24(S), 25-epoxycholesterol, and 24(S)-hydroxycholesterol (6–8), as natural ligands for the nuclear liver X-receptor α (LXRα; NR1H3 from the Nuclear Receptors Nomenclature Committee, 1999) (9), suggesting that LXRα is important in cholesterol homeostasis. Mice lacking LXRα indeed lose the natural resistance to a diet rich in cholesterol, being unable to adequately regulate the conversion of cholesterol into bile acids (10).
In parallel to LXRα, another orphan receptor, LXRβ (NR1H2; from Nuclear Receptors Nomenclature Committee, 1999) (9) was cloned and characterized (11–14). While the expression of LXRα is limited to organs such as liver, intestine, kidney, adipose tissue, and adrenals (15, 16), LXRβ is expressed in all tissues thus far analyzed (11–14). LXRα and LXRβ share a high degree of amino acid similarity (78%) (17) and have thus been proposed to be paralogues. Recently, a similar function for both LXRs has been shown in the regulation of ABCA1 and ABCG1, two transmembrane proteins mediating efflux of phospholipids and cholesterol from macrophages and enterocytes (18–22). Due to the similar ligand-binding ability as well as their specificity for endogenous oxysterols, a function of both LXRs in the regulation of hepatic cholesterol and bile acid metabolism has been suggested (6, 7).
With the aim to determine if LXRβ has a regulatory function in the response to cholesterol challenge, as described for LXRα (10), mouse lines deficient for the LXRβ or LXRα genes were generated and challenged with dietary cholesterol. Our results show that, in contrast to LXRα-mutant mice, LXRβ-deficient mice maintained their natural resistance to dietary cholesterol. This indicates that LXRβ does not have a role comparable to that of LXRα in hepatic cholesterol metabolism.
Methods
Production of LXR-deficient mice.
For the LXRβ-targeting vector a 6.1-kb EcoRI-ApaI genomic DNA fragment (European Molecular Biology Laboratory [EMBL] accession number AJ132602) (17), cloned into pBluescript, was used (Figure 1a). The DNA fragment between the BglII and NcoI sites, located in intron 4, was replaced by a 1.1-kb loxP cassette isolated from pGEM-30 (23). The AvrII site in intron 6 was used for insertion of a 1.4-kb SalI-XbaI loxP-flanked neo cassette isolated from pL2neo (24). The unique ApaI site at the 3′ end of the genomic insert was used for linearization of the vector before electroporation.
Figure 1.
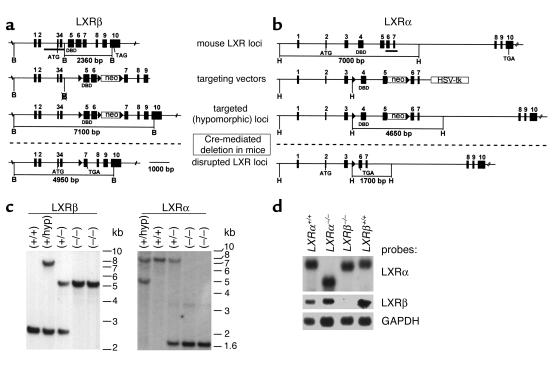
Strategy for the creation of LXRβ- and LXRα-deficient mice using a Cre/loxP system. (a and b) The genomic organization of LXRβ and LXRα, the corresponding targeting constructs containing the loxP sites, the predicted homologous recombinant alleles, and the LXR locus after Cre-mediated deletion are shown. Filled boxes indicate exons, arrows loxP sites, open boxes neomycin resistance and thymidine kinase genes. Filled bars indicate probes used for Southern blot analyses. H, HindIII; B, BglII; DBD, DNA-binding domain; ATG, translational start codon; TAG/TGA, stop codons. (c) Southern blot analysis. DNA was prepared from mouse tails and digested with BglII or HindIII as described in Methods. (d) Northern blot analysis. Poly A(+)-enriched RNA was extracted from a pool of livers (five females) from each respective group. Hybridization was performed by using full-length LXRα and LXRβ cDNAs as probes.
For the LXRα-targeting vector a 6.9-kb NcoI-NdeI genomic DNA fragment (EMBL accession number AJ132599) (17) was cloned into pGEM-5 (Figure 1b). The AflII site in intron 3 was used to insert a 153-bp cassette containing a single loxP site obtained from pGEM-30. A recreated HindIII site at the 5′ junction served as a restriction site for diagnostic Southern blot analysis (Figure 1, b and c). The SmaI site in intron 5 was used to insert a blunt-ended 1.3-kb XhoI fragment containing the neo cassette and a loxP site. For negative selection, a herpes simplex virus thymidine kinase cassette (HSV-tk) derived from pIC19R/MC1-TK (25) was cloned downstream of the short arm of the targeting vector. A unique BamHI site at the 3′ end of the thymidine kinase cassette was used to linearize the targeting vector.
R1 embryonic stem (ES) cells established from inbred 129/Sv embryos (26) were electroporated with 20 μg of the respective linearized targeting vector. After 48 hours, positive clones for either LXRβ or LXRα were selected with G418 or with G418 plus ganciclovir, respectively. Resistant ES cell clones were screened for hypomorphic (hyp) alleles using PCR. Homologous recombination was confirmed by Southern blot analysis (not shown). Recombinant ES-cell clones were injected into C57BL/6 blastocysts. Mating of chimeric males with C57BL/6 females led to the integration of the hypomorphic LXR alleles into the germ line. Disruption of the LXR alleles was performed by Cre-assisted deletion in vivo by mating LXRhyp/hyp and LXR+/hyp mice with cre-transgenic deleter mice (with hybrid C57BL/6-129/Sv genetic background) as described previously (27). Subsequent matings of offspring served to segregate the desired LXR+/– mutations from the cre transgene. Starting from this population of LXRα+/– and LXRβ+/– founder animals, we first crossed heterozygous LXR+/– animals and later also homozygous LXR–/– mice, as well as their respective wild-type littermates. All animals used for experiments were maximally third-generation descendants of the original heterozygous (+/–) founder animals. All mice used had a mixed genetic background based on C57BL/6 and 129/Sv strains. During establishment of all LXR-deficient mouse lines, genotypes were determined by Southern blot analysis of tail DNA (Figure 1c) and digested with HindIII (LXRα) or BglII (LXRβ). After colonies had been established, a PCR approach was used for genotyping.
Experimental procedure.
Mice from the two different knockout lines and corresponding control lines (designated LXRα+/+ and LXRβ+/+), derived by intercrossing wild-type littermates for each knockout, were studied in two separate experiments performed at different times. Mice were maintained on a 12-hour light/12-hour dark cycle and fed a low-fat rodent diet (diet R36; Lactamin AB, Vodstena, Sweden) ad libitum. Twelve- to 16-week old mice were challenged with R36 chow enriched with 2% cholesterol (AnalyCen Nordic AB, Lidköping, Sweden) for 28 days. In a separate experiment, serum cholesterol and triglyceride levels from the distinct LXRα+/+ and LXRβ+/+ control mice fed standard chow for 18 months were compared. No differences were observed (data not shown). Experiments were approved by the Karolinska Institutional Animal Care and Use Committee.
Chemical analysis of serum and tissue.
After 5-hour fasting, blood was drawn from mice by cardiac puncture under light methoxyflurane anesthesia. Animals were sacrificed by cervical dislocation, and tissues were immediately collected for further analyses. Blood was transferred into tubes, and serum was collected by centrifugation. Total cholesterol in serum was determined individually using a Monarch automated analyzer (ILS Laboratories Scandinavia AB, Sollentuna, Sweden) on fresh serum. Size fractionation of lipoproteins was performed on 10 μl of pooled sera from each group using a fast-performance liquid chromatography (FPLC) column (30 × 0.32 cm Superose 6B; Amersham Pharmacia Biotech, Uppsala, Sweden), and cholesterol content in lipoproteins were determined online using a commercially available reagent (Boehringer Mannheim Biochemicals Inc., Mannheim, Germany). Absorbency was monitored at 500 nM and collected using EZ Chrom software package (Scientific Software Inc., Pleasanton, California, USA). Hepatic lipids were extracted according to the methods of Folch et al. (28) and analyzed for total cholesterol using commercially available kits (Boehringer Mannheim Biochemicals Inc.). Oxysterols (24- and 27-hydroxycholesterol) in pooled serum samples were determined by isotope-dilution mass spectrometry using deuterium-labeled internal standards as described previously (29). Alanine and aspartate aminotransferase activities were measured in pooled sera according to the International Federation of Clinical Chemistry and Laboratory Medicine (30) using reagents from Roche Diagnostics (Basel, Switzerland) and a Hitachi 917 automatic analyzer (Hitachi Ltd., Naka, Japan).
Northern analysis.
Northern analysis was performed on 2–10 μg poly A+ mRNA, extracted from pooled livers of five female mice from each group. The cDNA probes were generated by RT-PCR with murine RNA from liver as previously described (10, 31) or using the following primers: cholesterol 7α-hydroxylase (Cyp7a), 5′-AGATTCTCCCCTTGGGACGTTTTCCTG-3′ and 5′-AACGCTCAGCAGTCGTTACATCATCCAGT-3′; oxysterol 7α-hydroxylase (Cyp7b), 5′-GCTCCCTACAGAGCCGCCAGCTCGTC-3′ and 5′-ACGGAGCTCAGCACATGCAGCCTTACTCTG-3′; sterol 27-hydroxylase (Cyp27), 5′-GGACAGCAGTGGTACCATCTGC-3′ and 5′-CCAGTTATTCATGTATCGCTTCC-3′; farnesyl diphosphate (FPP) synthase, 5′-GGTCCTGGAGTACAACACTGTAGGAGGCAA-3′ and 5′-TGGCAGCCGCGATAGGCAGGTAGAAAGAGT-3′.
The RT-PCR products were cloned into T-tailed pBluescript plasmid (Stratagene, La Jolla, California, USA) modified as described previously (32). The sequences of the different clones were confirmed using the BigDye Terminator Cycle Sequencing Ready Reaction Kit (Perkin Elmer Applied Biosystems, Warrington, United Kingdom). Plasmids containing probes for stearoyl CoA-desaturase-1 (SCD-1), fatty acid synthase (FAS,) and sterol 12α-hydroxylase (Cyp8b1) were kindly provided by Stefan Alexson (Karolinska Institutet, Huddinge, Sweden), the probe for SREBP-2 by Christine Sadek (Karolinska Institutet, Huddinge, Sweden). Probes were labeled with [α-32P] dCTP using random prime-labeling system (Amersham Pharmacia Biotech, Uppsala, Sweden). After hybridization, signals on Northern blot filters were quantified by an image analyzer (Fuji Photo Film Co., Stockholm, Sweden) and standardized according to GAPDH mRNA expression. The data shown resulted from two or more independent determinations.
Statistics.
Data are presented as mean plus SEM. The significance of differences between groups was tested by two-way ANOVA followed, when appropriate, by post hoc comparisons of group means according to honestly significant difference methods (Statistica software; Stat Soft Inc., Tulsa, Oklahoma, USA) (33). The authenticity of the absence of significant differences was determined by post hoc power calculation, when suitable.
Results
LXR-β and LXRα-deficient mice were created according to the strategy shown in Figure 1. We first generated mutant mice by flanking two exons encoding the DNA-binding domain and the hinge region of the respective LXR genes by loxP sites (Figure 1, a and c). Mice homozygous for these hypomorphic LXRβ or LXRα alleles (hyp/hyp) were viable, and no apparent phenotype was observed. Both LXR genes were successively disrupted by in vivo Cre-mediated deletion of the loxP-flanked gene segments. Southern blot analyses proved the proper homologous recombination (Figure 1c). Northern blot hybridizations of hepatic mRNA was performed to control for the presence of the right-truncated mRNA transcripts (Figure 1d). Unexpectedly, no LXRβ transcripts were detected in LXRβ–/– mice. However, in LXRα–/– mice the expected truncated LXRα transcript lacking the 500 nucleotides, corresponding to exons 4 and 5, was formed (Figure 1d).
LXRβ–/– mice were fertile and could produce offspring. No macroscopic phenotype was observed under standard conditions.
Since LXRβ and LXRα bind the same oxysterols with similar affinity (6, 7) and since Peet et al. (10) have shown that LXRα-deficient mice lose their natural resistance to dietary cholesterol, we addressed the question of whether LXRβ-deficient mice respond to cholesterol feeding in a similar way. Male and female LXRβ–/– mice and their corresponding wild-types were challenged with 2% cholesterol-enriched diet for 4 weeks. In a separate experiment LXRα–/– and their wild-types were treated similarly.
On a standard diet, no differences in the macroscopic appearance of the livers of LXRβ–/– and wild-type controls were seen. Surprisingly, livers from LXRβ–/– mice on 2% cholesterol were also similar to those from the corresponding wild-type controls (not shown). As expected, the livers of LXRα–/– mice on 2% cholesterol showed a dramatic change in color and an increase in size (Figure 2). Chemical analysis of hepatic extracts confirmed that the physiological resistance to dietary cholesterol was preserved in LXRβ–/– mice. An increase in hepatic total cholesterol of a magnitude similar to that of wild-type controls (less than twofold) was observed in LXRβ–/– mice after cholesterol feeding (LXRβ–/– vs. wild-type, not significant; Power >95%; Figure 3a), whereas a fivefold increase was found in LXRα–/– mice (Figure 3b).
Figure 2.
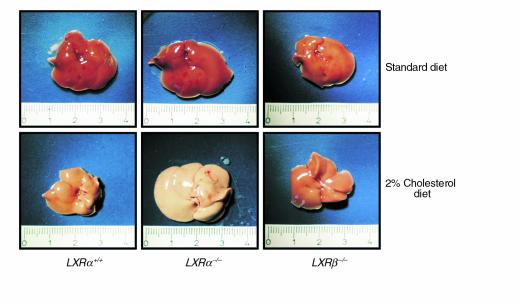
Macroscopic appearance of livers from LXRα+/+, LXRα–/–, and LXRβ–/– mice. Animals were fed a standard rodent diet or a 2% cholesterol-enriched diet for 28 days. Livers were removed immediately after the animals were sacrificed.
Figure 3.
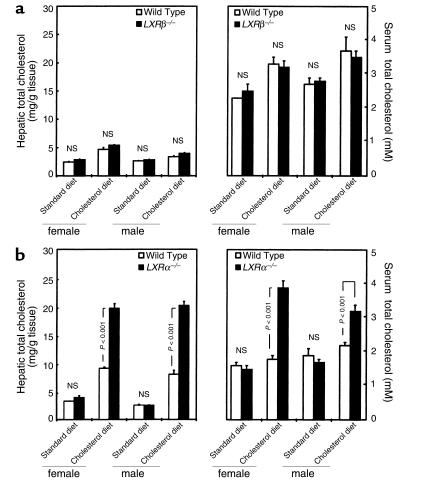
(a and b) Total cholesterol content in hepatic lipid extracts and in serum. Animals were fed a standard rodent diet or 2% cholesterol-enriched diet for 28 days. Livers were removed immediately after the animals were sacrificed. Blood was obtained by cardiac puncture and serum was collected by centrifugation. Total cholesterol was determined enzymatically in hepatic lipid extracts and serum from ten animals per experimental group. All values are expressed as mean + SEM. P values are versus wild-type. The results from two independent analyses are shown in a and b. NS, not significant.
Analysis of serum cholesterol again showed that LXRβ–/– mice responded in the same way as wild-type controls after dietary cholesterol (LXRβ–/– vs. wild-type, not significant; Power >95%; Figure 3a). This finding was further confirmed by comparable plasma lipoprotein profiles in these two categories of animals (Figure 4). In LXRα–/– mice an increase in serum cholesterol was found in response to cholesterol feeding (Figure 3b), especially LDL, as seen upon analysis of lipoprotein profiles (Figure 4).
Figure 4.
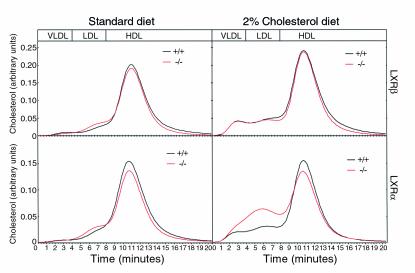
Serum lipoprotein profiles after separation by FPLC in LXRβ–/– (top) and LXRα–/– (bottom) mice. Animals were fed standard rodent diet (left) or challenged with 2% cholesterol (right) for 28 days. Ten microliters of pooled serum from each group was directly separated on a FPLC Superose 6B column. Cholesterol content was determined using the commercially available kit MPR 21442350 (Roche Molecular Biochemicals, Indianapolis, Indiana, USA) that was mixed online with the separated lipoproteins at a flow rate of 40 + 40 μl/min. The mixture was passed over a 37°C reaction coil and adsorbance was monitored every 20 seconds at 500 nm. The adsorbance profiles from two independent analyses are shown in the upper and lower panels.
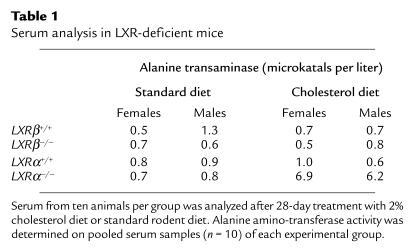
Dietary cholesterol did not increase serum alanine amino transferase (ALAT) activity in LXRβ–/– mice. LXRα–/– mice showed a distinct increase in serum ALAT activity after cholesterol feeding (Table 1).
Table 1.
Serum analysis in LXR-deficient mice
Recently, alternative pathways for the conversion of oxysterols into bile acids have been described, starting with hydroxylation in the 27 position of cholesterol in the liver and extrahepatic organs (34–36) or hydroxylation of cholesterol in the 24 position occurring in the brain (36, 37). Since 24-hydroxycholesterol has been demonstrated to be a potent ligand for both LXRs in vitro (6, 7), we wanted to investigate if disruption of the LXR genes would affect this pathway. Therefore, we determined the serum levels of 24- and 27-hydroxycholesterol in pooled samples from both LXR-mutant mouse lines. In LXRβ–/– mice, 24- and 27-hydroxycholesterol levels differed from those of wild-type controls. Conversely, an increased serum concentration of 24- and particularly 27-hydroxycholesterol was found in response to cholesterol feeding in LXRα–/– mice (Figure 5).
Figure 5.
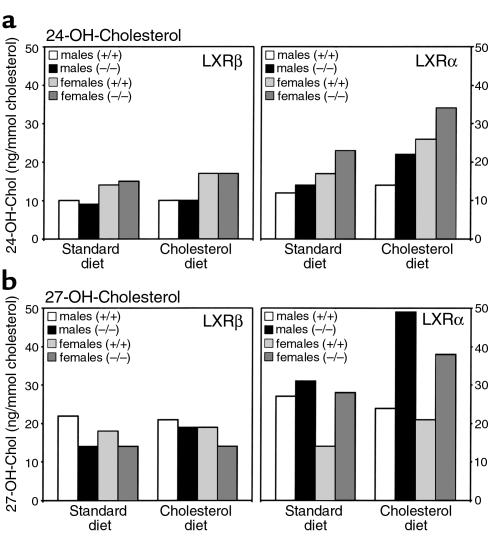
Measurement of 24-hydroxycholesterol (24-OH-Chol) (a) and of 27-hydroxycholesterol (27-OH-Chol) (b) in serum of LXRα–/– or LXRβ–/– mice and their respective wild-type controls. All values were obtained from pooled serum of the same animals as in Figure 3.
Analysis of mRNA expression of genes involved in the regulation of cholesterol, fatty acid, and bile acid metabolism was performed to identify alterations in both LXR mutants. Northern blot hybridizations were done with mRNA extracted from pooled livers of female mice. Signals were quantitated and related to GAPDH mRNA expression.
In LXRβ–/– mice on a standard diet, an increase in 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase (1.4-fold), FPP synthase (1.4-fold), and squalene synthase (1.6-fold) was observed as compared with wild-type controls (Figure 6). Differences in hepatic expression of HMG-CoA reductase, FPP synthase, and squalene synthase mRNA were not observed in cholesterol-fed LXRβ mutants and LXRα mutants when fed with both diets.
Figure 6.
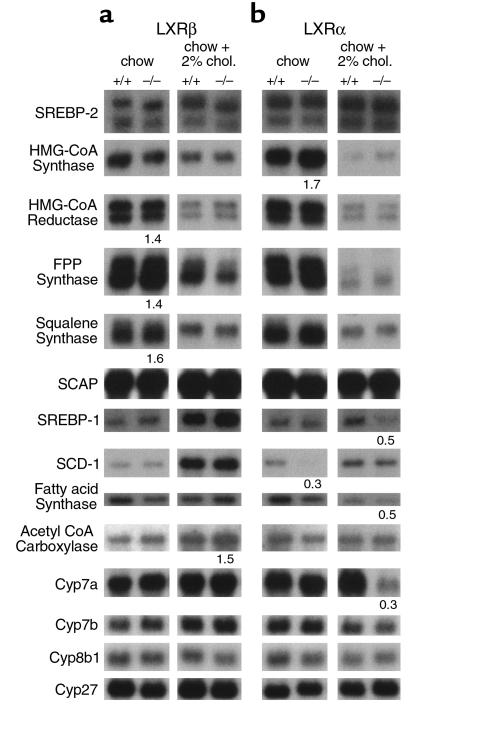
Expression of various lipid-regulating genes and of genes coding for enzymes in the neutral and acidic bile acid pathway in LXRβ–/– mice (a), LXRα–/– mice (b), and corresponding wild-type controls assayed by Northern blot analysis. Used were 2 or 5 μg/lane poly A+-enriched RNA, extracted from a pool of livers from five female mice fed as indicated. Filters were hybridized with [α-32P] dCTP-labeled cDNA probes, generated as described in Methods. Signals were standardized to the corresponding GAPDH expression. Data show the fold change compared with the respective wild-type control group fed the same diet. The results are representative of two or more independent determinations.
We next addressed the question of whether the expression of the lipogenic genes SCD-1, FAS, acetyl CoA carboxylase, and the transcriptional regulator ADD-1/SREBP-1 was altered by inactivation of the LXRβ gene. Northern blot analysis showed that the mRNA levels of these genes were not altered in animals on standard diet. After cholesterol feeding, a 1.5-fold increased mRNA level of acetyl CoA carboxylase was observed in LXRβ–/– mice (Figure 6). Disruption of the LXRα gene resulted in a downregulation of SCD-1, while expression of FAS, acetyl CoA carboxylase, and ADD-1/SREBP-1 remained unchanged. Cholesterol feeding reduced SREBP-1 and FAS mRNA levels in LXRα–/– mice compared with their wild-types (Figure 6).
In addition, we determined the hepatic mRNA expression of some selected genes important in the neutral and acidic biosyntheses of bile acids: Cyp7a, Cyp7b, Cyp8b1, and Cyp27. LXRβ-mutant mice fed either with standard chow or 2% cholesterol showed no differences in the expression levels of these genes (Figure 6). In LXRα–/– mice on either diet, the expression levels of Cyp7b, Cyp8b1, and Cyp27 were similar to those of wild-type animals. In contrast to what was observed in LXRβ–/– mice, cholesterol feeding reduced the mRNA level of Cyp7a in the liver of LXRα–/– mice (Figure 6).
Discussion
In this report we compare the function of the LXR receptors in hepatic cholesterol metabolism in vivo. We have focused particularly on responses after cholesterol feeding. We generated two mutant mouse lines bearing targeted disruptions of LXRα or LXRβ. Similar to what was described in a previous report on LXRα-deficient mice (10), we found LXRβ–/– mice to be fertile and show no apparent abnormal phenotypes. LXRβ–/– mice maintained a normal response following dietary cholesterol. Both on standard diet and on cholesterol-enriched diet, livers of LXRβ–/– mice preserved a normal appearance. After cholesterol challenge, hepatic cholesterol concentrations increased in a way similar to their wild-type controls. This was associated with normal serum cholesterol levels and with normal lipoprotein profiles. These results are therefore in striking contrast to the data obtained from cholesterol-fed LXRα–/– mice. As reported previously (10) and as confirmed in this study, a dramatic increase in hepatic cholesterol content and a pronounced elevation of serum cholesterol levels, especially in the LDL fraction, were indeed observed in this animal model.
In females of both wild-type and LXRβ–/– mice, a trend toward a larger increase in hepatic cholesterol levels than in corresponding male mice was observed in response to cholesterol feeding. This is in agreement with data from Turley et al. (38), showing sex-related differences in response to cholesterol feeding in 129/Sv and C57BL/6 inbred mice. A larger bile acid pool size and a consequently increased intestinal cholesterol absorption have been proposed to be responsible for a more pronounced increase in hepatic cholesterol concentration in female mice on cholesterol feeding (38). Such a sex-related difference in hepatic cholesterol was not observed in LXRα–/– mouse mutants following cholesterol feeding, probably since deletion of the LXRα gene resulted in such extensive metabolic perturbations that possible sex-related differences would not be apparent.
High levels of serum 27-hydroxycholesterol following cholesterol feeding in LXRα–/– mice represent another marked difference between LXRα–/– and LXRβ–/– mice. The observation of unchanged mRNA levels for Cyp27 and Cyp8b1 indicate that a new dynamic equilibrium between cholesterol as substrate and its oxysterol products may have been reached in LXRα–/– mice. The level of 27-hydroxycholesterol in the circulation is dependent upon three factors: the activity of the synthesizing enzyme sterol 27-hydroxylase, the activity of the metabolizing enzyme oxysterol 7α-hydroxylase, and the level of cholesterol. The level of cholesterol is of importance since 27-hydroxycholesterol is transported in the same lipoproteins as cholesterol, and there is a relatively constant ratio between cholesterol and 27-hydroxycholesterol levels in humans (39). In LXRα–/– mice fed high cholesterol, not only the absolute but also the cholesterol-related levels of 27-hydroxycholesterol were elevated. Thus the observed effect cannot be a dilution effect only. Since our results excluded a downregulation of hepatic Cyp7b expression, decreased metabolism of 27-hydroxycholesterol cannot be the explanation for the increase. An increased formation of 27-hydroxycholesterol is thus a probable explanation for the increased level of these oxysterols in the circulation. Furthermore, since the LXRα-deficient mice did not have an increased expression of the Cyp27 gene, an increased formation of 27-hydroxycholesterol likely is due to an increased enzyme activity, possibly resulting from the higher availability of the substrate, cholesterol. It must be assumed that the mitochondrial sterol 27-hydroxylase is not normally saturated with its substrate. The situation may be similar for oxysterol 24(S) hydroxycholesterol.
To explain the abnormal response of LXRα–/– mice to cholesterol feeding, it has been proposed that the normal increase in bile acid synthesis — secondary to an upregulation of Cyp7a expression — is greatly reduced in these animals (10). In cholesterol-fed LXRβ–/– mice, the mRNA levels of Cyp7a, Cyp7b, and Cyp8b1, as well as the enzymatic activity of CYP7A (data not shown), were comparable to those of wild-type controls, indicating a normal hepatic bile acid metabolism. Our results are, however, in disagreement with published data showing an increase of Cyp7a gene expression after cholesterol feeding (40), since this was not observed in either wild-type controls or in LXRβ–/– mice. Furthermore, in cholesterol-fed LXRα–/– mice, Cyp7a mRNA expression was reduced in contrast to was what found by Peet et al. (10). A possible explanation for this discrepancy could be that animals were fasted 5 hours before sacrifice, since fasting reduces bile acid synthesis in rodents (41, 42).
The reduced Cyp7a mRNA expression in LXRα–/– mice when challenged with dietary cholesterol was not accompanied by a decreased Cyp27 mRNA expression, although a coordinated regulation of Cyp27 and Cyp7a genes by bile acids has been observed previously both in vitro and in vivo in the rat (43, 44). Recently, it has been demonstrated that bile acids can signal via the nuclear receptor FXR (45–47), and that this receptor is responsible for at least part of the negative feedback regulation of CYP7A by bile acids (47). If bile acids control the expression of Cyp27 through binding to FXR is still unknown. Nevertheless, an interesting possibility is that bile acids might regulate the expression of both Cyp27 and Cyp7a through FXR, whereas cholesterol would only control the expression of Cyp7a (via LXRα) but not of Cyp27. This notion is also supported by the observations on the dietary regulation of CYP7A and CYP27 in normal mouse liver (48).
Compared with their wild-type controls, LXRβ–/– mice on a normal diet show an increase in the mRNA levels of HMG-CoA reductase, FPP synthase, and squalene synthase, three genes encoding enzymes involved in cholesterol biosynthesis. These increases occurred despite normal expression of the genes for SCAP and the SREBPs, proteins known to regulate the transcription of the former three genes (49, 50). However, whether fed a standard or cholesterol-enriched diet, LXRβ-deficient male mice did not show an altered enzymatic activity of hepatic HMG-CoA reductase (data not shown).
In our study we observed variations in serum and hepatic cholesterol levels between the two wild-type control groups. The knockout mice and their corresponding wild-types were descendants from different progenitors, both having mixed C57BL/6-129/Sv backgrounds. Variations of the genetic background might influence the expression of genes involved in the regulation of cholesterol metabolism. Differences in cholesterol concentrations in hepatic tissue and plasma have been shown previously among inbred mouse strains, especially between C57BL/6 and 129/Sv mice (51). The partial recombinant inbred breeding as used in our study may accentuate these strain-specific differences. A previous study from Jolley et al. (52) demonstrated that cholesterol absorption differs among mice having pure C57BL/6 and 129/Sv background (52), possibly leading to a variability in cholesterol absorption in mice with mixed C57BL/6 and 129/Sv background. However, both C57BL/6 and 129/Sv mice respond to dietary cholesterol with increased plasma and hepatic cholesterol levels as well as increased bile acid synthesis, albeit with different magnitude (52). Furthermore, it should be noted that our data are generated in two independent experiments, performed at different times. Cholesterol homeostasis is regulated in a complex way. Therefore, the identification of factors responsible for variations in plasma and hepatic cholesterol levels observed in different experiments is very difficult, unless appropriate studies are performed. In an additional control experiment in which animals were sacrificed and serum determinations carried out at the same time, no differences in serum cholesterol of LXRα+/+ and LXRβ+/+ mice were observed (data not shown). Thus, the maintained resistance to cholesterol feeding in LXRβ–/– mice is less likely caused by the variability of the mixed genetic background of the progenitor, but is presumably due to the presence of an intact LXRα gene in these mice.
LXRβ, similar to LXRα, appears to be involved in the regulation of cholesterol metabolism in peripheral tissues, since both LXRs seem to have a critical role in the regulation of ABCA1 and ABCG1 (18–22). Preliminary data on cholesterol-challenged double-LXRα/LXRβ-mutant mice obtained by mating of mice with either the LXRα+/–/LXRβ–/– or LXRα–/–/LXRβ–/– genotype, revealed no obvious additive or synergistic effect compared with LXRα–/– alone (data not shown). This further supports the idea of no major overlap function of the LXRs in the regulation of hepatic cholesterol metabolism. Since appropriate control mice were not available, additional experiments should be performed to confirm these findings.
In conclusion, our data indicate that LXRβ, compared to LXRα, does not have the same important function in regulating the hepatic response to cholesterol challenge in mice, albeit subtle effects on the expression of enzymes involved in lipid metabolism were observed. Our findings are particularly interesting since both LXRs have a high degree of amino acid identity (17), similar ligand-binding affinity to endogenous oxysterols (6, 7), and redundant functions in macrophages and enterocytes (19, 21).
Acknowledgments
We thank Helena Sundén and Annika Samuelsson for ES-cell culture, and José Inzunza for blastocyst injections and implantations; Ralf Kühn and Klaus Rajewsky (Cologne, Germany) for the generous gift of pGEM30, pL2neo, and pIC-Cre vectors; Stefan Alexson and Christine Sadek for Northern probes; Anne Plück and Kristina Vintersten (Transgenic Core Facility, EMBL, Heidelberg, Germany) for delivery and implantation of cryo-preserved deleter mouse embryos, and Christine Sadek and Thomas Günther for critical reading of the manuscript. This work was funded by the Swedish Medical Research Council (13X-2819, 03X-3141, and 03X-7137), the Tore Nilson, Ruth and Bertil Julin, Fredrik and Ingrid Thuring, Ax:son Johnson, Swedish Heart-Lung foundations, the Foundation of Strategic Research, and KaroBio AB (Sweden).
Footnotes
S. Alberti and D. Feltkamp’s present address is: University of Tübingen, Institute for Cell Biology, Department of Molecular Biology, Tübingen, Germany.
References
- 1.Gordon T, Kannel WB, Castelli WP, Dawber TR. Lipoproteins, cardiovascular disease, and death. The Framingham study. Arch Intern Med. 1981;141:1128–1131. [PubMed] [Google Scholar]
- 2.Keys A. Coronary heart disease, serum cholesterol, and the diet. Acta Med Scand. 1980;207:153–160. doi: 10.1111/j.0954-6820.1980.tb09697.x. [DOI] [PubMed] [Google Scholar]
- 3.Angelin B. 1994 Mack-Forster Award Lecture. Review. Studies on the regulation of hepatic cholesterol metabolism in humans. Eur J Clin Invest. 1995;25:215–224. doi: 10.1111/j.1365-2362.1995.tb01552.x. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- 5.Accad M, Farese RV., Jr Cholesterol homeostasis: a role for oxysterols. Curr Biol. 1998;8:R601–R604. doi: 10.1016/s0960-9822(98)70386-0. [DOI] [PubMed] [Google Scholar]
- 6.Lehmann JM, et al. Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J Biol Chem. 1997;272:3137–3140. doi: 10.1074/jbc.272.6.3137. [DOI] [PubMed] [Google Scholar]
- 7.Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. An oxysterol signalling pathway mediated by the nuclear receptor LXR alpha. Nature. 1996;383:728–731. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- 8.Forman BM, Ruan B, Chen J, Schroepfer J, Evans RM. The orphan nuclear receptor LXRα is positively and negatively regulated by distinct products of mevalonate metabolism. Proc Natl Acad Sci USA. 1997;94:105888–105893. doi: 10.1073/pnas.94.20.10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.A unified nomenclature system for the Nuclear Receptor Superfamily. Cell. 1999;97:161–163. doi: 10.1016/s0092-8674(00)80726-6. [DOI] [PubMed] [Google Scholar]
- 10.Peet DJ, et al. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell. 1998;93:693–704. doi: 10.1016/s0092-8674(00)81432-4. [DOI] [PubMed] [Google Scholar]
- 11.Shinar DM, et al. NER, a new member of the gene family encoding the human steroid hormone nuclear receptor. Gene. 1994;147:273–276. doi: 10.1016/0378-1119(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 12.Seol W, Choi HS, Moore DD. Isolation of proteins that interact specifically with the retinoid X receptor: two novel orphan receptors. Mol Endocrinol. 1995;9:72–85. doi: 10.1210/mend.9.1.7760852. [DOI] [PubMed] [Google Scholar]
- 13.Song C, Kokontis JM, Hiipakka RA, Liao S. Ubiquitous receptor: a receptor that modulates gene activation by retinoic acid and thyroid hormone receptors. Proc Natl Acad Sci USA. 1994;91:10809–10813. doi: 10.1073/pnas.91.23.10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teboul M, et al. OR-1, a member of the nuclear receptor superfamily that interacts with the 9-cis-retinoic acid receptor. Proc Natl Acad Sci USA. 1995;92:2096–2100. doi: 10.1073/pnas.92.6.2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Willy PJ, et al. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995;9:1033–1045. doi: 10.1101/gad.9.9.1033. [DOI] [PubMed] [Google Scholar]
- 16.Apfel R, et al. A novel orphan receptor specific for a subset of thyroid hormone-responsive elements and its interaction with the retinoid/thyroid hormone receptor subfamily. Mol Cell Biol. 1994;14:7025–7035. doi: 10.1128/mcb.14.10.7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alberti S, Steffensen KR, Gustafsson J-Å. Structural characterization of the mouse nuclear oxysterol receptor genes LXRα and LXRβ. Gene. 2000;243:93–103. doi: 10.1016/s0378-1119(99)00555-7. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz K, Lawn RM, Wade DP. ABC1 gene expression and ApoA-I-mediated cholesterol efflux are regulated by LXR. Biochem Biophys Res Commun. 2000;274:794–802. doi: 10.1006/bbrc.2000.3243. [DOI] [PubMed] [Google Scholar]
- 19.Venkateswaran A, et al. Human white/murine ABC8 mRNA levels are highly induced in lipid-loaded macrophages. A transcriptional role for specific oxysterols. J Biol Chem. 2000;275:14700–14707. doi: 10.1074/jbc.275.19.14700. [DOI] [PubMed] [Google Scholar]
- 20.Klucken J, et al. ABCG1 (ABC8), the human homolog of the Drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport. Proc Natl Acad Sci USA. 2000;97:817–822. doi: 10.1073/pnas.97.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Repa JJ, et al. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science. 2000;289:1524–1529. doi: 10.1126/science.289.5484.1524. [DOI] [PubMed] [Google Scholar]
- 22.Costet P, Luo Y, Wang N, Tall AR. Sterol-dependent transactivation of the ABC1 promoter by the liver X Receptor/Retinoid X receptor. J Biol Chem. 2000;275:28240–28245. doi: 10.1074/jbc.M003337200. [DOI] [PubMed] [Google Scholar]
- 23.Gu H, Zou YR, Rajewsky K. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 1993;73:1155–1164. doi: 10.1016/0092-8674(93)90644-6. [DOI] [PubMed] [Google Scholar]
- 24.Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–106. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- 25.Mansour SL, Thomas KR, Capecchi MR. Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature. 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 26.Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Folch J, Lees M, Stanley GHS. A simple method for the isolation and purification of total lipids from animals tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 29.Dzeletovic S, Breuer O, Lund E, Diczfalusy U. Determination of cholesterol oxidation products in human plasma by isotope dilution-mass spectrometry. Anal Biochem. 1995;225:73–80. doi: 10.1006/abio.1995.1110. [DOI] [PubMed] [Google Scholar]
- 30.Bergmeyer HU, Horder M, Rej R. IFCC expert panel on enzymes. Part 2. J Clin Chem Clin Biochem. 1986;24:497–510. [PubMed] [Google Scholar]
- 31.Shimano H, et al. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J Clin Invest. 1996;98:1575–1584. doi: 10.1172/JCI118951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marchuk D, Drumm M, Saulino A, Collins FS. Construction of T-vectors, a rapid and general system for direct cloning of unmodified PCR products. Nucleic Acids Res. 1991;19:1154. doi: 10.1093/nar/19.5.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Snedecor, G.W., and Cochran, W.G. 1980. Statistical methods. Seventh edition. The Iowa State University Press. Ames, Iowa, USA. 507 pp.
- 34.Stravitz RT, et al. Regulation of sterol 27-hydroxylase and an alternative pathway of bile acid biosynthesis in primary cultures of rat hepatocytes. J Steroid Biochem Mol Biol. 1996;57:337–347. doi: 10.1016/0960-0760(95)00282-0. [DOI] [PubMed] [Google Scholar]
- 35.Axelson M, Sjövall J. Potential bile acid precursors in plasma: possible indicators of biosynthetic pathways to cholic and chenodeoxycholic acids in man. J Steroid Biochem. 1990;36:631–640. doi: 10.1016/0022-4731(90)90182-r. [DOI] [PubMed] [Google Scholar]
- 36.Björkhem I, Diczfalusy U, Lütjohann D. Removal of cholesterol from extrahepatic sources by oxidative mechanisms. Curr Opin Lipidol. 1999;10:161–165. doi: 10.1097/00041433-199904000-00010. [DOI] [PubMed] [Google Scholar]
- 37.Lütjohann D, et al. Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci USA. 1996;93:9799–9804. doi: 10.1073/pnas.93.18.9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turley SD, Schwarz M, Spady DK, Dietschy JM. Gender-related differences in bile acid and sterol metabolism in outbred CD-1 mice fed low- and high-cholesterol diets. Hepatology. 1998;28:1088–1094. doi: 10.1002/hep.510280425. [DOI] [PubMed] [Google Scholar]
- 39.Harik-Khan R, Holmes RP. Estimation of 26-hydroxycholesterol in serum by high-performance liquid chromatography and its measurement in patients with atherosclerosis. J Steroid Biochem. 1990;36:351–355. doi: 10.1016/0022-4731(90)90228-k. [DOI] [PubMed] [Google Scholar]
- 40.Rudling M. Hepatic mRNA levels for the LDL receptor and HMG-CoA reductase show coordinate regulation in vivo. J Lipid Res. 1992;33:493–501. [PubMed] [Google Scholar]
- 41.Björkhem, I. 1985. Mechanism of bile acid synthesis in mammalian liver. In New comprehensive biochemistry. Volume 12. Elsevier Science Publishing Co. Amsterdam, The Netherlands. 231–278.
- 42.Myant NB, Mitropoulos KA. Cholesterol 7 alpha-hydroxylase. J Lipid Res. 1977;18:135–153. [PubMed] [Google Scholar]
- 43.Twisk J, et al. Heterogeneous expression of cholesterol 7 alpha-hydroxylase and sterol 27-hydroxylase genes in the rat liver lobulus. J Clin Invest. 1995;95:1235–1243. doi: 10.1172/JCI117773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Twisk J, et al. Structural aspects of bile acids involved in the regulation of cholesterol 7 alpha-hydroxylase and sterol 27-hydroxylase. Eur J Biochem. 1995;228:596–604. doi: 10.1111/j.1432-1033.1995.0596m.x. [DOI] [PubMed] [Google Scholar]
- 45.Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3:543–553. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- 46.Parks DJ, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 47.Makishima M, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 48.Schwarz M, Lund EG, Russell DW. Two 7 alpha-hydroxylase enzymes in bile acid biosynthesis. Curr Opin Lipidol. 1998;9:113–118. doi: 10.1097/00041433-199804000-00006. [DOI] [PubMed] [Google Scholar]
- 49.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 50.Hua X, Nohturfft A, Goldstein JL, Brown MS. Sterol resistance in CHO cells traced to point mutation in SREBP cleavage-activating protein. Cell. 1996;87:415–426. doi: 10.1016/s0092-8674(00)81362-8. [DOI] [PubMed] [Google Scholar]
- 51.Kirk EA, et al. Hyper- and hypo-responsiveness to dietary fat and cholesterol among inbred mice: searching for level and variability genes. J Lipid Res. 1995;36:1522–1532. [PubMed] [Google Scholar]
- 52.Jolley CD, Dietschy JM, Turley SD. Genetic differences in cholesterol absorption in 129/Sv and C57BL/6 mice: effect on cholesterol responsiveness. Am J Physiol. 1999;276:G1117–G1124. doi: 10.1152/ajpgi.1999.276.5.G1117. [DOI] [PubMed] [Google Scholar]