

Abstract
Rationale: Asthma is a syndrome whose common pathogenic expression is inflammation of the airways. Plasminogen plays an important role in cell migration and is also implicated in tissue remodeling, but its role in asthma has not been defined.
Objectives: To test whether plasminogen is a critical component in the development of asthma.
Methods: We used a mouse model of ovalbumin-induced pulmonary inflammation in Plg+/+, Plg+/−, and Plg−/− mice.
Measurements and Main Results: The host responses measured included lung morphometry, and inflammatory mediators and cell counts were assessed in bronchoalveolar lavage fluid. Bronchoalveolar lavage demonstrated a marked increase in eosinophils and lymphocytes in ovalbumin-treated Plg+/+ mice, which were reduced to phosphate-buffered saline–treated control levels in Plg+/− or Plg−/− mice. Lung histology revealed peribronchial and perivascular leukocytosis, mucus production, and increased collagen deposition in ovalbumin-treated Plg+/+ but not in Plg+/− or Plg−/− mice. IL-5, tumor necrosis factor-α, and gelatinases, known mediators of asthma, were detected in bronchoalveolar lavage fluid of ovalbumin-treated Plg+/+ mice, yet were reduced in Plg−/− mice. Administration of the plasminogen inhibitor, tranexamic acid, reduced eosinophil and lymphocyte numbers, mucus production, and collagen deposition in the lungs of ovalbumin-treated Plg+/+ mice.
Conclusions: The decreased inflammation in the lungs of Plg−/− mice and its blockade with a plasminogen inhibitor indicate that plasminogen plays an important role in orchestrating the asthmatic response and suggests that plasminogen may be a therapeutic target for the treatment of asthma.
Keywords: lung, knockout mice, pulmonary inflammation, fibrinolysis
AT A GLANCE COMMENTARY
Scientific Knowledge on the Subject
Asthma is a syndrome whose common pathogenic expression is inflammation of the airways. Plasminogen plays an important role in cell migration and is also implicated in tissue remodeling, but its role in asthma has not been defined.
What This Study Adds to the Field
The decreased inflammation in the lungs of Plg−/− mice and its blockade with a plasminogen inhibitor indicate that plasminogen plays an important role in asthma and suggest that plasminogen may be a therapeutic target for the treatment of the disease.
Asthma is a complex response in which the genetic background and the immune system interact with environmental factors to elicit the overt manifestations of the disease (1). Inflammatory cells, mainly T lymphocytes, monocytes/macrophages, mast cells, and eosinophils, are recruited into the lungs where they act as the primary mediators in the acute- and late-phase asthmatic responses (1). As a consequence of the chronic inflammation, lung morphology may become altered even in milder forms of the disease (2, 3). Reduction in inflammatory cell migration could potentially ameliorate the severity of asthma.
Inflammatory cell migration depends on the activity of the plasminogen system. Plasminogen is the precursor of the active serine protease plasmin. This conversion is induced by the plasminogen activators urokinase (uPA) or tissue plasminogen activator (tPA), and plasmin activity is tightly regulated by multiple inhibitors and modulators (reviewed in Reference 4). Plasmin has broad substrate recognition. Its capacity to efficiently cleave fibrin establishes its role as the primary fibrinolytic enzyme. In addition, plasmin degrades a variety of extracellular matrix (ECM) proteins (5, 6) and can activate a number of the matrix metalloproteinases (MMPs) (7), which extends its capacity to turnover the ECM and thus facilitate inflammatory cell migration.
Studies conducted in plasminogen (Plg)-deficient mice have demonstrated significant suppression of inflammatory cell recruitment in response to a variety of experimental stimuli (8, 9). In another study, marked alteration in inflammatory cell recruitment into the lungs of Plg−/− mice suppressed the development of tissue damage in a model of lung injury (10). Nevertheless, even though an organ-specific relationship of plasminogen to lung pathology is emerging, the linkage of plasminogen to bronchial asthma is almost nonexistent.
Several clinical studies have demonstrated activation of the blood coagulation and plasminogen systems in patients with asthma (11). Such associations can arise secondarily to primary pathogenic events. Recent studies suggest more causal roles of the plasminogen system in the pathogenesis of asthma. Eotaxin, a potent eosinophil chemotactic factor, was more effective in attracting eosinophils from subjects with asthma than from normal subjects; and this migration was plasminogen- and MMP-dependent (12). This plasminogen-dependent mechanism may contribute to the inflammatory response and early histological changes in the airways of patients with asthma.
The goal of these studies was to test the hypothesis that plasminogen plays a significant role in the pathogenesis of asthma by influencing inflammatory cell recruitment and subsequent manifestations of asthma such as increased collagen and mucus hypersecretion. We tested this hypothesis using an ovalbumin (OVA) sensitization and challenge model of asthma (reviewed in Reference 13) in mice with reduced (Plg+/−) or absent (Plg−/−) plasminogen and their wild-type (WT) littermates. In addition, we manipulated the function of plasminogen in vivo to determine if it may represent a therapeutic target for the treatment of asthma. Some of the results of these studies have been previously reported in the form of an abstract (14).
METHODS
Animals
Plg−/− mice were developed and characterized previously (15). The mice were in a 50/50 J129/C57BL/6 mixed background or bred for seven generations into a C57BL/6 background. The mice used were littermates, produced by mating of heterozygous (Plg+/−) mice. Immunocompetent, nonirradiated female BALB/c mice, 6 to 8 weeks old, from the Jackson Laboratory (Bar Harbor, ME) were used for airway hyperresponsiveness (AHR) studies. Details can be found in the online supplement. All in vivo manipulations were performed under protocols approved by the Institutional Animal Care and Use Committee of the Cleveland Clinic.
OVA Model
A murine model of lung eosinophilia was used as previously described (16). Mice were sensitized with an intraperitoneal injection of OVA (0.1 mg/400 μl phosphate-buffered saline [PBS]/mouse; Sigma, St. Louis, MO) on Day 1 followed by exposure to aerosolized antigen (2% OVA for 5 min on Day 8 and 1% OVA for 20 min on Days 15–21). Control mice received PBS. To evaluate cellular recruitment over time, animals were killed at 3 hours post-treatment on Days 15 and 18, and at 1, 3, and 6 hours post-treatment on Day 21.
Leukocyte Recruitment
Bronchoalveolar lavage (BAL) was performed as described (10) by infusing 1 ml of PBS via a tracheal cannula. After centrifugation, the BAL fluid was frozen for cytokine and MMP measurements. BAL cells were cleared of erythrocytes and counted, and differentials were performed using Wright stain (see details in the online supplement).
Histology
After BAL, the lungs were prepared for histological examination. Five-micron paraffin sections were stained with hematoxylin and eosin. Collagen deposition and mucin production were assessed from adjacent tissue sections stained with Mason's trichrome or periodic acid-Schiff (PAS), respectively, by quantitative morphometric techniques (described in the online supplement). A Sircol (Biocolor Ltd., Newtownabbey, Northern Ireland) assay was used to quantify new collagen deposition in lung homogenates.
Tranexamic Acid Treatment
Plg+/+ mice received the OVA treatment outlined above. From Days 18 to 21, the mice had free access to water containing tranexamic acid (TXA) (trans-4-aminomethylcyclohexane carboxylic acid [AMCA, 50 mg/ml; Sigma, St. Louis, MO]). Animals were killed at 3 hours post-treatment on Day 21, and leukocyte differentials and histology were performed.
Cytokine mRNA Expression and Release
RNA protection assays on mRNA extracted from whole lungs were performed using the Riboquant RNase protection assay mouse cytokine kits (mCK-1 and mCK-5c; BD Biosciences, San Diego, CA) following the manufacturer's instructions. Type 1 and type 2 cytokines in the BAL fluid were analyzed using the Mouse Th1/Th2 Cytokine Cytometric Bead Array kit (BD Biosciences) according to the manufacturer's instructions. In a separate assay, transforming growth factor (TGF)-β1 was assayed using the immunoassay kit (MB100) from R&D Systems, Inc. (Minneapolis, MN). For monocyte chemotactic protein 1 (JE/MCP-1) and IL-13, ELISA kits from R&D Systems, Inc., were used. Details are provided in the online supplement.
Zymographic and Western Blot Analysis of MMPs
MMP-2 and MMP-9 in BAL fluid were detected by gelatin zymography and Western blot as detailed in the online supplement.
Measurement of AHR in Plg-deficient and WT Mice
C57Bl/6 and J129/C57BL/6 Plg+/+ and Plg−/− mice received the OVA treatment outlined above. On Day 21, 3 hours after treatment, animals were anesthetized by intraperitoneal injection with pentobarbital and placed on a rodent ventilator inside a body plethysmography chamber. After a stable baseline pressure was established, mice were given a dose of methacholine, 411 μg/kg in a volume of 40–55 μl. Resistance and compliance were determined using Buxco whole body plethysmography and Biosystems XA software (Buxco Electronics, Wilmington, NC).
Measurement of AHR in TXA-treated BALB/c Mice
Standard induction of allergic airway disease was performed as previously described (17). In brief, BALB/c mice were immunized by intraperitoneal injection with OVA (10 μg, precipitated in Al[OH]3). Two weeks later, a series of eight daily inhalations (40 min/d) were started, with mice placed as a group in a chamber kept saturated with nebulized OVA solution (1% wt/vol in sterile PBS). OVA-sensitized mice were divided into two groups: one group (OVA/TXA) received OVA plus TXA treatment as described above; the other group (OVA/H2O) received OVA and no TXA. On Day 22, AHR was measured as described above on both groups and naive (untreated) mice were used as controls.
Statistical Analysis
Values were expressed as means ± SEM. Comparisons were made using the Student t test and analysis of variance when more than two values were compared. The Mann-Whitney test (unpaired, nonparametric, two-tailed) was used for AHR comparisons. p values less than 0.05 were considered significant.
RESULTS
Plasminogen Deficiency Reduces Leukocyte Recruitment into the Lungs
The OVA sensitization/aerosol challenge model of asthma was used in Plg−/− and Plg+/− mice and their WT (Plg+/+) littermates. BAL was performed to identify and quantify the different cell types present. Compared with the PBS-treated mice, OVA caused an increase in total leukocytes in the airways of Plg+/+ mice on Days 18 and 21 but not on Day 15 (Figure 1). On Day 21, the increase in recruited cells in the OVA-treated animals was approximately fivefold greater than in the PBS-treated mice. In contrast, no increase was observed in the OVA-treated compared with the PBS-treated Plg−/− mice (Figure 1). Of note, the number of leukocytes in the PBS-treated Plg+/+ and Plg−/− mice was similar. Although total leukocytes from OVA-treated Plg+/− mice increased significantly over PBS-treated and Plg−/− mice (280.3 ± 26.7 × 103 cells [n = 16] vs. 137.5 ± 12.5 × 103 cells [n = 4], p = 0.02; and 131.4 ± 21.2 × 103 cells [n = 9], p = 0.0009, in PBS-treated and Plg−/− mice, respectively], they were reduced fourfold compared with their WT counterparts (797.5 ± 85.7 × 103 cells [n = 8], p = 0.0001) 3 hours after treatment on Day 21. These latter results suggest that even a partial reduction of systemic plasminogen levels, as in the Plg+/− mice (15) has a profound effect on leukocyte recruitment to the lungs in this model.
Figure 1.

Timeline of total leukocyte counts in bronchoalveolar lavage fluid of Plg+/+ (left) and Plg−/− (right) mice 3 hours after phosphate-buffered saline (PBS) or ovalbumin (OVA) treatment on Days 15 (open bars), 18 (striped bars), and 21 (solid bars). Values are expressed as mean ± SEM; n = 3–9 mice/group. OVA induced an increase in leukocyte counts in Plg+/+ (§p = 0.0001 vs. PBS), but not in Plg−/− mice (‡p = 0.8 vs. PBS).
To determine which leukocyte populations were affected by plasminogen deficiency, differentials were performed on BAL cells. Eosinophils increased in the OVA-treated Plg+/+ mice more than 500-fold over their PBS-treated counterparts (Figure 2). In sharp contrast, mice with partial (Plg+/−) or complete absence (Plg−/−) of plasminogen failed to show any increase in eosinophils (Table 1; Figure 2). This effect was not restricted only to eosinophils; lymphocyte and neutrophil recruitment was significantly suppressed in both Plg+/− and Plg−/− mice compared with the Plg+/+ mice at Day 21, 3 hours post–OVA challenge. In contrast, there was no significant increase in the total number of macrophages in the airways of OVA-treated Plg+/+ or Plg−/− mice on Day 21 at 3 hours after treatment compared with their PBS-treated counterparts (Figure 2). Importantly, reduction of pulmonary inflammation due to lack of plasminogen is not restricted by strain background (see the online supplement).
Figure 2.

Differential cell counts in bronchoalveolar lavage fluid from Plg+/+, Plg+/− and Plg−/− mice 3 hours after ovalbumin (OVA) challenge on Day 21. Values are expressed as mean ± SEM; n = 8–16 mice/group. OVA induced an increase in neutrophils (open bars), lymphocytes (dotted bars), and eosinophils (solid bars) in Plg+/+ but not in Plg+/− or Plg−/− mice (p < 0.001 by analysis of variance). There was no difference in the macrophage (striped bars) counts between the genotypes.
TABLE 1.
KINETICS OF LEUKOCYTE RECRUITMENT TO THE AIRWAYS AFTER OVALBUMIN TREATMENT ON DAY 21
| 1 h
|
3 h
|
6 h
|
||||
|---|---|---|---|---|---|---|
| Lymph (× 103) | Eos (× 103) | Lymph (× 103) | Eos (× 103) | Lymph (× 103) | Eos (× 103) | |
| Plg+/+ | 41.8 ± 24 | 86.7 ± 40.9 | 47.2 ± 11.7 | 571.9 ± 78.5 | 22.7 ± 16.7 | 78.9 ± 76.2 |
| Plg+/− | N/D | N/D | 6.6 ± 4.2 | 17.2 ± 9.9 | 1.4 ± 1.4 | 0 |
| Plg−/− | 0.9 ± 0.4 | 0 | 1.5 ± 0.6 | 12.3 ± 9.2 | 3.5 ± 1.1 | 5.8 ± 5.2 |
Definition of abbreviations: Eos = eosinophils; Lymph = lymphocytes; N/D = not determined.
n = 8–16 mice/group.
To determine if the lack of increase in eosinophils and lymphocytes in the Plg+/− and Plg−/− mice was due to delayed kinetics, we also studied leukocyte recruitment at Day 21, 6 hours after treatment. At this time point, eosinophils and lymphocytes had begun to decrease in Plg+/+ mice (Table 1). However, eosinophil and lymphocyte recruitment remained suppressed in the Plg+/− and Plg−/− mice (Table 1), showing that the recruitment of these cells was not simply delayed.
The extent and anatomic location of leukocyte infiltrates were determined in hematoxylin-and-eosin–stained sections of lungs taken at Day 21, 3 hours after OVA or PBS challenge. The airways from Plg+/+ and Plg−/− mice exposed to PBS showed similar and normal lung histology (Figures 3A and 3C). OVA induced widespread peribronchiolar and perivascular inflammation, which was primarily eosinophilic (Figure 3B, inset) in Plg+/+ mice compared with their PBS-treated counterparts (Figures 3A and 3B). In contrast to Plg+/+ mice, a substantial reduction in the number of leukocytes was observed in the same areas of lungs from Plg+/− (data not shown) and Plg−/− (Figure 3D and inset) mice. Instead, the lungs of these mice appeared to be similar to their PBS-treated controls (Figure 3C).
Figure 3.

Effects of phosphate-buffered saline (PBS) and ovalbumin (OVA) 3 hours after challenge on Day 21in the lungs of Plg+/+ and Plg−/− mice. Representative photomicrographs of hematoxylin-and-eosin–stained lung sections are shown; n = 3–9 mice/group. PBS treatment had no effect on the lungs of either Plg+/+ (A) or Plg−/− (C) mice. However, OVA induced peribronchial and perivascular leukocytosis in Plg+/+ mice (B), but not in Plg−/− (D) mice. Original magnifications for A–D are ×100. Higher magnification (×400) insets in B and D highlight the differences in leukocytosis in the peribronchial areas between the genotypes and the eosinophilic nature of the infiltrate in OVA-treated Plg+/+ mice. Bars = 137 μm.
Reduced Collagen and Mucus Production in Plasminogen Knockout Mice
Changes in collagen levels, both measured and by histopathology, goblet cell hyperplasia, and mucus production are noted early during the acute phase of OVA antigen exposure (18–20). To compare the early histological changes induced by OVA challenge, lung sections from Plg+/+ and Plg−/− mice were stained with Masson trichrome (for collagen deposition) and PAS (for goblet cell hyperplasia and increased mucus production). In lung tissue from PBS-treated mice of both genotypes, collagen deposition (stained blue) was detected around blood vessels but not in the airways as expected (see Figure 4A for Plg+/+). However, the lungs from OVA-treated Plg+/+ mice displayed dense deposition of collagen in large portions of the peribronchial area (Figure 4B and inset). This was not observed in Plg−/− mice, which showed limited collagen deposition (Figure 4C and inset). Quantification of the intensity of the Masson trichrome staining in lung sections indicated a 33% increase in collagen deposition in the OVA-treated Plg+/+ lungs compared with those treated only with PBS. In contrast, OVA-treated Plg−/− lungs showed no increase in staining intensity compared with their PBS-treated counterparts, and a 34% reduction in staining intensity compared with the OVA-treated Plg+/+ lungs (Figure 4G). Deposition of new collagen, assessed by Sircol assay, was also significantly increased in OVA-treated Plg+/+ lungs over those treated only with PBS, whereas OVA-treated Plg−/− lungs showed no increase over their PBS-treated counterparts (Figure 4H). As assessed by PAS staining, OVA treatment caused an increase in goblet cells and mucus production in the bronchial epithelium of Plg+/+ (Figure 4E and inset) but not in Plg−/− mice (Figure 4F and inset). PAS staining of the lung of OVA-treated Plg−/− mice resembled the PAS staining of lungs of PBS-treated mice (Figure 4D). Quantification of PAS staining verified a significant increase in mucus production in OVA-treated Plg+/+ lungs compared with their PBS-treated counterparts and OVA-treated Plg−/− lungs (Figure 4I). Therefore, plasminogen deficiency inhibits the early increase in collagen deposition, goblet cell hyperplasia, and mucus production, hallmarks of asthmatic lungs.
Figure 4.

Effects of phosphate-buffered saline (PBS) and ovalbumin (OVA) 3 hours after challenge on Day 21 in the lungs of Plg+/+ and Plg−/− mice. Representative photomicrographs of Masson trichrome (A–C)– and periodic acid-Schiff (PAS) (D–F)–stained lung sections are shown; n = 3–9 mice/group. PBS treatment had no effect on the lungs of Plg+/+ mice (A and D). OVA treatment resulted in increased deposition of collagen (blue) in large portions of the peribronchial area (B) as well as increased goblet cells and mucus production (magenta) in the bronchial epithelium (E) of Plg+/+ mice. These changes were not observed in the lungs of OVA-treated Plg−/− mice (C and F, respectively). Original magnifications for A–F are ×100. Higher magnification (×400) insets in B versus C and E versus F highlight the difference in collagen deposition and goblet cells and mucus production, respectively, between the genotypes. Bars = 137 μm. (G) Quantification of Masson trichrome–stained lung sections revealed a statistically significant increase (p = 0.009) in collagen deposition in OVA-treated Plg+/+ mice (n = 20) over their PBS counterparts (n = 11) and the PBS- (n = 9, p = 0.01) or OVA-treated Plg−/− mice (n = 10, p = 0.05). (H) Sircol assay using lung homogenates also showed a significant increase of new collagen in OVA-treated Plg+/+ mice (n = 4) over their PBS counterparts (n = 3, p = 0.05) and the PBS- (n = 3, p = 0.04) or OVA-treated Plg−/− mice (n = 3, p = 0.01). (I) Quantification of PAS-stained lungs sections revealed a significant increase of mucus production in OVA-treated Plg+/+ mice (n = 29) over their PBS counterparts (n = 16, p = 0.0001) and OVA-treated Plg−/− mice (n = 16, p = 0.001). SAL = PBS-treated; WT = wild-type.
TXA, a Plasminogen Inhibitor, Reduces Pulmonary Inflammation in Plg+/+ Mice
Given the dramatic reduction in inflammation in Plg−/− mice, we next asked if suppression of plasminogen in WT mice could alter cellular recruitment and histological changes caused by OVA treatment. C57/BL/6 mice, subjected to the same regimen of OVA sensitization and challenge as above, were administered TXA in their drinking water from Days 18 to 21. The amount of TXA used in the water is well tolerated and leads to blood levels of greater than 200 μM, which are sufficient to inhibit functions mediated by the lysine binding sites of plasminogen (21). WT mice treated with OVA/TXA had a reduced total leukocyte recruitment compared with those that received only OVA (Figure 5A). The inhibition was to the level of cells present in the BAL of PBS-treated control animals. Examination of the individual cell types showed that TXA treatment significantly reduced lymphocyte and eosinophil recruitment (Figure 5B), whereas the number of macrophages remained the same. As an important control, TXA caused no additional reduction in the total number of leukocytes in Plg−/− mice (Figure 5A), establishing specificity to the effect of TXA. Histological examination of lungs from OVA/TXA-treated WT mice showed reduced leukocytosis in the perivascular and peribronchial areas (Figure 6C) compared with WT mice that received only OVA (Figure 6A). Because TXA does not affect circulating blood cell numbers (unpublished data), its effect is exerted at the level of cell recruitment and/or migration into the lungs. In addition, TXA reduced the amount of collagen deposited in the subepithelial areas (Figure 6D) compared with treatment with OVA alone (Figure 6B). These data show that reduction of functional plasminogen is effective in reducing the pulmonary inflammation in this model of asthma.
Figure 5.

Effect of tranexamic acid (TXA) treatment on leukocyte recruitment to the lungs of C57BL/6 Plg+/+ and Plg−/− mice. (A) Total leukocyte counts in the bronchoalveolar lavage fluid of Plg+/+ (open bars) and Plg−/− (solid bar) mice after phosphate-buffered saline (PBS) or ovalbumin (OVA) treatment. Wild-type mice treated with OVA/TXA had reduced total leukocyte recruitment compared with those that received only OVA. The inhibition was to the level of cells present in the BAL of PBS-treated controls. TXA caused no additional reduction in leukocytes in Plg−/− mice. (B) Differential cell counts in BAL fluid showed that TXA (Solid bars) treatment significantly reduced lymphocyte and eosinophil recruitment while the number of macrophages remained the same as in mice treated only with OVA (open bars). Values are expressed as mean ± SEM; n = 3–9 mice/group.
Figure 6.

Effects of tranexamic acid (TXA) treatment on lung histology in ovalbumin (OVA)-treated Plg+/+ C57BL/6 mice 3 hours after challenge on Day 21. Representative photomicrographs of hematoxylin-and-eosin (A and C)– and Masson trichrome (B and D)–stained lung sections are shown (original magnification, ×100); n = 3–9 mice/group. TXA treatment reduced leukocytosis in the perivascular and peribronchial areas (C) compared with wild-type mice that received only OVA (A). TXA also reduced collagen deposition in the subepithelial areas (D) compared with treatment with OVA alone (B). Bars = 137 μm.
Reduced Gelatinase Protein and Activity in OVA-treated Plg Knockout Mice
Recent studies have shown that WT mice increase their MMP-9 activity after OVA treatment and that cellular infiltration into the lungs after OVA is reduced in MMP-9 knockout mice (22, 23). Because plasminogen is a known activator of MMP-9 (7), we determined whether the absence of plasminogen had an effect on the expression and activation of MMP-9 and another 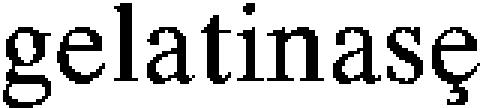 MMP-2, in this model. BAL fluid from Plg+/+ and Plg−/− mice collected 3 hours after OVA or PBS treatment on Days 15 and 18, and 1 and 3 hours after treatment on Day 21, was analyzed by zymography and Western blot. MMP-2 and MMP-9 proenzymes showed low basal levels at all times studied in PBS-treated Plg+/+ and Plg−/− mice (see Figures 7A and 7B for Day 21, 3 hours). OVA treatment increased MMP-2 protein and activity at 1 hour post-treatment on Day 21 in Plg+/+ mice, with maximal activity observed at 3 hours on this day (Figures 7A and B). This was not the case in Plg−/− mice; levels of MMP-2 protein were the same as in PBS-treated mice (Figure 7B), and enzyme activity did not increase as much as in the Plg+/+ mice (Figure 7A). The results were corroborated by ELISA, which measured total MMP-2 in BAL. OVA increased MMP-9 in both genotypes by Day 21 at 3 hours, but higher levels were observed in WT mice (Figure 7A). These data indicate that absence of plasminogen reduces the levels and activities of these gelatinases in response to OVA challenge, which could contribute to the reduced leukocyte levels and histological changes observed in these mice.
MMP-2, in this model. BAL fluid from Plg+/+ and Plg−/− mice collected 3 hours after OVA or PBS treatment on Days 15 and 18, and 1 and 3 hours after treatment on Day 21, was analyzed by zymography and Western blot. MMP-2 and MMP-9 proenzymes showed low basal levels at all times studied in PBS-treated Plg+/+ and Plg−/− mice (see Figures 7A and 7B for Day 21, 3 hours). OVA treatment increased MMP-2 protein and activity at 1 hour post-treatment on Day 21 in Plg+/+ mice, with maximal activity observed at 3 hours on this day (Figures 7A and B). This was not the case in Plg−/− mice; levels of MMP-2 protein were the same as in PBS-treated mice (Figure 7B), and enzyme activity did not increase as much as in the Plg+/+ mice (Figure 7A). The results were corroborated by ELISA, which measured total MMP-2 in BAL. OVA increased MMP-9 in both genotypes by Day 21 at 3 hours, but higher levels were observed in WT mice (Figure 7A). These data indicate that absence of plasminogen reduces the levels and activities of these gelatinases in response to OVA challenge, which could contribute to the reduced leukocyte levels and histological changes observed in these mice.
Figure 7.

Matrix metalloproteinase (MMP)-2 and MMP-9 expression in bronchoalveolar lavage (BAL) fluid from Plg+/+ and Plg−/− mice collected 3 hours after ovalbumin (OVA) or phosphate-buffered saline (PBS) treatment on Day 21. BAL fluid was analyzed by gelatin zymography (A) and Western blot (B). MMP-2 and MMP-9 proenzymes showed low basal expression in PBS-treated Plg+/+ (lanes 1–2) and Plg−/− (lane 3 in A, lanes 5 and 6 in B) mice. OVA treatment increased MMP-2 pro- and active enzymes in Plg+/+ (lane 2 in A and lanes 3 and 4 in B) but not in Plg−/− mice, in which levels of MMP-2 were the same as in PBS-treated mice (lane 4 in A and lanes 7 and 8 in B). OVA increased pro–MMP-9 in both genotypes, but higher levels were observed in wild-type mice (lane 2 in A and lanes 3 and 4 in B). In addition, the active form of MMP-9 was observed only in wild-type mice.
Reduced Cytokines in Plg-deficient Mice after OVA Treatment
Because leukocyte recruitment and histological changes observed in lungs after OVA treatment have been shown to be cytokine dependent (reviewed in Reference 24), we studied a variety of Th1 and Th2 cytokines to understand whether Plg deficiency altered their expression. RNA protection assays on RNA extracted from whole lungs of both genotypes were performed using a mouse cytokine kit, and no difference in the mRNA expression levels of a variety of cytokines were found, including RANTES (regulated upon activation, T-cell expressed and secreted), macrophage inflammatory protein (MIP)-1α, MIP-1β, MIP-2, interferon inducible protein (IP)-10, MCP-1, T-cell activation gene (TCA)-3, eotaxin, IL-2, IL-4, IL-5, IL-6, IL-9, IL-10, IL-13, and IL-15, as well as IFN-γ between OVA-treated Plg+/+ and Plg−/− mice. In addition, a mouse cytometric bead array was used to quantify cytokine protein expression in the BAL at the same time points as MMPs were measured (see above). Levels for IFN-γ, IL-2, IL-4, and IL-13 were similar among the genotypes at the time points studied. OVA caused a significant increase in IL-5 levels in Plg+/+ mice over those observed in the PBS-treated counterparts at all time points studied (Table 2). IL-5 levels were higher in OVA-treated Plg+/+ compared with Plg−/− mice (Table 2). At all time points studied, OVA treatment significantly increased the levels of tumor necrosis factor (TNF)-α in WT mice over those found in their PBS-treated counterparts or the PBS- or OVA-treated Plg−/− mice (Table 2). These data indicate that OVA induced expression of multiple cytokines, including TNF-α and IL-5, in WT mice, but not in mice with Plg deficiency.
TABLE 2.
TIME COURSE OF CYTOKINE EXPRESSION IN BRONCHOALVEOLAR LAVAGE FLUID
| Genotype/Days of and Hours after Treatment | TNF-α (pg/ml) | IL-5 (pg/ml) |
|---|---|---|
| Day 15/3 h | ||
| Plg+/+-PBS | 4.6 ± 0.7 | 0 |
| Plg+/+-OVA | 26.2 ± 6 | 12 ± 7.9 |
| Plg−/−-OVA | 11.4 ± 8 | 1.5 ± 0.8 |
| Day 18/3 h | ||
| Plg+/+-PBS | 14.2 ± 2.3 | 0 |
| Plg+/+-OVA | 35.6 ± 9.9 | 14 ± 6.3 |
| Plg−/−-OVA | 16.9 ± 10.1 | 2.2 ± 1.7 |
| Day 21/1 h | ||
| Plg+/+-PBS | 11.3 ± 11.3 | 0 |
| Plg+/+-OVA | 57.1 ± 20 | 0 |
| Plg−/−-OVA | 6.7 ± 2.7 | 0 |
| Day 21/3 h | ||
| Plg+/+-PBS | 0.7 ± 0.7 | 0 |
| Plg+/+-OVA | 85.1 ± 13.3 | 39.9 ± 23.3 |
| Plg−/−-OVA | 26.6 ± 10 | 1.6 ± 0.5 |
Definition of abbreviations: OVA = ovalbumin; PBS = phosphate-buffered saline; TNF-α = tumor necrosis factor α.
n = 3–6 mice/group.
In separate assays, we measured the levels of MCP-1, the mouse counterpart of MCP-1, in the BAL of PBS- and OVA-challenged mice because reduced levels of this chemotactic factor are observed in association with a blunted inflammatory response to biomaterial implants in Plg−/− mice compared with WT littermates (21). Levels of JE, measured by immunoassay, were undetectable (sensitivity of the assay was ∼ 15 pg/ml) or very low (< 20 pg/ml) in BAL from Day 15, at 1 or 3 hours, or Day 21, at 1 or 3 hours, and were not different in the two genotypes. Plasmin is an activator of latent TGF-β1 (25, 26). TGF-β1 levels change during OVA challenge and the growth factor may influence the response (27). We measured the levels of both total and active TGF-β1 in the BAL from Day 15, at 1 or 3 hours, or Day 21, at 1 or 3 hours, and lung extracts from Day 21, at 3 hours Active TGF-β1 was undetectable in lung homogenates at this time point. The BAL levels of TGF-β1 showed great variability among the mice of the same genotype, consistent with the report of Kumar and colleagues (27), and differences between the two genotypes were not significant (p > 0.1). Within the BAL of an individual mouse, 5 to 10% of the total TGF-β1 was active, and this level of activation was observed in both genotypes.
TXA Reduces AHR in OVA-treated BALB/c Mice
To determine if Plg deficiency also affected AHR, PBS- or OVA-treated Plg+/+ and Plg−/− mice were subjected to methacholine in a body plethysmography chamber. Lung resistance in the OVA-treated C57Bl/6 Plg+/+ mice did not increase over that of their PBS-treated counterparts (2.895 ± 0.53 vs. 2.483 ± 0.41 cm H2O/ml/s) to the levels expected for that dose of methacholine in other WT mice (see Reference 17). Likewise, lung resistance in OVA-treated C57Bl/6 Plg−/− mice was not higher than that of their PBS-treated counterparts (2.061 ± 0.07 vs. 2.5 ± 0.43 cm H2O/ml/s). Measurements of lung resistance in Plg+/+ and Plg−/− mice of the 50/50 J129/C57BL/6 background resulted in similar findings (not shown).
BALB/c mice are frequently used for evaluation of AHR. Therefore, OVA-sensitized BALB/c mice were treated with the plasminogen inhibitor TXA to determine its effect in AHR. Lung resistance in mice treated with OVA alone (OVA/H2O) increased significantly over that observed in naive mice in response to methacholine (Figure 8). TXA treatment significantly reduced lung resistance in OVA-treated mice (OVA/TXA) to levels observed in the naive mice (Figure 8).
Figure 8.

Effects of tranexamic acid (TXA) treatment on airway hyperreactivity in ovalbumin (OVA)-treated BALB/c mice. Mean ± SEM lung resistance values after methacholine injection. The means were derived from eight individual mice, in two separate experiments, for each data point. Mice treated with OVA/TXA had reduced lung resistance compared with those that received only OVA. The inhibition was to the level observed in the naive mice.
DISCUSSION
Asthma is an inflammatory disorder of the airways. T cells are key to the initiation of the response (reviewed in Reference 1); and eosinophils induce many of the pathological changes in the lungs (18) and correlate with disease severity (28). Because plasminogen plays a key role in inflammatory cell migration (8, 9, 29), we compared the response of Plg+/+ and Plg−/− mice in a widely used OVA challenge model to induce allergic pulmonary inflammation. Our major findings are as follows:
Plasminogen is key for leukocyte recruitment into the peribronchial and perivascular areas and airways of the lungs.
Plasminogen is centrally involved in early histological changes observed in asthma, including collagen deposition in the peribronchial areas and mucus metaplasia.
A mechanism for involvement of plasminogen in the pathogenesis of asthma may involve its regulation of specific MMPs and cytokines.
Inhibition of plasminogen function decreases pulmonary inflammation.
OVA sensitization and challenge induced the characteristic increase in total leukocytes in the airways of Plg+/+ mice compared with PBS-treated animals, peaking at Day 21, at 3 hours after treatment. This response was greatly diminished in Plg+/− mice and was absent in Plg−/− mice. Plg+/− mice contain half the amount of blood plasminogen compared with WT mice (15), but recruitment of most cell types to OVA was decreased by more than 75%. This dramatic effect of partial plasminogen reduction is more pronounced than in other models (8, 15, 30) and bodes well if plasminogen were to be a therapeutic target in asthma. The specific cell types most affected by plasminogen deficiency were lymphocytes and eosinophils. This effect was not simply a delay in inflammatory cell response because both Plg+/− and Plg−/− mice failed to show any increase in these cell types between Day 21, at 3 hours, and Day 21, at 6 hours after treatment, times at which their recruitment is maximal (16). The suppression of eosinophil recruitment is particularly noteworthy because of the causative role of these cells in the pathology of asthma (28, 31, 32). Not only were leukocytes diminished in the airways but also in the peribronchial and perivascular areas of the lungs of Plg+/− and Plg−/− mice. These areas are known to be important in the inflammatory reaction associated with lung injury (33).
Although, to our knowledge, our study is the first to directly implicate plasminogen in the pathogenesis of asthma, multiple studies suggest a relationship between the plasminogen system and the disease. For example, the urokinase plasminogen activator (uPA)/plasminogen have been implicated in the extravasation of T cells and the uPA receptor was identified as a T-lymphocyte activation antigen (34). A variety of agonists (35–37) induce uPA-dependent plasminogen activation of eosinophils. Also, inhibition of uPA, MMPs, or Plg/plasmin decreases in vitro eosinophil adhesion (38) or transmigration (12, 37). More recently, associations between allelic variants of uPA and asthma phenotypes, including AHR, were observed in three independent Canadian population samples (39). Together, these previous findings consistently show a key role for the plasminogen system in asthma.
OVA-treated Plg+/+ mice contained increased collagen in the peribronchial areas, an early histological change in the lungs of mice during the acute phase in several OVA models (18–20) and of patients with newly diagnosed or mild asthma (2, 3). Collagen hyperplasia has been reported in patients that die of asthma (40), and a direct correlation has been found between the severity of asthma and the amount of subepithelial collagen (41, 42). In the Plg+/− and Plg−/− mice, increased collagen deposition was absent. Also absent in these mice were goblet cell hyperplasia and mucus production, two other features in lungs of OVA-treated mice. Given the dramatic decrease in the early histological changes in mice with reduced plasminogen, the role that plasminogen plays in airway remodeling in chronic antigen exposure models will be explored in future studies.
The histological differences observed between Plg+/+ and Plg−/− mice may be a consequence of diminished inflammatory cell recruitment. In human subjects with asthma and murine models of asthma, inflammatory cell levels correlate with collagen levels (41, 43, 44). However, plasminogen activator inhibitor 1 (PAI-1)–deficient mice showed no difference in leukocyte recruitment into the lungs in response to OVA challenge, but their collagen content was still decreased (45), consistent with a significant role of the plasminogen system in policing ECM turnover.
Our results show that OVA treatment induced a significant increase in gelatinase levels and activities in OVA-sensitized WT but not in Plg−/− mice. This difference may be central to the suppression of the asthmatic response in Plg−/− mice because MMP inhibitors reduced lymphocyte and eosinophil infiltration into the lungs of OVA-challenged mice (46). MMP-9 is believed to play a key role in cell trafficking and inflammation through the degradation of basement membrane type IV collagen (reviewed in Reference 47). MMP-9 is overexpressed by eosinophils in bronchial tissues of individuals with asthma (48, 49) and the number of eosinophils correlates with mucosal expression of MMP-9 (50, 51). Thus, decreased MMP-9 activity may underlie many of the differences between Plg−/− and Plg+/+ mice in the OVA challenge model. However, airway wall thickness was not different between WT and MMP-9−/− mice (22), whereas it was dramatically reduced in Plg−/− mice. Moreover, the pattern of recruitment of specific cell types in the OVA challenge model in MMP-9−/− mice differs from that we observed in the Plg−/− mice (22, 52). Clearly, the contributions of plasminogen to lung pathology extend beyond its regulation of MMP-9. Differences in the effect of MMP-9 and plasminogen deficiency might be due to reduction of MMP-2 in Plg−/− mice. Soluble MMP-2 and its activator, membrane type 1 matrix metallo protease (MMT1)-MMP, are increased in the sputum and BAL fluid of patients with asthma (53) and are coexpressed in various cells in the lung (54).
Reduction in cytokine expression also may contribute to the differential response of Plg+/+ and Plg−/− mice to OVA challenge. TNF-α levels were reduced in Plg−/− mice, and this cytokine plays an important role in lung fibrosis (reviewed in References 55 and 56). Chemokine induction of MMP-9 promotes cellular influx to the airways of OVA-treated mice (57), and TNF-α regulates MMP-9 expression in monocytes (58) and eosinophils (59). Thus, TNF-α could be involved in both cellular recruitment and tissue destruction by increasing MMP activity. This interrelationship may be reciprocal because several MMPs regulate TNF-α activity (reviewed in Reference 55). In addition to TNF-α, differential levels of IL-5 were found between Plg+/+ and Plg−/− mice. IL-5 is a key regulatory cytokine for eosinophil responses (reviewed in Reference 24). Treatment of OVA-sensitized mice with inactive IL-5 showed no lung eosinophilia and very little inflammation and lung damage (60). With the very profound effect of plasminogen deficiency on the OVA response, a combination of these mechanisms may come into play. Although IL-13 has been shown to be important in mucus cell metaplasia in allergic airway disease (61), we did not find difference in the mRNA or protein levels of this cytokine in the BAL fluid of OVA-challenged Plg+/+ and Plg−/− mice. We also found no relationship between BAL levels of JE and TGF-β1, either in an active or latent form. Thus, whereas these mediators are candidates to influence cellular recruitment in a plasminogen-dependent fashion (21, 62), there was no evidence for their differential expression in OVA-treated Plg−/− and Plg+/+ mice.
Although the above discussion emphasizes the role of plasminogen in regulating MMP and cytokine production as potential mechanisms underlying its participation in pulmonary inflammation, other mechanisms cannot be excluded. For example, the absence of plasminogen may limit the capacity of inflammatory cells to migrate through the ECM and reach the lungs. Thus, some of the changes in MMP and cytokine levels may be secondary to the reduction in cellular recruitment in Plg−/− mice.
WT mice given TXA after sensitization but during challenge with OVA showed reduced cellular recruitment and pulmonary changes. This effect was not observed in Plg−/− mice, demonstrating specificity. Interestingly, even late after challenge (Days 18–21), TXA blocked the inflammatory response and tissue changes induced by OVA. These experiments were performed in C57BL/6 mice as well as in the 50/50 J129/C57BL/6 mice (not shown). Our efforts to measure AHR in these backgrounds were unsuccessful, possibly due to the presence of the C57BL/6 background. C57BL/6 mice are resistant to AHR and require a particular regimen of OVA treatment to induce this response (60). BALB/c mice, in which the response to OVA is more TH2 driven, are more sensitive to AHR. When OVA-sensitized BALB/c mice were treated with TXA, lung resistance was significantly reduced. Further studies of the effects of TXA on the AHR in this background at varying methacholine doses and on different induction regimens will be needed to more fully understand the role of plasminogen and its inhibition on physiology.
In summary, we have shown that plasminogen is important in the recruitment of inflammatory cells into the lung and related early pulmonary changes in a mouse model of OVA-induced pulmonary inflammation. This profound effect of plasminogen on the pathogenesis of asthma may depend on its capacity to regulate multiple events, which contribute to the response, including the generation of TNF-α, IL-5, and certain MMPs. In addition, a plasminogen inhibitor suppresses the inflammation, early histological changes, and AHR in this murine model of asthma, suggesting that plasminogen may be an effective therapeutic target in the treatment of the disease.
Supplementary Material
Acknowledgments
The authors thank Carla Drumm, Shiying Wang, Irina Polyakova, Stephen D. Moeller, and Amit Vasanji for technical assistance.
Supported by National Institutes of Health grant HL-17964.
This article has an online supplement, which is accessible from this issue's table of contents at www.atsjournals.org
Originally Published in Press as DOI: 10.1164/rccm.200609-1345OC on May 31, 2007
Conflict of Interest Statement: None of the authors has a relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Djukanovic R, Holgate ST. An atlas of asthma. New York: Parthenon Publishing; 1999.
- 2.Li X, Wilson JW. Increased vascularity of the bronchial mucosa in mild asthma. Am J Respir Crit Care Med 1997;156:229–233. [DOI] [PubMed] [Google Scholar]
- 3.Barbato A, Turato G, Baraldo S, Bazzan E, Calabrese F, Tura M, Zuin R, Beghe B, Maestrelli P, Fabbri LM, et al. Airway inflammation in childhood asthma. Am J Respir Crit Care Med 2003;168:798–803. [DOI] [PubMed] [Google Scholar]
- 4.Castellino FJ, Ploplis VA. Structure and function of the plasminogen/plasmin system. Thromb Haemost 2005;93:647–654. [DOI] [PubMed] [Google Scholar]
- 5.Bonnefoy A, Legrand C. Proteolysis of subendothelial adhesive glycoproteins (fibronectin, thrombospondin, and von Willebrand factor) by plasmin, leukocyte cathepsin G, and elastase. Thromb Res 2000;98:323–332. [DOI] [PubMed] [Google Scholar]
- 6.Liotta LA, Goldfarb RH, Brundage GP, Siegal GP, Terranova V, Garbisa S. Effect of plasminogen activator (urokinase), plasmin, and thrombin on glycoprotein and collagenous components of basement membrane. Cancer Res 1981;41:4629–4636. [PubMed] [Google Scholar]
- 7.Lijnen HR. Molecular interactions between the plasminogen/plasmin and matrix metalloproteinase systems. Fibrinolysis Proteolysis 2000;14:175–181. [Google Scholar]
- 8.Ploplis VA, French EL, Carmeliet P, Collen D, Plow EF. Plasminogen deficiency differentially affects recruitment of inflammatory cell populations in mice. Blood 1998;91:2005–2009. [PubMed] [Google Scholar]
- 9.Plow EF, Ploplis VA, Carmeliet P, Collen D. Plasminogen and cell migration in vivo. Fibrinolysis Proteolysis 1999;13:49–53. [Google Scholar]
- 10.Swaisgood CM, French EL, Noga C, Simon RH, Ploplis VA. The development of bleomycin-induced pulmonary fibrosis in mice deficient for components of the fibrinolytic system. Am J Pathol 2000;157:177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Banach-Wawrzenczyk E, Dziedziczko A, Rosc D. Fibrinolysis system in patients with bronchial asthma. Med Sci Monit 2000;6:103–107. [PubMed] [Google Scholar]
- 12.Ferland C, Guilbert M, Davoine F, Flamand N, Chakir J, Laviolette M. Eotaxin promotes eosinophil transmigration via the activation of the plasminogen-plasmin system. J Leukoc Biol 2001;69:772–778. [PubMed] [Google Scholar]
- 13.Lloyd CM, Gutierrez-Ramos JC. Animal models to study chemokine receptor function: in vivo mouse models of allergic airway inflammation. Methods Mol Biol 2004;239:199–210. [DOI] [PubMed] [Google Scholar]
- 14.Swaisgood CM, Swaidani S, Erzurum S, Aronica MA, Plow EF. Plasminogen regulation of pathogenesis of asthma [abstract]. Am J Respir Crit Care Med 2004;169:A81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ploplis VA, Carmeliet P, Vazirzadeh S, Van Vlaenderen I, Moons L, Plow EF, Collen D. Effects of disruption of the plasminogen gene in mice on thrombosis, growth and health. Circulation 1995;92:2585–2593. [DOI] [PubMed] [Google Scholar]
- 16.Gonzalo J-A, Lloyd CM, Kremer L, Finger E, Martinez-A. C, Siegelman MH, Cybulsky M, Gutierrez-Ramos J-C. Eosinophil recruitment to the lung in a murine model of allergic inflammation: the role of T cells, chemokines, and adhesion receptors. J Clin Invest 1996;98:2332–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aronica MA, McCarthy S, Swaidani S, Mitchell D, Goral M, Sheller JR, Boothby M. Recall helper T cell response: T helper 1 cell-resistant allergic susceptibility without biasing uncommitted CD4 T cells. Am J Respir Crit Care Med 2004;169:587–595. [DOI] [PubMed] [Google Scholar]
- 18.Humbles AA, Lloyd CM, McMillan SJ, Friend DS, Xanthou G, McKenna EE, Ghiran S, Gerard NP, Yu C, Orkin SH, et al. A critical role for eosinophils in allergic airways remodeling. Science 2004;305:1776–1779. [DOI] [PubMed] [Google Scholar]
- 19.Reinhardt AK, Bottoms SE, Laurent GJ, McAnulty RJ. Quantification of collagen and proteoglycan deposition in a murine model of airway remodelling. Respir Res 2005;6:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tanaka H, Masuda T, Tokuoka S, Takahashi Y, Komai M, Nagao K, Nagai H. Time course study on the development of allergen-induced airway remodeling in mice: the effect of allergen avoidance on established airway remodeling. Inflamm Res 2002;51:307–316. [DOI] [PubMed] [Google Scholar]
- 21.Busuttil SJ, Ploplis VA, Castellino FJ, Tang L, Eaton JW, Plow EF. A central role for plasminogen in the inflammatory response to biomaterials. J Thromb Haemost 2004;2:1798–1805. [DOI] [PubMed] [Google Scholar]
- 22.Cataldo DD, Tournoy KG, Vermaelen K, Munaut C, Foidart JM, Louis R, Noel A, Pauwels RA. Matrix metalloproteinase-9 deficiency impairs cellular infiltration and bronchial hyperresponsiveness during allergen-induced airway inflammation. Am J Pathol 2002;161:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vermaelen KY, Cataldo D, Tournoy K, Maes T, Dhulst A, Louis R, Foidart JM, Noel A, Pauwels R. Matrix metalloproteinase-9-mediated dendritic cell recruitment into the airways is a critical step in a mouse model of asthma. J Immunol 2003;171:1016–1022. [DOI] [PubMed] [Google Scholar]
- 24.Renauld JC. New insights into the role of cytokines in asthma. J Clin Pathol 2001;54:577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lyons RM, Keski-Oja J, Moses HL. Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J Cell Biol 1988;106:1659–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taipale J, Koli K, Keski-Oja J. Release of transforming growth factor-beta 1 from the pericellular matrix of cultured fibroblasts and fibrosarcoma cells by plasmin and thrombin. J Biol Chem 1992;267:25378–25384. [PubMed] [Google Scholar]
- 27.Kumar RK, Herbert C, Foster PS. Expression of growth factors by airway epithelial cells in a model of chronic asthma: regulation and relationship to subepithelial fibrosis. Clin Exp Allergy 2004;34:567–575. [DOI] [PubMed] [Google Scholar]
- 28.Bousquet J, Chanez P, Lacoste JY, Barneon G, Ghavanian N, Enander I, Venge P, Ahlstedt S, Simony-Lafontaine J, Godard P, et al. Eosinophilic inflammation in asthma. N Engl J Med 1990;323:1033–1039. [DOI] [PubMed] [Google Scholar]
- 29.Ploplis VA, Castellino FJ. Nonfibrinolytic functions of plasminogen. Methods 2000;21:103–110. [DOI] [PubMed] [Google Scholar]
- 30.Swaisgood CM, Schmitt D, Eaton D, Plow EF. In vivo regulation of plasminogen function by plasma carboxypeptidase B. J Clin Invest 2002;110:1275–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Bourne PM. Allergen-induced airway hyperresponsiveness. J Allergy Clin Immunol 1988;81:119–127. [DOI] [PubMed] [Google Scholar]
- 32.Lee SA, Fitzgerald SM, Huang SK, Li C, Chi DS, Milhorn DM, Krishnaswamy G. Molecular regulation of interleukin-13 and monocyte chemoattractant protein-1 expression in human mast cells by interleukin-1β. Am J Respir Cell Mol Biol 2004;31:283–291. [DOI] [PubMed] [Google Scholar]
- 33.Pabst R, Tschernig T. Perivascular capillaries in the lung: an important but neglected vascular bed in immune reactions? J Allergy Clin Immunol 2002;110:209–214. [DOI] [PubMed] [Google Scholar]
- 34.Nykjaer A, Moller B, Todd RF III, Christensen T, Andreasen PA, Gliemann J, Petersen CM. Urokinase receptor: an activation antigen in human T lymphocytes. J Immunol 1994;152:505–516. [PubMed] [Google Scholar]
- 35.Dahl R, Venge P. Enhancement of urokinase-induced plasminogen activation by the cationic protein of human eosinophil granulocytes. Thromb Res 1979;14:599–608. [DOI] [PubMed] [Google Scholar]
- 36.Schweizer RC, Kessel-Welmers BA, Warringa RA, Maikoe T, Raaijmakers JA, Lammers JW, Koenderman L. Mechanisms involved in eosinophil migration: platelet-activating factor-induced chemotaxis and interleukin-5-induced chemokinesis are mediated by different signals. J Leukoc Biol 1996;59:347–356. [DOI] [PubMed] [Google Scholar]
- 37.Guilbert M, Ferland C, Bosse M, Flamand N, Lavigne S, Laviolette M. 5-Oxo-6,8,11,14-eicosatetraenoic acid induces important eosinophil transmigration through basement membrane components: comparison of normal and asthmatic eosinophils. Am J Respir Cell Mol Biol 1999;21:97–104. [DOI] [PubMed] [Google Scholar]
- 38.Brooks AM, Bates ME, Vrtis RF, Jarjour NN, Bertics PJ, Sedgwick JB. Urokinase-type plasminogen activator modulates airway eosinophil adhesion in asthma. Am J Respir Cell Mol Biol 2006;35:503–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Begin P, Tremblay K, Daley D, Lemire M, Claveau S, Salesse C, Kacel S, Montpetit A, Becker A, Chan-Yeung M, et al. Association of urokinase-type plasminogen activator with asthma and atopy. Am J Respir Crit Care Med 2007;175:1109–1116. [DOI] [PubMed] [Google Scholar]
- 40.Gabbrielli S, Di Lollo S, Stanflin N, Romagnoli P. Myofibroblast and elastic and collagen fiber hyperplasia in the bronchial mucosa: a possible basis for the progressive irreversibility of airway obstruction in chronic asthma. Pathologica 1994;86:157–160. [PubMed] [Google Scholar]
- 41.Cho SH, Seo JY, Choi DC, Yoon HJ, Cho YJ, Min KU, Lee GK, Seo JW, Kim YY. Pathological changes according to the severity of asthma. Clin Exp Allergy 1996;26:1210–1219. [DOI] [PubMed] [Google Scholar]
- 42.Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M. Airway structural alterations selectively associated with severe asthma. Am J Respir Crit Care Med 2003;167:1360–1368. [DOI] [PubMed] [Google Scholar]
- 43.Minshall EM, Leung DY, Martin RJ, Song YL, Cameron L, Ernst P, Hamid Q. Eosinophil-associated TGF-β1 mRNA expression and airways fibrosis in bronchial asthma. Am J Respir Cell Mol Biol 1997;17:326–333. [DOI] [PubMed] [Google Scholar]
- 44.Tanaka H, Masuda T, Tokuoka S, Komai M, Nagao K, Takahashi Y, Nagai H. The effect of allergen-induced airway inflammation on airway remodeling in a murine model of allergic asthma. Inflamm Res 2001;50:616–624. [DOI] [PubMed] [Google Scholar]
- 45.Oh CK, Ariue B, Alban RF, Shaw B, Cho SH. PAI-1 promotes extracellular matrix deposition in the airways of a murine asthma model. Biochem Biophys Res Commun 2002;294:1155–1160. [DOI] [PubMed] [Google Scholar]
- 46.Kumagai K, Ohno I, Okada S, Ohkawara Y, Suzuki K, Shinya T, Nagase H, Iwata K, Shirato K. Inhibition of matrix metalloproteinases prevents allergen-induced airway inflammation in a murine model of asthma. J Immunol 1999;162:4212–4219. [PubMed] [Google Scholar]
- 47.Atkinson JJ, Senior RM. Matrix metalloproteinase-9 in lung remodeling. Am J Respir Cell Mol Biol 2003;28:12–24. [DOI] [PubMed] [Google Scholar]
- 48.Ohno I, Ohtani H, Nitta Y, Suzuki J, Hoshi H, Honma M, Isoyama S, Tanno Y, Tamura G, Yamauchi K, et al. Eosinophils as a source of matrix metalloproteinase-9 in asthmatic airway inflammation. Am J Respir Cell Mol Biol 1997;16:212–219. [DOI] [PubMed] [Google Scholar]
- 49.Han Z, Junxu, Zhong N. Expression of matrix metalloproteinases MMP-9 within the airways in asthma. Respir Med 2003;97:563–567. [DOI] [PubMed] [Google Scholar]
- 50.Hoshino M, Nakamura Y, Sim J, Shimojo J, Isogai S. Bronchial subepithelial fibrosis and expression of matrix metalloproteinase-9 in asthmatic airway inflammation. J Allergy Clin Immunol 1998;102:783–788. [DOI] [PubMed] [Google Scholar]
- 51.Wenzel SE, Balzar S, Cundall M, Chu HW. Subepithelial basement membrane immunoreactivity for matrix metalloproteinase 9: association with asthma severity, neutrophilic inflammation, and wound repair. J Allergy Clin Immunol 2003;111:1345–1352. [DOI] [PubMed] [Google Scholar]
- 52.McMillan SJ, Kearley J, Campbell JD, Zhu XW, Larbi KY, Shipley JM, Senior RM, Nourshargh S, Lloyd CM. Matrix metalloproteinase-9 deficiency results in enhanced allergen-induced airway inflammation. J Immunol 2004;172:2586–2594. [DOI] [PubMed] [Google Scholar]
- 53.Cataldo D, Munaut C, Noel A, Frankenne F, Bartsch P, Foidart JM, Louis R. MMP-2- and MMP-9-linked gelatinolytic activity in the sputum from patients with asthma and chronic obstructive pulmonary disease. Int Arch Allergy Immunol 2000;123:259–267. [DOI] [PubMed] [Google Scholar]
- 54.Maisi P, Prikk K, Sepper R, Pirila E, Salo T, Hietanen J, Sorsa T. Soluble membrane-type 1 matrix metalloproteinase (MT1-MMP) and gelatinase A (MMP-2) in induced sputum and bronchoalveolar lavage fluid of human bronchial asthma and bronchiectasis. APMIS 2002;110:771–782. [DOI] [PubMed] [Google Scholar]
- 55.Fowlkes JL, Winkler MK. Exploring the interface between metallo-proteinase activity and growth factor and cytokine bioavailability. Cytokine Growth Factor Rev 2002;13:277–287. [DOI] [PubMed] [Google Scholar]
- 56.Winkler MK, Fowlkes JL. Metalloproteinase and growth factor interactions: do they play a role in pulmonary fibrosis? Am J Physiol Lung Cell Mol Physiol 2002;283:L1–11. [DOI] [PubMed] [Google Scholar]
- 57.Das AM, Ajuebor MN, Flower RJ, Perretti M, McColl SR. Contrasting roles for RANTES and macrophage inflammatory protein-1 alpha (MIP-1 alpha) in a murine model of allergic peritonitis. Clin Exp Immunol 1999;117:223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robinson SC, Scott KA, Balkwill FR. Chemokine stimulation of monocyte matrix metalloproteinase-9 requires endogenous TNF-alpha. Eur J Immunol 2002;32:404–412. [DOI] [PubMed] [Google Scholar]
- 59.Schwingshackl A, Duszyk M, Brown N, Moqbel R. Human eosinophils release matrix metalloproteinase-9 on stimulation with TNF-alpha. J Allergy Clin Immunol 1999;104:983–989. [DOI] [PubMed] [Google Scholar]
- 60.Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J Exp Med 1996;183:195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elias JA, Lee CG, Zheng T, Ma B, Homer RJ, Zhu Z. New insights into the pathogenesis of asthma. J Clin Invest 2003;111:291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gonzalo JA, Lloyd CM, Wen D, Albar JP, Wells TN, Proudfoot A, Martinez A, Dorf M, Bjerke T, Coyle AJ, et al. The coordinated action of CC chemokines in the lung orchestrates allergic inflammation and airway hyperresponsiveness. J Exp Med 1998;188:157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.