

Abstract
The importance of arachidonic acid metabolites (termed eicosanoids), particularly those derived from the COX-1 and COX-2 pathways (termed prostanoids), in platelet homeostasis has long been recognized. Thromboxane is a potent agonist, whereas prostacyclin is an inhibitor of platelet aggregation. In contrast, the effect of prostaglandin E2 (PGE2) on platelet aggregation varies significantly depending on its concentration. Low concentrations of PGE2 enhance platelet aggregation, whereas high PGE2 levels inhibit aggregation. The mechanism for this dual action of PGE2 is not clear. This study shows that among the four PGE2 receptors (EP1–EP4), activation of EP3 is sufficient to mediate the proaggregatory actions of low PGE2 concentration. In contrast, the prostacyclin receptor (IP) mediates the inhibitory effect of higher PGE2 concentrations. Furthermore, the relative activation of these two receptors, EP3 and IP, regulates the intracellular level of cAMP and in this way conditions the response of the platelet to aggregating agents. Consistent with these findings, loss of the EP3 receptor in a model of venous inflammation protects against formation of intravascular clots. Our results suggest that local production of PGE2 during an inflammatory process can modulate ensuing platelet responses.
Introduction
Platelets, upon activation by specific agonists, play a critical role in hemostasis by contributing to thrombus formation. In addition, a role for platelets in the pathogenesis of a number of cardiovascular events, such as myocardial infarction and stroke, is recognized. In many such cases, the underlying vascular disease, including atherosclerosis and vasculitis, has an important inflammatory component that might affect platelet function.
Arachidonic acid released upon cell activation by phospholipases is converted by PGH/G synthase 1 and 2, commonly referred to as cyclooxygenases COX-1 and COX-2, into PGH2. The latter is further metabolized to form PGE2, PGF2α, PGD2, prostacyclin (PGI2) and thromboxane (TXA2). COX-1 and COX-2 metabolites are termed prostanoids, and the particular prostanoid produced varies depending on cell type. Platelets contain high levels of thromboxane synthase and thus produce primarily TXA2. In healthy vessels, low levels of prostanoids are produced by both the endothelium and the underlying smooth muscle cells (1). The amount of prostanoids produced is determined at least in part by the activity of COX-1 and COX-2 (2, 3). During an inflammatory response, a number of stimuli including IFN-1β, TNF-α, and bacterial lipopolysaccharide can induce isolated cells to express COX-2 (4). IFN-1β has been shown to increase expression of COX-2 in both human saphenous vein and internal mammary arteries in organ culture (2). Furthermore COX-2 expression in vivo has been reported to be higher in atherosclerotic lesions (which contains macrophages, smooth muscle cells, or endothelial cells) than in healthy vessels (5). PGE2 is the predominant prostaglandin released in response to inflammatory mediators by primary cultures of vascular smooth muscle cells (2) and by macrophages (6, 7).
The potential for COX metabolites of arachidonic acid to serve as agonists for platelet function is well known. For example, TXA2, the principal metabolite of arachidonic acid in platelets, is a potent stimulator of platelet aggregation (8), whereas prostacyclin, the principal cyclooxygenase product of vascular endothelium, inhibits platelet activation. However, the role of other COX metabolites, such as PGE2, in the regulation of platelet function remains unclear.
The actions of PGE2 as well as other prostanoids are mediated through binding to specific high-affinity receptors that belong to the large family of G protein–associated receptors. Unlike the other prostanoids, for which a single receptor has been identified, PGE2 binds with similar affinity to four receptors: EP1, EP2, EP3, and EP4 (9). The EP3 receptor is unique in that multiple isoforms have been identified. All of the prostanoid receptors identified to date are highly conserved across species (the sequence homology between human and mouse receptors is in the range of 80–90%) (9), and multiple isoforms of EP3 are found in all species examined to date. In the mouse, three isoforms of the EP3 receptor have been identified (10). EP3 isoforms differ in the primary sequence of the cytoplasmic domain and, as a consequence, differ in their coupling efficiency with various G proteins. EP3 receptors have been shown to couple to Gi, Gs, and Gq proteins (11, 12). Therefore it is likely that physiological consequences of activation of the EP3 receptor may vary substantially depending on the cell type examined.
In addition to revealing the expression of the thromboxane TP and the prostacyclin IP receptors, RT-PCR analysis of RNA prepared from human platelets indicates that platelets express at least two EP receptors, including three of the six human EP3 isoforms and EP4 (10). The expression of numerous EP receptors coupled to different second messenger systems provides an explanation for the often contradictory actions of PGE2 in physiological responses. Generation of mice deficient in each of the EP receptors has provided a direct approach for identifying both the receptors through which PGE2 activates various cells and the contribution of these pathways to physiological responses in the animal. In this report, using a series of mice deficient in EP, IP, and TP receptors, we identified the prostanoid receptors and the mechanism through which PGE2 regulates platelet physiology in vitro and in vivo. Taken together, these studies support a model in which local production of PGE2 during inflammatory responses dramatically enhances platelet aggregation by opposing increases in cAMP, such as those mediated by prostacyclin, and hence increasing the sensitivity of platelets to aggregating agents.
Methods
Animal welfare.
The use of experimental animals was in accordance with the guidelines of the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
Mice.
Generation of Ep2–/– (13), Ep3–/– (14), Ep4–/– (15), and Ip–/– (16) mice used in this study has been reported previously. The Ep1 null mutation was introduced into the DBA/1Lac background by targeted disruption of this gene using an embryonic stem cell line derived directly from the DBA/1Lac mouse strain. EP2-, EP3-, and IP-deficient mice were maintained in our animal facility on 129 background, EP1-deficient mice on DBA background, and EP4-deficient mice on mixed background. Eight- to twenty-week-old male and female mice were used for experiments, and wild-type littermate animals were used as controls.
Platelet aggregation.
Whole blood was collected from the heart of mice under pentobarbital anesthesia into heparinized (10 U/ml) syringes, except when stated that blood was collected on 3.8% sodium citrate. Blood was pooled from four to seven animals and centrifuged at 100 g for 15 minutes, enabling separation of platelet-rich plasma (PRP). Platelet-poor plasma was obtained by centrifugation of the remaining blood at 2,000 g for 15 minutes. Aggregation tests were performed in an optical Chronolog aggregometer (Chrono-Log Corp., Havertown, Pennsylvania, USA), using a volume of 250 μl of PRP adjusted to 500,000 platelets per μl, with platelet-poor plasma as diluent. Platelet-poor plasma also served as a 100% reference for aggregation.
Determination of cytosolic [Ca2+].
After collecting blood in 0.35% (wt/vol) sodium citrate, PRP was prepared and centrifuged at 500 g for 10 minutes. Platelets were resuspended in Ca2+-free Tyrode’s buffer containing 0.35% (wt/vol) albumin. Washed platelets were incubated with Indo-1AM (2 μg ml–1) for 45 minutes at ambient temperature in the dark. After centrifugation, platelets were resuspended in Ca2+-free Tyrode’s buffer, and 1 mM CaCl2 was added before the experiment. Cytosolic calcium was measured using a Perkin-Elmer 650-40 fluorometer (Perkin-Elmer Corp., Norwalk, Connecticut, USA).
Measurement of adenylyl cyclase activity in platelets.
Washed platelets (1.5 × 107 ml–1) were stimulated with 10 μM PGE2 (dinoprostone; Cayman Chemical, Ann Arbor, Michigan, USA) or 7 nM PGI2 (epoprostenol sodium salt; Cayman Chemical) at 37°C. These PGE2 or PGI2 concentrations were previously determined to yield values in the middle of the cAMP standard curve. The reaction was stopped by 1:5 (vol/vol) perchloric acid (20%) at 15 seconds. Samples were incubated for 20 minutes on ice and centrifuged at 2,000 g for 15 minutes at 4°C. A total of 10 M KOH was added, and precipitating KClO4 was pelleted by centrifuging at 2,000 g for 15 minutes at 4°C. Perchloric acid was removed using a mix of tri-n-octylamine and 1,1,2-trichloro-trifluoroethane. The amount of cAMP in the supernatant was determined using an enzyme immunoassay kit and following the manufacturer’s instructions (Amersham Pharmacia Biotech, Little Chalfont, United Kingdom).
Bleeding time measurement.
The tails of restrained mice were placed under a heating lamp at controlled temperature (37°C). The tail was cut off at 1 cm from the tip, and emerging blood was blotted every 15 seconds without touching the wound. Bleeding time was measured until bleeding had ceased for 30 seconds (17). In a second experiment, mice were placed in a restraining device and their tails were secured in a vertical position. A 3- to 5-mm segment of the tail was transected, and the remaining length was immersed into 37°C sterile isotonic saline. Bleeding time was measured from transection time until bleeding could no longer be detected.
Analysis of thrombus formation.
Mice (20–25 g) were anesthetized with pentobarbital and the jugular veins exposed by a cervical incision. A strip of Parafilm (American National Can, Chicago, Illinois, USA) was slipped under the vein and 100% ethanol, 30 μl of arachidonic acid (free acid, 50 mg/ml in ethanol), or eicosatrienoic acid (Cayman Chemical) was applied to the vein. After 1 minute, the Parafilm was removed, the wound was closed, and the mice were allowed to recover from anesthesia. After 3 hours, the vein was exposed again, and the presence of a thrombus was determined under magnification. The persistence of blood flow was examined by cutting the proximal end of the vein just upstream of the ligation of the jugular confluent. Thrombosis was scored from 0 to 4 as proposed previously (18).
Results
Although PGE2 (10–7 to 10–3 M) alone has no effect on platelet aggregation ex vivo, this lipid mediator can stimulate or inhibit platelet aggregation triggered by other agonists, including collagen, ADP, and thromboxane and its analogue U46619. The particular effect of PGE2 on platelet aggregation is determined primarily by its local concentration. Thus, as shown in Figure 1 and consistent with previous observations (19–21), PGE2 added at concentrations below 10–5 M does not induce platelet aggregation but enhances aggregation induced by U46619, ADP, or collagen. Conversely, at concentrations greater than 10–4 M, PGE2 markedly inhibits the ability of these agonists to promote aggregation.
Figure 1.
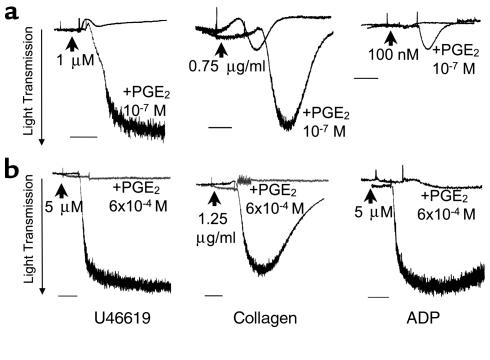
The dual effect of PGE2 on platelet aggregation. (a) Wild-type platelets are exposed to a low concentration of U46619 (1 μM), collagen (0.75 μg/ml), or ADP (100 nM) in the absence or presence of 10–7 M PGE2. At these low levels of stimulation, these agonists do not induce aggregation, except in the presence of PGE2. (b) Higher PGE2 concentrations inhibit the full aggregation induced by 5 μM U46619, 1.25 μg/ml collagen, or 5 μM ADP. Scale bars represent 1 minute. These experiments were repeated three times, and representative traces are shown. The mean of the maximal aggregation was calculated for samples treated with PGE2 and the aggregating agent and for samples treated with the aggregating agent alone. In all cases, the maximal aggregation was significantly higher in the PGE2-treated samples (P < 0.01; unpaired t test).
PGE2 mediates its biologic actions by binding to one of four different cell-surface receptors, termed EP1, EP2, EP3, and EP4 (9, 22, 23). This raises the possibility that the two opposing actions of PGE2 on platelet function might be mediated by different EP receptors. To determine whether this is the case, we examined the effects of low (10–7 M) and high (10–4 M) concentrations of PGE2 on the function of platelets that were deficient in each of the four EP receptors.
As shown in Figure 2, low concentration of PGE2 enhanced U46619-induced aggregation of platelets from EP1-, EP2-, or EP4-deficient mice, and the proaggregatory action of PGE2 on these platelets was essentially identical to that observed with wild-type platelets. In contrast, 10–7 M PGE2 did not enhance aggregation of platelets obtained from EP3-deficient mice. This indicates that the ability of PGE2 to augment platelet aggregation is mediated primarily by stimulation of the EP3 receptor.
Figure 2.
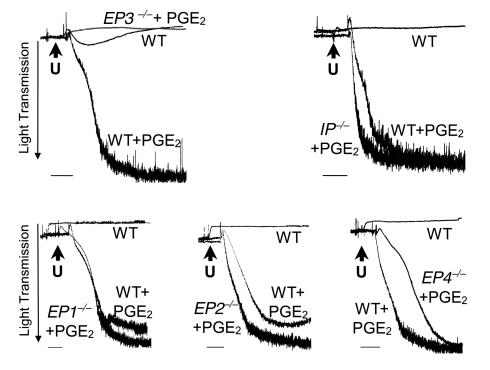
The PGE2-induced potentiation of aggregation is mediated by the EP3 receptor. Comparison of PGE2 (10–7 M) mediated potentiation of aggregation induced by U46619 (1 μM; indicated by the letter “U”) treatment of wild-type platelets (WT) and platelets deficient for each of the prostaglandin receptors. PGE2-induced potentiation is observed in all the samples, with the exception of those lacking the EP3 receptor. Bars = 1 minute. Similar results were obtained on three consecutive experiments, and representative traces are shown. The maximal aggregation induced by PGE2 treatment of EP receptor-deficient platelets in each experiment was calculated, and the mean was compared with similarly treated wild-type platelets. A significant difference (unpaired t test; P < 0.01) was observed only on comparison of the EP3-deficient platelet with wild-type controls.
We then examined the mechanism by which high concentrations of PGE2 inhibit platelet aggregation. To determine the role of the EP receptors in the inhibitory effect of PGE2 at higher concentrations (10–4 M), we again isolated platelets from mice deficient in each of the four PGE2 receptors and strain-matched control animals. As shown in Figure 3, 6 × 10–4 M PGE2 completely inhibited the ability of ADP to induce platelet aggregation of wild-type platelets (Figure 3a) and of platelets deficient in each of the EP receptors (Figure 3d).
Figure 3.
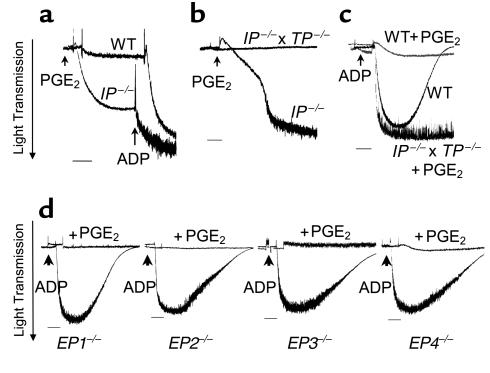
The PGE2-induced inhibition of aggregation is mediated by the IP receptor. (a) PGE2 (10–4 M) alone is sufficient to induce aggregation of IP-deficient platelets, but not wild-type platelets. Further aggregation observed upon addition of ADP reveals that the response to PGE2 in IP-deficient platelets is submaximal. (b) PGE2 (10–4 M) induced aggregation of IP-deficient platelets. The failure to observe aggregation of platelets deficient in both the IP and the TP receptors under similar conditions suggests that high concentrations of PGE2 induced aggregation through the TP receptor. (c) PGE2 (6 × 10–4 M) fails to abolish ADP (5 μM) induced aggregation in platelets lacking both the IP and TP receptors. (d) Comparison of aggregation induced by 5 μM ADP in the presence and absence of 6 × 10–4M PGE2, in platelets deficient in each of the EP receptors. Bars = 1 minute. Three experiments were carried out, and representative traces from these experiments are shown. Differences observed on comparison of the mean maximal aggregation for the various genotypes (a–c) or between PGE2-treated and untreated samples (d) are significant (P < 0.01; unpaired t test).
As already discussed here, activation of the IP receptor has long been recognized to exert a strong inhibitory effect on platelet aggregation (24). We therefore carried out experiments to determine whether this receptor might contribute to PGE2-mediated inhibition. As depicted in Figure 3, 10–4 M PGE2 alone induced aggregation in IP-deficient platelets, unmasking for the first time a third activity of PGE2 on platelets (Figure 3a). Moreover, the addition of ADP further stimulated this aggregation response.
Given that the inhibitory effect of prostacyclin in platelets might be countered by thromboxane, among other mediators, we considered the possibility that high concentrations (>10–4 M) of PGE2 might indirectly or directly activate the receptor for thromboxane (TP). To prevent thromboxane production, platelets were treated with the COX-inhibitor indomethacin. Under these conditions, 10–4 M PGE2 alone still induced aggregation (data not shown). This result suggests that the response of platelets to PGE2 results from a direct and nonspecific activation of the TP receptor. To address this question, we generated mice with combined deficiency of both the IP and TP receptors. As shown in Figure 3b, in contrast to platelets that are deficient in IP receptors alone, PGE2 failed to induce aggregation of IP-deficient platelets that also lacked the TP receptor. This provides direct evidence that at concentrations above 10–4 M, PGE2 can activate the TP receptor, and in the absence of the IP receptor this activation leads to platelet aggregation. The observation that PGE2 alone was sufficient to induce aggregation only in the absence of the IP receptor suggests that the IP receptor mediates the inhibitory effect of high concentrations of PGE2 on aggregation of wild-type platelets. To examine this possibility, we tested the ability of PGE2 to inhibit ADP-induced aggregation of platelets deficient in both IP and TP receptors. In this case, 10–4 M PGE2 did not inhibit aggregation in response to ADP (Figure 3c). Taken together, these data indicate that PGE2 mediates its inhibitory effects on platelet aggregation through activation of the IP receptor.
We next examined the signaling mechanism used by PGE2 to mediate these various effects on platelet physiology. Among the EP receptors, the EP3 receptor is unique in that multiple EP3 isoforms are generated by alternative splicing, and these isoforms couple to different G proteins (11, 12, 25). These include Gq, which drives mobilization of internal calcium stores (26), and Gs or Gi, which regulates intracellular levels of cAMP. We first determined whether treatment of platelets with PGE2 alters intracellular calcium levels (Figure 4a). Exposure of wild-type platelets to concentrations of PGE2 from 10–5 to 10–7 M had no effect on intracellular calcium levels, whereas subsequent stimulation of these same platelets with U46619 induced a brisk calcium signal. In contrast, 10–4 M PGE2 increased calcium levels in IP-deficient platelets under the same experimental condition. However, this calcium signal was not observed in platelets deficient for both IP and TP receptors that were exposed to 10–4 M PGE2. These data suggest that PGE2-induced calcium signaling in IP-deficient platelets is due to activation of the thromboxane receptor.
Figure 4.
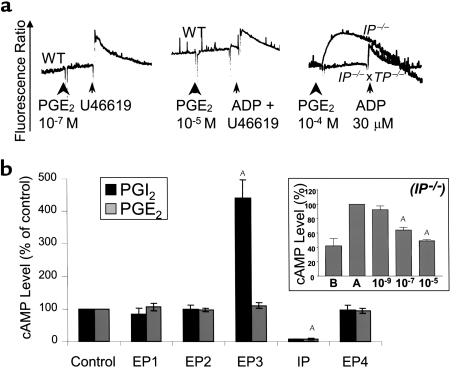
Effects of PGE2 on cytosolic calcium and cAMP levels. (a) Absence of internal calcium mobilization upon treatment of wild-type platelets with 10–7 and 10–5 M PGE2. Change in fluorescence upon subsequent addition of 10 μM U46619 demonstrated that these platelets were able to mobilize their internal calcium stores. After exposure to 10–5 M PGE2, platelets did not respond strongly to 30 μM ADP because of the inhibitory effect of high PGE2 concentration on calcium mobilization. PGE2 (10–4 M) induced calcium mobilization in IP-deficient, but not in IP- and TP-deficient platelets. Experiments examining calcium mobilization in wild-type platelets were repeated five times. Experiments examining calcium mobilization in Ip–/– and Ip–/– × Tp –/– platelets were repeated four times, and similar results were observed in all experiments. A representative trace is shown. (b) Effects of 5 × 10–5 M PGE2 and 7 × 10–9 M PGI2 on accumulated cAMP production in each of the receptor-deficient mouse lines compared with their age- and strain-matched controls. Values were normalized to the cAMP level obtained in appropriate control animals (DBA for Ep1–/–, 129 for Ep2–/–, Ep3–/–, and Ip–/–, and mixed background for Ep4–/– mice), and the bars represent the mean of the percent change observed in four experiments (each bar graph represents the values obtained from 16 mice). Error bars = SEM. ASignificant difference: P < 0.01 (ANOVA test, and Dunnett as post test). Inset: effect of increasing concentration of PGE2 on cAMP level elevated by treatment of platelets of IP-deficient platelets with 10–5 M adenosine. Bar A (inset), the cAMP level in platelets treated with 10–5 M adenosine was set at 100%; Bar B (inset), cAMP level in untreated platelets. The remaining bars indicate the percentage of maximal cAMP observed in platelets treated with 10–5 M adenosine and the indicated amount of PGE2 (10–9, 10–7, and 10–5 M). Three experiments were carried out. Error bars = SEM. ASignificant difference: P < 0.01 (ANOVA test, and Dunnett as post test).
Previous studies have suggested that the intracellular level of cAMP in platelets plays a key role in determining their propensity for aggregation (27). For example, inhibition of platelet aggregation by Gs-coupled receptors such as the IP receptor is associated with increased intracellular cAMP levels (20). Thus, we reasoned that if the inhibitory actions of higher concentrations of PGE2 are mediated by the activation of the IP receptor, they should be associated with stimulation of intracellular cAMP. We therefore determined whether, similarly to prostacyclin, PGE2 modifies platelet aggregation by altering cAMP levels. As seen in Figure 4b, and consistent with the results already described here, loss of the EP1, EP2, or EP4 receptor did not alter intracellular cAMP levels when compared with strain- and age-matched controls. In contrast, EP3-deficient platelets exposed to 5 × 10–5 M PGE2 elicited a marked elevation of intracellular cAMP, and IP-deficient platelets failed to increase intracellular cAMP in response to PGE2 or to prostacyclin. These results support a model in which PGE2 alters platelet function through inhibition of adenylate cyclase activity, and not through a direct mobilization of intracellular calcium. We next determined whether PGE2 stimulation of the EP3 receptor could inhibit cAMP levels induced by agents in addition to prostacyclin. Platelets express the adenosine A2a receptor, and activation of this Gs coupled receptor has been shown to be responsible for adenosine-mediated increases in cAMP levels (28). As shown in Figure 4 (inset), PGE2 treatment decreased cAMP levels in platelets pretreated with adenosine in a dose-dependent manner. We suggest that the concentration of PGE2 in the local environment modifies the intracellular level of cAMP, which in turn determines the response of the platelets to aggregating agents. Current literature suggests that the intracellular concentration of cAMP determines the extent of PKA activation, which is one pathway for regulating the availability of cytosolic calcium and phosphorylation of myosin (29–32). Consistent with this model, platelets isolated from wild-type mice partially aggregated in response to 5 μM ADP in the presence of 8 × 10–5 M PGE2. Under the same conditions, platelets from Ip–/– × Tp–/– mice (low cAMP level) fully aggregated, whereas platelets lacking Ep3–/– receptors (high cAMP level) did not respond (Figure 5a).
Figure 5.
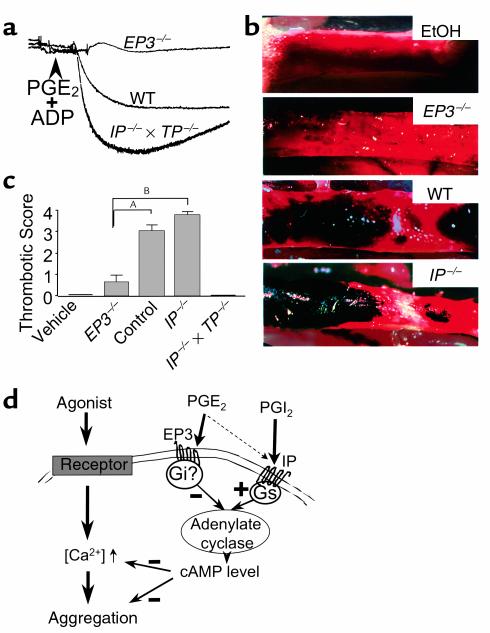
Ex vivo and in vivo implications for PGE2 modulation of cAMP platelet content. (a) Wild-type platelets collected on sodium citrate exposed to 8 × 10–5 M PGE2, then treated with 5 μM ADP did not display a full aggregation (intermediate trace). The PGE2 concentration used was similar to that used for studies described in Figure 4b. In the same experimental conditions, Ep3–/– platelets, which have higher intracellular cAMP levels, do not aggregate (upper trace). By contrast, platelets lacking the receptor for prostacyclin and thus containing lower cAMP levels aggregated maximally (lower trace). Similar results were obtained in two consecutive experiments. (b) Photomicrographs of venous thrombosis in vivo, induced by periadventitial application of arachidonic acid. Shown are the effect of vehicle (ethanol, EtOH), and the effect induced by arachidonic acid in EP3-deficient, wild-type, and IP-deficient mice. ×15. (c) Thrombotic scores in EP3-deficient, wild-type, IP-deficient, and IP- and TP-deficient mice. Thrombosis was scored as follows: 0, no apparent thrombus; 1, small and isolated thrombus; 2, mural thrombi; 3, partially occlusive thrombi; 4, occlusive thrombi. The scores were 0 in wild-type veins treated with vehicle (n = 6) or in Ip–/– × Tp–/– veins (n = 10). Errors bars = SEM. Data were analyzed using a Kruskal-Wallis test followed by Dunn’s tests. This showed significant differences between Ep3–/– mice (0.60 ± 0.34; n = 14), and control (3.00 ± 0.32; n = 18) or Ip–/– mice (3.75 ± 0.17; n = 16). AP < 0.01. BP < 0.001. (d) Proposed model for the role of cAMP in platelets exposed to prostaglandins. PGE2 preferentially stimulates the EP3 receptor, resulting in a decrease in adenylate cyclase activity and opposing the stimulatory effect induced by the IP receptor. The resulting cAMP level in platelets affects both calcium mobilization and aggregation induced by these agents.
We next sought to determine whether the role of the EP3 receptor in modifying the response of platelets could be observed in vivo. We first examined the effect of loss of EP3 on bleeding time. Using two different protocols, we found no difference in the bleeding time between wild-type and EP3-deficient mice when measured by section of 1 cm of the tail (7.09 ± 4.09 minutes [n = 7] vs. 8.17 ± 1.44 minutes [n = 10]) or when measured by section of 3 mm of the tail immersed in saline (70 ± 18 seconds [n = 5] vs. 84 ± 19 seconds [n = 5]). We reasoned that the lack of any apparent role for EP3 receptors in primary hemostasis might reflect low levels of PGE2 in these conditions. Furthermore, we suggest that the ability of the EP3 receptor to modulate platelet function might be more prominent in situations such as in inflammatory lesions or conditions in which PGE2 production is stimulated. We further reasoned that PGE2-mediated modulation of platelet function might be particularly important in veins, given that after exposure to arachidonic acid, veins produce PGE2 in excess of PGI2 (33). To mimic such conditions, we induced thrombosis by application of arachidonic acid to the periadventitial region of the vein. Within 3 hours in wild-type mice, periadventitial delivery of arachidonic acid caused significant intravascular clot formation without major alteration of blood flow (Figure 5, b and c). In this model, thrombus formation results from stimulation of the COX pathways. The inability of eicosatrienoic acid, a 20-carbon lipid with a chemical structure very similar to arachidonic acid, to induce thrombosis suggests that thrombus formation in this model depends on formation of metabolites of arachidonic acid (data not shown). Also, thrombus formation could be blocked by indomethacin treatment, which suggests that arachidonic acid induces thrombosis through the production of prostanoids (data not shown). To further characterize this model, we examined thrombus formation in mice lacking the TP and IP receptors: no thrombi were observed in these animals after periadventitial delivery of arachidonic acid. Thrombus formation was enhanced in IP-deficient mice, leading to total obstruction and interruption of vascular flow. In contrast, thrombus formation was almost completely abolished in EP3-deficient mice. The absence of thrombosis in the EP3-deficient mice suggests that PGE2 produced during inflammatory processes may enhance thrombosis by activating the EP3 receptor, thereby opposing prostacyclin-mediated increases in cAMP. In this model, the opposing effects of PGE2 and PGI2 might modulate the amplitude of the platelet response to proaggregatory agents, such as thromboxane and ADP (Figure 5c).
Discussion
Using a series of mice deficient in prostanoid receptors, we show that the reported ability of PGE2 to inhibit platelet activation does not result from activation of the Gs-coupled EP4 or EP2 receptors. In fact, this activity can be attributed to activation of the prostacyclin receptor at high PGE2 concentrations. In contrast, the ability of PGE2 to potentiate platelet activation is mediated by activation of a PGE2 receptor, the EP3 receptor. In addition, we show that PGE2 is unable to induce aggregation alone and potentiates platelet activation induced by other agonists through regulation of intracellular cAMP levels. Although EP3-deficient mice do not display increased bleeding times in a venous model of arachidonic acid induced inflammation, we show that mice deficient in the EP3 receptor are resistant to thrombus formation.
The ability of agents that increase intracellular cAMP levels to inhibit platelet aggregation has long been recognized. The ability of PGE2 and PGE1 to inhibit platelet aggregation was believed to be due to activation of adenylate cyclase by these agents (19, 20). Cloning and characterization of the PGE2 receptors revealed three receptors capable of coupling to Gs proteins: the EP2 receptor, the EP4 receptor, and some isoforms of the EP3 receptor (9). This raised the possibility that these receptors might mediate the inhibitory action of PGE2. We show here, however, that in mouse platelets the inhibitory actions of PGE2 are not affected by loss of the EP2, EP4, or EP3 receptor. Only loss of the IP receptor affected the ability of PGE2 to inhibit platelet aggregation. To observe this difference, it was first necessary to generate platelets that were deficient in both the IP- and the TP-receptors, owing to the unexpected observation that high concentrations of PGE2 are able to activate the TP receptor. In the absence of inhibitory IP-mediated signals, PGE2 activation of the TP receptor leads to platelet aggregation. Based on the concentration needed for the PGE2-induced inhibitory effect (>10–4 M), this pathway is not expected to contribute extensively to the regulation of platelet activation in vivo.
The physiologically relevant action of PGE2 on platelet aggregation is likely to be related to the ability of low concentrations of PGE2 (<10–6 M) to increase markedly the sensitivity of platelets to aggregating agents. We show here that this action is dependent on expression of the EP3 receptor. As already discussed here, the EP3 receptor is capable of coupling to a number of different G proteins. Our studies show that in platelets, EP3 receptor activation does not lead to measurable changes in intracellular calcium. Instead, consistent with the coupling of the EP3 receptor to Gi, exposure of Ep3–/– platelets to PGE2 leads to an IP-mediated increase in intracellular level of cAMP. Our demonstration that PGE2 potentiates platelet aggregation by decreasing cAMP level through EP3 provides an explanation for previous pharmacological studies showing that in some circumstances, PGE2 could decrease cAMP levels (34). Our results also are consistent with the suggestions that two separate receptors coupled to adenylate cyclase are activated in the response to PGE1, a common ligand for IP and EP (20, 35, 36). In summary, PGE2 can now be added to a growing number of mediators, including epinephrine and ADP, which modulate platelet aggregation by regulating intracellular levels of cAMP. It is likely that these pathways provide a means of linking this homeostatic response to other ongoing physiological responses in the organism.
Pharmacological studies suggest that the response of human and mouse platelets to prostanoids are very similar. For example, in platelets of both species, prostacyclin is a potent inhibitor of platelet aggregation including the response to thromboxane. Similar to our results with mouse platelets, PGE2 has been shown to enhance the response of human platelets to aggregating agents in vitro (19, 20, 37). PCR analysis of RNA prepared from purified platelets indicates that human platelets express high levels of both EP3 and the Gs-coupled EP4 receptors, leading to speculation that these receptors might mediate the opposing actions of PGE2 (10). Although our studies do not identify a role for the EP4 receptor in cAMP production, they clearly show that activation of the EP3 receptor opposes the actions of the Gs-coupled IP receptors in platelets. In both human and mouse platelets, the primary stimulus for increases in cAMP is likely to be through activation of IP. The ability of EP3 to oppose the protective functions of IP and enhance platelet aggregation should have a potent impact on thrombus formation in diseases.
While circulating levels of prostanoids are extremely low in healthy individuals (38), the local concentration of PGE2 can dramatically increase in inflammatory states. For example, the local production of PGE2 was shown in vitro to increase more than 30-fold in aortoiliac occlusive disease (39). It is, therefore, plausible that chronically inflamed vessels produce sufficient quantities of PGE2 to activate EP3 receptors on platelets. In this environment, the intracellular events triggered by activation of the EP3 receptor may enhance platelet aggregation by opposing the effects of PGI2 and enhancing the effects of primary aggregating agents such as ADP. EP3 receptor activation may therefore contribute to the thrombosis observed in pathological states such as vasculitis and atherosclerosis.
Acknowledgments
The authors thank V.A. Wagoner for assistance with animal husbandry, M. Verghese for assistance in the measurement of calcium, S. Tilley for the EP2-deficient mice, and J. Snouwaert for reviewing the manuscript. This work was supported in part by a grant from the NIH (HL-58554 to B.H. Koller) and by a grant from the American Heart Association (9920376U to J.-E. Fabre).
References
- 1.Smith WL. Prostaglandin biosynthesis and its compartmentation in vascular smooth muscle and endothelial cells. Annu Rev Physiol. 1986;48:251–262. doi: 10.1146/annurev.ph.48.030186.001343. [DOI] [PubMed] [Google Scholar]
- 2.Bishop-Bailey D, et al. Induction of cyclooxygenase-2 in human saphenous vein and internal mammary artery. Arterioscler Thromb Vasc Biol. 1997;17:1644–1648. doi: 10.1161/01.atv.17.9.1644. [DOI] [PubMed] [Google Scholar]
- 3.Bishop-Bailey D, Pepper JR, Larkin SW, Mitchell JA. Differential induction of cyclooxygenase-2 in human arterial and venous smooth muscle: role of endogenous prostanoids. Arterioscler Thromb Vasc Biol. 1998;18:1655–1661. doi: 10.1161/01.atv.18.10.1655. [DOI] [PubMed] [Google Scholar]
- 4.Mitchell JA, Larkin S, Williams TJ. Cyclooxygenase-2: regulation and relevance in inflammation. Biochem Pharmacol. 1995;50:1535–1542. doi: 10.1016/0006-2952(95)00212-x. [DOI] [PubMed] [Google Scholar]
- 5.Stemme V, Swedenborg J, Claesson H, Hansson GK. Expression of cyclo-oxygenase-2 in human atherosclerotic carotid arteries. Eur J Vasc Endovasc Surg. 2000;20:146–152. doi: 10.1053/ejvs.2000.1145. [DOI] [PubMed] [Google Scholar]
- 6.Brock T, McNish R, Peters-Golden M. Arachidonic acid is preferentially metabolized by cyclooxygenase-2 to prostacyclin and prostaglandin E2. J Biol Chem. 1999;274:11660–11666. doi: 10.1074/jbc.274.17.11660. [DOI] [PubMed] [Google Scholar]
- 7.Matsumoto H, et al. Concordant induction of prostaglandin E2 synthase with cyclooxygenase-2 leads to preferred production of prostaglandin E2 over thromboxane and prostaglandin D2 in lipopolysaccharide-stimulated rat peritoneal macrophages. Biochem Biophys Res Commun. 1997;230:110–114. doi: 10.1006/bbrc.1996.5894. [DOI] [PubMed] [Google Scholar]
- 8.Thomas D, et al. Coagulation defects and altered hemodynamic response in mice lacking receptors for thromboxane A2. J Clin Invest. 1998;102:1994–2001. doi: 10.1172/JCI5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Narumiya S, Sugimoto A, Ushikubi F. Prostanoid receptors: structures, properties, and functions. Physiol Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- 10.Paul BZS, Ashby B, Sheth SB. Distribution of prostaglandin IP and EP receptor subtypes and isoforms in platelets and human umbilical artery smooth muscle cells. Br J Haematol. 1998;102:1204–1211. doi: 10.1046/j.1365-2141.1998.00910.x. [DOI] [PubMed] [Google Scholar]
- 11.Irie A, et al. Third isoform of the prostaglandin E-receptor EP3 subtype with different C-terminal tail coupling to both stimulation and inhibition of adenylate cyclase. Eur J Biochem. 1993;217:313–318. doi: 10.1111/j.1432-1033.1993.tb18248.x. [DOI] [PubMed] [Google Scholar]
- 12.Irie A, Segi E, Sugimoto Y, Ichikawa A, Negishi M. Mouse prostaglandin E receptor EP3 subtype mediates calcium signals via Gi in cDNA-transfected Chinese hamster ovary cells. Biochem Biophys Res Comm. 1994;204:303–309. doi: 10.1006/bbrc.1994.2460. [DOI] [PubMed] [Google Scholar]
- 13.Tilley S, et al. Reproductive failure and reduced blood pressure in mice lacking the EP2 prostaglandin E2 receptor. J Clin Invest. 1999;103:1539–1545. doi: 10.1172/JCI6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fleming EF, et al. Urinary concentrating function in mice lacking EP3 receptors for prostaglandin E2. Am J Physiol. 1998;275:F955–F961. doi: 10.1152/ajprenal.1998.275.6.F955. [DOI] [PubMed] [Google Scholar]
- 15.Nguyen M, et al. The prostaglandin receptor EP4 triggers remodelling of the cardiovascular system at birth. Nature. 1997;390:78–81. doi: 10.1038/36342. [DOI] [PubMed] [Google Scholar]
- 16.Egan K, Austin S, Smyth E, FitzGerald G. Accelerated atherogenesis in prostacyclin receptor deficient mice. Circulation. 2000;102(Suppl. 2):176. (Abstr.) [Google Scholar]
- 17.Dejana E, Callioni A, Quintana A, de Gaetano G. Bleeding time in laboratory animals. II. A comparison of different assay conditions in rats. Thromb Res. 1979;15:191–197. doi: 10.1016/0049-3848(79)90064-1. [DOI] [PubMed] [Google Scholar]
- 18.Murata T, et al. Altered pain perception and inflammatory response in mice lacking prostacyclin receptor. Nature. 1997;388:678–681. doi: 10.1038/41780. [DOI] [PubMed] [Google Scholar]
- 19.Shio H, Ramwell P. Effect of prostaglandin E2 and aspirin on the secondary aggregation of human platelets. Nature. 1972;236:45–46. doi: 10.1038/newbio236045a0. [DOI] [PubMed] [Google Scholar]
- 20.Gray J, Heptinstall S. Interactions between prostaglandin E2 and inhibitors of platelet aggregation which act through cyclic AMP. Eur J Pharmacol. 1991;194:63–70. doi: 10.1016/0014-2999(91)90124-9. [DOI] [PubMed] [Google Scholar]
- 21.Vezza R, Roberti R, Nenci GG, Gresele P. Prostaglandin E2 potentiates platelet aggregation by priming protein kinase C. Blood. 1993;82:2704–2713. [PubMed] [Google Scholar]
- 22.Coleman R, Smith W, Narumya S. Classification of prostanoid receptors: properties, distribution, and structure of the receptor and their subtypes. Pharmacol Rev. 1994;46:205–228. [PubMed] [Google Scholar]
- 23.Armstrong R. Platelet prostanoid receptors. Pharmacol Ther. 1996;72:171–191. doi: 10.1016/s0163-7258(96)00103-9. [DOI] [PubMed] [Google Scholar]
- 24.Bunting S, Gryglewski R, Moncada S, Vane JR. Arterial walls generate from prostaglandin endoperoxides a substance (prostaglandin X) which relaxes strips of mesenteric and coeliac arteries and inhibits platelet aggregation. Prostaglandins. 1976;12:897–913. doi: 10.1016/0090-6980(76)90125-8. [DOI] [PubMed] [Google Scholar]
- 25.Schmid A, Thierauch K, Schleuning W, Dinter H. Splice variants of the human EP3 receptor for prostaglandin E2. Eur J Biochem. 1995;228:23–30. doi: 10.1111/j.1432-1033.1995.tb20223.x. [DOI] [PubMed] [Google Scholar]
- 26.Offermanns S, Toombs CF, Hu YH, Simon MI. Defective platelet activation in Gαq-deficient mice. Nature. 1997;389:183–186. doi: 10.1038/38284. [DOI] [PubMed] [Google Scholar]
- 27.Weber A, Hohlfeld T, Schror K. cAMP is an important messenger for ADP-induced platelet aggregation. Platelets. 1999;10:238–241. doi: 10.1080/09537109976086. [DOI] [PubMed] [Google Scholar]
- 28.Ledent C, et al. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature. 1997;388:674–678. doi: 10.1038/41771. [DOI] [PubMed] [Google Scholar]
- 29.Hettasch JM, Le Breton GC. Modulation of Ca2+ fluxes in isolated platelet vesicles: effects of cAMP-dependent protein kinase and protein kinase inhibitor on Ca2+ sequestration and release. Biochim Biophys Acta. 1987;931:49–58. doi: 10.1016/0167-4889(87)90049-8. [DOI] [PubMed] [Google Scholar]
- 30.Enouf J, et al. Relationship between cAMP and Ca2+ fluxes in human platelet membranes. Biochimie. 1987;69:297–304. doi: 10.1016/0300-9084(87)90020-4. [DOI] [PubMed] [Google Scholar]
- 31.Enouf J, Giraud F, Bredoux R, Bourdeau N, Levy-Toledano S. Possible role of a cAMP-dependent phosphorylation in the calcium release mediated by inositol 1,4,5-trisphosphate in human platelet membrane vesicles. Biochim Biophys Acta. 1987;928:76–82. doi: 10.1016/0167-4889(87)90087-5. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura K, Kimura M, Aviv A. Role of cyclic nucleotides in store-mediated external Ca2+ entry in human platelets. Biochem J. 1995;310:263–269. doi: 10.1042/bj3100263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skidgel R, Printz M. PGI2 production by rat blood vessels: diminished prostacyclin formation in veins compared to arteries. Prostaglandins. 1978;16:1–16. doi: 10.1016/0090-6980(78)90196-x. [DOI] [PubMed] [Google Scholar]
- 34.Andersen N, Eggerman L, Harker L, Wilson C, Biswanath D. On the multiplicity of platelet prostaglandin receptors. I. Evaluation of competitive antagonism by aggregometry. Prostaglandins. 1980;19:713–735. doi: 10.1016/0090-6980(80)90170-7. [DOI] [PubMed] [Google Scholar]
- 35.Ashby B. Cyclic AMP turnover in response to prostaglandins in intact platelets: evidence for separate stimulatory and inhibitory prostaglandin receptors. Second Messengers Phosphoproteins. 1988;12:45–47. [PubMed] [Google Scholar]
- 36.Ashby B. Interactions among prostaglandin receptors. Receptor. 1994;4:31–42. [PubMed] [Google Scholar]
- 37.MacIntyre D, Gordon J. Calcium-dependent stimulation of platelet aggregation by PGE2. Nature. 1975;258:337–339. doi: 10.1038/258337a0. [DOI] [PubMed] [Google Scholar]
- 38.FitzGerald GA, Brash AR, Falardeau P, Oates JA. Estimated rate of prostacyclin secretion into the circulation of normal man. J Clin Invest. 1981;68:1272–1275. doi: 10.1172/JCI110373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reilly J, Miralles M, Wester W, Sicard G. Differential expression of prostaglandin E2 and interleukin-6 in occlusive and aneurysmal aortic disease. Surgery. 1999;126:624–628. [PubMed] [Google Scholar]