

Abstract
Experimental autoimmune encephalomyelitis (EAE) is a T cell–mediated disease initiated by antigen-specific CD4+ T cells. Signaling through CD28 is a critical second signal for activation of T cells, and CD28 knockout (CD28KO) mice have been reported to be resistant to induction of EAE. We now report that CD28KO mice have no intrinsic defect in mediating disease, because they developed EAE after passive transfer of primed T cells. After immunization, peripheral T cells from CD28KO mice were primed and developed memory phenotype, but had decreased antigen-specific IFN-γ production as compared with cells from wild-type (WT) animals. Reimmunization of CD28KO mice brought out clinical disease and increased IFN-γ production in vitro. Pathologically, there were cellular infiltrates in the central nervous system, in contrast to single-immunized mice. We show furthermore that blocking B7-1 or CTLA4, but not B7-2, in CD28KO mice induces disease after a single immunization. Thus, EAE can be induced in animals lacking CD28-dependent costimulation, suggesting that alternative costimulatory pathways were used. Blocking the OX40-OX40L costimulatory pathway differentially affected disease induction in CD28KO mice as compared with WT controls. Our data show that EAE may develop in the absence of CD28 T-cell costimulation. These findings have implications for therapies aimed at blocking costimulatory signals in autoimmune diseases.
Introduction
Activation of mature T lymphocytes requires antigen recognition and costimulatory signals, but the requirements for T-cell anergy and apoptosis are more controversial. Earlier reports suggested that antigen recognition in the absence of second signals induces a state of unresponsiveness (1, 2). However, recent reports indicate that secondary signaling through costimulatory molecules may be involved in the induction of T-cell anergy. CTLA4 (CD152), a homologue of CD28 that binds with higher affinity to B7 molecules, functions as a negative regulator of immune responses (3, 4). CTLA4 ligation results in inhibition of IL-2 production and a consequent arrest in cell cycle progression from G0/G1, rather than induction of apoptotic cell death (5, 6). Recent data also indicate that negative T-cell signaling through CTLA4 plays an important role in the induction of peripheral tolerance to nominal antigens in vivo (7–9). We have shown recently that CTLA4 signaling is required for the induction of acquired thymic tolerance (10). B7-CD28 interactions play a pivotal role in the development and maintenance of T-cell responses (11, 12). Blocking CD28-B7 interactions protects animals from experimental autoimmune encephalomyelitis (EAE) (13–15) and other autoimmune diseases (16, 17). However, the role of B7-1 versus B7-2 blockade remains controversial. Administration of anti–B7-1 Ab was reported to protect from EAE, while administration of anti–B7-2 increased disease severity (18, 19). Anti–B7-1 administration was also associated with prevention of epitope spread (20). In our study, anti–B7-1 administration worsens EAE in the Lewis rat (21) and (PL/J×SJL) F1 mice (22), and expression of B7-1 in the central nervous system (CNS) correlates with episodes of remission (23). CD28-deficient mice have been reported to be resistant to EAE (24–26). We wanted to examine the mechanisms of resistance to disease since priming of T cells was reported to occur normally in these mice (26). The TNF family of receptors includes several members that have been implicated as having costimulatory function, specifically the CD40-CD154 (27) and the OX40-OX40L (28) receptor pairs. OX40L has been reported to provide a potent costimulatory signal to T cells and to synergize with B7 (29). Cells isolated from the CNS of animals with EAE express high levels of OX40 (30, 31). Similarly, CD40L is expressed in the CNS of EAE mice and multiple sclerosis (MS) patients (32), and blockade of the CD40-CD154 pathway protects from EAE (22, 32).
In this report we investigated the conditions leading to disease development in CD28 knockout (CD28KO) mice and the alternative costimulatory pathways that may be used to induce EAE in these mice.
Methods
Mice.
Female CD28KO mice on a C57BL/6 background were obtained from The Jackson Laboratory (Bar Harbor, Maine, USA). Wild-type C57BL/6 sex- and age-matched controls were also obtained from The Jackson Laboratory. The mice were maintained at our facility and used at 6–8 weeks of age.
EAE induction with myelin oligodendrocyte glycoprotein.
Myelin oligodendrocyte glycoprotein peptide 35-55 (MOG 35-55) (M-E-V-G-W-Y-R-S-P-F-S-R-O-V-H-L-Y-R-N-G-K) corresponding to mouse sequence was synthesized by QCB Inc., Division of BioSource International (Hopkinton, Massachusetts, USA), and purified by HPLC. Peptide purity was greater than 99% after HPLC. Mice were immunized subcutaneously in the flanks with 150–200 μg of MOG peptide in 0.1 ml PBS and 0.1 ml CFA containing 0.4 mg Mycobacterium tuberculosis (H37Ra; Difco Laboratories, Detroit, Michigan, USA) and intraperitoneally injected with 200 ng pertussis toxin (List Biological Laboratories Inc., Campbell, California, USA) on the day of immunization and 48 hours later. A second immunization was performed when indicated, 14 days after the first, consisting of 150 μg of MOG peptide in CFA and pertussis administration on the day of injection and 48 hours later. EAE was scored as described previously (23): 0, no disease; 1, limp tail or isolated weakness of gait without limp tail; 2, partial hind leg paralysis; 3, total hind leg or partial hind and front leg paralysis; 4, total hind leg and partial front leg paralysis; 5, moribund or dead animal.
Costimulatory blockade of EAE with CTLA4Ig, anti–B7-1, anti–B7-2, and anti-CTLA4 Ab’s.
Anti–B7-1 (CD80) mAb 1G10 and anti–B7-2 (CD86) mAb 2D10 were a kind gift from Roche Research Group (Nutely, New Jersey, USA). CTLA4IgY100F is a mutant form of CTLA4Ig that binds and blocks B7-1 (21, 22) (a kind gift of Robert Peach, Bristol-Myers-Squibb, Hillside, New Jersey, USA). These Ab’s were administered at a dose of 500 μg on the day of immunization and 200 μg on days 2, 4, 6, and 8. Control animals received an equal dose of control Ig (clone B7), which was a kind gift of Robert Peach. Anti-CTLA4 Ab from ascites of a B-cell hybridoma (clone 4F10; kindly provided by Jeffrey Bluestone, Diabetes Center, University of California at San Francisco, San Francisco, California, USA) (3) was administered at 500 μg on days 0, 2, 4, 6, 8, 10, and 12 after immunization. Anti–OX-40L Ab, clone RM134L, has been characterized previously (33) and was administered at a dose of 500 μg on day 0 and 250 μg on days 2, 4, and 6 after immunization. Anti-CD40L Ab from ascites of MR1 B-cell hybridoma, a kind gift of Randy Noelle (Department of Microbiology, Dartmouth Medical School, Lebanon, New Hampshire) (34), was administered at a dose of 500 μg on day 0 and 250 μg on days 2, 4, and 6.
Adoptive transfer.
C57BL/6 mice were immunized once with MOG 35-55 peptide as described above, and spleens were harvested 14 days later. A single-cell suspension was prepared from the spleens, and 100 × 106 splenocytes were injected intraperitoneally into CD28KO mice. The recipient CD28KO mice were then immunized with MOG peptide as described above, 1 day after adoptive transfer. Pertussis (200 ng) was administered intraperitoneally the day of immunization and 48 hours later.
Generation of primed T cells and cell culture.
Mice were immunized in one hind footpad and in both flanks with an emulsion of 100 μl of CFA and 100 μl PBS containing 100 μg of MOG p35-55. A single-cell suspension was prepared from the inguinal and the draining popliteal lymph nodes or spleens. The cells were cultured in 96-well plates (Corning-Costar Corp., Cambridge, Massachusetts, USA). Media used for proliferation and cytokine assays consisted of serum-free DMEM (BioWhittaker Inc., Walkersville, Maryland, USA) containing 75 mM/ml L-glutamine, 100 U/ml penicillin and streptomycin, 1 ml/100 ml of a 100× concentrated nonessential amino acid solution, 0.1 mM HEPES/ml, 1 mM/ml sodium pyruvate (all from BioWhittaker Inc.), and 0.05 mM/ml 2-mercaptoethanol (Sigma Chemical Co., St. Louis, Missouri, USA). Cells were incubated at 37°C in humidified air containing 6% CO2.
Proliferation assay.
Cells were cultured at 2 × 106 cells/ml and 200 μl/well with antigen. After 48 hours of culture 1 μCi 3H-thymidine (NEN Life Science Products Inc., Boston, Massachusetts, USA) was added in 20 μl of media to each well for another 16 hours. Cells were harvested on filter mats, dried, and counted.
Cytokine ELISA.
For cytokine assays, cells were cultured at 2 × 106 cells/ml in 200 μl media with antigen. Supernatants for IL-2, IL-10, and IFN-γ ELISA were collected after 48 hours of culture. Quantitative ELISAs for IL-2, IL-10, and IFN-γ were performed using paired Ab’s and recombinant cytokines from PharMingen (San Diego, California, USA), according to manufacturer’s recommendations.
Enzyme-linked immunosorbent spot assay.
For 24 hours, 4 × 106 cells per well were incubated with antigen in U-bottom plates. The cells were then counted, resuspended, and serially diluted from a concentration of 4 × 106/ml down to 3 × 104/ml, then added to nitrocellulose plates (Millipore Corp., Bedford, Massachusetts, USA). The plates were coated with 50 μl primary IFN-γ Ab (clone R4-6A2; Endogen Inc., Woburn, Massachusetts, USA) at a concentration of 5 μg/ml. One hundred microliters of the appropriate concentrations of antigen or mitogen were added to the nitrocellulose enzyme-linked immunosorbent spot (ELISPOT) plates. The cells were incubated for an additional 18 hours at 37°C. After washing three times, 50 μl/well of biotinylated IFN-γ secondary Ab (clone XMG1.2; Endogen Inc.) was added to the plates at a concentration of 2 μg/ml diluted in 1% BSA/PBS for 5 hours at room temperature. After washing, 50 μl of alkaline phosphatase (E-2636; Sigma Chemical Co.) was added to the plates at a dilution of 1:1000 in 0.05% Tween/PBS for 2 hours at room temperature. Plates were washed twice with wash buffer and then twice in PBS. Fifty microliters of BCIP/NBT (FASTR B-5655; Sigma Chemical Co.) solution was added for 5–20 minutes until blue-colored spots developed. Plates were washed with distilled water. Spots were counted using a Zeiss Stemi 100 dissecting microscope.
Anti-MOG Ab assay.
Serum samples were obtained from blood samples of immunized or naive mice and stored at –20°C. MOG 35-55 peptide was dissolved at 1 μg/ml in NaHCO3 buffer 0.1 M, pH 9.6. The peptide was coated on a 96-well Nunc-Immuno Plate (Nalge-Nunc International, Roskilde, Denmark) at 50 μg/well for 3 hours at 37°C. The plates were blocked overnight with 2% BSA (wt/vol) in PBS at 4°C. The serum samples (diluted 150 in PBS) were added, then serially diluted on the ELISA plate. The plates were incubated for 3 hours at 37°C, then washed three times, and horse anti-mouse IgG biotinylated Ab (Vector Laboratories, Burlingame, California, USA) was added (concentration 1:1000 in PBS). After 1-hour incubation at 37°C, streptavidin-horseradish peroxidase (PharMingen) diluted at a concentration of 1:2000 was added for 30 minutes at room temperature, then washed. Tetramethylbenzidine microwell peroxidase substrate (Kirkegaard and Perry Laboratories, Gaithersburg, Maryland, USA) was used to develop the plates, and the reaction was stopped with TMB Stop solution (Kirkegaard & Perry Laboratories), and read at 450 nm. Serum samples from immunized animals were compared to a naive serum standard.
FACS staining of lymphocytes.
Splenocytes from immunized mice were separated and resuspended in PBS to a concentration of 107 cells/ml. Aliquots (100 μl) were plated in a 96-well plate. The cells were treated with Fc Block (PharMingen) at a concentration of 1 μg/106 cells for 5 minutes on ice. The cells were then stained with Ab’s to memory markers and counterstained with Ab’s to CD4. Ab’s used were rat anti-mouse CD44 IgG2b phycoerythrin (PE), rat anti-mouse CD62L IgG2a PE, rat anti-mouse CD45RB PE, and rat anti-mouse CD4 IgG2a CyChrome (all from PharMingen). Appropriate isotype controls were used. The cells were incubated on ice for 30 minutes and then washed twice with 1% FBS in PBS, and resuspended for analysis on a FACScan (Becton Dickinson Immunocytometry Systems, San Jose, California, USA) equipped with CellQuest software (Becton Dickinson Immunocytometry Systems).
Immunohistology and histopathology.
Spinal cords and brains were collected on day 17–21 after immunization from two to four mice in each experimental group. Spinal cord tissues were embedded in OCT, quick-frozen in liquid nitrogen, and kept at –70°C until sectioning. Cryostat sections (10 μm) of spinal cords were fixed with acetone or 4% paraformaldehyde. The sections were stained using the avidin-biotin technique (VectaStain Elite kit; Vector Laboratories), visualized with diamino-benzidine (Vector Laboratories), and counterstained in hematoxylin. Isotype-matched Ig and omission of the primary Ab served as negative controls. Each specimen was evaluated at three different levels of sectioning at least. The whole tissue section (a longitudinal spinal cord section) was evaluated for a given cellular marker at magnification ×40.
Results
CD28KO mice are resistant to the induction of EAE but have normally primed T cells.
We tested the ability of CD28KO animals to develop actively induced EAE by immunization with MOG 35-55 peptide. As seen in Figure 1, CD28KO mice immunized with MOG 35-55 peptide are resistant to EAE, unlike wild-type (WT) C57BL/6 mice. Only 4 of 23 animals developed disease in the CD28KO group, compared with 23 of 23 in the WT controls (P < 0.0001 by Fisher exact test). The CD28KO animals that developed EAE had a very mild disease that remained significantly less severe than the WT mice (Figure 1). Increasing the dose of peptide from 200 μg to 600 μg/immunization did not change the disease resistance (not shown). This natural resistance of CD28KO to actively induced EAE is consistent with published data from other investigators (24–26).
Figure 1.
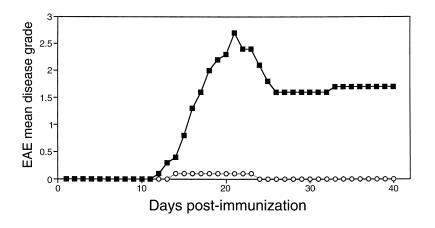
EAE is suppressed in CD28KO mice. A representative experiment showing disease induction in C57BL/6 WT mice (filled squares) and CD28KO mice (open circles). Mice were immunized with MOG p35-55 and graded for disease daily. The mean daily grade for each group (n = 5 mice per group) is shown. The incidence was five of five in the WT and one in five in the CD28KO mice; the mean maximal grade was 2.7 in WT and 0.1 in CD28KO (P < 0.0001).
In vitro proliferative responses of splenocytes from WT and CD28KO mice showed a comparable degree (P value NS) of in vitro proliferation to MOG 35-55 peptide (stimulation index = 3 for CD28KO mice vs. 4.3 for WT, with a background proliferation of 5346 ± 452), indicating that T cells from CD28KO animals can be primed in vivo.
CD28KO mice develop EAE after adoptive transfer with splenocytes from WT immunized mice.
We tested the ability of CD28KO to develop passive disease. Primed splenocytes from immunized WT mice were adoptively transferred to CD28KO mice that were then immunized with MOG peptide (Figure 2). CD28KO mice developed EAE after transfer of WT splenocytes. The disease severity correlated with the number of transferred splenocytes. Adoptive transfer of splenocytes from CD28KO mice did not transfer disease. These data confirm that the natural resistance of CD28KO mice to EAE is due to a defect in the function of primed T cells.
Figure 2.
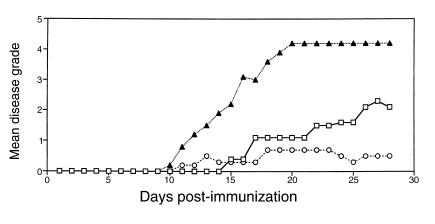
Adoptive transfer of WT splenocytes into CD28KO mice restores EAE. Splenocytes from C57BL/6 WT mice or CD28KO mice were transferred to CD28KO mice. On day –1, 10 × 106 WT splenocytes (open squares), 100 × 106 WT splenocytes (filled triangles), or 100 × 106 CD28KO splenocytes (open circles) were transferred, and all recipients were immunized with MOG p35-55 on day 0. The mean daily score for each group (n = 5 mice per group) is shown.
CD28KO mice generate memory cells after immunization.
Some reports suggest that naive T cells are more dependent on costimulation than memory cells, although naive T cells may be primed by increasing the strength of signal 1 (35). To investigate whether development of memory markers (36) was impaired in CD28KO mice, we harvested splenocytes from WT or CD28KO mice on day 14 after immunization. Figure 3 shows that the expression of CD44hi, CD62Llo, and CD45RBlo was comparable between splenocytes of WT and CD28KO mice across various antigen concentrations. These data establish that T cells from primed CD28KO mice can develop normal memory phenotype in vivo.
Figure 3.
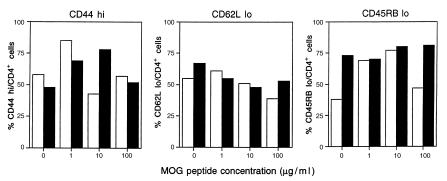
Similar expression of memory cell markers on CD4+ cells from CD28KO and WT mice. Quantitation of memory cell markers on CD4+ cells from C57BL/6 WT mice (open bars) and CD28KO mice (filled bars). Splenocytes were harvested on day 14 after immunization with MOG p35-55 and cultured for 72 hours in the presence of MOG p35-55 (1, 10, or 100 μg/ml). Cells were stained and analyzed by FACS for expression of CD44, CD62 ligand, and CD45RB. All cells were counterstained for the CD4 marker, and the number of cells expressing the given memory marker per number of CD4 cells is shown as a percentage value.
CD28KO mice generate an anti–MOG 35-55 peptide Ab response with a slightly delayed peak titer compared with WT mice.
Previous studies had shown a critical role of anti-MOG Ab’s in augmentation of demyelination and worsening of EAE (37). In addition, CD28 signaling functions to augment T cell–dependent B-cell growth and immunoglobulin secretion (38). We therefore investigated whether anti-MOG Ab production was impaired in the CD28KO mice. The relative Ab titers from immunized CD28KO mice sera were similar to those from WT mice on day 24 and 45 after immunization, but were significantly lower on day 20 (WT mean titer = 1/1828, CD28KO mean titer 1/168; P = 0.0003). This finding suggests that the production of Ab in the CD28KO mice was slightly delayed compared with WT mice.
Cells from CD28KO mice have decreased production of MOG-induced IFN-γ in vitro and a decreased frequency of MOG-specific IFN-γ–producing cells.
In vitro culture of primed splenocytes from CD28KO and WT mice with MOG 35-55 peptide revealed decreased production of IFN-γ in the supernatants of cultures from CD28KO mice (Figure 4a). Furthermore, we found a significantly reduced number of MOG 35-55 peptide–specific IFN-γ–positive cells as measured by ELISPOT in the splenocytes of CD28KO mice (Figure 4b). These data establish the specific defect in CD28KO lymphocytes in response to antigen.
Figure 4.
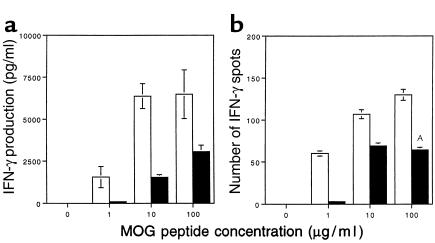
Decreased IFN-γ production by MOG-reactive splenocytes in CD28KO mice. (a) IFN-γ production was measured by ELISA in the supernatants of splenocytes harvested on day 14 from C57BL/6 (open bars) or CD28KO mice (filled bars), after 48 hours of culture with MOG p35-55 at concentrations of 1, 10, or 100 μg/ml. IFN-γ production was significantly greater in the cultures from WT mice at all concentrations of MOG p35-55 (P < 0.005). (b) MOG p35-55–specific IFN-γ–producing cells were measured by ELISPOT in cultures of splenocytes from C57BL/6 WT (open bars) or CD28KO mice (filled bars). The y axis represents the number of positive cells per 2 × 105 cells plated. The frequency of IFN-γ–producing cells was significantly higher in WT cultures at the 100 μg/ml antigen dose (AP = 0.006).
CD28KO mice develop EAE after a booster immunization.
Immunization followed by a second round of immunization on day 14 induced disease in CD28KO mice that was indistinguishable from the disease in WT mice except for the day of onset, which was delayed to day 29 from the initial immunization or 15 days after the second immunization (Table 1). A booster injection of pertussis alone or CFA alone failed to induce EAE in CD28KO mice after single immunization with MOG peptide (not shown), indicating the necessity for re-exposure to the antigen. Double immunization and restoration of disease susceptibility were associated with increased IFN-γ production in vitro to the level seen in cells from WT mice and an increase in the frequency of MOG-specific IFN-γ–producing cells (Figure 5, a and b). More importantly, using immunohistology cellular infiltrates are observed invading the parenchyma after double immunization similar to WT mice, whereas in CD28KO mice immunized once the infiltrates are restricted to the meninges and within blood vessels (Figure 6). This may be the first report indicating that T cells lacking CD28 can induce autoimmune disease in CD28KO mice.
Table 1.
Disease characteristics in WT and CD28KO mice after double immunization
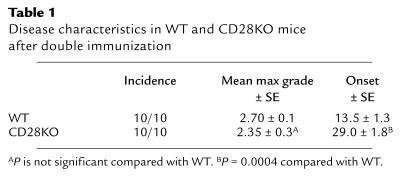
Figure 5.
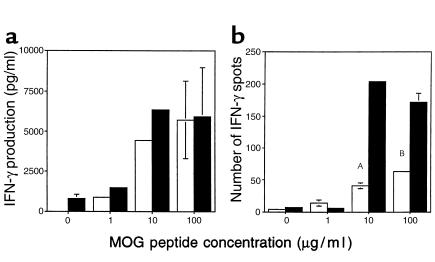
Increased IFN-γ production by MOG-reactive splenocytes in double-immunized CD28KO mice. (a) IFN-γ production was measured by ELISA in the supernatants of splenocytes harvested on day 14 from single-immunized C57BL/6 (open bars) or harvested on day 28 from double-immunized CD28KO mice (filled bars), after 48 hours’ culture with MOG p35-55 at concentrations of 1, 10, or 100 μg/ml. IFN-γ production was similar in both groups with no significant differences. (b) MOG p35-55–specific IFN-γ–producing cells were measured by ELISPOT in cultures of splenocytes from single-immunized C57BL/6 WT (open bars) or double-immunized CD28KO mice (filled bars). The frequency of IFN-γ–producing cells was significantly greater in cultures from double-immunized CD28KO (AP = 0.0009, BP = 0.009).
Figure 6.
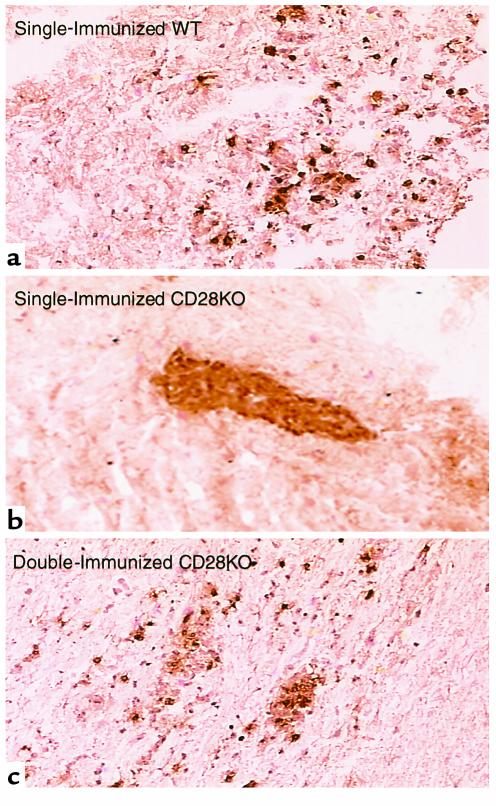
The reduction of CNS parenchymal inflammatory infiltration in single-immunized CD28KO mice is reversed in double-immunized CD28KO mice. Microphotograph (×40) of representative spinal cord section from (a) single-immunized WT mice day 17; (b) single-immunized CD28KO mice day 17; (c) double-immunized CD28KO mice day 35. Sections are immunohistochemically stained for CD4+ cells, which display a brown color. Cellular infiltrates are seen invading the brain parenchyma in WT (a) and double-immunized (c) mice, while the cells remain within blood vessels in the single-immunized CD28KO mice (b).
Blocking the CTLA4-B7-1 pathway is permissive for EAE induction.
Several recent reports suggest that negative signaling through CTLA4 (CD152) is an important mechanism in physiologic termination of immune responses (4, 39) and in tolerance induction (7–10). Indeed, in EAE CTLA4 signal blockade can worsen disease (40). Thus, we treated CD28KO mice with either anti–B7-1 or anti–B7-2 mAb at the time of immunization. Since these mice lack CD28, anti-B7 Ab’s should block a negative signal delivered through CTLA4. As seen in Table 2, administration of anti–B7-1, but not anti–B7-2, mAb reversed the disease resistance in CD28KO mice. Treatment with CTLA4IgY100F, a mutant form of CTLA4Ig that selectively binds B7-1 (21, 22, 41, 42), also reversed the disease resistance. Consistent with the restoration of disease susceptibility in anti–B7-1 mAb-treated mice, IFN-γ production and the frequency of IFN-γ–producing cells were increased in vitro (Figure 7), and immunohistology of the CNS shows infiltration of cells in the parenchyma (Figure 8a). These data suggest that natural resistance to EAE in CD28KO is at least in part due to a negative regulatory signal provided by B7-1 through CTLA4. However, it is possible, although unlikely, that anti–B7-1 therapy may have resulted in signaling of B7-1 on antigen-presenting cells (APCs), leading to T-cell activation. To differentiate these two possibilities, we treated CD28KO mice with a blocking anti-CTLA4 mAb (5, 10). As shown in Table 2, anti–CTLA4-treated mice developed significant disease as compared with CD28KO mice. In addition, similar to our findings with anti–B7-1 Ab, IFN-γ production was increased as measured by ELISA and ELISPOT in the anti–CTLA4-treated mice compared with control CD28KO mice. Furthermore, there were significant infiltrates into the CNS parenchyma of the treated mice (Figure 8b).
Table 2.
Effects of B7 blockade on disease in CD28KO mice
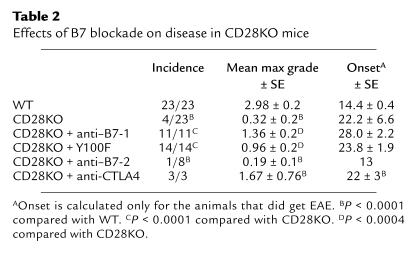
Figure 7.
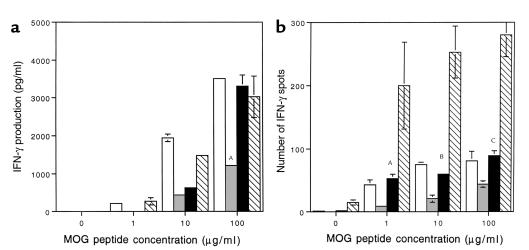
Increased IFN-γ production by MOG-reactive splenocytes in anti–B7-1–treated and anti-CTLA4–treated CD28KO mice. (a) IFN-γ production was measured by ELISA in the supernatants of splenocytes harvested on day 35 from C57BL/6 (open bars), CD28KO mice (gray bars), CD28KO mice treated with anti–B7-1 Ab (black bars), and CD28KO mice treated with anti-CTLA4 Ab (hatched bars). Cells were incubated with MOG p35-55 at concentrations of 1, 10, or 100 μg/ml. IFN-γ production was significantly increased in the anti–B7-1–treated CD28KO group (AP = 0.00006) and the anti-CTLA4–treated CD28KO group (P < 0.00009) compared with control Ig–treated CD28KO at the highest concentration of MOG. (b) MOG p35-55–specific IFN-γ–producing cells were measured by ELISPOT in cultures of splenocytes from C57BL/6 WT (open bars), or CD28KO mice (gray bars), or CD28KO mice treated with anti–B7-1 Ab (black bars), or CD28KO mice treated with anti-CTLA4 Ab (hatched bars), and harvested on day 35. The frequency of IFN-γ–producing cells was significantly greater in cultures from CD28KO mice treated with anti–B7-1 Ab (AP = 0.0007, BP = 0.0005, CP = 0.0011) and those treated with anti-CTLA4 than control CD28KO mice (P < 0.00001 at all concentrations of MOG peptide).
Figure 8.
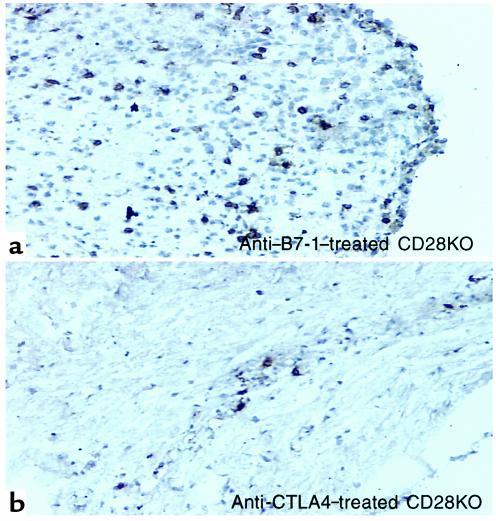
Increased inflammatory infiltration in the CNS of anti–B7-1–treated and anti–CTLA4-treated CD28KO mice. Microphotograph (×40) of representative spinal cord section from (a) CD28KO mice treated with anti–B7-1 Ab, or (b) CD28KO mice treated with anti-CTLA4, at the peak of disease day 35. The section is immunohistochemically stained for CD4+ cells, which display a brown color. Cellular infiltrates are seen invading the brain parenchyma.
The OX40-OX40L pathway is an alternative activation pathway in CD28KO mice.
The TNF-TNF receptor superfamily of molecules has been reported to have functional costimulatory capacity. The best characterized are the CD40-CD154 (34) and, more recently, the OX40-OX40L (43) pathways. Both of these pathways have been reported to synergize with the CD28-mediated costimulation, but at the same time to mediate costimulatory signals independent of CD28 (29, 33, 44). Therefore, we investigated whether these pathways play a role in mediating CD28-independent EAE in double-immunized animals. We treated WT and CD28KO double-immunized mice with anti-CD154 or anti-OX40L mAb. We found that while anti-CD154 protected WT as well as CD28KO double-immunized mice, anti-OX40L protected only the CD28KO mice from developing disease (Figure 9). These data suggest that the OX40-OX40L pathway may be a unique alternative costimulatory pathway for mediating autoimmune responses in CD28KO mice.
Figure 9.
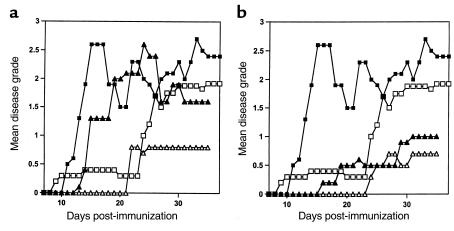
Anti-OX40L reverses EAE induced in double-immunized CD28KO mice but not in WT mice. Disease course of double-immunized CD28KO mice treated with anti-OX40L (a) or anti-CD40L (b). Mice were immunized with MOG p35-55 and graded for disease daily. The mean daily grade for each group (n = 3–10 mice per group) is shown. (a) A representative experiment showing the disease course in C57BL/6 WT mice (filled squares) or in WT mice treated with anti-OX40L Ab (filled triangles) compared with double-immunized CD28KO mice (open squares) and double-immunized CD28KO mice treated with anti-OX40L Ab (open triangles). The mean maximal disease grade in WT mice was 2.7 ± 0.1; in WT treated with anti-OX40L Ab it was 2.75 ± 0.3. The mean maximal grade in double-immunized CD28KO mice was 2.35 ± 0.3, and in those treated with anti-OX40L Ab it was 0.83 ± 0.6 (P = 0.001 compared with untreated double-immunized CD28KO). (b) A representative experiment showing the disease course in C57BL/6 WT mice (filled squares) or in WT mice treated with anti-CD40L Ab (filled triangles) compared with double-immunized CD28KO mice (open squares) and double-immunized CD28KO mice treated with anti-CD40L Ab (open triangles). The mean maximal disease grade in untreated WT mice was 2.7 ± 0.1, whereas in WT treated with anti-CD40L it was 1.08 ± 1.1 (P = 0.03). The mean maximal grade in double-immunized CD28KO mice was 2.35 ± 0.3 compared with those treated with anti-CD40L (0.67 ± 1.15; P = 0.0008 compared with untreated double-immunized CD28KO mice).
Discussion
Signaling through CD28 provides the T cell with an important second signal for activation. Blockade of the CD28-B7 pathway has been shown to inhibit T-cell activation in vitro (2, 45) and prevent autoimmune diseases in vivo (13–17). Furthermore, in EAE the presence of an intact CD28 signaling pathway was reported to be essential for disease induction (24–26, 46). In this report, we found that EAE can be induced in a CD28-independent manner.
Our data show that CD28KO mice have no intrinsic defect in developing EAE, because they develop clinical disease after adoptive transfer of WT T cells. Priming of T cells in the CD28KO mice is not impaired, as evidenced by normal in vitro proliferation to the priming antigen. The requirement for CD28 signaling to activate naive T cells remains controversial. Some studies have shown that CD28 signaling is required for naive T-cell activation (47–49), while other studies suggest that this requirement can be bypassed by increased antigen signal strength (35, 50) or through interaction with other costimulatory molecules such as ICAM-1 (51). In the CD28KO mice we were unable to induce disease by increasing antigen dose. Furthermore, the development of memory phenotype was unaffected by the absence of CD28 signaling. Since the appearance of a “mature” phenotype is dependent on progression of the cells through cell division (52–54), our finding of memory phenotype is consistent with the normal proliferative response to MOG peptide in vitro. The absence of costimulatory signals blocks clonal expansion of antigen-specific cells in vivo (55, 56). In the absence of CD28 signaling, IL-2 production, as well as IFN-γ production, is impaired (55). A high T-cell receptor signal strength can increase the number of cells going into cell cycle and improve IL-2 secretion, but in the absence of costimulation, IFN-γ remains impaired (56). In the CD28KO mice we found decreased secretion of IFN-γ in vitro and decreased precursor frequency of IFN-γ–producing cells consistent with this paradigm.
Double immunization reverses the resistance to EAE and increases the precursor frequency of MOG-specific IFN-γ–producing cells. This is probably mediated by recruiting naive CD4+ T cells into the effector pool, thus reaching a threshold frequency of MOG-specific IFN-γ–secreting cells that is permissive for disease induction. Therefore, a second immunization may result in upregulation of alternative costimulatory signaling pathways, such as OX40-OX40L (57). Indeed, OX40 and OX40L are expressed in the CNS during EAE, and administration of OX40Ig during the first relapse blocked the induction of EAE in SJL/J mice (31). Our data show that OX40-OX40L blockade ameliorated disease in double-immunized CD28KO mice, but not in single-immunized C57BL/6 WT mice, suggesting that the OX40-OX40L may serve as an alternative costimulatory pathway in mediating CD28-independent autoimmune responses. This does not exclude other costimulatory pathways from playing an important role in CD28-independent immune responses. Indeed, recent data by Gaglia et al. show that ICAM-1 may costimulate T cells in the absence of CD28 (51). Our data with CD40-CD154 blockade, which was protective in both WT and CD28KO animals, suggest that this pathway may also contribute to CD28-independent costimulation. Our findings with OX40-OX40L blockade, however, support a unique role for this pathway in CD28-independent costimulation in EAE.
An interesting aspect of EAE induction in CD28KO mice is the accumulation of cells in the meninges and within blood vessels, as we show here and as was previously described (58). In addition, delayed-type hypersensitivity responses to the immunizing antigen were impaired in these mice (26), suggesting a problem with trafficking of antigen-specific cells. Production of C-C chemokines is enhanced by CD28-B7 ligation (59), and one can hypothesize that IFN-γ–dependent chemokines such as IP-10 may be decreased in the CD28KO mice, thus contributing to the disease resistance. CD28 may play a direct role in the penetration of vessels through interaction of CD28 on the T cell and B7 molecules on the endothelial vessels, as has been demonstrated in a vascular coculture model (60). Alternatively, CD28 may indirectly induce the expression of adhesion molecules and chemokines important in cell adhesion and penetration. We are currently investigating these possibilities.
A critical and novel finding in our studies is the reversal of disease resistance by B7-1, but not by B7-2, blockade. B7-1 has a higher tendency to dimerize than B7-2; therefore the avidity of B7-1 to CTLA4 is higher than that of B7-2 (61), which favors the formation of very stable inhibitory signaling complexes. In addition, previous studies using B7-1 transgenic mice suggest that B7-1 may contribute to the downregulation of T-cell immune response (62). Our findings are consistent with previous reports showing that transplant rejection was accelerated by blocking CTLA4 in CD28KO mice (63). Our data that disease sensitivity was restored by CTLA4-B7-1 blockade supports the hypothesis that the T-cell response is complex and is dependent on the integration of positive and negative signals delivered through CD28 and CTLA4, respectively, such that in the absence of CD28 signaling CTLA4 predominates. Another interesting observation is that B7-2 blockade was ineffective in reversing disease resistance, confirming previous observations that B7-1 may be the dominant ligand for CTLA4 (21, 64, 65).
In summary, this is the first report indicating that autoimmune responses can occur in the absence of CD28 costimulation. Novel T-cell costimulatory pathways of the TNF-TNF receptor superfamily of molecules, such as the OX40-OX40L pathway, may play an important role in mediating CD28-independent costimulation and autoimmune disease in vivo. Furthermore, our data support the notion that in the absence of CD28 costimulation, a negative regulatory signal provided by interaction of B7-1 with CTLA4 predominates. Since strategies to block CD28-B7 T-cell costimulation are being tested currently in a variety of autoimmune diseases in humans (66), one needs to be aware of the conditions and mechanisms whereby an autoimmune response may occur even in the absence of CD28 costimulation. Therefore, our results have relevant clinical implications for treatment of autoimmune diseases with agents that block costimulatory signals.
Acknowledgments
This work is supported by research grants from the National Multiple Sclerosis Society (RG-2589 to S.J. Khoury and FG-1287 to T. Chitnis) and the NIH (AI-40945 to S.J. Khoury and PO1 AI-41521 to M.H. Sayegh). N. Najafian is a fellow of the National Kidney Foundation.
Footnotes
Tanuja Chitnis and Nader Najafian contributed equally to this work.
References
- 1.Mueller DL, Jenkins MK, Schwartz RH. Clonal expansion versus functional clonal inactivation: a costimulatory signalling pathway determines the outcome of T cell antigen receptor occupancy. Annu Rev Immunol. 1989;7:445–480. doi: 10.1146/annurev.iy.07.040189.002305. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz RH. A cell culture model for T lymphocyte clonal anergy. Science. 1990;248:1349–1356. doi: 10.1126/science.2113314. [DOI] [PubMed] [Google Scholar]
- 3.Walunas TL, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. [PubMed] [Google Scholar]
- 4.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walunas TL, Bakker CY, Bluestone JA. CTLA-4 ligation blocks CD28-dependent T cell activation. J Exp Med. 1996;183:2541–2550. doi: 10.1084/jem.183.6.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krummel MF, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med. 1996;183:2533–2540. doi: 10.1084/jem.183.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perez VL, et al. Induction of peripheral T cell tolerance in vivo requires CTLA-4 engagement. Immunity. 1997;6:411–417. doi: 10.1016/s1074-7613(00)80284-8. [DOI] [PubMed] [Google Scholar]
- 8.Bluestone JA. Is CTLA-4 a master switch for peripheral T cell tolerance? J Immunol. 1997;158:1989–1993. [PubMed] [Google Scholar]
- 9.Samoilova EB, et al. CTLA-4 is required for the induction of high dose oral tolerance. Int Immunol. 1998;10:491–498. doi: 10.1093/intimm/10.4.491. [DOI] [PubMed] [Google Scholar]
- 10.Issazadeh S, Zhang M, Sayegh MH, Khoury SJ. Acquired thymic tolerance: role of CTLA4 in the initiation and maintenance of tolerance in a clinically relevant autoimmune disease model. J Immunol. 1999;162:761–765. [PubMed] [Google Scholar]
- 11.Schwartz RH. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell. 1992;71:1065–1068. doi: 10.1016/s0092-8674(05)80055-8. [DOI] [PubMed] [Google Scholar]
- 12.Linsley PS. The CD28/CTLA-4:B7 receptor system in experimental autoimmune encephalomyelitis. J Clin Invest. 1995;95:2429–2430. doi: 10.1172/JCI117940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perrin PJ, et al. Role of B7:CD28/CTLA-4 in the induction of chronic relapsing experimental allergic encephalomyelitis. J Immunol. 1995;154:1481–1490. [PubMed] [Google Scholar]
- 14.Khoury SJ, et al. CD28-B7 costimulatory blockade by CTLA4Ig prevents actively induced experimental autoimmune encephalomyelitis and inhibits Th1 but spares Th2 cytokines in the central nervous system. J Immunol. 1995;155:4521–4524. [PubMed] [Google Scholar]
- 15.Cross AH, et al. Long-term inhibition of murine experimental autoimmune encephalomyelitis using CTLA-4-Fc supports a key role for CD28 costimulation. J Clin Invest. 1995;95:2783–2789. doi: 10.1172/JCI117982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tang A, Judge TA, Nickoloff BJ, Turka LA. Suppression of murine allergic contact dermatitis by CTLA4Ig. Tolerance induction of Th2 responses requires additional blockade of CD40-ligand. J Immunol. 1996;157:117–125. [PubMed] [Google Scholar]
- 17.Finck BK, Linsley PS, Wofsy D. Treatment of murine lupus with CTLA4Ig. Science. 1994;265:1225–1227. doi: 10.1126/science.7520604. [DOI] [PubMed] [Google Scholar]
- 18.Kuchroo VK, et al. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell. 1995;80:707–718. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- 19.Racke MK, et al. Distinct roles for B7-1 (CD-80) and B7-2 (CD-86) in the initiation of experimental allergic encephalomyelitis. J Clin Invest. 1995;96:2195–2203. doi: 10.1172/JCI118274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miller SD, et al. Blockade of CD28/B7-1 interaction prevents epitope spreading and clinical relapses of murine EAE. Immunity. 1995;3:739–745. doi: 10.1016/1074-7613(95)90063-2. [DOI] [PubMed] [Google Scholar]
- 21.Gallon L, et al. Differential effects of B7-1 blockade in the rat experimental autoimmune encephalomyelitis model. J Immunol. 1997;159:4212–4216. [PubMed] [Google Scholar]
- 22.Schaub M, et al. Costimulatory signal blockade in murine relapsing experimental autoimmune encephalomyelitis. J Neuroimmunol. 1999;96:158–166. doi: 10.1016/s0165-5728(99)00022-3. [DOI] [PubMed] [Google Scholar]
- 23.Issazadeh S, Navikas V, Schaub M, Sayegh M, Khoury S. Kinetics of expression of costimulatory molecules and their ligands in murine relapsing experimental autoimmune encephalomyelitis in vivo. J Immunol. 1998;161:1104–1112. [PubMed] [Google Scholar]
- 24.Oliveira-dos-Santos AJ, et al. CD28 costimulation is crucial for the development of spontaneous autoimmune encephalomyelitis. J Immunol. 1999;162:4490–4495. [PubMed] [Google Scholar]
- 25.Chang TT, Jabs C, Sobel RA, Kuchroo VK, Sharpe AH. Studies in B7-deficient mice reveal a critical role for B7 costimulation in both induction and effector phases of experimental autoimmune encephalomyelitis. J Exp Med. 1999;190:733–740. doi: 10.1084/jem.190.5.733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Girvin AM, et al. A critical role for B7/CD28 costimulation in experimental autoimmune encephalomyelitis: a comparative study using costimulatory molecule-deficient mice and monoclonal antibody blockade. J Immunol. 2000;164:136–143. doi: 10.4049/jimmunol.164.1.136. [DOI] [PubMed] [Google Scholar]
- 27.Hollenbaugh D, Ochs HD, Noelle RJ, Ledbetter JA, Aruffo A. The role of CD40 and its ligand in the regulation of the immune response. Immunol Rev. 1994;138:23–37. doi: 10.1111/j.1600-065x.1994.tb00845.x. [DOI] [PubMed] [Google Scholar]
- 28.Chen AI, et al. OX40-ligand has a critical costimulatory role in dendritic cell:T cell interactions. Immunity. 1999;11:689–698. doi: 10.1016/s1074-7613(00)80143-0. [DOI] [PubMed] [Google Scholar]
- 29.Gramaglia I, Weinberg AD, Lemon M, Croft M. OX-40 ligand: a potent costimulatory molecule for sustaining primary CD4 T cell responses. J Immunol. 1998;161:6510–6517. [PubMed] [Google Scholar]
- 30.Weinberg AD, et al. Target organ-specific up-regulation of the MRC OX-40 marker and selective production of Th1 lymphokine mRNA by encephalitogenic T helper cells isolated from the spinal cord of rats with experimental autoimmune encephalomyelitis. J Immunol. 1994;152:4712–4721. [PubMed] [Google Scholar]
- 31.Weinberg AD, Wegmann KW, Funatake C, Whitham RH. Blocking OX-40/OX-40 ligand interaction in vitro and in vivo leads to decreased T cell function and amelioration of experimental allergic encephalomyelitis. J Immunol. 1999;162:1818–1826. [PubMed] [Google Scholar]
- 32.Gerritse K, et al. CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci USA. 1996;93:2499–2504. doi: 10.1073/pnas.93.6.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Akiba H, et al. CD28-independent costimulation of T cells by OX40 ligand and CD70 on activated B cells. J Immunol. 1999;162:7058–7066. [PubMed] [Google Scholar]
- 34.Noelle RJ, et al. A 39-kDa protein on activated helper T cells binds CD40 and transduces the signal for cognate activation of B cells. Proc Natl Acad Sci USA. 1992;89:6550–6554. doi: 10.1073/pnas.89.14.6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manickasingham SP, Anderton SM, Burkhart C, Wraith DC. Qualitative and quantitative effects of CD28/B7-mediated costimulation on naive T cells in vitro. J Immunol. 1998;161:3827–3835. [PubMed] [Google Scholar]
- 36.Swain SL, et al. From naive to memory T cells. Immunol Rev. 1996;150:143–167. doi: 10.1111/j.1600-065x.1996.tb00700.x. [DOI] [PubMed] [Google Scholar]
- 37.Linington C, Engelhardt B, Kapocs G, Lassman H. Induction of persistently demyelinated lesions in the rat following the repeated adoptive transfer of encephalitogenic T cells and demyelinating antibody. J Neuroimmunol. 1992;40:219–224. doi: 10.1016/0165-5728(92)90136-9. [DOI] [PubMed] [Google Scholar]
- 38.Klaus SJ, et al. Costimulation through CD28 enhances T cell-dependent B cell activation via CD40-CD40L interaction. J Immunol. 1994;152:5643–5652. [PubMed] [Google Scholar]
- 39.Karandikar NJ, Vanderlugt CL, Walunas TL, Miller SD, Bluestone JA. CTLA-4: a negative regulator of autoimmune disease. J Exp Med. 1996;184:783–788. doi: 10.1084/jem.184.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perrin PJ, Maldonado JH, Davis TA, June CH, Racke MK. CTLA-4 blockade enhances clinical disease and cytokine production during experimental allergic encephalomyelitis. J Immunol. 1996;157:1333–1336. [PubMed] [Google Scholar]
- 41.Peach RJ, et al. Complementarity determining region 1 (CDR1)- and CDR3-analogous regions in CTLA-4 and CD28 determine the binding to B7-1. J Exp Med. 1994;180:2049–2058. doi: 10.1084/jem.180.6.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khoury SJ, et al. Ex vivo treatment of antigen-presenting cells with CTLA4Ig and encephalitogenic peptide prevents experimental autoimmune encephalomyelitis in the Lewis rat. J Immunol. 1996;157:3700–3705. [PubMed] [Google Scholar]
- 43.Weinberg AD, Vella AT, Croft M. OX-40: life beyond the effector T cell stage. Semin Immunol. 1998;10:471–480. doi: 10.1006/smim.1998.0146. [DOI] [PubMed] [Google Scholar]
- 44.Griggs ND, et al. The relative contribution of the CD28 and gp39 costimulatory pathways in the clonal expansion and pathogenic acquisition of self-reactive T cells. J Exp Med. 1996;183:801–810. doi: 10.1084/jem.183.3.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Linsley PS, Ledbetter JA. The role of the CD28 receptor during T cell responses to antigen. Annu Rev Immunol. 1993;11:191–212. doi: 10.1146/annurev.iy.11.040193.001203. [DOI] [PubMed] [Google Scholar]
- 46.Perrin PJ, June CH, Maldonado JH, Ratts RB, Racke MK. Blockade of CD28 during in vitro activation of encephalitogenic T cells or after disease onset ameliorates experimental autoimmune encephalomyelitis. J Immunol. 1999;163:1704–1710. [PubMed] [Google Scholar]
- 47.Van Parijs L, Ibraghimov A, Abbas AK. The roles of costimulation and Fas in T cell apoptosis and peripheral tolerance. Immunity. 1996;4:321–328. doi: 10.1016/s1074-7613(00)80440-9. [DOI] [PubMed] [Google Scholar]
- 48.Perrin PJ, Lovett-Racke A, Phillips SM, Racke MK. Differential requirements of naive and memory T cells for CD28 costimulation in autoimmune pathogenesis. Histol Histopathol. 1999;14:1269–1276. doi: 10.14670/HH-14.1269. [DOI] [PubMed] [Google Scholar]
- 49.McAdam AJ, Farkash EA, Gewurz BE, Sharpe AH. B7 costimulation is critical for antibody class switching and CD8(+) cytotoxic T-lymphocyte generation in the host response to vesicular stomatitis virus. J Virol. 2000;74:203–208. doi: 10.1128/jvi.74.1.203-208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dubey C, Croft M, Swain SL. Naive and effector CD4 T cells differ in their requirements for T cell receptor versus costimulatory signals. J Immunol. 1996;157:3280–3289. [PubMed] [Google Scholar]
- 51.Gaglia JL, et al. Intercellular adhesion molecule 1 is critical for activation of CD28-deficient T cells. J Immunol. 2000;165:6091–6098. doi: 10.4049/jimmunol.165.11.6091. [DOI] [PubMed] [Google Scholar]
- 52.Hasbold J, Lyons AB, Kehry MR, Hodgkin PD. Cell division number regulates IgG1 and IgE switching of B cells following stimulation by CD40 ligand and IL-4. Eur J Immunol. 1998;28:1040–1051. doi: 10.1002/(SICI)1521-4141(199803)28:03<1040::AID-IMMU1040>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 53.Hodgkin PD, Lee JH, Lyons AB. B cell differentiation and isotype switching is related to division cycle number. J Exp Med. 1996;184:277–281. doi: 10.1084/jem.184.1.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gett AV, Hodgkin PD. Cell division regulates the T cell cytokine repertoire, revealing a mechanism underlying immune class regulation. Proc Natl Acad Sci USA. 1998;95:9488–9493. doi: 10.1073/pnas.95.16.9488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Judge TA, et al. The in vivo mechanism of action of CTLA4Ig. J Immunol. 1996;156:2294–2299. [PMC free article] [PubMed] [Google Scholar]
- 56.Gudmundsdottir H, Wells AD, Turka LA. Dynamics and requirements of T cell clonal expansion in vivo at the single-cell level: effector function is linked to proliferative capacity. J Immunol. 1999;162:5212–5223. [PubMed] [Google Scholar]
- 57.Kaleeba JA, Offner H, Vandenbark AA, Lublinski A, Weinberg AD. The OX-40 receptor provides a potent co-stimulatory signal capable of inducing encephalitogenicity in myelin-specific CD4+ T cells. Int Immunol. 1998;10:453–461. doi: 10.1093/intimm/10.4.453. [DOI] [PubMed] [Google Scholar]
- 58.Perrin PJ, Lavi E, Rumbley CA, Zekavat SA, Phillips SM. Experimental autoimmune meningitis: a novel neurological disease in CD28-deficient mice. Clin Immunol. 1999;91:41–49. doi: 10.1006/clim.1998.4684. [DOI] [PubMed] [Google Scholar]
- 59.Herold KC, et al. Regulation of C-C chemokine production by murine T cells by CD28/B7 costimulation. J Immunol. 1997;159:4150–4153. [PubMed] [Google Scholar]
- 60.Denton MD, Geehan CS, Alexander SI, Sayegh MH, Briscoe DM. Endothelial cells modify the costimulatory capacity of transmigrating leukocytes and promote CD28-mediated CD4(+) T cell alloactivation. J Exp Med. 1999;190:555–566. doi: 10.1084/jem.190.4.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ikemizu S, et al. Structure and dimerization of a soluble form of B7-1. Immunity. 2000;12:51–60. doi: 10.1016/s1074-7613(00)80158-2. [DOI] [PubMed] [Google Scholar]
- 62.Sethna MP, van Parijs L, Sharpe AH, Abbas AK, Freeman GJ. A negative regulatory function of B7 revealed in B7-1 transgenic mice. Immunity. 1994;1:415–421. doi: 10.1016/1074-7613(94)90072-8. [DOI] [PubMed] [Google Scholar]
- 63.Lin H, et al. Cytotoxic T lymphocyte antigen 4 (CTLA4) blockade accelerates the acute rejection of cardiac allografts in CD28-deficient mice: CTLA4 can function independently of CD28. J Exp Med. 1998;188:199–204. doi: 10.1084/jem.188.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lenschow DJ, et al. Differential effects of anti-B7-1 and anti-B7-2 monoclonal antibody treatment on the development of diabetes in the nonobese diabetic mouse. J Exp Med. 1995;181:1145–1155. doi: 10.1084/jem.181.3.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Judge TA, et al. The role of CD80, CD86, and CTLA4 in alloimmune responses and the induction of long-term allograft survival. J Immunol. 1999;162:1947–1951. [PubMed] [Google Scholar]
- 66.Sayegh MH. Finally, CTLA4Ig graduates to the clinic. J Clin Invest. 1999;103:1223–1225. doi: 10.1172/JCI6952. [DOI] [PMC free article] [PubMed] [Google Scholar]