

Abstract
Fluorescent nucleotide base analogs can serve as sensitive probes of the local structure and chemical environment of the base within a nucleic acid sequence. A significant strength of these base analogs is their similarity in molecular constitution and chemical properties to natural bases. While chemical synthesis has afforded the ability to generate oligonucleotides in good yield with sequence-specific incorporation of fluorescent base analogs, this method is limited in practice to the synthesis of relatively small RNAs of less than ~ 80 nucleotides. Since most RNAs of biological interest are greater than 80 nucleotides in length, methods for synthesizing these larger RNAs in good yield, while maintaining the ability to site-specifically incorporate base analogs that allow for fluorescence measurements, could be of broad interest. Here we describe an approach for synthesis of large RNA molecules (>100 nt) that uses T4 RNA ligase to segmentally join a sequence fragment of an RNA, chemically synthesized with a fluorescent base analog, with the remaining unmodified portion of the RNA oligonucleotide, synthesized through in vitro transcription with T7 polymerase. This method is demonstrated through synthesis of packaging sequences (Ψ-site) derived from HIV-1 genomic RNA leader sequence (~ 120 nt) with the fluorescent base analog, 2-aminopurine (2-AP), selectively incorporated into the dimerization initiation site (DIS) stem-loop sequence. Using 2-AP fluorescence, RNA conformational changes associated with the formation of non-covalent DIS mediated Ψ-site dimers have been analyzed.
Keywords: fluorescence, base analog, 2-aminopurine, HIV-1, genomic RNA, dimerization, maturation
1. Introduction
In the human immunodeficiency virus type-1 (HIV-1), genomic RNA is packaged as a homodimer that is linked non-covalently through a 5′-leader sequence (Figure 1) called the dimer linkage structure (DLS). Proper packaging of genomic RNA as a homodimer appears functionally relevant in a number of important events in the retroviral life cycle, including recombination and reverse transcription,1 translation of the gag gene,2 and selective encapsidation.3–5 RNA genome dimerization in HIV-1 is initiated by the dimerization initiation site (DIS) sequence which forms a highly conserved 35-nucleotide stem-loop and is located within the DLS.6–11 The DIS loop contains an auto-complementary hexanucleotide sequence, which is found most often to be either GUGCAC (subtype A or Mal variant) or GCGCGC (subtype B or Lai variant), together with highly conserved 5′ and 3′ flanking purine nucleotides (Figure 1). Previous studies have shown that sequences derived from the DLS can spontaneously form homodimers in vitro through a loop-loop kissing interaction and that isolated DIS stem-loops will also self-associate to form loop-loop kissing dimer complexes.1,11–14 Moreover, the DIS stem-loop has been found to convert from a metastable kissing to an extended duplex dimer conformation in a structural rearrangement that is catalyzed by the HIV-1 nucleocapsid protein (NCp7) and suggested to be associated with maturation of the budded viral particle.
Figure 1.
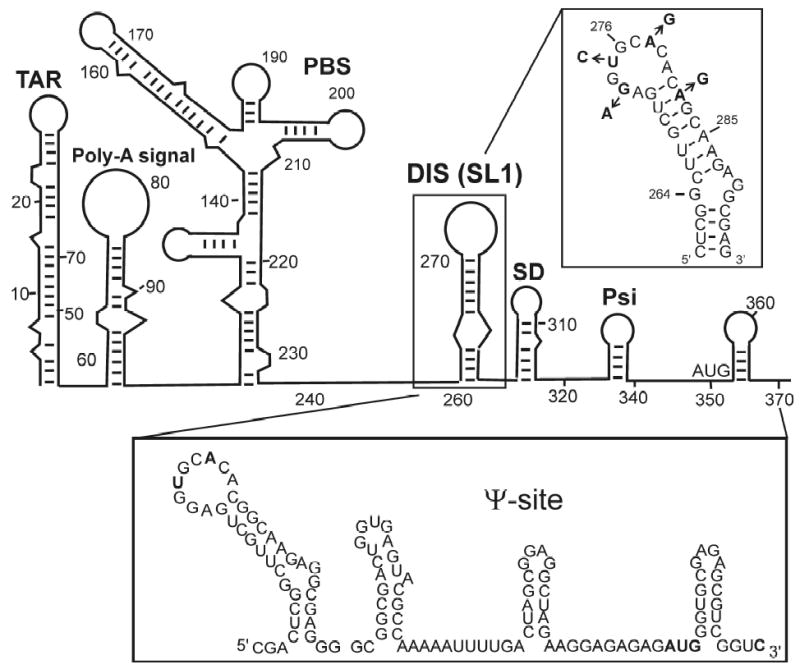
(A) Proposed secondary structure of the 5′-leader sequence of the HIV-1 genomic RNA, with the DIS (SL1) stem-loop highlighted. [Boxed above] RNA sequence and secondary structure of the DIS (SL1) stem-loop found in the HIV-1 subtype-A isolate. The DIS loop contains a highly conserved hexanucleotide palindrome sequence and flanked by conserved purine nucleotides. Positions in the sequences which differ for the subtype-B isolate of HIV-1, a second major strain of the virus, are indicated by arrows and substituted nucleotides. [Boxed below] RNA sequence and secondary structure of the Ψ-site packaging sequence found in the HIV-1 subtype-A isolate.
By exploiting the exquisite dependence of the quantum yield of the highly fluorescent, commercially available, nucleotide base analog, 2-aminopurine (2-AP), to its local structural and chemical environment, we have designed fluorescence methods to directly detect RNA structural changes associated with dimerization of the HIV-1 DIS stem-loop.14,15 The sensitivity of the quantum yield of 2-AP to its local environment (Figure 2) has been used in a number of studies as an intrinsic probe of changes in RNA conformation associated with macromolecular interactions and/or chemical reactions.16 For example, 2-AP fluorescence has been applied to characterize RNA conformational changes associated with peptide binding17,18 and ribozyme cleavage.19–21 2-AP fluorescence methods have also been used to characterize binding of small molecule inhibitors and Rev peptide to the HIV-1 rev responsive element (RRE),22,23 aminoglycoside and Tat peptide binding to the HIV-1 trans activator region (TAR),24 aminoacridine binding to an RNA tetraloop,25 aminoglycoside binding to prokaryotic and eukaryotic ribosomal RNA sequences,26,27 and inhibition of hammerhead ribozymes.28 In general, 2-AP is a non-perturbing substitution since it is similar in structure to adenine (6-aminopurine) and can form a wobble base pair with uridine or thymine. 2-AP is usually highly quenched when it is stacked with other bases, but increases as much as 100-fold when fully exposed to solvent.16,29 In our studies, the range observed for the quantum yield (ΦF) of 2-AP bases incorporated within RNA oligonucleotides has been found to be about an order of magnitude smaller likely due to the fact that 2-AP is efficiently quenched in the context of an oligonucleotide sequence even in ‘solvent exposed’ regions of the structure. Nonetheless, the sensitivity of 2-AP’s quantum yield to its microenvironment allows binding to be detected through direct contact, as well as indirectly through conformational changes in the RNA that may accompany macromolecular interactions or chemistry.
Figure 2.
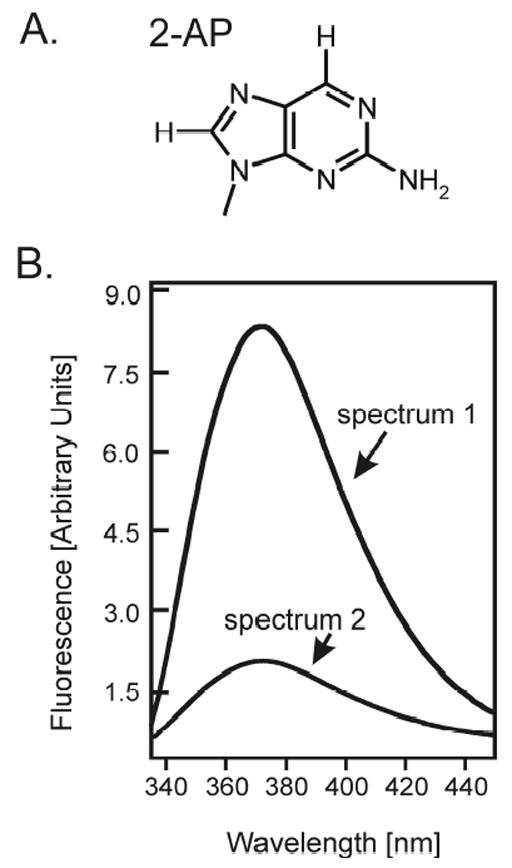
(A) Structure of the fluorescent nucleotide base analog 2-aminopurine (2-AP). (B) The quantum yield (ΦF) of 2-AP fluorescence is strongly dependent on the microenvironment of the base. If 2-AP is exposed to aqueous solvent, as is often the case when it is found in a bulged or unstructured loop region of secondary structure, it is observed to be highly fluorescent (spectrum 1); while if it is in a hydrophobic environment, as is the case when it is stacked in single or double stranded helical regions of secondary structure, its fluorescence emission is severely quenched (spectrum 2).
To use fluorescence methods to detect with high-sensitivity and selectivity the HIV-1 DIS loop-loop kissing dimer formation and the structural rearrangement of the DIS kissing dimer catalyzed by the HIV-1 NCp7 protein (Figure 3A), DIS stem-loops have been designed to contain 2-AP at positions in the sequence which provided unambiguous fluorescence probes for the formation of each of the two structural conformations of the DIS dimer in solution (Figure 3B, C). It should be noted that since these two DIS stem-loop dimers are the same molecular weight and roughly similar in global structure, they can not be readily distinguished using methods, like native polyacrylamide gel electrophoresis (PAGE) or gel filtration, which exploit size and/or shape differences between macromolecules and macromolecular complexes. Nonetheless, DIS loop-loop kissing and extended duplex dimers can be distinguished using native PAGE by exploiting the difference in the Mg+2 dependence of the gel stability of the two dimer forms.36 While both loop-loop kissing and extended duplex dimers are observed as stable complexes on native gels run with Mg+2 in the gel buffers, only the extended duplex is observed as a stable complex on native gels run without Mg+2. Using 2-AP labeled DIS stem-loops, fluorescence-detected methods have been used to determine the molecular mechanism of HIV-1 DIS structural isomerization and address the specific role of NCp7 in the process. In particular, fluorescence equilibrium and kinetic binding constants for DIS dimerization, as well as NCp7 activated rates of DIS structural conversion from kissing to extend duplex dimer could be measured.14 Using these methods, it has also been demonstrated15 that the rate of NCp7 catalyzed maturation of the DIS kissing dimer is directly correlated with an observed proton-coupled conformational dynamics localized around the DIS loop purine bases, where NCp7 is found to convert the dynamic A272 (pKa ~ 6.5) protonated state with a faster rate. In general, these studies have revealed how specific base protonation could modulate local RNA structure and thereby accelerate the protein chaperone mediated structural rearrangement of a kinetically trapped RNA state.
Figure 3.
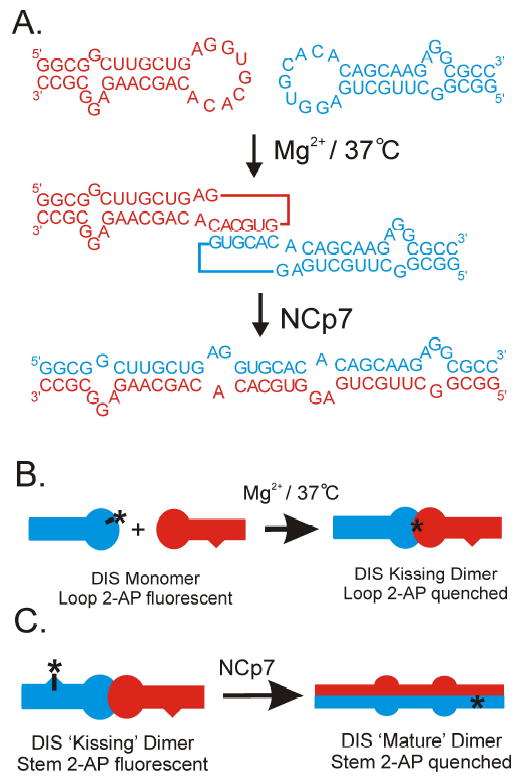
(A) Schematic of the intermolecular base pair interactions which form the basis for the DIS homodimeric loop-loop kissing complex and the structural conversion of the DIS homodimer from loop-loop kissing to extended duplex conformations. Note that one of the two identical hairpin sequences is shown in red and the other in blue. Formation of the DIS loop-loop kissing homodimer is salt dependent, with complexes formed at physiological temperature (37 °C) with divalent metals, like Mg2+, found to be kinetically trapped. At 37 °C, the conversion of Mg2+ stabilized DIS homodimer from kissing loop to extended duplex conformations requires the HIV-1 protein NCp7, which functions as a nucleic acid chaperone in stimulating and facilitating the RNA structural rearrangement. (B) Incorporation of a 2-AP, which is indicated schematically by an asterisk, within the complementary loop sequence of the DIS stem-loop allows the kissing interaction to be specifically monitored as a function of quenching of the fluorescence of the 2-AP base as it forms a stacked, base pair in the loop-loop helix. (C) Similarly, the stem strand exchange associated with conversion of the DIS stem-loop from kissing to extended duplex dimer can be monitored as a function of the quenching of a highly fluorescent bulged 2-AP again shown as an asterisk in the DIS kissing dimer that is engineered to forms a stacked, base pair in the intermolecular stem helix of the extended duplex dimer.
While RNA structure is often found to be modular and isolated 2-AP labeled DIS stem-loops have provided a wealth of information about structural and dynamic changes in this stem-loop associated with DIS dimerization and NCp7 conversion of the DIS dimer from kissing to extended duplex forms, measurement of DIS mediated interactions in the context of the larger 5′-leader sequences would allow more detailed questions about this interaction to be addressed. For example, it is of interest to determine if the flanking HIV-1 leader sequence influences kinetic binding rates for the DIS stem-loop in kissing dimer formation and/or the thermodynamic stability of the DIS dimer linkage. It is also still not known how the structural conversion of the DIS dimer linkage from kissing to extended duplex dimer is accomplished by NCp7 in the context of the full 5′-leader sequence, if there are any long-range interactions between DIS and other elements in the 5′-leader sequence that effect the DIS conversion and what effects the DIS conversion may have on the global structure of the 5′-leader sequence. To study DIS structure and interactions within the context of larger HIV-1 leader sequences, here we describe the initial synthesis of packaging or Ψ-site sequences from HIV-1 with 2-AP probes incorporated as fluorescent probes for the detection of DIS mediated dimerization and NCp7 chaperone-assisted conversion of the DIS dimer.
2. Results and Discussion
To extend the application of fluorescent nucleotide base analog probes to studies of large RNA sequences, such as those derived from the leader sequence of HIV-1, T4 RNA ligase has been employed to couple chemically synthesized, sequence fragments containing 2-AP bases, with unmodified RNA sequences to produce large RNA oligonucleotides (>100 nt) that are site-specifically labeled with the fluorescent base analog. Using reaction conditions optimized for the joining of single stranded RNAs,30,31 T4 RNA ligase has previously been used to segmentally couple RNA sequences for x-ray crystallography32 and heteronuclear NMR studies.33 Here, this approach of enzymatic ligation has been adopted to generate the HIV-1 packaging sequences (nt 258–370), which is a sequence element at the 3′-end of the 5′-leader sequence (nt 1–370) of HIV-1, that contain single site 2-AP labeled DIS (SL1) stem-loops. In addition to the DIS stem-loop, the Ψ-Site sequence includes sequences downstream of the 5′ splice donor site (SL3 and a GA-rich region) that have been suggested to be involved in genomic RNA dimerization based on mutation and deletion analysis.34
2.1 Strategy for the synthesis of HIV-1 Ψ-Site sequences with 2-AP labeled DIS stem-loops
T4 RNA ligase can catalyze the joining of a 3′-donor RNA having a 5′ terminal monophosphate and a 5′-acceptor RNA terminating in a 3′-hydroxyl (Figure 4A). The ligation reaction is ATP-dependent and results in a product that is coupled via a standard 3′, 5′ phosphodiester linkage. The catalytic efficiency of T4 RNA ligase depends on the structure of the 5′- and 3′-ends of the RNA substrates and is most efficient at joining single stranded RNAs which are unstructured at the point of ligation. To avoid intramolecular ligation and/or the formation of unwanted by-products in the reaction, the donor and acceptor RNA fragments need to be synthesized or modified to contain the appropriate terminal hydroxyls or phosphorylation. Briefly, the 5′-acceptor RNA should contain terminal hydroxyls at both the 5′- and 3′-ends of the sequence; while, the 3′-donor RNA should be phosphorylated at both ends of the oligonucleotide. The requirements for the 5′-acceptor RNA to contain terminal hydroxyls on both the 5′- and 3′-ends of the sequence for the T4 RNA ligation reaction are met without further modification by RNA synthesized using standard phosphoramidite chemistry. Chemical synthesis generates RNA oligonucleotides with terminal hydroxyls on both the 5′- and 3′-ends of the oligonucleotide. Thus, chemically synthesized DIS stem-loop oligonucleotides, with 2-AP bases incorporated within the sequence, and appropriate single stranded ends could be used directly in the T4 RNA ligation reaction (Figure 4B). For the 3′-donor RNA, the sequence fragment was generated using in vitro transcription with T7 RNA polymerase. To obtain a 3′-donor RNA fragment with the appropriate termini, the transcription reaction was primed using GMP so that RNA synthesis initiates at the 5′-end with a 5′-monophosphate. Further, the RNA transcript was designed to contain a cis-acting 3′-hammerhead ribozyme which self-cleaves concurrently with RNA transcription to produce a 2′,3′ phosphodiester linkage at the 3′-end.33,35 The 2′,3′ phosphodiester effectively blocks the 3′-end of the RNA so that it can not be joined to another sequence by T4 RNA ligase. Thus, in vitro transcription of a 3′-donor fragment that includes the truncated Ψ-site sequence and a 3′-hammerhead ribozyme (Figure 4C) using T7 polymerase and primed by GMP could be used to generate a 3′ truncated Ψ-site fragment suitable for use in the T4 RNA ligation reaction without further modification. It should be noted, however, that use of the 3′-hammerhead ribozyme results in some sequence restrictions at the 3′-end of the oligonucleotide due to requirements for high-efficiency ribozyme cleavage (Figure 4C). Specifically, the use of the 3′-hammerhead requires the RNA sequence to terminate with UX, where X = rA, rU, rC. As an alternative to a hammerhead ribozyme, a hairpin ribozyme can be used at the 3′-end which has sequence requirements on the 5′ side of the cleavage site that are less stringent.35
Figure 4.
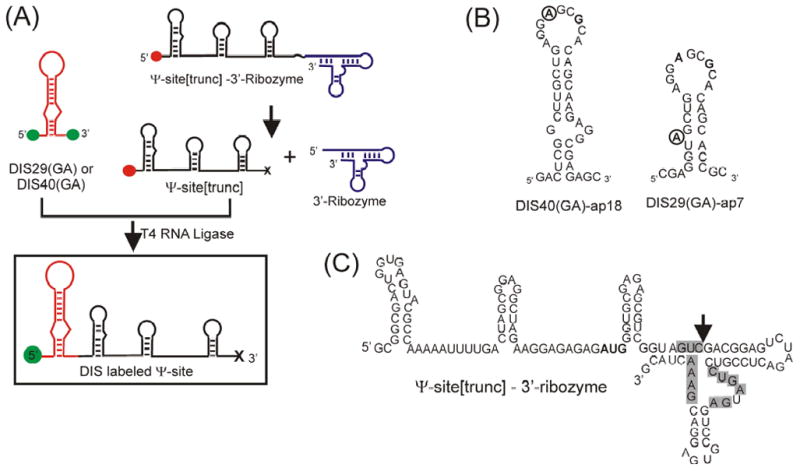
(A) General synthetic strategy for the generation of DIS labeled Ψ-site sequences. Green filled circles indicate terminal hydroxyls, red filled circles indicate terminal monophosphates and the ‘x’ indicates the terminal 2′, 3′ cyclic phosphodiester resulting from the ribozyme cleavage reaction. (B) RNA sequence and secondary structure of the DIS stem-loop constructs [DIS40(GA)-ap18 and DIS29(GA)-ap7] designed to form heterodimer complexes. Point mutations in the hexanucleotide sequence [U275 → A275 and C278 → G278] to form the DIS(GA) stem-loop which destroy the palindromic nature of the sequence and promote heterodimer formation are bolded. The position of the 2-AP substitution in the sequence is circled. (C) Sequence and secondary structure of the RNA construct containing the truncated Ψ-site sequence and a 3′ cis-acting hammerhead ribozyme. The sequence positions required for ribozyme activity are shadowed in grey and the cleavage site is indicated by an arrow.
The 5′ DIS acceptor and 3′ truncated Ψ-site donor RNA sequence fragments prepared through chemical and enzymatic protocols, respectively, were separately purified before use in the ligation reaction. For each ligation reaction, optimal conditions were initially screened using a series of small 10 μl scale reactions, focusing on customizing reagent concentrations and buffer conditions previously reported to be the most critical for individual reaction yields.31 For the DIS/Ψ-site(trunc) ligations, the concentration and molar ratios of RNA donor and acceptor fragments and the concentration of PEG were found to have the largest impact on yields of the ligation reactions. In this respect, the phenomena of steric exclusion which results from addition of PEG to the reaction mixture, as well as cation-stimulated aggregation, have previously been found to have dramatic effects on ligation yields of single stranded RNA substrates using T4 RNA ligase.31 The optimized concentrations for these components of the reaction were found to be 2.5 nmol of 3′ DIS fragment and 5.0 nmol of 5′ Ψ-site(trunc) fragment per 1 ml of reaction and 15% PEG8000. After incubation at 22 °C for 4 hour, approximately 50% of the RNA fragments were found to be ligated using these optimized concentrations and experimental conditions as described in the Experimental section. After heat inactivation of T4 RNA ligase, the products were purified from the remaining fragments using preparative scale denaturing PAGE (Figure 5).
Figure 5.
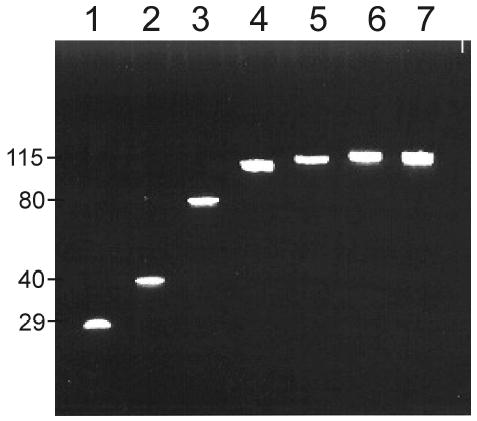
A denaturing polyacrylamide gel of the RNA ligation reactant and product sequences post-purification. Lane 1. DIS29(AG)-ap7; Lane 2. DIS40(AG)-ap18; Lane 3. Ψ-site(trunc), Lane 4. Ψ-Site[DIS29(GA)-ap7]; Lane 5. Ψ-Site[DIS40(GA)-ap18]; Lane 6. Ψ-Site[DIS40(GA)]; Lane 7. Ψ-Site[DIS40(UC)]. Note that the band observed for the Ψ-Site[DIS29(GA)-ap7] ligation product (Lane 4) has a slightly faster mobility due to its size difference relative to the unlabeled Ψ-Sites (Lanes 6 & 7) and the Ψ-Site[DIS40(GA)-ap18] ligation product (Lane 5). Sequence lengths are indicated on the left side of the gel.
2.2 Sequence design of the DIS stem-loops
As previously described,14,15 the DIS stem-loops used in this study are designed to form heterodimeric kissing dimers through point mutations in the loop palindromic sequence [U275 to A275 and C278 to G278 to form the DIS(GA) stem-loop and U275 to C275 and C278 to U278 to form the complementary DIS(UC) stem-loop]. These altered sequences exhibit wild-type kissing dimer properties, while exclusively forming hetero- rather than homodimer complexes.1,14,36 Since the native DIS stem-loops can form dimers to various extents and affinities depending on RNA concentrations and experimental conditions, heterodimeric forming DIS stem-loops have been employed in the experiments to allow a finer control over the aggregation state of the DIS stem-loops (e.g. monomer versus dimer), as well as facilitate quantitative analysis of the binding reactions. To obtain a fluorescent probe for the DIS conformational changes associated with dimerization and NCp7 mediated maturation (Figure 6A), 2-AP has been inserted at two specific positions in the DIS(GA) stem-loop: one construct with 2-AP substituted at the A275 loop position [so called DIS40(GA)-ap18] and a second with 2-AP inserted as a single bulged nucleotide in the stem [so called DIS29(GA)-ap7]. In addition, these DIS stem-loops have been designed with both 5′ and 3′ single stranded overhanging sequences to make them more ideal substrates for T4 RNA ligase.
Figure 6.
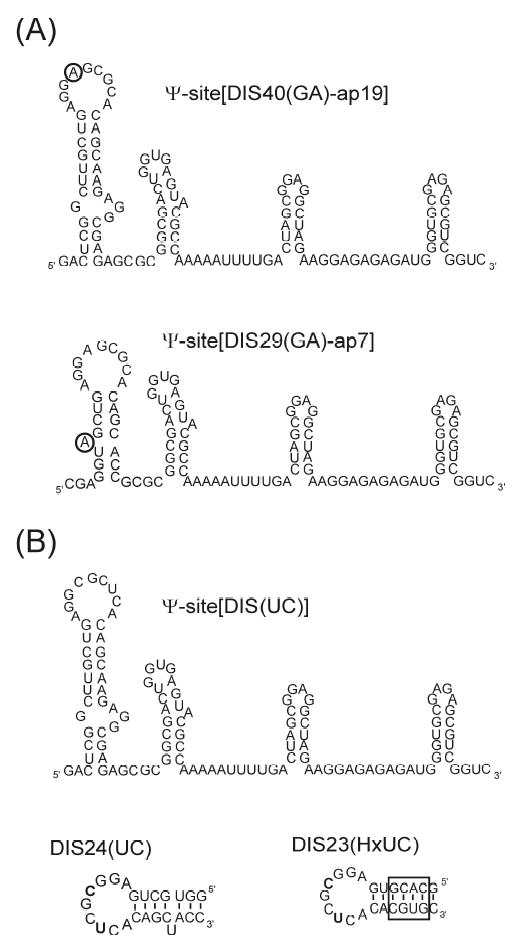
(A) RNA sequence and secondary structure of the Ψ-Site constructs, Ψ-Site[DIS40(GA)-ap18] and Ψ-Site[DIS29(GA)-ap7] synthesized using the T4 RNA ligation method with 2-AP bases incorporated into the DIS stem-loop sequences for fluorescence studies. The position of the 2-AP substitution in the sequence is circled. (B) RNA sequence and secondary structure of the unlabeled ΨSite construct, Ψ-Site[DIS(UC)] and DIS stem-loops, DIS24(UC) and DIS23(HxUC) designed with DIS loop sequences complementary to the 2-AP labeled Ψ-Site constructs to promote formation of heterodimeric loop-loop kissing complexes. The sequence for DIS23(HxUC), which contains a stem sequence which is inverted relative to the wild-type so that it can not form a stable extended duplex structure, is shown with the exchanged stem sequence boxed.
In addition to an unmodified Ψ-site[DIS40(UC)] construct, two unmodified DIS(UC) stem-loops have also been chemically synthesized (Figure 6B), with hexanucleotide loop sequences complementary to the DIS(GA) stem-loops: one construct with a stem sequence complementary to DIS29(GA)-7ap, that can be used to specifically probe the DIS structural isomerization [DIS24(UC)] and a second construct with an exchanged stem sequence that is capable of forming only the kissing dimer [DIS23(HxUC)]. These stem-loops allow the measurement of dimer formation and NCp7 mediated structural conversion between full length Ψ-site[DIS(GA)] packaging RNAs and isolated DIS(UC) stem-loops. To confirm independently of the fluorescence measurements that no structural perturbation resulted from substitution of 2-AP probes in the DIS(GA) stem-loop, native gel electrophoresis analysis was performed using the 2-AP constructs. For all 2-AP DIS(GA) RNA constructs used in this study, stable complexes could be observed under identical native gel conditions used to observe the wild-type RNA complexes (data not shown).
2.3 Fluorescence detection of Ψ-Site[DIS] kissing dimer formation
Using the 2-AP labeled Ψ-Site[DIS40(GA)-ap18] construct (Figure 7A), changes in 2-AP fluorescence emission could be used to monitor DIS kissing dimer formation. Titration of the Ψ-Site[DIS40(GA)-ap18], substituted with 2-AP at position A275 in the loop with either the unmodified, complementary Ψ-Site[DIS40(UC)] or the DIS(UC) stem-loops [DIS24(UC) or DIS23(HxUC)], results in an approximately 3-fold decrease in 2-AP fluorescence emission (Figure 7B). The direction and magnitude of this fluorescence change indicates that the 2-AP substituted base becomes significantly stacked upon binding, which is consistent with 2-AP·U base pairing that would accompany the formation of the DIS kissing complex loop-loop helix. A similar quenching behavior of loop substituted 2-AP probes has previously been observed upon formation of kissing complexes between DIS RNA stem-loops,14 as well as in the formation of kissing complexes between stem-loops derived from antisense RNAs which regulate ColE1 plasmid replication.37 As observed with the isolated DIS stem-loops, Mg+2-stabilized DIS kissing dimer linkages were found to form kinetically trapped complexes. In addition, Ψ-Site constructs were also observed to associate as kissing dimers in the presence of monovalent cations, such as Na+, in concentrations of 100 to 300 mM under standard buffer conditions. Using 2-AP fluorescence quenching measurements, heterodimeric DIS kissing complexes formed by Ψ-Site[DIS40(GA)-ap18] and Ψ-Site[DIS40(UC)], in standard buffer containing 5 mM MgCl2, were observed to have an apparent equilibrium binding constant (KD ~ 200 ± 26 nM), which is slightly weaker than previously measured for the isolated DIS stem-loops (KD ~ 1.3 ± 0.7 nM).14 Similarly (Figure 7B, inset), formation of the heterodimeric DIS kissing complex formed between Ψ-Site[DIS40(GA)-ap18] and the DIS24(UC) stem-loop could be detected using 2-AP fluorescence and was found to have an apparent equilibrium binding constant (KD ~ 30 ± 1 nM). This measured binding affinity is closer to the KD observed previously for the isolated DIS stem-loops14 and taken together with the Ψ-site kissing dimer measurements, may suggest an entropic cost to organizing these larger sequences which results in the lower apparent binding constants.
Figure 7.
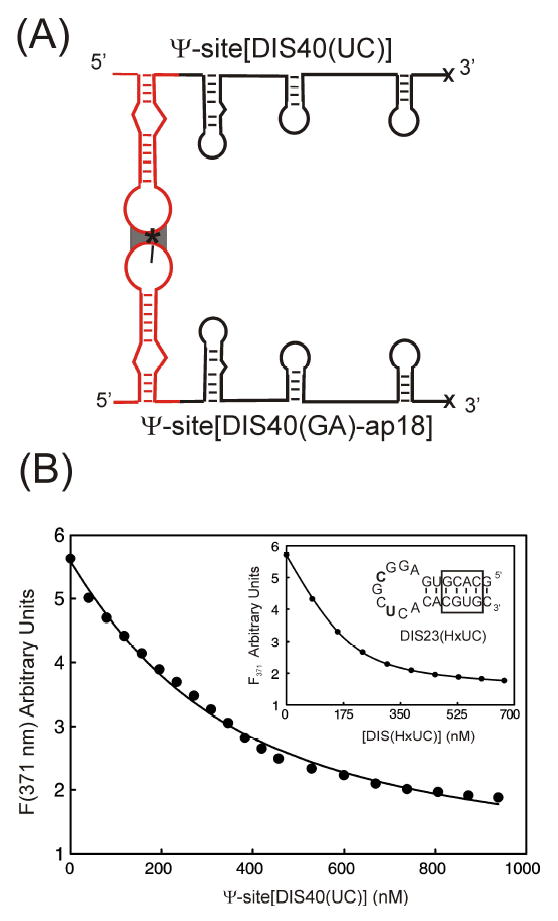
(A) Schematic of the formation of the Ψ-Site[DIS40(GA)-ap18] and Ψ-Site[DIS40(UC)] intermediate dimer via the DIS(SL1) loop-loop kissing interaction. The location of the 2-AP probe on the DIS stem-loop is indicated by an asterisk. Note that only the dimerization mediated via DIS, which is detected by the 2-AP probe, is shown and that additional contacts may also occur between other RNA sequences within the Ψ-Site. (B) Plot of the fluorescence decrease at 371 nm as a function of total Ψ-Site[DIS40(UC)] concentration for a titration using 200 nM Ψ-Site[DIS40(GA)-ap18]. The solid curve is fit (KD = 200 ± 23 nM) using a single site equilibrium binding equation. [Inset] Plot of the fluorescence decrease at 371 nm as a function of total DIS(HxUC) concentration for a titration using 200 nM Ψ-Site[DIS40(GA)-ap18]. The solid curve is fit (KD = 30 ± 1 nM) using a single site equilibrium binding equation.) The sequence for DIS23(HxUC) is shown in the inset, with the altered palidromic nucleotide positions U275 and C278 bolded and exchanged stem sequence boxed.
2.4 Measurement of NCp7 chaperone-assisted maturation of Ψ-Site[DIS] kissing dimmers
As in previous studies using isolated DIS stem-loop,14,15 NCp7 catalyzed structural isomerization of the DIS kissing dimer linkage could be measured by following the time-dependent decrease in fluorescence emission as the Ψ-Site[DIS29(GA)-ap7]·Ψ-Site[DIS40(UC)] kissing dimer is converted to an extended duplex (Figure 8). The Ψ-Site[DIS29(GA)-ap7] construct, which is capable of forming heterodimeric Ψ-Site[DIS29(GA)-ap7]·Ψ-Site[DIS40(UC)] kissing dimer, contains a 2-AP probe inserted into the stem sequence such that it formed a single nucleotide bulge. The fluorescence of the 2-AP probe at this position in DIS stem-loop in the Ψ-Site[DIS(GA)] construct was found to be relatively high and confirmed that the 2-AP is indeed in an unpaired bulged state. Upon formation of a DIS mediated kissing dimer with the Ψ-Site[DIS40(UC)] in standard buffer containing 5 mM MgCl2, the fluorescence intensity of the 2-AP probe of Ψ-Site[DIS29(GA)-ap7] was found to initially increase slightly but then remained constant. Addition of NCp7 to the DIS kissing complex preformed between the Ψ-Site[DIS29(GA)-ap7] and Ψ-Site[DIS40(UC)] results in a time-dependent 3-fold decrease in 2-AP fluorescence emission (Figure 8B). Even though the DIS stem-loop in the Ψ-Site[DIS40(UC)] does not form a 2-AP·U base pair in the mature duplex DIS dimer with the 2-AP base from Ψ-Site[DIS29(GA)-ap7] when the stem strands are exchanged, the 2-AP base can adopt a stacked conformation within an intermolecular helix formed by the exchanged DIS stem strands in the mature duplex form. The direction and magnitude of the observed fluorescence change indicates that the 2-AP substituted base becomes significantly stacked with time, which is consistent with 2-AP stacking that would accompany exchange of the stem strands during the isomerization process. Similarly (Figure 8B, inset), addition of NCp7 to DIS kissing complexes preformed between the Ψ-Site[DIS29(GA)-ap7] and the DIS24(UC) stem-loop results in a time-dependent 3-fold decrease in 2-AP fluorescence emission. For these constructs, DIS24(UC) stem-loop contains a bulged U base which is capable of forming a 2-AP·U base pair in the mature duplex DIS dimer with the 2-AP base from Ψ-Site[DIS29(GA)-ap7] if the stem strands are exchanged. The direction and magnitude of the observed fluorescence change therefore again indicates 2-AP base pairing and stacking that would accompany exchange of the stem strands during the isomerization process. In this context, it should be noted that the 2-AP fluorescence response of NCp7 addition to kissing complexes formed between DIS stem-loops with mismatched stem sequences incapable of forming an exchanged intermolecular helix in the mature duplex dimer has been measured to confirm that the NCp7 dependent quenching of 2-AP in these reactions is due to stem strand exchange associated with DIS conversion.14 Overall, the results from experiments using Ψsite/Ψ-site dimers and Ψ-site/DIS stem-loop complexes suggest that NCp7 acts by catalyzing a similar exchange of DIS stem strands in the context of the Ψ-site packaging sequence as has been previously observed with isolated DIS stem-loops.14,15
Figure 8.
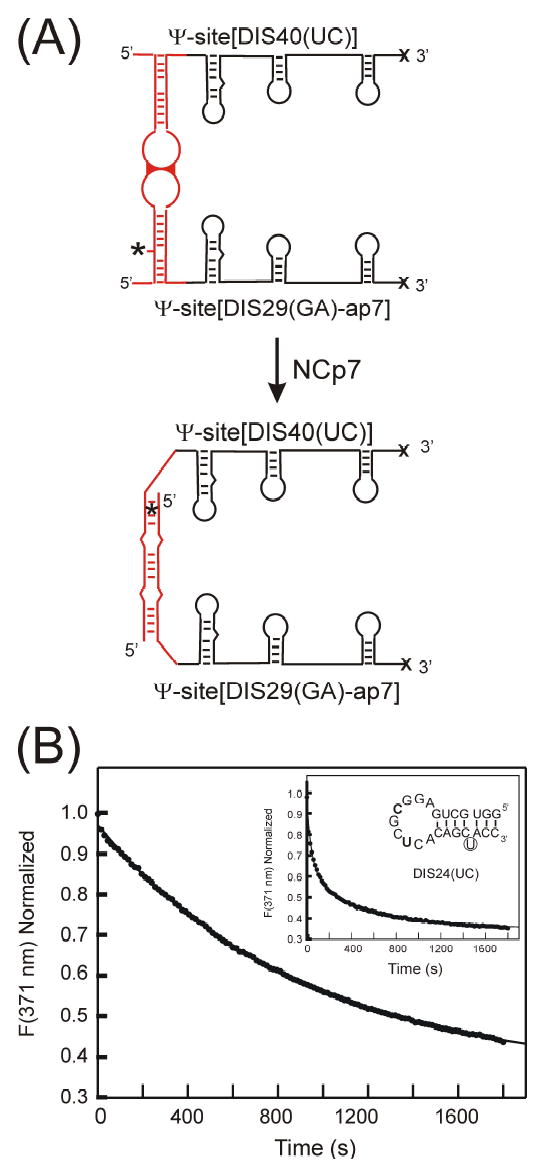
(A) Schematic of the structural conversion of the Ψ-Site[DIS29(GA)-ap7]·Ψ-Site[DIS40(UC)] homodimer from a kissing to extended duplex conformation. Note that only the structural rearrangement of DIS, which is detected by the 2-AP probe, is shown and that additional structural changes may also occur. The location of the 2-AP probe on the DIS stem-loop is indicated by an asterisk. (B) Maturation of the Ψ-Site[DIS29(GA)-ap7]·Ψ-Site[DIS40(UC)] kissing complex monitored as a function of quenching of 2-AP fluorescence emission after addition of NCp7 at pH 6.0. Plot of the fluorescence decrease at 371 nm (filled circles) as a function of time after a ~ 20-fold excess of NCp7 protein is added to 100 nM of the Ψ-Site[DIS29(GA)-ap7]·Ψ-Site[DIS40(UC)] kissing complex preformed in the presence of 5 mM MgCl2. The structural isomerization rate was fit using a first order rate constant equation (kconv = 0.066 min−1). Structural isomerization rates (kconv = 0.066 ± 0.001 min−1, karr = 0.006 ± 0.01 min−1) were fit using a double exponential rate equation. [Inset] Maturation of the Ψ-Site[DIS29(GA)-ap7]·[DIS24(UC)] kissing complex monitored as a function of quenching of 2-AP fluorescence emission after addition of NCp7 at pH 6.0. Plot of the fluorescence decrease at 371 nm (filled circles) as a function of time after a ~ 10-fold excess of NCp7 protein is added to 100 nM of the Ψ-Site[DIS29(GA)-ap7]·[DIS24(UC)] kissing complex preformed in the presence of 5 mM MgCl2. The sequence for DIS24(UC) stem-loop shown in the inset, with altered palindromic nucleotide positions C275 and U278 bolded and bulged U base in the stem sequence circled. Structural isomerization rates (kconv = 1.31 ± 0.05 min−1, karr = 0.12 ± 0.005 min−1) were fit using a double exponential rate equation.
3. Conclusion
Non-natural fluorescent nucleotide analogs have found widespread use as probes for detecting and monitoring structure, dynamics and interactions of nucleic acids in solution.1 The use of fluorescent nucleotide base analogs as structural probes, however, has been limited owing to the practical size limit for chemical synthesis of RNA (≤80 nt). To address this issue, here we have described the use of the T4 RNA ligase to couple chemically synthesized 2-AP labeled sequence fragments with unlabeled RNA sequences to produce large RNA oligonucleotides site-specifically labeled with the nucleotide base analog. Using this approach, HIV-1 packaging sequences (~ 120 nt) have been synthesized with 2-AP labeled DIS (SL1) stem-loops that allow DIS mediated dimerization of the packaging sequence and HIV-1 NCp7 mediated structural rearrangements of the DIS dimer linkage to be directly measured using fluorescence methods. Methods described here for synthesis of 2-AP labeled Ψ-site constructs should be generally applicable to the synthesis of any large RNA sequence with site specific incorporation of other fluorescent base analogs that are available in phosphoramidite form. As such, this synthetic approach opens the possibility for using fluorescent base analogs more broadly in larger biological RNAs whose size currently precludes direct synthesis via chemical methods.
4. Experimental2
4.1 Materials
T4 RNA ligase, T4 DNA ligase, EcoR1 and BamH1 enzymes were all purchased from New England BioLabs (Ipswich, MA). All other buffers and reagents were of the highest quality commercially available and were used without further purification.
4.2 Chemical Synthesis of DIS (SL1) stem-loops
2-aminopurine 2′-O-methyl riboside containing DIS stem-loop RNA oligonucleotides were synthesized on an Applied Biosystems 390 synthesizer (Perkin-Elmer, Forest City, CA) using standard phosphoramidite chemistry.38 The nucleoside phophoramidites were purchased from Glen Research (Sterling, VA). Note that for this study, the palindromic hexanucleotide DIS loop sequence has been altered to create two unique DIS stem-loops, so called DIS(GA) and DIS(UC), which associate at nanomolar concentrations as hetero- rather than homo-dimers, as has been previously demonstrated.1,14,36 2-AP labels were inserted at two specific positions in the DIS(GA) stem-loop: one construct with 2-AP substituted at the A18 loop position [DIS40(GA)-ap18] and a second with 2-AP inserted as a single bulged nucleotide in the stem [DIS29(GA)-ap7]. Both DIS stem-loops contain 5′ and 3′ single stranded sequence regions and hydroxyl-termini to make them ideal substrates for T4 RNA ligase. These two constructs are designed to probe kissing versus mature duplex dimer formation, respectively. Two unlabeled DIS(UC) stem-loops have also been chemically synthesized, with hexanucleotide loop sequences complementary to the DIS(GA) stem-loops: one construct with a stem sequence complementary to DIS29(GA)-7ap, that can be used to specifically probe the DIS structural isomerization [DIS24(UC)] and a second construct with an exchanged stem sequence that is capable of forming only the kissing dimer [DIS23(HxUC)].
All chemically synthesized RNA oligonucleotides were deprotected using standard protocols and purified using preparative-scale denaturing polyacrylamide gel electrophoresis (PAGE), recovered by electrophoretic elution and then desalted and exchanged into Standard buffer (1 mM Cacodylate [pH=6.5], 25 mM NaCl) using an Amicon Centriplus concentrator (Millipore, Bedford, MA). RNA concentrations were determined by measuring the absorbance at 260nm using the following extinction coefficients: [DIS40(GA)-ap18], 393.1 mM−1 cm−1; [DIS29(GA)-ap7], 280.9 mM−1 cm−1; DIS23(HxUC), 214.4 mM−1 cm−1; DIS24(UC), 223.5 mM−1 cm−1.
4.3 Enzymatic synthesis of Ψ-Site and truncated Ψ-Site sequences
Using enzymatic methods, heterodimer DIS versions of the Ψ-Site (Ψ-Site[DIS(GA)] and Ψ-Site[DIS(UC)]) have been synthesized using T7 polymerase and linearlized plasmid DNA templates. The plasmid DNA templates were constructed using a set of four synthetic oligonucleotides which contained the T7 promoter sequence followed the Ψ-Site sequence and restrictions sites for BamHI and EcoR1 on the 5′- and 3′-ends of the oligonucleotides sequences, respectively. Synthetic oligonucleotides were ligated using T4 DNA ligase and cloned into a pUC18 vector between BamHI and EcoR1 restriction sites using standard molecular biology protocols. The plasmid containing template was amplified in E. coli and purified using a plasmid MEGA kit (Qiagen, Calencia, CA) which yielded ~2.5 milligrams of DNA per preparation. The plasmid DNA was linearized with the restriction enzyme, EcoR1 and purified using a plasmid MAXI column (Qiagen, Calencia, CA) prior to use as a template in the in vitro transcription reaction using T7 RNA polymerase. The optimal in vitro transcription reaction conditions used for RNA synthesis are shown in Table I.
Table I.
In vitro Transcription Reaction Conditions
| 50 ug/ml | plasmid template |
| 40 mM | Tris-HCl [pH 8.3] |
| 5 mM | DTT |
| 1 mM | Spermidine |
| 0.01 % | Triton X-100 |
| 25 mM | MgCl2 |
| 4 mM | NTP |
| 0.1 mg/ml | T7 RNA polymerase |
For synthesis of the construct containing a truncated Ψ-Site sequence (Ψ-Site[trunc]) and a cis-acting 3′-hammerhead ribozyme, a DNA fragment (150 nt total) was inserted into a pUC18 vector again using restriction sites, BamHI and EcoR1, and standard molecular biology protocols. The DNA fragment contained the T7 promoter sequence, an 80 nucleotide sequence (296nt - 371nt) of the Ψ-Site and a 3′ cis-acting hammerhead ribozyme. Linearlized template DNA was prepared by EcoR1 restriction enzyme digestion of the plasmid, purified using a plasmid MAXI column (Qiagen, Calencia, CA) and used as a template in the in vitro transcription reactions essentially as described above. To prime the reaction with GMP, concentrations of 8 mM GMP and 2 mM GTP were used respectively, instead of 4 mM GTP as described above, so that the 5′-end of the transcript contained a monophosphate group.39 Under the reaction conditions used for RNA synthesis, cleavage by the cis-acting hammerhead ribozyme occurs simultaneously with RNA synthesis. Both RNA transcription and ribozyme cleavage reactions were stopped by addition of EDTA after 4 hours.
All in vitro transcribed RNA oligonucleotides were purified using preparative-scale denaturing polyacrylamide gel electrophoresis (PAGE), recovered by electrophoretic elution and exchanged into Standard buffer (1 mM Cacodylate [pH=6.5], 25 mM NaCl) using an Amicon Centriplus concentrator (Millipore, Bedford, MA). RNA concentrations were determined by measuring the absorbance at 260nm using the following extinction coefficients: Ψ-site[DIS40(GA)], 1162.3 mM−1 cm−1; Ψ-site[DIS40(UC)], 1155.3 mM−1 cm−1; Ψ-site[trunc], 817.1 mM−1 cm−1.
4.4 Ligation of DIS with the truncated Ψ-Site
Purified DIS stem-loops were ligated with the truncated form of the Ψ-Site (Ψ-Site[trunc]) using T4 RNA ligase to form the two Ψ-Site[DIS(GA)] constructs with site specific incorporation of 2-AP: Ψ-Site[DIS29(GA)-ap7] and Ψ-Site[DIS40(GA)-ap18]. For the two ligation reactions, the conditions used per 1 milliliter of reaction are shown in Table II. The optimal ligation efficiency for each reaction was determined by altering the following reaction conditions: (1) the molar ratio of donor Ψ-site[trunc] and acceptor (DIS) RNA fragment concentrations which was varied from 1:1 to 1:5 and (2) PEG concentration which was varied from 3% to 20%. Using a molar ratio of donor (trunc Ψ-site) and acceptor (DIS) of 1:2 and a PEG concentration of 15 %, an approximately 50 % yield of ligated product could be achieved, which corresponds to 1 to 1.5 ng of product per ml of reaction.
Table II.
T4 RNA Ligation Reaction Conditions
| 2.5 nanomoles | Donor RNA (trunc Ψ-site) |
| 5.0 nanomoles | Acceptor RNA (DIS(GA)) |
| 15% | PEG8000 |
| 10 mM | MgCl2 |
| 50 mM | Tris-HCl (pH 7.5) |
| 100 μM | ATP |
| 10 μM | Bovine Serume Albumin (BSA) |
| 1.0 mM | Hexammine cobalt chloride (HCC) |
| 1600 Units | T4 RNA ligase |
Full-length product was separated from unreacted Ψ-site[trunc] and DIS hairpin sequences using preparative-scale denaturing polyacrylamide gel electrophoresis (PAGE), recovered by electrophoretic elution and then desalted and exchanged into standard buffer (1 mM Cacodylate [pH=6.5], 25 mM NaCl) using an Amicon centriplus concentrator (Millipore, Bedford, MA). RNA concentrations were determined by measuring the absorbance at 260nm using the following extinction coefficients: Ψ-site[DIS40(GA)-ap18], 1168 mM−1 cm−1; Ψ-site[DIS29(GA)-ap7], 11061 mM−1 cm−1.
4.5 Denaturing Polyacrylamide Gel analysis
Denaturing polyacrylamide gel electrophoresis (PAGE) was used to analysis the progress of the RNA ligation reactions and the purity of the products after post-purification. Denaturing polyacrylamide gels were cast using 12% (19:1) acrylamide and 8M Urea, a running buffer of 90 mM Tris HCl [pH 7.5], 90 mM Boric Acid, 1.0 mM EDTA (TBE buffer) and run in a Hoefer minigel apparatus for ~ 1 hour at 25 V/cm. 10 μl of ~ 5μM sample concentration in 50 % Formamide: 50 % TBE were loaded in each lane and the bands were visualized using ethidium bromide staining.
4.6 NCp7 expression in E.coli and purification
The HIV-1 NCp7 protein was expressed and purified using a protocol previously described.40
4.7 Native Gel analysis of Ψ-Site[DIS] dimer formation
Formation of kissing dimer complexes with Ψ-Site constructs containing 2-AP was assayed using native PAGE. Monomer versus dimer states of these constructs can be readily observed using gel methods, while the two dimer forms (kissing versus mature) can be distinguished based on thermal stability.1,41,42 RNA was visualized by ethidium bromide staining. Binding reactions (10μl) were performed in a buffer containing 90 mM Tris HCl [pH 7.5], 90 mM Boric Acid, 1.0 mM MgCl2 (1 × TBM). Samples were incubated for 10 min, then 10μl were loaded on a non-denaturing 15% polyacrylamide gel (75:1 acrylamide:bisacrylamide) and run in 1 × TBM at 4 °C at 10V/cm using a BioRad Minigel apparatus.
4.6 Fluorescence Detection of DIS kissing dimer formation
The fluorescence emission spectra of RNA oligonucleotide samples (200 nM) selectively labeled with 2-AP were measured at 25 °C on a SPEX Fluoromax-3 spectrofluorometer (Instruments SA, Edison, NJ) using a 0.3 cm square cuvette in 150 μL of standard buffer solution with 5 mM MgCl2. Emission spectra were recorded over the wavelength range of 330 to 450 nm with an excitation wavelength of 310 nm and a spectral band pass of 5 nm. The equilibrium binding constant (KD) for DIS loop-loop kissing dimer formation was determined by following the decrease in fluorescence at 371 nm as a fixed concentration of the fluorescent Ψ-site[DIS40(GA)-18ap] was titrated with increasing amounts of either the Ψ-site[DIS40(UC)] or DIS23(HxUC) complement. The RNA-RNA interaction in each case was fit using a single site equilibrium binding equation using the program Sigma Plot. For example, single site binding of DIS23(HxUC) to Ψ-site[DIS40(GA)-18ap] was fit using equation 1 as follows,
where Fo and Ff are the initial and final fluorescence intensities, respectively, [DIS(GA)]tot is the total Ψ-site[DIS40(GA)-18ap] concentration, and [DIS(UC)]tot is the total concentration of DIS23(HxUC).
4.7 Kinetic Measurement of NCp7 Catalyzed Structural Conversion of the DIS dimmer
NCp7 catalyzed structural isomerization of DIS was measured at 25 °C using the SPEX Fluoromax-3 spectrofluorometer (Instruments SA, Edison, NJ) by following the decrease in fluorescence emission at 371 nm as the kissing dimer linkage in the Ψ-site[DIS29(GA)-7ap]·Ψ-site[DIS40(UC)] and the Ψ-site[DIS29(GA)-7ap]·DIS24(UC) complexes is converted to the extended duplex conformation. In both experiments, the preformed kissing dimer complex was first formed in Standard buffer solution at pH 6.0 in the presence of 5 mM MgCl2, then mixed manually with NCp7 and rapidly inserted into the spectrofluorometer for fluorescence measurements. The concentration of the kissing complex in each case was 100 nM and that of the NCp7 protein was 1.1 μM and 2.2 μM, respectively, for the Ψsite/stem-loop and Ψ-site/Ψ-site complexes. The time course of the conversion of the DIS kissing dimer was fit using the equation, Ft = F1 exp(−kconv t) + F2 exp(−karr t) + C, where kconv is the observed isomerization rate for conversion of the DIS kissing to extended mature duplex dimer and karr is attributed to the rate of rearrangement of the 2-AP probe subsequent to its stacking in the duplex conformation.
Acknowledgments
We thank F. Song (CARB) for oligonucleotide synthesis, T. Bradrick (CARB) for assistance with the fluorescence data analysis and D. Brabazon (Loyola College) for careful reading of the manuscript. This work is supported by NIH Grant GM59107 awarded to J.P.M. Fluorescence instrumentation is supported in part by the National Institute of Standards and Technology.
Footnotes
Although the major nucleotide bases are essentially non-fluorescent, there are a few naturally occurring fluorescent bases, such as wyosine (Yt),43 which is found in the anticodon region of tRNAPhe. The rarity of naturally occurring fluorescent bases, however, limits their use in fluorescence studies.
Certain commercial equipment, instruments, and materials are identified in this paper in order to specify the experimental procedure. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the material or equipment identified is necessarily the best available for the purpose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Paillart JC, Marquet R, Skripkin E, Ehresmann C, Ehresmann B. Biochimie. 1996;78:639–653. doi: 10.1016/s0300-9084(96)80010-1. [DOI] [PubMed] [Google Scholar]
- 2.Miele G, Mouland A, Harrison GP, Cohen E, Lever AML. J Virol. 1996;70:944–951. doi: 10.1128/jvi.70.2.944-951.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McBride MS, Panganiban AT. J Virol. 1996;70:2963–2973. doi: 10.1128/jvi.70.5.2963-2973.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Skripkin E, Paillart JC, Marquet R, Ehresmann B, Ehresmann C. Proc Natl, Acad Sci USA. 1994;91:4945–4949. doi: 10.1073/pnas.91.11.4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laughrea M, Shen N, Jette L, Wainberg MA. Biochemistry. 1999;38:226–234. doi: 10.1021/bi981728j. [DOI] [PubMed] [Google Scholar]
- 6.Muriaux D, Girard PM, Bonnetmathoniere B, Paoletti J. J Biol Chem. 1995;270:8209–8216. doi: 10.1074/jbc.270.14.8209. [DOI] [PubMed] [Google Scholar]
- 7.Berkhout B, vanWamel JLB. J Virol. 1996;70:6723–6732. doi: 10.1128/jvi.70.10.6723-6732.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clever JL, Wong ML, Parslow TG. J Virol. 1996;70:5902–5908. doi: 10.1128/jvi.70.9.5902-5908.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haddrick M, Lear AL, Cann AJ, Heaphy S. J Mol Biol. 1996;259:58–68. doi: 10.1006/jmbi.1996.0301. [DOI] [PubMed] [Google Scholar]
- 10.Laughrea M, Jette L. Biochemistry. 1996;35:9366–9374. doi: 10.1021/bi960395s. [DOI] [PubMed] [Google Scholar]
- 11.Laughrea M, Jette L. Biochemistry. 1996;35:1589–1598. doi: 10.1021/bi951838f. [DOI] [PubMed] [Google Scholar]
- 12.Skripkin E, Paillart JC, Marquet R, Blumenfeld M, Ehresmann B, Ehresmann C. J Biol Chem. 1996;271:28812–28817. doi: 10.1074/jbc.271.46.28812. [DOI] [PubMed] [Google Scholar]
- 13.Paillart JC, Westhof E, Ehresmann C, Ehresmann B, Marquet R. J Mol Biol. 1997;270:36–49. doi: 10.1006/jmbi.1997.1096. [DOI] [PubMed] [Google Scholar]
- 14.Rist MJ, Marino JP. Biochemistry. 2002;41:14762–14770. doi: 10.1021/bi0267240. [DOI] [PubMed] [Google Scholar]
- 15.Mihailescu MR, Marino JP. Proc Natl Acad Sci U S A. 2004;101:1189–1194. doi: 10.1073/pnas.0307966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rist M, Marino JP. Curr Org Chem. 2001;6:775–793. [Google Scholar]
- 17.Barrick JE, Takahashi TT, Balakin A, Roberts RW. Methods. 2001;23:287–293. doi: 10.1006/meth.2000.1139. [DOI] [PubMed] [Google Scholar]
- 18.Austin RJ, Xia TB, Ren JS, Takahashi TT, Roberts RW. Biochemistry. 2003;42:14957–14967. doi: 10.1021/bi0351312. [DOI] [PubMed] [Google Scholar]
- 19.Menger M, Tuschl T, Eckstein F, Porschke D. Biochemistry. 1996;35:14710–14716. doi: 10.1021/bi960440w. [DOI] [PubMed] [Google Scholar]
- 20.Harris DA, Rueda D, Walter NG. Biochemistry. 2002;41:12051–12061. doi: 10.1021/bi026101m. [DOI] [PubMed] [Google Scholar]
- 21.Jeong S, Sefcikova J, Tinsley RA, Rueda D, Walter NG. Biochemistry. 2003;42:7727–7740. doi: 10.1021/bi034627g. [DOI] [PubMed] [Google Scholar]
- 22.Lacourciere KA, Stivers JT, Marino JP. Biochemistry. 2000;39:5630–5641. doi: 10.1021/bi992932p. [DOI] [PubMed] [Google Scholar]
- 23.DeJong ES, Chang CE, Gilson MK, Marino JP. Biochemistry. 2003;42:8035–8046. doi: 10.1021/bi034252z. [DOI] [PubMed] [Google Scholar]
- 24.Bradrick TD, Marino JP. RNA. 2004;10:1459–1468. doi: 10.1261/rna.7620304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yan ZH, Baranger AM. Bioorg Med Chem Lett. 2004;14:5889–5893. doi: 10.1016/j.bmcl.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 26.Kaul M, Barbieri CM, Pilch DS. J Am Chem Soc. 2004;126:3447–3453. doi: 10.1021/ja030568i. [DOI] [PubMed] [Google Scholar]
- 27.Kaul M, Barbieri CM, Pilch DS. J Mol Biol. 2005;346:119–134. doi: 10.1016/j.jmb.2004.11.041. [DOI] [PubMed] [Google Scholar]
- 28.Kirk SR, Luedtke NW, Tor Y. Bioorg Med Chem. 2001;9:2295–2301. doi: 10.1016/s0968-0896(01)00123-7. [DOI] [PubMed] [Google Scholar]
- 29.Millar DP. Curr Opin Struct Biol. 1996;6:322–326. doi: 10.1016/s0959-440x(96)80050-9. [DOI] [PubMed] [Google Scholar]
- 30.Romaniuk PJ, Uhlenbeck OC. Method Enzymol. 1983;100:52–59. doi: 10.1016/0076-6879(83)00045-2. [DOI] [PubMed] [Google Scholar]
- 31.Tessier DC, Brousseau R, Vernet T. Anal Biochem. 1986;158:171–178. doi: 10.1016/0003-2697(86)90606-8. [DOI] [PubMed] [Google Scholar]
- 32.Sherlin LD, Bullock TL, Nissan TA, Perona JJ, Lariviere FJ, Uhlenbeck OC, Scaringe SA. RNA. 2001;7:1671–1678. [PMC free article] [PubMed] [Google Scholar]
- 33.Kim I, Lukavsky PJ, Puglisi JD. J Am Chem Soc. 2002;124:9338–9339. doi: 10.1021/ja026647w. [DOI] [PubMed] [Google Scholar]
- 34.Russell RS, Hu J, Beriault V, Mouland AJ, Laughrea M, Kleiman L, Wainberg MA, Liang C. J Virol. 2003;77:84–96. doi: 10.1128/JVI.77.1.84-96.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Price SR, Ito N, Outbridge C, Avis JM, Nagai K. J Mol Biol. 1995;249:398–408. doi: 10.1006/jmbi.1995.0305. [DOI] [PubMed] [Google Scholar]
- 36.Takahashi KI, Baba S, Chattopadhyay P, Koyanagi Y, Yamamoto N, Takaku H, Kawai G. RNA. 2000;6:96–102. doi: 10.1017/s1355838200991635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rist MJ, Marino JP. Nucleic Acids Res. 2001;29:2401–2408. doi: 10.1093/nar/29.11.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beaucage SL, Caruthers MH. Tetrahedron Lett. 1981;22:1859. [Google Scholar]
- 39.Milligan JF, Uhlenbeck OC. Method Enzymol. 1989;180:51. doi: 10.1016/0076-6879(89)80091-6. [DOI] [PubMed] [Google Scholar]
- 40.Amarasinghe GK, De Guzman RN, Turner RB, Chancellor KJ, Wu ZR, Summers MF. J Mol Biol. 2000;301:491–511. doi: 10.1006/jmbi.2000.3979. [DOI] [PubMed] [Google Scholar]
- 41.Huthoff H, Berkhout B. RNA. 2001;7:143–157. doi: 10.1017/s1355838201001881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huthoff H, Berkhout B. Biochemistry. 2002;41:10439–10445. doi: 10.1021/bi025993n. [DOI] [PubMed] [Google Scholar]
- 43.Plavec J, Chattopadhyaya J. Tetrahedron. 1996;52:1597–1608. [Google Scholar]