

Abstract
Churchill is a zinc-containing protein that is involved in neural induction during embryogenesis. At the time of its discovery, it was thought on the basis of sequence alignment to contain two zinc fingers of the C4 type. Further, binding of an N-terminal GST-Churchill fusion protein to a particular DNA sequence was demonstrated by immunoprecipitation selection assay, suggesting that Churchill may function as a transcriptional regulator by sequence-specific DNA binding. We show by NMR solution structure determination that, far from containing canonical C4 zinc fingers, the protein contains three bound zinc ions in novel coordination sites, including an unusual binuclear zinc cluster. The secondary structure of Churchill is also unusual, consisting of a highly solvent exposed single-layer β-sheet. Hydrogen-deuterium exchange and backbone relaxation measurements reveals that Churchill is unusually dynamic on a number of time scales, with the exception of regions surrounding the zinc coordinating sites, which serve to stabilize the otherwise unstructured N-terminus and the single-layer β-sheet. No binding of Churchill to the previously-identified DNA sequence could be detected, and extensive searches using DNA sequence selection techniques could find no other DNA sequence that was bound by Churchill. Since the N-terminal amino acids of Churchill form part of the zinc-binding motif, the addition of a fusion protein at the N-terminus causes loss of zinc and unfolding of Churchill. This observation most likely explains the published DNA-binding results, which would arise due to non-specific interaction of the unfolded protein in the immunoprecipitation selection assay. Since Churchill does not appear to bind DNA, we suggest that it may function in embryogenesis as a protein-interaction factor.
INTRODUCTION
Gastrulation is a process during the early stages of vertebrate embryonic development, in which the embryo undergoes an intricate cellular reorganization under the guidance of a distinct group of cells collectively called the organizer.1-3 During this process, surface cells of the embryo internalize and form three distinct germ layers, endoderm, mesoderm, and ectoderm. The ectoderm layer further gives rise to epidermis and neural tissues. The classic ‘default model’ mechanism of neural induction postulated that ectodermal cells have a predisposition for forming neural tissues, but are inhibited from doing so by bone morphogenetic protein (BMP) signaling. At a certain point in development, BMP antagonists are secreted from the organizer allowing the proximal ectodermal cells to undergo differentiation into neural cells.4 On the other hand, recent evidence from animal models has shown that inhibition of BMP signaling alone is not sufficient for neural induction.5-9 The fibroblast growth factor family of proteins (FGFs) has been directly implicated in both mesoderm formation10 and neural induction,11-13 and is required in combination with additional signaling events to ensure a neural fate.7,11 In addition, cells must be exposed to the organizer or FGF derived signals for several hours before becoming sensitized to BMP inhibitors and initiating neural cell formation.11,13,14
Churchill (ChCh), a putative zinc finger protein, was discovered in a differential screen for neural inducing factors present in chick embryos after several hours of signaling from the organizer.14 Sequence alignment of ChCh predicted the presence of two CCCC motif zinc fingers (Figure 1).14 ChCh was identified as a late FGF response gene that is upregulated within 4-5 hours of signaling from both the organizer and FGF and shows no indication of down-regulation in the presence of BMP.14 C-terminal fusions of VP16-activator and engrailed repressor (EnR) domains to ChCh showed repression of targets of FGF signaling in mesoderm formation in the case of VP16 but not EnR, suggesting a role for ChCh in transcriptional regulation. Further suggesting the ability of ChCh to function as a transcriptional regulatory protein, a 6 base pair DNA binding consensus sequence (CGGG(G/A/T)(G/A/C)) was identified utilizing an N-terminal GST-ChCh fusion in an immunoprecipitation DNA selection assay and confirmed by electrophoretic mobility gel shift assay (EMSA).14
Figure 1.

Alignment of the ChCh sequence from selected species indicating the high level of sequence homology amongst vertebrates, Homo sapiens (human), Canis familiaris (dog), Rattus norvegicus (rat), Mus musculus (mouse), Sus scrofa (pig), Bos taurus (cow), Takifugu rubripes (fugu), Xenopus laevis (xenopus), Danio rerio (zebrafish), Gallus gallus (chicken). Sequence identity is indicated by yellow (Cys), green (aliphatic), blue-green (aromatic), red (acidic), blue (basic), beige (P, G, S, T, N, Q, A). The CXXC motifs are shown by brackets at the bottom of the figure, and the additional completely conserved Cys and His residues are shown by asterisks. Secondary structure elements (β-strands) identified in the NMR analysis and structure determination are shown above the sequence.
Here we describe the structure of ChCh, obtained in solution by NMR. The protein does not contain the predicted two C4 zinc fingers, but consists of a highly unusual solvent-exposed β-sheet, stabilized by the coordination of three zinc ions, including a zinc binuclear cluster. We also find no evidence that ChCh binds to DNA. Based on the non-canonical structure and the lack of interaction with DNA, we propose that ChCh is not a DNA binding transcription factor but probably functions in neural induction through interactions with other proteins involved in transcriptional regulation.
RESULTS
Design of Protein Constructs
Initial NMR analysis of full-length ChCh (residues 1-112; ChCh112) yielded a well-dispersed HSQC spectrum indicating the presence of a well-folded protein. After several days, however, the protein was apparently truncated by 4 residues at the C-terminus (Thr109 to Phe112) as indicated by mass spectrometry analysis. The C-terminal residues are unstructured; a new construct eliminating the five C-terminal residues (ChCh107) also gives a well-dispersed NMR spectrum that is superimposable with the full-length ChCh112 construct. The N-terminal methionine is cleaved during expression of the recombinant protein in E. coli; this was also confirmed by mass spectrometric analysis of both full-length ChCh112 (12,901 Da vs. 13,033 Da) and ChCh107 (12,290 Da vs. 12,422 Da). Cleavage of the N-terminal methionine residue is common in both prokaryotes and eukaryotes,15 and it has been well established that the propensity for N-terminal methionine cleavage is increased when the adjacent amino acid is one of the seven residues having sidechains with the smallest radii of gyration (glycine, alanine, serine, cysteine, threonine, proline, and valine).16 The highly conserved Cys2 is involved in zinc coordination, which stabilizes the N-terminus of ChCh.
Zinc Coordination
The sequence alignment of Figure 1 shows the four conserved CXXC motifs that formed the basis of the “two zinc finger” ChCh model, but ESI-MS analysis of ChCh107 indicated the presence of three zinc atoms rather than the expected two (see Supplementary Material Figure S1). A closer inspection of the sequence alignment reveals the presence of three completely conserved histidine residues, His59, His66 and His71, and an additional conserved cysteine residue, Cys17, giving four potential ligands for the third zinc. In order to identify the zinc ligands, single amino acid mutants were prepared. The majority of these mutants, including C5H, H66C, H66A, and C30H, either failed to express or were aggregated or unfolded by NMR. Interestingly, mutation of Cys17 had variable consequences: a C17S mutant was aggregated/unfolded by NMR, while the spectra of C17A and C17V showed that they were folded, but with multiple additional signals for several residues in the N-terminal loop region. These results are consistent with the NOESY data, which show that Cys17 is not in close proximity to any other zinc ligands, which gives a shortage of one ligand for three normally coordinated zinc ions, and suggests that the zinc coordination sphere of ChCh is unusual.
Metal coordination causes recognizable changes in the NMR spectrum of a histidine side chain; ligand histidines can be identified and their tautomeric states assigned from 2JHN 15N HMQC,17 13C HMQC,18 and 3D 13C HMQC-NOESY19 spectra. The nitrogen atoms that are coordinated to zinc show characteristic chemical shift values near 215 ppm that can be utilized to assign zinc coordination via the δ1 or ε2 positions in the histidine ring by inspection of the variation in intensity of the two-bond and three-bond scalar couplings from the non-exchangeable protons, Hδ2 and Hε1. For ChCh, these experiments showed unequivocally that His59, His66, and His79 are zinc ligands, coordinating the zinc atom through Nε2 for His59 and His66 and Nδ1 for His71. The non-conserved His65 is not involved in zinc atom coordination, as indicated by significant line broadening of the resonances in comparison to the zinc coordinating histidine atoms and the large pH dependence of its proton and nitrogen chemical shifts.
NMR Spectroscopy
The NMR spectra of ChCh107, exemplified by the 1H-15N HSQC (Figure 2), are well dispersed and allow for nearly complete resonance assignments. The amide proton of Arg36 was broadened beyond detection, but the Hα and all side-chain resonances were readily assigned. The aromatic ζ protons of Phe27 and Phe38 were not assigned due to overlap of both proton and carbon resonances in the aromatic region. Secondary structural elements for ChCh, consisting of 5 β-strands, were initially identified by deviations in backbone chemical shifts from random coil values. An antiparallel topology of the five-stranded β-sheet was discerned from the NOESY data, which showed the expected short cross-strand distances with strong Hα(i) to Hα(j), medium Hα(i) to HN(j+1) and medium HN(i) to HN(j) intensities. All four of the C-X-X-C sequences show chemical shift deviations consistent with rubredoxin turns, which are often seen in zinc finger proteins.20 Between the first two N-terminal C-X-X-C sequences, there are no regular secondary structural elements apparent in the chemical shift data. The third C-X-X-C sequence appears in the longer histidine-rich loop between β2 and β3, and the final C-X-X-C sequence is part of a short loop between β4 and β5. The asymmetric distribution of the zinc binding C-X-X-C motifs and the absence of any helical content in the ChCh secondary structure are inconsistent with the predicted14 presence of two C4 type zinc fingers.
Figure 2.

600 MHz 1H-15N HSQC spectrum of ChCh107 showing selected assignments. The broadened cross peaks for residues 18 and 24 are indicated with blue labels, and the approximate position of the weak broadened cross peak of Gly21 is indicated. Side chain guanidinium cross peaks (folded) are indicated in red, and side chain carboxamide resonances of Asn and Gln are indicated by a horizontal line and italicized label. A fully labeled spectrum is shown in the Supplementary Material (Figure S2).
Structure Determination by NMR
NOE cross peaks from the 3D 15N NOESY-HSQC21 and 3D 13C HMQC-NOESY19 spectra were used to generate 2,351 distance restraints through automated assignment methods.22 Initial structures were generated without zinc ligand bond restraints. Additional restraints for dihedral angles from chemical shift and scalar coupling data were used in refinement cycles with the program AMBER23 to generate a well defined ensemble structure with a backbone root mean square deviation of 0.14 Å from the lowest energy structure. A superposition of the 20 lowest-energy structures of ChCh is shown in Figure 3(a) and a ribbon representation of the lowest energy structure is depicted in Figure 3(b). Structural statistics for the ChCh ensemble are summarized in Table 1. As predicted from the spectroscopic evidence, ChCh does not contain canonical C4 zinc finger motifs; rather, the five strands of the antiparallel β-sheet dominate the structure. The single layer β-sheet is solvent-exposed on both sides and is partly stabilized by cross-strand interactions mediated by the zinc coordination sites. The first strand, β1, begins with Met39 and runs to Glu49 with residues Thr42 and Asn43 forming a β-bulge. The second strand, β2, begins with Glu52 and ends with the zinc ligand Cys61. The third strand, β3, begins with the zinc ligand His66, and runs to Met78 with residues Val67 and Ile68 forming a second β-bulge. The remaining β-strands occur without breaks, including β4 which extends from Phe81 to the zinc ligand Cys88, and β5 which encompasses residues Lys93 to Ser99. As in other zinc finger proteins, coordination to zinc ions in ChCh appears to serve a primarily structural function that is essential to maintain the folded conformation. The N-terminal region forms an irregular structure extending from Cys2 to Thr38, which is held together by an unusual Cys6His-Zn2 binuclear cluster bridged by Cys30 (Figure 4). The binuclear cluster includes ligands from the first two zinc coordination sites comprising Cys2, Cys5, Cys30 and His66 for the first zinc ion and of Cys30, Cys33, Cys61 and Cys64 for the second zinc ion. The cluster effectively links the N-terminal region of ChCh to strands β2 and β3 in the β-sheet. The central section of the N-terminal loop crosses a portion of strands β3-β5, though there is a surprising paucity of stabilizing hydrophobic interactions. The binding site for the third zinc, which is ligated by His59, His71, Cys88 and Cys91, effectively cross-links four of the five strands of the β-sheet, with strands β2 and β3 providing both histidine ligands and the rubredoxin turn between strands β4 and β5 contributing both cysteine ligands.
Figure 3.

Solution structure of ChCh107. (a) Stereo representation of a superposition of the heavy atoms for residues 2-99 of the 20 lowest energy structures. Residues at the ends of the β-strands are indicated. (b) Ribbon diagram of the lowest energy refined structure shown in the same orientation with zinc ligands and β-strands labeled. The backbone is shown in coral, zinc in purple, cysteine side chain zinc ligands in yellow, histidine side chain zinc ligands in blue. The unliganded His65 and Cys17 are shown in light blue and green respectively. The figure was prepared with MOLMOL.74
Table 1.
Structural Statistics for the 20 Lowest Energy Structures of ChCh107
| NMR Restraints for AMBER | |
| Total restraints | 2483 |
| NOE distance restraints | 2351 |
| Intra-residue | 429 |
| Sequential | 550 |
| Medium range | 813 |
| Long range | 1344 |
| Hydrogen Bonds | 0 |
| Zinc ligand bonds | 12 |
| Dihedral angle restraints | 132 |
| φ | 31 |
| Ψ | 30 |
| χ1 | 71 |
| AMBER Energies and Violations | |
| Mean AMBER energy (kcal/mol) | -4974 |
| Mean number of NOE violations > 0.1 Å | 3.55 |
| Maximum NOE violation (Å) | 0.15 |
| Maximum torsion angle violation (°) | 0.84 |
| Mean restraint violation energy (kcal/mol) | 21.6 |
| Average deviation from idealized bond lengths (Å) | 0.01 |
| Average deviation from idealized bond angles (°) | 3.1 |
| Average RMSD from Lowest Energy Structure (Å) | |
| ChCh107 (2-99) backbone atoms | 0.14 ± 0.05 |
| ChCh107 (2-99) all heavy atoms | 0.73 ± 0.07 |
| Ramachandran Statistics | |
| Residues in most favored regions | 76.2% |
| Residues in additionally allowed regions | 21.5% |
| Residues in generously allowed regions | 0.9% |
| Residues in disallowed regions | 1.4% |
Figure 4.

Lowest energy refined structure of ChCh showing the disposition of ligands around the three zinc atoms. The figure was prepared with MOLMOL.74
Dynamics Analysis of ChCh
The backbone dynamics of ChCh107 were assessed by measuring the 15N spin relaxation parameters, R1, R2 and the steady-state heteronuclear 15N NOE ([1H]-15N NOE). The results of the relaxation analysis are summarized in Figure 5. Only well-resolved resonances were used for the analysis and, of the 101 assigned backbone amides in ChCh, 16 were excluded due to severe spectral overlap and 3 due to significant line broadening. From the [1H]-15N NOE data, the backbone of ChCh appears to be relatively rigid on the ps-ns time scale, with increased mobility in the β-turns between strands β1 and β2 (Glu49-Gly51) and β3 and β4 (Met78-Phe81) and in the unstructured C-terminal region (Ile100-Gln107). Interestingly, although the first 38 N-terminal residues of ChCh do not form regular secondary structure, the [1H]-15N NOE values indicate little mobility, except for a slight NOE decrease for residues 16-20 located in the center of the loop that covers strands β3-β5. The R1 and R2 values are in general consistent with the [1H]-15N NOE, although they suggest that strands β1 and β2 (residues 39-61) experience increased mobility compared to strands β3-β5, as evidenced by a larger variability in both the R1 and R2 values for the residues in these regions. This is most likely due to the fact that strands β1 and β2 are more solvent exposed and interact only peripherally with the N-terminal loop. In addition, several residues in the center of the N-terminal loop have above average R2 values (e.g. Thr16, Asn20, Ser22 and Phe23), suggesting contributions to R2 from μs-ms fluctuations. These findings are consistent with resonance broadening for residues between 18 and 24, observable in the HSQC spectrum (Figure 2). Residues in this region are likely engaged in a conformational exchange process.
Figure 5.
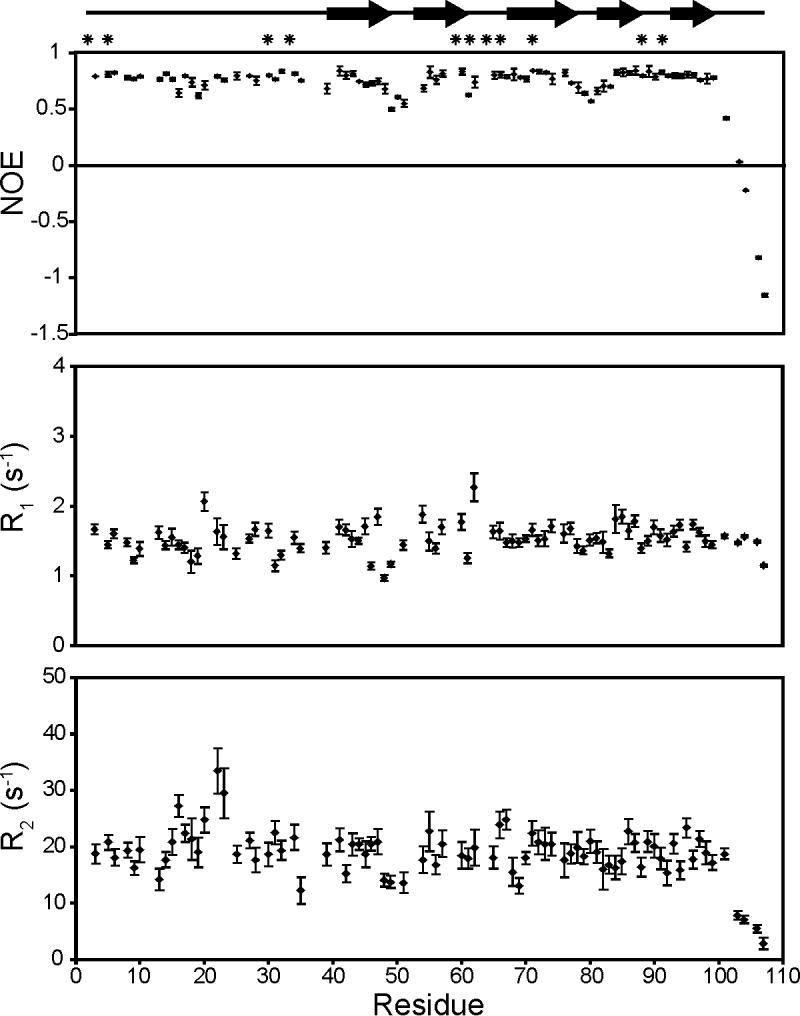
Plot of R1, R2 and the [1H]-15N NOE per-residue for ChCh107. The location of the β-strands is indicated at the top of the figure. The positions of the zinc ligands are indicated by asterisks at the top of the figure. Error bars denote standard deviations.
To characterize the dynamic processes of ChCh on the μs-ms timescale, 15N relaxation dispersion curves for each residue were measured using the constant time relaxation compensated CPMG experiment,24,25 in which the effective transverse relaxation rate constant measured as a function of the B1 field strength is separated into contributions resulting from conformational exchange processes (Rex) and all other transverse relaxation processes (R0).26 These experiments indicate that an unusually large number of resonances in ChCh experience conformational exchange: 66 out of the 101 assigned backbone amides of ChCh have a measurable dispersion profile.
It is not clear from analysis of the R2 dispersion data whether the protein is experiencing a single global conformational exchange process or multiple distinct exchange processes. On the one hand, all of the residues in ChCh experiencing exchange contributions to the relaxation could be fit globally to a two-site exchange model (kex 3570 ± 75 s-1, pb 1.8 ± 0.2%). In all cases the value of kex was significantly greater than the chemical shift difference between the two states (Δω) indicating a fast exchange process on the NMR chemical shift time scale.25,27 On the other hand, a single global process that gives rise to all of the observed exchange contributions may not be a good model for the slow time scale motions of ChCh. Some of the residues that exhibit exchange contributions to the relaxation fit better locally to equations describing the limit of fast exchange, in which the relaxation rate constant is derived from a single population-averaged resonance.26 These residues are mostly localized to the more flexible regions of the protein including the center of the N-terminal loop and strands β1 and β2. The remainder of the resonances fit better locally to equations describing a two-site exchange process, as judged by F-test analysis. In addition, there appear to be inconsistencies in the exchange behavior in a given region of the protein, with the residues experiencing exchange distributed unevenly around regions of no conformational exchange. We have therefore analyzed the R2 dispersion data by local fitting to fast or two-site exchange models as appropriate.
Figure 6 shows representative dispersion curves of residues that were fit locally to a fast or two-site exchange model, and a mapping of the various Rex contributions onto the ChCh structure. The majority of the residues that exhibit exchange contributions are localized to the center of the N-terminal loop, as anticipated from the standard backbone dynamics experiments, as well as a large portion of the β-sheet. Three of the N-terminal loop residues, Gly21, Ser22, and Leu24 could not be analyzed due to significant broadening of the amide resonances. For the remaining residues, the values of Rex vary considerably, with the highest values observed for solvent-exposed residues in the central portion of the N-terminal loop and strands β1 and β2 (Figure 6). Interestingly, three residues in strands β3 and β4 (Tyr73, Thr85 and Met86) localized below the N-terminal loop also had Rex values > 10 s-1, indicating that these residues are sensitive, via side chain contacts, to the conformational fluctuations in the N-terminal loop.
Figure 6.

Relaxation dispersion data for ChCh107. (Left) Representative 15N R2 dispersion curves: Rex > 10 s-1 (Phe23), 5 < Rex < 10 s-1 (Thr42), Rex < 5 s-1 (Arg70). The relaxation dispersion data were collected and analyzed simultaneously at 500 MHz (black) and 800 MHz (red). (Right) Location of residues for which conformational exchange was observed. The backbone is colored gray, yellow, orange or red for residues experiencing no measurable conformational exchange, Rex values < 5 s-1, between 5 < Rex < 10 s-1, or Rex > 10 s-1, respectively. The location of the amides of residues 73, 85 and 86 are indicated. The zinc atoms (gold) and corresponding zinc ligands are indicated (black) for clarity. The figure was prepared with MOLMOL.74
The most striking feature in the dispersion analysis of ChCh is the lack of any observable exchange processes for the zinc ligands in the zinc binuclear cluster and the surrounding amino acids, aside from Cys64. These data correlate with hydrogen/deuterium (H/D) exchange studies. The majority of the ChCh amide protons exchange with deuterium within a matter of minutes, which is not surprising given that the central structural element is a solvent exposed, single-layer β-sheet. Only 22 amides are protected from deuterium exchange, all of which belong to zinc ligands and a few neighboring residues. From the protection factors, it was estimated that the average ΔG for residues in and around the zinc binuclear cluster is 6.3 kcal/mol, indicating a high level of stability in this region. The amide proton of Cys64 is an exception, showing no measurable deuterium exchange protection, together with an observable Rex contribution. On the other hand, the amides of the zinc ligands in the HHCC zinc site exhibit deuterium exchange protection but show only small R2 dispersion. The residues in and around the HHCC zinc site have an average ΔG of 5.2 kcal/mol, which again indicates a high level of stability. Since the HHCC zinc ligands are spread over strands β2-β5, it does not seem unreasonable that these residues would experience a conformational exchange process consistent with the motions of the β-sheet. Nonetheless, the H/D exchange data indicate that this conformational exchange process does not disrupt the hydrogen bonding network for the residues in and around the HHCC zinc site. These findings imply that the zinc binuclear cluster and the HHCC zinc site provide significant stabilization to the N-terminal loop and the solvent exposed β-sheet, respectively.
Mutagenesis to Explore Specific Stabilizing Interactions
Beneath Cys17 in the ChCh structure there is an extensive solvent-exposed hydrophobic region spanning strands β3-β5, including Tyr73, Phe75, Ile77, Tyr84, Ile98 and Ile100. Analysis of the multiple populations observed upon mutation of Cys17 to alanine or valine reveals that the majority of perturbed residues are localized to neighboring residues in the center of the N-terminal loop and residues directly below the loop in strands β3-β5, indicating a possible disruption in side chain contacts between the N-terminal loop and the β-sheet. To probe the sources of the stability of the unusual single-layer β-sheet, the hydrophobic residues in contact with Cys17 were substituted individually with alanines. The stability of the ChCh mutants was determined qualitatively since the unfolding of ChCh is irreversible: the protein does not refold after denaturation and loss of zinc, significantly decreasing the accuracy of standard thermal denaturation experiments. For F75A, I77A, Y84A, I98A, and I100A, the HSQC spectra were superimposable with that of wild-type ChCh, showing only localized chemical shift perturbations for residues in close proximity to the mutation sites. In contrast, the HSQC spectrum of Y73A was similar to that observed for C17A and C17V, where the majority of the protein was folded but several residues also exhibited multiple populations. The ChCh solution structure shows the β-CH2 group of Cys17 packed against the Tyr73 aromatic ring. Cysteine-aromatic interactions can result in significant stabilization of proteins (∼ 2 kcal).28 Stabilization to this extent appears to require a close interaction between the aromatic ring and the sulfur, which does not appear to be the case for Cys17 of ChCh. Instead, the β-methylene group points towards Tyr73 and the -SH group sits in a hydrophobic pocket between strands β3 and β4. The side chain rotamer population of Cys17 was determined directly from the NMR data: Cys17 is unambiguously in the -60 (gauche-) rotamer as shown in the structures, consistent with the measured side chain coupling constants, and intra- and inter-residue NOEs. This interaction seems to be important to the stability of ChCh, since mutation of either Cys17 or Tyr73 results in severe perturbations to the backbone structure. These findings, which are consistent with the dynamics analysis above, indicate that while the central portion of the N-terminal loop can experience local structural fluctuations, the loop serves to stabilize strands β3-β5 through interactions between the side chains of Cys17 and Tyr73.
Experiments to Detect DNA Binding by ChCh
Published immunoprecipitation DNA selection experiments14 identified a putative double-stranded DNA sequence (consensus CGGG(G/A/T)(G/A/C) and its complementary strand) as the specific binding target of ChCh. Utilizing native ChCh112 or ChCh107 under optimized buffer conditions, we were unable to detect binding to representative examples of the consensus sequence (GCCCGGGAAGCC, or GCGCGGGAGGCC) by either NMR spectroscopy or EMSA. The NMR experiments indicated that there was no line broadening or chemical shift perturbation for either the amide resonances of ChCh or the imino resonances of the DNA. The HSQC spectrum of ChCh remained identical to that in Figure 2, indicating that it remained correctly folded, but did not bind this DNA sequence. In the EMSA experiments, we observed no DNA binding with increasing protein concentration up to 10 μM. The crucial difference between our ChCh constructs and those used in the published study is that the published immunoprecipitation DNA selection assay and EMSA results were obtained using ChCh fused at the N-terminus to GST.14 We observe by NMR that an N-terminal modification of ChCh (GB1-ChCh112 or GB1-ChCh107) by fusion to a tag consisting of the B1 domain of streptococcal protein G, which is frequently used to enhance the solubility of recombinant proteins,29 destabilizes the ChCh structure and results in irreversible aggregation/unfolding (Figure 7a). The solution NMR structure of ChCh further suggests that an N-terminal fusion would severely disrupt the structural integrity of the binuclear zinc cluster that begins at Cys2 and thereby compromise the folded structure of the protein. On the other hand, the addition of a GB1 fusion tag to the flexible C-terminal tail of ChCh (ChCh112-GB1 or ChCh107-GB1) does not perturb the structure and results in a well folded protein (Figure 7b). Based on these findings, it is likely that the weak DNA binding observed in the previously published work may represent a non-specific interaction mediated by the unfolded GST-ChCh.
Figure 7.

Overlay HSQC spectra of ChCh107 (black) with (a) GB1-ChCh107 (red) and (b) ChCh107-GB1 (red). The addition of an N-terminal fusion to ChCh results in a severe disruption of the secondary structure resulting in an aggregation/unfolding of the protein (a), while a fusion to the unstructured C-terminal tail results in a well-folded protein (b). The well dispersed cross peaks (indicated by an overlaid gray dot) in the spectrum of ChCh107-GB1 arise from the GB1 tag.
Our findings indicate that a sequence-specific interaction between ChCh and the published double-stranded DNA sequence is unlikely. Nevertheless, the evidence presented by Sheng et al.14 that ChCh has transcriptional regulatory function appears convincing. These experiments were conducted in vivo utilizing VP16 and EnR domain C-terminal fusions,14 which based on our findings should result in a properly folded and active protein. We reasoned that, if ChCh functions as a transcription factor, but does not bind the specific DNA sequence determined by the reported immunoprecipitation DNA selection and EMSA assays, it may bind to some other sequence. Consequently we undertook extensive DNA selection and amplification assays to identify the specific ChCh DNA recognition sequence under conditions where the protein was properly folded.
DNA Selection Assays
The method of DNA selection utilized for these studies was the restriction endonuclease protection, selection and amplification (REPSA) assay.30 The major advantage of REPSA over other selection assays such as SELEX is there is no need to isolate bound DNA from free DNA left in solution.31 This is due to the use of IIS restriction endonucleases (IISRE), which cleave indiscriminately at a site distant from their recognition sequence. Interpolation of a randomized DNA sequence between two such restriction sites will allow the detection of sequences that are specifically targeted by the protein of interest. Any sequence that is protected by protein binding will not be degraded by the endonuclease, and can be recovered for later analysis: no modifications to the protein are required to separate free from bound DNA. The DNA selection experiments were conducted utilizing a standard protein concentration of 10 nM. After several rounds of selection with folded ChCh, there was only a slight DNA enrichment observable over background levels and no consensus sequence emerged from sequencing analysis of the selected DNA pool. The REPSA experiments were also conducted under the same conditions utilizing GB1-ChCh (to test for enhanced interaction with unfolded ChCh) and ChCh-GB1 (which should remain folded); these constructs yielded similar results to ChCh. The validity of these REPSA results for ChCh was verified by control experiments utilizing a non-DNA binding domain of the CREB-binding protein, KIX, and the DNA binding Wilms’ tumor suppressor zinc finger protein (WT1). REPSA experiments with KIX gave no consensus DNA sequence, while the analysis with WT1 selected the published DNA consensus sequence,32-34 indicating that the REPSA experiments were working correctly.
To account for the possibility that ChCh may bind its consensus DNA sequence with low affinity, the selection experiments were repeated with 500 nM concentration of ChCh, and again after several rounds of selection only a slight DNA enrichment over background levels was detected. Under these conditions, however, the sequencing analysis of the selected DNA pool resulted in the emergence of very weakly conserved consensus sequence, GCCG(G/T). NMR titration of ChCh with double stranded oligonucleotides containing the potential DNA target (GGTTGCTAGCCGGGGCCTTAG) showed absolutely no interaction, again as evidenced by the lack of line broadening or chemical shift perturbations for the amide resonances of ChCh or the imino resonances of the DNA. It should be noted that a major disadvantage of the REPSA methodology is that certain experimental conditions, such as high concentrations of MgCl2 (5 mM), are required to maintain the activity of the IIS restriction enzymes, which is the foundation of the selection protocol. This can be potentially problematic, as MgCl2 has been shown to interfere with the ability of some zinc finger proteins to coordinate their nucleic acid binding sequences.35 We therefore repeated the NMR ChCh/DNA binding studies under a variety of conditions, altering the ionic strength (0 - 100 mM NaCl), pH (6.8 - 7.5) and the concentration of divalent ions (such as Mg2+ and Zn2+), all with the same negative result. In addition, the single-stranded forward or reverse DNA fragments were titrated independently with ChCh, once again resulting in no observable binding interaction. Since NMR is sensitive enough to detect even very weak (μM - mM) binding interactions, we conclude that ChCh does not bind directly to DNA. If ChCh does function as a transcriptional activator, as proposed in the literature14, it must do so through interactions with other unidentified proteins.
DISCUSSION
Stabilization of the N-terminal Loop
The irregular N-terminal region of ChCh extends from the first to the second C-X-X-C sequences with few stabilizing interactions except for those provided by zinc coordination. Zinc binuclear clusters are not common in proteins, but there are several well-known examples, including the zinc finger binuclear cluster in the transcriptional activator, GAL436 and the dimerization domain of RAG1 which directly participates in V(D)J recombination for antigen binding T cell receptors and immunoglobulins.37 The binuclear cluster of ChCh is atypical: whereas GAL4, for example, contains a Cys6Zn2 cluster with two shared cysteine ligands, which are the first residues in each of two symmetric C-X-X-C sequences, the binuclear cluster in ChCh only shares a single cysteine ligand and includes a histidine residue as a ligand to the first zinc ion. A single shared cysteine ligand is also seen in RAG1, which contains two different zinc finger domains, a canonical C2H2 zinc finger and a RING domain. The apparent function of the binuclear clusters of both RAG1 and ChCh is to stabilize a large region of irregular structure on the N-terminal side of the shared zinc ligand. Aside from the shared cysteine in the binuclear cluster, there is no sequence homology or structural similarity between ChCh and RAG1.
Several hydrophobic and hydrogen-bonding interactions contribute to the stability of the N-terminal loop and connect it to the β-sheet. The side chains of Val6 and Tyr10 have favorable hydrophobic interactions with Leu89, while the backbone amides and carbonyls of the extended region from Glu7 to Asn12 form hydrogen bonds into the core of the ChCh structure, specifically with the backbone of the core residues Leu89 and Lys93 (Figure 8a). Several hydrophobic side chains (Phe27, Met39, Ile68, Ala69 and Leu90) fill the pocket between Leu89 and the zinc sites. The backbone of Lys8 makes two hydrogen bonds to the side chain of Asn26, which appears to play an essential role in joining together both ends of this N-terminal loop and bridging to the core structure at Leu90, all through hydrogen bonds with the asparagine side chain. At the end of the extended region is a tight turn involving Gly14 and Asn15, which is stabilized by a hydrogen bond between the carbonyl oxygen of Arg13 and the amide proton of Thr16. The end of the N-terminal loop region is stabilized by interactions with Asn26 and hydrophobic interactions with Phe27, which is buried within the zinc binding loops of the binuclear cluster. Mutagenesis of Asn26 results in a construct that is not expressed by E. coli, suggesting that the contribution of the Asn26 interactions provides important stabilization of the N-terminal loop.
Figure 8.

Stabilizing interactions of the N-terminal loop with the β-sheet (a) hydrophobic interactions between Val6 and Tyr10 of the N-terminal loop (green) with the side chain of Leu89 on the surface of the β-sheet (light blue). Also shown are hydrogen bonding interactions between the backbones of the loop and strand β5 (Arg13 HN-Lys93CO; Lys93 HN- Pro11 and Tyr10 CO; Tyr10 HN-Leu89 CO) (b) hydrophobic interactions of the Cys17 side chain with aromatic groups of the β-sheet. The figure was prepared with MOLMOL.74
The central portion of the N-terminal loop lies across the single-layer β-sheet, with few hydrophobic, electrostatic or hydrogen bonding interactions, aside from the interactions of Cys17. The completely conserved Cys17, located in the center of the N-terminal loop, is buried in a hydrophobic core of the β-sheet in the vicinity of the side chains of Met86, Tyr73 and Tyr84 (Figure 8b), and is not available for zinc coordination or disulfide formation. The position of Cys17 in the structure is consistent with the results of mutagenesis: C17S is not folded, while C17A and C17V are folded, though of lower stability than wild-type protein, as indicated by the presence of multiple species in solution. We conclude that a cysteine is the most favorable residue at position 17, with optimum size and hydrophobicity for interactions in this site. A similar situation has been observed for the three free cysteines of the α subunit of E. coli tryptophan synthase.38 Mutation of the hydrophobic groups that contact Cys17 indicates that this side chain makes a specific hydrophobic contact with an aromatic group at position 73. These results explain the sequence conservation of this residue, even though it is not a zinc ligand.
Stabilizing Interactions in the Single Layer β-sheet
The zinc coordination sites and the hydrophobic core interact with only one end of the five-stranded anti-parallel β-sheet, leaving the majority of the sheet exposed to solvent. Both sides of the single-layer β-sheet of ChCh have an unusually large number of exposed hydrophobic residues, which are intermixed with several acidic amino acids (Figure 9). The aliphatic regions of the long acidic side chains contribute to the hydrophobic packing, while the hydrophilic ends facilitate the stability of the exposed β-sheet in an aqueous environment. A similar stabilizing interaction has been observed for the Arabidopsis WRKY DNA binding domain, in which a solvent exposed β-sheet is stabilized by the hydrophobic packing of several tyrosine residues and is maintained by a significant number of basic residues.39 In addition, studies of the solvent exposed central β-sheet in OspA have identified cross-strand hydrophobic interactions as a primary stabilizing factor, with additional contributions from an optimal arrangement of inter-strand ionic salt bridges and hydrogen bonding interactions.40-43 The cross strand hydrophobic interactions in ChCh include packing between the aliphatic part of the Lys44 side chain and the aromatic group of Tyr57, which in turn packs against the imidazole group of the His71 ligand. On this face of the sheet, an entire complement of hydrophobic residues stretches across all five strands, with hydrophobic interactions between the side chains of Leu46, Val55 and Tyr73, (Figure 9a), as well as the Met86 and Ala94 side chains, which lie between the β-sheet and the N-terminal loop. Strands β3, β4 and β5 are further stabilized by cross-strand hydrophobic interactions between the aromatic rings of Phe75 and Tyr84, which pack against the aliphatic side chains of Ile77, Gln82, Ile98, and Ile100 forming another stretch of interacting residues running across the three strand width of the β-sheet at this position. The type II’ β-turn involving Asp79 and Glu80 is further stabilized by hydrophobic interactions between Met78 and Phe81. Similarly, on the reverse side of the β-sheet (Figure 9b), two pairs of cross-strand interactions between lysine methylenes and aliphatic methyl groups are seen between Lys93 and Leu87 as well as Lys47 and Ile54. There are a number of exposed hydrophobic groups on this side (residues 54, 60, 67, 78, 81, 87, 89), which are also interspersed with long-chain charged groups, primarily acidic, which further serve to ensure the water solubility of ChCh.
Figure 9.

Exposed hydrophobic and charged groups on the exposed faces of the ChCh β-sheet. (a) front face (corresponding to the view in Figure 3); (b) back face (turned 180° about the vertical axis). For each view, the left figure shows hydrophobic side chains (green), acidic side chains (red) and basic side chains (blue), the middle panel shows electrostatic charge mapped on the surface, and the right panel shows exposed hydrophobic side chains (green) mapped onto the surface. The figure was prepared with MOLMOL.74
Cross-strand salt bridge interactions may also play a role in maintaining the rigidity of the β-sheet, through specific inter-strand interactions augmenting the backbone hydrogen-bonding network. The β-sheet of ChCh lacks the combined salt bridge and hydrophobic interactions that are prevalent in OspA through the cross strand pairing of long chain charged glutamate and arginine residues.40 This is due to the distinctive segregation of the charged residues in the ChCh structure, where the negatively charged side chains are localized to the bottom half of the β-sheet, while the majority of the positive charge is localized around the zinc binuclear cluster (Figure 9). Nonetheless, there are a few potential salt bridge interactions and hydrogen bonding interactions between side chains in ChCh. The negative side chain of Asp58 on the β2 strand is sandwiched between the positive side chain of Arg70 of the β3 strand and the polar side chain of Asn43 of the β1 strand with potential hydrogen bonding interactions between the Hε of Arg70 and the amide side chain protons of Asn43. Cross-strand hydrogen bonding interactions between side chains are also possible for Ser45 in the β1 strand and Thr56 in the β2 strand as well as for Thr74 in the β3 strand and Thr85 in the β4 strand.
It is important to note that the majority of the surface of ChCh is negatively charged (Figure 9), which would lower the probability of its binding to DNA. Most DNA-binding proteins contain ample surface positive charge. Indeed, the only other example of a structural motif that in any way resembles ChCh, the WRKY protein of Arabidopsis,39 has a highly positively charged surface, consistent with its demonstrated DNA-binding function. The comparisons with OspA, WRKY and RAG1 demonstrate that the ChCh structure is unique among proteins so far described: neither BLAST44 nor DALI45 register any similar structures that are not species variants of ChCh itself.
Role of ChCh in Neural Induction
ChCh plays an important role in the signaling and transcriptional events leading to the development of neural tissue.14 The specific role of ChCh appears to be to up-regulate the Smad-interacting protein1 (Sip1),14 which is an essential protein in neural induction.46 Suppression of ChCh expression results in abrogation of Sip1 expression and an observable decrease in the development of a neural plate in the chick embryo, implying that Sip1 is a direct regulatory target of ChCh. Based on these findings, it was proposed that ChCh is a key factor in the cellular decision to adopt a mesodermal or neural fate, exerted through the actions of its regulatory target gene Sip1.14 Suppression of the POU91 protein in Xenopus, an upstream regulator of ChCh expression, results in the loss of Sip1 expression and leads to impaired embryonic neural development.47 Addition of ectopic ChCh or Sip1, can rescue the null-POU91 phenotype, further implicating ChCh as an upstream regulator of Sip1 expression.47
While there is convincing evidence that ChCh is an essential upstream regulator of Sip1,14,47 our experiments indicate that it does not function as a direct DNA-binding transcriptional regulator. Aspects of the previously published studies are consistent with our findings. The published studies utilizing the VP16 and EnR ChCh C-terminal fusion were conducted in vivo, and under these conditions it cannot be determined whether ChCh is a direct transcriptional activator of Sip1, or whether it participates in other key interactions essential for transcriptional activation of Sip1. In fact, the authors point out that the ChCh-VP16 fusion was more effective at down-regulating FGF signaling targets than wild-type ChCh, indicating that ChCh may require a co-factor for its function.14 Furthermore, the solution structure of ChCh does not resemble any of the known zinc finger motifs that have been implicated in nucleic acid recognition. The large number of exposed hydrophobic side chains, as well as the extensive regions of negative charge (Figure 9), suggests that ChCh is better suited for protein/protein interactions rather than specific DNA recognition. Zinc finger proteins are known to participate in a variety of cellular activities including development, differentiation, and tumor suppression and though originally determined to be DNA binding proteins, there is increasing evidence that zinc finger proteins also recognize RNA and participate in protein-protein and protein-lipid interactions.48 Based on our analysis we propose that ChCh does not bind directly to DNA, but rather interacts with an unidentified protein partner to mediate a key step in the pathway that leads to the transcriptional activation of Sip1 and consequently, the onset of neural induction in the developing embryo. More studies will be needed to confirm this hypothesis and to ascertain the identity of the ChCh interacting partner(s).
MATERIALS AND METHODS
Sample Preparation
ChCh112 and ChCh107 were amplified by PCR from a human liver cDNA library and cloned into the expression vector pET21a (Novagen). All site-directed mutants were designed using standard methods. GB1-ChCh107 and ChCh107-GB1 were generated by cloning the ChCh107 gene into a pET-21a expression vector modified to include a GB1 fusion and thrombin cleavage linker at the N- or C-terminus, respectively. All constructs were over-expressed after transformation into the BL21(DE3) strain of Escherichia coli (Stratagene) harboring an additional pUBS520 vector containing the dnaY gene (tRNAArgAGA/AGG). Starter cultures of 100 ml LB containing carbenicillin (100 μg/ml) and kanamycin (30 μg/ml) were inoculated with a single colony of transformed cells and grown over night to an OD600 of 1.0 at 37°C. Starter cultures were diluted 1:40 in 1 L M9 minimal media (6.8 g Na2HPO4, 3.0 g KH2PO4, 0.5 g NaCl, 1 mM MgCl2, 100 μM CaCl2, 100 μg/ml carbenicillin, 30 μg/ml kanamycin, 10 ml 100x BME vitamin mix (Sigma), 4 g D(+)-glucose or 2.5 g 13C glucose, 0.5 g NH4Cl, and 0.5 g (NH4)2(SO4)) and grown at 37° C to an OD600 of 0.8-0.9. The cells were induced with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) and 150 μM ZnSO4 and incubated at 15°C for 12 hours. Cells were harvested by centrifugation at 5,000 xg for 20 min at 4°C.
Cell pellets were resuspended in lysis buffer (25 mM Tris (pH 7.5), 40 mM NaCl, 20 mM DTT, 1 mM phenylmethylsulfonyl fluoride) and sonicated with a Branson Sonifier at 4°C for 7 cycles of pulsing for 1 min and incubating for 2 min. After centrifugation, the lysate was diluted 1:1 with ion exchange buffer (25 mM Tris pH 7.5, 40 mM NaCl, 10 mM DTT) and loaded onto 2 consecutive 5 ml HiTrap Q ion exchange columns (Amersham Pharmacia). ChCh was eluted using a linear salt gradient. Pure fractions were pooled, concentrated, purified further by gel filtration (25 mM Tris pH 7.5, 100 mM NaCl, 2 mM DTT) using a Sephacryl S100HR XK26 column (Amersham Pharmacia), and concentrated to 1 mM. Protein purity was verified by SDS-PAGE and MALDI mass spectrometry. ESI-MS analysis of ChCh was performed in 10 mM ammonium acetate.
For NMR analysis, ∼1 mM ChCh107 was exchanged into NMR buffer (25 mM Tris pH 6.8, 100 mM NaCl, 10 mM DTT, and 10% 2H2O), transferred to an NMR tube, overlayed with argon and flame sealed to prevent oxidation. For R2 dispersion experiments, 0.8 mM ChCh107 was exchanged into a modified NMR buffer (20 mM d11-Tris pH 7.28, 50 mM NaCl, 10 mM d6-DTT, 2 mM NaN3, 0.2 mM DSS and 10% 2H2O), split into two 500 μl aliquots, and transferred to NMR tubes that were overlaid with argon and flame sealed. Flame sealing was necessary to minimize air oxidation of the sample, to maximize long-term stability: all samples of ChCh were kept under reducing conditions, which do not interfere with zinc binding, but does prevent oxidation of free cysteine residues that can lead to aggregation and precipitation of the protein.
NMR Spectroscopy
NMR spectra were acquired at 25°C on Bruker DMX, DRX and Avance spectrometers at 500, 600, 750, 800 and 900 MHz. Sspectra were processed with NMRPipe49 and analyzed with NMRView.50 Backbone and side chain assignments were correlated back to the amide 15N HSQC,51 aliphatic 13C ct-HSQC,52 and aromatic 13C HMQC.18 Backbone assignments were derived from HNCO,53 HNCA, HNCACB,54 CBCA(CO)NH,55 HBHA(CBCACO)NH,51 and (HCA)CO(CA)NH.56 Side chain protons and carbons were assigned from (H)C(CO)NH,57 HCCH-COSY,58 CBCGCD and CBCGCE.59 Coupling constants for χ1 torsion angle restraints were derived from both HNHB60 and HACAHB-COSY.61 The protein backbone chemical shifts were used to generate ϕ and Ψ dihedral angle restraints with TALOS.62 Distances restraints were derived from 2D NOESY,63 3D 15N NOESY-HSQC,21 and 3D 13C HMQC-NOESY19 spectra.
The T1, T2 and [1H]-15N NOE data for ChCh107 were collected on a Bruker DRX-600 spectrometer utilizing the sensitivity-enhanced experiments described by Farrow et al.64 and water flip-back pulses to achieve water suppression.65 The probe temperature was calibrated at 25 °C using methanol. 15N R2 relaxation dispersion rates were measured on Bruker DRX-800 and Avance-500 MHz spectrometers equipped with z-gradient cryoprobes, utilizing a constant time relaxation-compensated CPMG pulse sequence.24,25 The probe temperature was calibrated to 35 °C on each instrument with methanol. All relaxation data sets were collected with 2048 direct complex points and 256 indirect complex points. The direct carrier frequency was set to the water resonance with a spectral width of 16 ppm, while the indirect carrier frequency was set to 120 ppm with a spectral width of 38 ppm. T1 experiments were collected with 16 scans and a repetition delay of 3T1 s, while the T2 experiments were acquired with 16 scans. The [1H]-15N NOE spectra were acquired using 16 scans with or without a proton saturation period of 3 s. R2 dispersion experiments were acquired with 8 scans, a constant CPMG period of 40 ms and a recycle delay of 3 s.
R2 dispersion curves were generated by sampling over 18 (1/τcp) values: 100, 200, 300, 400, 400, 500, 600, 700, 800, 1000, 1200, 1200, 1400, 1600, 1800, 2000, 2500, 3000, 3500, 4000, and 4000 s-1. Duplicate reference spectra in which the CPMG pulse period is omitted66 were also collected at each field. For the T1, T2 and R2 dispersion data sets, points were acquired in a random order to avoid systematic errors, and duplicate data points were utilized as an estimation of the experimental uncertainty. The [1H]-15N NOE data was collected in triplicate, and for each set, the saturated and unsaturated data were collected in an interleaved manner.
Amide exchange was initiated by taking a 1 mM lyophilized, protonated ChCh107 NMR sample (pH 6.8) and resuspending it in 2H2O. The dead-time between resuspension of the sample and data acquisition was 15 min. A series of 15N HSQC were recorded at 25 °C on a Bruker DRX-600 MHz spectrometer, with a total acquisition time of 30 min per experiment, for 36 h. The hydrogen exchange rates were determined by fitting the peak intensities with a nonlinear least squares single exponential decay function available in the program Curvefit.67 Protection factors and ΔGHX were calculated from the exchange rates68 taking into account corrections for nearest-neighbor effects.69 A table of exchange rates and protection factors is given in the Supplementary Material (Table S1).
Samples for DNA titrations were prepared by titrating double or single stranded DNA into a 65 - 130 μM sample of ChCh107 or ChCh107-Gb1 up to 1:1.4 [protein]:[DNA] ratio. Binding interactions were monitored using 1D 1H-NMR to observe the DNA imino proton resonances and 2D 1H-15N HSQC of the 15N labeled protein.
Structure Calculations
Nearly complete backbone and side chain assignments were used for automated assignment of hand picked NOESY cross peaks from 3D 15N NOESY-HSQC21 and 3D 13C HMQC-NOESY19 with CYANA 2.122 Stereo-specific assignments of methylene protons and pro-chiral methyls were determined from the NOESY spectra and the χ1 vicinal coupling spectra.70,71 Separate assignment lists were used for each NOESY dataset, with amide proton assignments removed from the list for data collected in 2H2O. The total number of NOESY cross peaks was 6,596 from both spectra. For each cross peak, the heteronuclear assignment derived from the parent HSQC or HMQC spectra was retained, while the indirect assignment of the NOE correlated proton was left undetermined prior to running the automated assignment procedure in CYANA 2.1. Chemical shift based restraints from TALOS62 were included only for regions of regular secondary structure. Zinc ligand restraints were not used during the automated assignment procedure. During the initial cycle of automated assignment, each NOESY cross peak was ambiguously assigned based on chemical shift with an average of 4.03 possible assignments per restraint and structures were calculated by torsion angle dynamics. Spurious NOESY cross peak assignments were removed during subsequent cycles of structure calculation and refinement of assignments through distance filtering based on the intermediate structure and network anchoring of related self-consistent restraint assignments.22 The final restraint list included 3,727 distances, which were subsequently used along with zinc ligand restraints to generate 200 starting structures in CYANA 2.1 following the REDAC strategy.72 Starting structures were further refined by molecular dynamics calculations with AMBER 8 under in vacuo conditions with reduced charges.23 The zinc ions were incorporated through covalent bonds with the ligand residues. Iterative refinement and editing of the distance restraints based on the NOESY spectra to remove incorrect and ambiguous assignments reduced the number of restraints to a qualified list of 2,351 distances. Final refinement in AMBER 8 incorporated the generalized Born solvation model during molecular dynamics calculations with the qualified restraints obtained from in vacuo calculations.23 Coordinates for the final 20 refined structures have been deposited in the Protein Data Bank (PDB code 2jox).
Analysis of Relaxation Data
The R1 and R2 rates were determined by fitting the exponential decay curves for T1 and T2 peak intensities for each residue using the program Curvefit.67 Decay curves were fit with a three-parameter fit I(t)=I∞ + I0e-t/T1,2 where I is the intensity, t is the CPMG pulse train delay or the longitudinal delay, and I0 is the initial peak intensity. Three-parameter fits were used for the fittings of both T1 and T2 data because each experiment sampled the full decay of the signals. Overlapped residues were not used in the analysis. The [1H]-15N NOE values were obtained from the ratio of peak intensities between the 1H saturated and unsaturated spectra. Standard errors for the relaxation data was assessed from repeated measurements for the heteronuclear NOE and for a sub-set of time points in the T1 and T2 decay curves. Due to the large proportion of residues with exchange terms, model-free calculations could not be performed reliably; the relaxation data were therefore evaluated qualitatively and more quantitative treatment was reserved for the relaxation dispersion measurements. A table of relaxation parameters is given in the Supplementary Material (Table S2).
Decay rates from the peak intensities of the R2 dispersion experiments at both fields were fit simultaneously with an in-house program to equations describing a fast or two-site exchange model.25,26,73 F-test criteria at the 99.9% confidence level were utilized to identify residues that had a statistical significance for adding an Rex term compared to fits with a straight line. All residues participating in a conformational exchange process were fit locally as well as globally to single kex and pa values. A table of the parameters obtained from these fits is given in the Supplementary Material (Table S3).
DNA Selection Assays
All DNA templates, primers and oligonucleotides were synthesized commercially (IDT, Inc.) and used without further purification. The sequences of the REPSA primers and the randomized pool are given in the Supplementary Material (Figure S3). The 12 bp randomized region of 62R12 was generated by machine mixing of A:T:C:G phosphoramidites. The single stranded selection oligonucleotide and corresponding primers (62AR and 62AL) were designed following strategies described for the REPSA DNA selection assay.30 The double stranded randomized pool (ds62R12) was synthesized using 2 cycles of PCR in a final volume of 50 μl with an amplification profile of 94°C for 1 min followed by 3 min at 50°C. The ds62R12 DNA was gel purified before use in the DNA selection assays. DNA oligonucleotides for EMSA or NMR analysis were purified utilizing pre-packed NAP-5 Sephadex G25 columns (GE Healthcare Life Sciences) to remove any residual salts and/or reaction impurities and then lyophilized. The complementary oligonucleotides were resuspended in appropriate buffer and stoichiometric amounts of each were combined, heated to 100° C for ∼2 minutes, and allowed to anneal by slow cooling to room temperature.
For each round of REPSA, 4.9 pM ds62R12 randomized DNA pool was incubated with 10 nM or 500 nM ChCh, 10 nM Gb1-ChCh, 10 nM ChCh-Gb1, 10 nM KIX, or 10 nM WT1 in 20 μL binding buffer (10 mM Tris pH 7.0, 20 mM KCl or 50 mM NaCl, 5 μM ZnSO4, 5 mM MgCl2, 1 mM DTT) at 25°C for 45 min. Unbound DNA was digested with 2 U FokI, or 3U of BpmI or HphI IIS restriction enzymes (New England Biolabs) for 30 min at 37°C. Cleavage resistant DNA was PCR amplified in 100 μl using an amplification profile of 94°C for 1 min followed by 3 min at 50°C for either 7 or 9 cycles. Aliquots of each reaction were analyzed by 8% non-denaturing PAGE to monitor the progress of the selection. Remaining reaction mixtures were purified by phenol extraction and the aqueous phase was subsequently ethanol precipitated. DNA pellets were resuspended in reaction buffer for use in the next round of REPSA. The binding, enzymatic cleavage, PCR amplification and purification were repeated 6 - 9 times. The final selected DNA pools were gel purified, ligated into the pCR2.1-TOPO cloning vector (Invitrogen), and transformed into One Shot TOP10 chemically competent cells (Invitrogen). Individual colonies were used to inoculate 5 ml overnight cultures in LB medium containing 100 μg/ml carbenicillin. The clones were mini-plasmid prepped (Qiagen) and sequenced (EtonBio).
Supplementary Material
ACKNOWLEDGEMENTS
The DNA selection experiments were done with the helpful advice of Dr. Joel Gottesfeld. We thank M. Zeeb, J. Wojciak and T. Nishikawa for many helpful discussions; T. Nishikawa and K. Sugase for generously donating control protein samples; J. Chung and G. Kroon for assistance with NMR experiments; and N. Greenman, J. Hosea and L. Tennant for expert technical assistance. This work was supported by grant GM36643 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Spemann H, Mangold H. Uber induktion von embryonanlagen durch implantation artfremender origanisatoran (Induction of embryonic primordia by implantation of organizers from a different species) Arch. Mikrosk. Anat. Entwiklungsmech. 1924;100:599–638. [Google Scholar]
- 2.Leptin M. Gastrulation movements: the logic and the nuts and bolts. Dev. Cell. 2005;8:305–320. doi: 10.1016/j.devcel.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 3.Solnica-Krezel L. Conserved patterns of cell movements during vertebrate gastrulation. Curr. Biol. 2005;15:R213–R228. doi: 10.1016/j.cub.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 4.Hemmati-Brivanlou A, Melton D. Vertebrate embryonic cells will become nerve cells unless told otherwise. Cell. 1997;88:13–17. doi: 10.1016/s0092-8674(00)81853-x. [DOI] [PubMed] [Google Scholar]
- 5.Streit A, Sockanathan S, Perez L, Rex M, Scotting PJ, Sharpe PT, Lovell-Badge R, Stern CD. Preventing the loss of competence for neural induction: HGF/SF, L5 and Sox-2. Development. 1997;124:1191–1202. doi: 10.1242/dev.124.6.1191. [DOI] [PubMed] [Google Scholar]
- 6.Wilson SI, Rydstrom A, Trimborn T, Willert K, Nusse R, Jessell TM, Edlund T. The status of Wnt signalling regulates neural and epidermal fates in the chick embryo. Nature. 2001;411:325–330. doi: 10.1038/35077115. [DOI] [PubMed] [Google Scholar]
- 7.Linker C, Stern CD. Neural induction requires BMP inhibition only as a late step, and involves signals other than FGF and Wnt antagonists. Development. 2004;131:5671–5681. doi: 10.1242/dev.01445. [DOI] [PubMed] [Google Scholar]
- 8.Delaune E, Lemaire P, Kodjabachian L. Neural induction in Xenopus requires early FGF signalling in addition to BMP inhibition. Development. 2005;132:299–310. doi: 10.1242/dev.01582. [DOI] [PubMed] [Google Scholar]
- 9.Stern CD. Neural induction: old problem, new findings, yet more questions. Development. 2005;132:2007–2021. doi: 10.1242/dev.01794. [DOI] [PubMed] [Google Scholar]
- 10.Wilson SI, Graziano E, Harland R, Jessell TM, Edlund T. An early requirement for FGF signalling in the acquisition of neural cell fate in the chick embryo. Curr. Biol. 2000;10:421–429. doi: 10.1016/s0960-9822(00)00431-0. [DOI] [PubMed] [Google Scholar]
- 11.Streit A, Berliner AJ, Papanayotou C, Sirulnik A, Stern CD. Initiation of neural induction by FGF signalling before gastrulation. Nature. 2000;406:74–78. doi: 10.1038/35017617. [DOI] [PubMed] [Google Scholar]
- 12.Bertrand V, Hudson C, Caillol D, Popovici C, Lemaire P. Neural tissue in ascidian embryos is induced by FGF9/16/20, acting via a combination of maternal GATA and Ets transcription factors. Cell. 2003;115:615–627. doi: 10.1016/s0092-8674(03)00928-0. [DOI] [PubMed] [Google Scholar]
- 13.Streit A, Lee KJ, Woo I, Roberts C, Jessell TM, Stern CD. Chordin regulates primitive streak development and the stability of induced neural cells, but is not sufficient for neural induction in the chick embryo. Development. 1998;125:507–519. doi: 10.1242/dev.125.3.507. [DOI] [PubMed] [Google Scholar]
- 14.Sheng G, dos Reis M, Stern CD. Churchill, a zinc finger transcriptional activator, regulates the transition between gastrulation and neurulation. Cell. 2003;115:603–613. doi: 10.1016/s0092-8674(03)00927-9. [DOI] [PubMed] [Google Scholar]
- 15.Varshavsky A. The N-end rule. Cell. 1992;69:725–735. doi: 10.1016/0092-8674(92)90285-k. [DOI] [PubMed] [Google Scholar]
- 16.Bradshaw RA, Brickey WW, Walker KW. N-terminal processing: the methionine aminopeptidase and N alpha-acetyl transferase families. Trends Biochem. Sci. 1998;23:263–267. doi: 10.1016/s0968-0004(98)01227-4. [DOI] [PubMed] [Google Scholar]
- 17.Pelton JG, Torchia DA, Meadow ND, Roseman S. Tautomeric states of the active-site histidines of phosphorylated and unphosphorylated IIIGlc, a signal-transducing protein from Escherichia coli, using two-dimensional heteronuclear NMR techniques. Protein Sci. 1993;2:543–558. doi: 10.1002/pro.5560020406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bax A, Griffey RH, Hawkins BL. Correlation of proton and nitrogen-15 chemical shifts by multiple quantum NMR. J. Magn. Reson. 1983;55:425–431. [Google Scholar]
- 19.Fesik SW, Zuiderweg ERP. Heteronuclear three-dimensional NMR spectroscopy. A strategy for the simplification of homonuclear two-dimensional NMR spectra. J. Magn. Reson. 1988;78:588–593. [Google Scholar]
- 20.Summers MF, South TL, Kim B, Hare DR. High-resolution structure of an HIV zinc fingerlike domain via a new NMR-based distance geometry approach. Biochemistry. 1990;29:329–340. doi: 10.1021/bi00454a005. [DOI] [PubMed] [Google Scholar]
- 21.Marion D, Driscoll PC, Kay LE, Wingfield PT, Bax A, Gronenborn AM, Clore GM. Overcoming the overlap problem in the assignment of 1H NMR spectra of larger proteins by use of three-dimensional heteronuclear 1H-15N Hartmann-Hahn-multiple quantum coherence and nuclear Overhauser-multiple quantum coherence spectroscopy: Application to Interleukin 1β. Biochemistry. 1989;28:6150–6156. doi: 10.1021/bi00441a004. [DOI] [PubMed] [Google Scholar]
- 22.Güntert P. Automated protein structure calculation with CYANA. Meth. Mol. Biol. 2004;278:353–378. doi: 10.1385/1-59259-809-9:353. [DOI] [PubMed] [Google Scholar]
- 23.Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KMJ, Onufriev A, Simmerling C, Wang B, Woods R. The Amber biomolecular simulation programs. J. Comput. Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Loria JP, Rance M, Palmer AG. A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. J. Am. Chem. Soc. 1999;121:2331–2332. [Google Scholar]
- 25.Tollinger M, Skrynnikov NR, Mulder FA, Forman-Kay JD, Kay LE. Slow dynamics in folded and unfolded states of an SH3 domain. J Am. Chem. Soc. 2001;123:11341–11352. doi: 10.1021/ja011300z. [DOI] [PubMed] [Google Scholar]
- 26.Palmer AG., III NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 27.Akke M. NMR methods for characterizing microsecond to millisecond dynamics in recognition and catalysis. Curr. Opin. Struct Biol. 2002;12:642–647. doi: 10.1016/s0959-440x(02)00369-x. [DOI] [PubMed] [Google Scholar]
- 28.Viguera AR, Serrano L. Side-chain interactions between sulfur-containing amino acids and phenylalanine in α-helices. Biochemistry. 1995;34:8771–8779. doi: 10.1021/bi00027a028. [DOI] [PubMed] [Google Scholar]
- 29.Koenig BW, Rogowski M, Louis JM. A rapid method to attain isotope labeled small soluble peptides for NMR studies. J. Biomol. NMR. 2003;26:193–202. doi: 10.1023/a:1023887412387. [DOI] [PubMed] [Google Scholar]
- 30.Hardenbol P, Wang JC, Van Dyke MW. Identification of preferred hTBP DNA binding sites by the combinatorial method REPSA. Nucleic Acids Res. 1997;25:3339–3344. doi: 10.1093/nar/25.16.3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hardenbol P, Van Dyke MW. Sequence specificity of triplex DNA formation: Analysis by a combinatorial approach, restriction endonuclease protection selection and amplification. Proc. Natl Acad. Sci U. S. A. 1996;93:2811–2816. doi: 10.1073/pnas.93.7.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamilton TB, Barilla KC, Romaniuk PJ. High affinity binding sites for the Wilms’ tumour suppressor protein WT1. Nucleic Acids Res. 1995;23:277–284. doi: 10.1093/nar/23.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakagama H, Heinrich G, Pelletier J, Housman DE. Sequence and structural requirements for high-affinity DNA binding by the WT1 gene product. Mol. Cell. Biol. 1995;15:1489–1498. doi: 10.1128/mcb.15.3.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamilton TB, Borel F, Romaniuk PJ. Comparison of the DNA binding characteristics of the related zinc finger proteins WT1 and EGR1. Biochemistry. 1998;37:2051–2058. doi: 10.1021/bi9717993. [DOI] [PubMed] [Google Scholar]
- 35.Lee BM, Xu J, Clarkson BK, Martinez-Yamout MA, Dyson HJ, Case DA, Gottesfeld JM, Wright PE. Induced Fit and “Lock and Key” Recognition of 5 S RNA by Zinc Fingers of Transcription Factor IIIA. J. Mol. Biol. 2006;357:275–291. doi: 10.1016/j.jmb.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 36.Marmorstein R, Carey M, Ptashne M, Harrison SC. DNA recognition by GAL4: structure of a protein-DNA complex. Nature. 1992;356:408–414. doi: 10.1038/356408a0. [DOI] [PubMed] [Google Scholar]
- 37.Bellon SF, Rodgers KK, Schatz DG, Coleman JE, Steitz TA. Crystal structure of the RAG1 dimerization domain reveals multiple zinc-binding motifs including a novel zinc binuclear cluster. Nat. Struct. Biol. 1997;4:586–591. doi: 10.1038/nsb0797-586. [DOI] [PubMed] [Google Scholar]
- 38.Hiraga K, Yutani K. Study of cysteine residues in the alpha subunit of Escherichia coli tryptophan synthase. 1 Role in conformational stability. Protein Eng. 1996;9:425–431. doi: 10.1093/protein/9.5.425. [DOI] [PubMed] [Google Scholar]
- 39.Yamasaki K, Kigawa T, Inoue M, Tateno M, Yamasaki T, Yabuki T, Aoki M, Seki E, Matsuda T, Tomo Y, Hayami N, Terada T, Shirouzu M, Tanaka A, Seki M, Shinozaki K, Yokoyama S. Solution structure of an Arabidopsis WRKY DNA binding domain. Plant Cell. 2005;17:944–956. doi: 10.1105/tpc.104.026435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pham TN, Koide A, Koide S. A stable single-layer β-sheet without a hydrophobic core. Nature Struct. Biol. 1998;5:115–119. doi: 10.1038/nsb0298-115. [DOI] [PubMed] [Google Scholar]
- 41.Nakagawa T, Shimizu H, Link K, Koide A, Koide S, Tamura A. Calorimetric dissection of thermal unfolding of OspA, a predominantly [beta]-sheet protein containing a single-layer [beta]-sheet. J. Mol. Biol. 2002;323:751–762. doi: 10.1016/s0022-2836(02)00974-9. [DOI] [PubMed] [Google Scholar]
- 42.Pawley NH, Koide S, Nicholson LK. Backbone Dynamics and Thermodynamics of Borrelia Outer Surface Protein A. J. Mol. Biol. 2002;324:991–1002. doi: 10.1016/s0022-2836(02)01146-4. [DOI] [PubMed] [Google Scholar]
- 43.Yan S, Gawlak G, Makabe K, Tereshko V, Koide A, Koide S. Hydrophobic Surface Burial Is the Major Stability Determinant of a Flat, Single-layer [beta]-Sheet. J. Mol. Biol. 2007;368:230–243. doi: 10.1016/j.jmb.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Altschul SF, Koonin EV. Iterated profile searches with PSI-BLAST--a tool for discovery in protein databases. Trends Biochem. Sci. 1998;23:444–447. doi: 10.1016/s0968-0004(98)01298-5. [DOI] [PubMed] [Google Scholar]
- 45.Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends Biochem. Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 46.Nitta KR, Tanegashima K, Takahashi S, Asashima M. XSIP1 is essential for early neural gene expression and neural differentiation by suppression of BMP signaling. Dev. Biol. 2004;275:258–267. doi: 10.1016/j.ydbio.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 47.Snir M, Ofir R, Elias S, Frank D. Xenopus laevis POU91 protein, an Oct3/4 homologue, regulates competence transitions from mesoderm to neural cell fates. EMBO J. 2006;25:3664–3674. doi: 10.1038/sj.emboj.7601238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Laity JH, Lee BM, Wright PE. Zinc finger proteins: new insights into structural and functional diversity. Curr. Opin. Struct. Biol. 2001;11:39–46. doi: 10.1016/s0959-440x(00)00167-6. [DOI] [PubMed] [Google Scholar]
- 49.Delaglio F, Grzesiek S, Vuister GW, Guang Z, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 50.Johnson BA, Blevins RA. NMRView: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:604–613. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 51.Grzesiek S, Bax A. Amino acid type determination in the sequential assignment procedure of uniformly 13C/15N-enriched proteins. J. Biomol. NMR. 1993;3:185–204. doi: 10.1007/BF00178261. [DOI] [PubMed] [Google Scholar]
- 52.Vuister GW, Bax A. Measurement of two-bond JCOHa coupling constants in proteins uniformly enriched with 13C. J. Biomol. NMR. 1992;2:401–405. doi: 10.1007/BF01874818. [DOI] [PubMed] [Google Scholar]
- 53.Kay LE, Xu GY, Yamazaki T. Enhanced-sensitivity triple-resonance spectroscopy with minimal H2O saturation. J. Magn. Reson. Series A. 1994;109:129–133. [Google Scholar]
- 54.Grzesiek S, Bax A. Correlating backbone amide and side chain resonances in larger proteins by multiple relayed triple resonance NMR. J. Am. Chem. Soc. 1992;114:6291–6293. [Google Scholar]
- 55.Grzesiek S, Bax A. Improved 3D triple-resonance NMR techniques applied to a 31 kDa protein. J. Magn. Reson. 1992;96:432–440. [Google Scholar]
- 56.Löhr F, Rüterjans H. Improved NMR experiments for the correlation of backbone-carbon with aliphatic-proton resonances in proteins. J. Magn. Reson. Series B. 1995;109:80–87. [Google Scholar]
- 57.Gardner KH, Konrat R, Rosen MK, Kay LE. An (H)C(CO)NH-TOCSY pulse scheme for sequential assignment of prototonated methyl groups in otherwise deuterated 15N, 13C-labeled proteins. J. Biomol. NMR. 1996;8:351–356. doi: 10.1007/BF00410333. [DOI] [PubMed] [Google Scholar]
- 58.Gehring K, Ekiel I. H(C)CH-COSY and (H)CCH-COSY experiments for 13C-labeled proteins in H2O solution. J. Magn. Reson. 1998;135:185–193. doi: 10.1006/jmre.1998.1543. [DOI] [PubMed] [Google Scholar]
- 59.Yamazaki T, Forman-Kay JD, Kay LE. Two-dimensional NMR experiments for correlating 13Cβ and 1Hδ/ε chemical shifts of aromatic residues in 13C-labeled proteins via scalar couplings. J. Am. Chem. Soc. 1993;115:11054–11055. [Google Scholar]
- 60.Archer SJ, Ikura M, Torchia DA, Bax A. An alternative 3D NMR technique for correlating backbone 15N with side chain Hβ resonances in larger proteins. J. Magn. Reson. 1991;95:636–641. [Google Scholar]
- 61.Grzesiek S, Kuboniwa H, Hinck AP, Bax A. Multiple-quantum line narrowing for measurement of Hα-Hβ J coupling in isotopically enriched proteins. J. Am. Chem. Soc. 1995;117:5312–5315. [Google Scholar]
- 62.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 63.Macura S, Ernst RR. Elucidation of cross-relaxation in liquids by two-dimensional N.M.R. spectroscopy. Mol. Phys. 1980;41:95–117. [Google Scholar]
- 64.Farrow NA, Muhandiram R, Singer AU, Pascal SM, Kay CM, Gish G, Shoelson SE, Pawson T, Forman-Kay JD, Kay LE. Backbone dynamics of a free and a phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry. 1994;33:5984–6003. doi: 10.1021/bi00185a040. [DOI] [PubMed] [Google Scholar]
- 65.Grzesiek S, Bax A. The importance of not saturating H2O in protein NMR. Application to sensitivity enhancement and NOE measurements. J. Am. Chem. Soc. 1993;115:12593–12594. [Google Scholar]
- 66.Mulder FAA, van Tilborg PJA, Kaptein R, Boelens R. Microsecond time scale dynamics in the RXR DNA-binding domain from a combination of spin-echo and off-resonance rotating frame relaxation measurements. J. Biomol. NMR. 1999;13:275–288. doi: 10.1023/a:1008354232281. [DOI] [PubMed] [Google Scholar]
- 67.Mandel AM, Akke M, Palmer AG. Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J. Mol. Biol. 1995;246:144–163. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- 68.Bai YW, Englander JJ, Mayne L, Milne JS, Englander SW. Thermodynamic parameters from hydrogen exchange measurements. Methods Enzymol. 1995;259:344–356. doi: 10.1016/0076-6879(95)59051-x. [DOI] [PubMed] [Google Scholar]
- 69.Bai Y, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bax A, Max D, Zax D. Measurement of long-range 13C-13C J coupling in a 20-kDa protein complex. J. Am. Chem. Soc. 1992;114:6923–6925. [Google Scholar]
- 71.Vuister GW, Wang AC, Bax A. Measurement of three-bond nitrogen-carbon J couplings in proteins uniformly enriched in 15N and 13C. J. Am. Chem. Soc. 1993;115:5334–5335. [Google Scholar]
- 72.Güntert P, Wüthrich K. Improved efficiency of protein structure calculations from NMR data using the program DIANA with redundant dihedral angle constraints. J. Biomol. NMR. 1991;1:447–456. doi: 10.1007/BF02192866. [DOI] [PubMed] [Google Scholar]
- 73.Davis DG, Perlman ME, London RE. Direct measurements of the dissociation-rate constant for inhibitor-enzyme complexes via the T1 rho and T2 (CPMG) methods. J. Magn Reson. B. 1994;104:266–275. doi: 10.1006/jmrb.1994.1084. [DOI] [PubMed] [Google Scholar]
- 74.Koradi R, Billeter M, Wüthrich K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.