

Summary
Nuclear receptors undergo ligand-dependent conformational changes that are required for corepressor-coactivator exchange, but whether there is an actual requirement for specific epigenetic landmarks to impose ligand dependency for gene activation remains unknown. Here we report an unexpected and general strategy that is based on the requirement for specific cohorts of inhibitory histone methyltransferases (HMTs) to impose gene-specific gatekeeper functions that prevent unliganded nuclear receptors and other classes of regulated transcription factors from binding to their target gene promoters and causing constitutive gene activation in the absence of stimulating signals. This strategy, based at least in part on an HMT dependent inhibitory histone code, imposes a requirement for specific histone demethylases, including LSD1, to permit ligand- and signal dependent activation of regulated gene expression. These events link an inhibitory methylation component of the histone code to a broadly used strategy that circumvents pathological constitutive gene induction by physiologically regulated transcription factors.
Introduction
Ligand-dependent activation of large transcriptional gene programs that are simultaneously regulated by nuclear receptors provides critical strategies for development and homeostasis of all metazoans, and its misregulation is associated with many types of disease. Regulated transcription by nuclear receptors is mediated by ligands binding to the C-terminal domain, thus causing conformational changes; these include a change in the position of the so-called AF2 helix, which favors association with specific coactivator complexes and functional conversion of the receptor to an activator (reviewed in Rosenfeld et al., 2006). Thus, when unliganded, nuclear receptors, such as the thyroid hormone (T3) and the retinoid acid (RA) receptors, act as repressors primarily by recruiting specific corepressor complexes via the “CoRNR” domain (Horleinet al., 1995; Chen and Evans, 1995; Heinzel et al., 1997; Privalsky, 2004), but, when liganded, they are functionally converted to activators by recruiting coactivator complexes. In addition, for many nuclear receptors, such as estrogen receptor α (ER α) and androgen receptor (AR), other signaling pathways can cause similar recruitment of coactivators and the consequent functional conversion to transcriptional activators even in the absence of ligand (Culig et al., 1994; Nazareth and Weigel, 1996; Zwijsenet al., 1997; Rogatsky et al., 1999; Ueda et al., 2002; Ogawa et al., 2004; Kim et al., 2005). Therefore, it is of particular interest to further explore the linkage between the recruitment of nuclear receptors and the coregulatory complexes that underlie ligand-dependent and -independent activation of transcriptional programs.
Most coregulatory complexes exhibit a diversity of enzymatic activities that can be divided into two generic classes: enzymes capable of remodeling the structure of the nucleosome in an ATP-dependent manner and enzymes capable of covalently modifying histone tails; this latter group includes acetylating and deacetylating activities (HATs and HDACs); methylating and demethylating activities (HMTs and HDMs); kinases and phosphatases; poly(ADP) ribosylases; and ubiquitin and SUMO ligases (reviewed in Narlikar et al., 2002; Rosenfeld et al., 2006). The histone code model (Strahl and Allis, 2000; Jenuwein and Allis, 2001) suggests that serial posttranslational histone modifications, such as acetylation, methylation, phosphorylation, and sumoylation, correlate with the specific activated or repressed status of the promoter (reviewed in Fischle et al., 2003; Peterson and Laniel, 2004;Margueron et al., 2005). Presumably, the deposition and removal of these marks correspond to a large variety of coregulatory complexes that act in a sequential and combinatorial fashion to ultimately determine spatial and temporal control of gene expression (reviewed in McKenna and O'Malley, 2002; Rosenfeld et al., 2006). One of these marks, the histone lysine methylation, was initially considered as a permanent posttranslational modification that exerts long-term epigenetic memory (reviewed in Kouzarides, 2002; Lachner and Jenuwein, 2002). However, recent data demonstrating the existence of lysine demethylase activities have dramatically challenged this model (Shi et al., 2004; Metzger et al., 2005; Tsukada et al.,2006; Yamane et al., 2006; Whetstine et al., 2006).
Histone lysine methylation has been extensively linked to both gene activation and gene repression events in euchromatic and heterochromatic regions (reviewed in Lachner and Jenuwein, 2002; Martin and Zhang, 2005). A large number of SET-domain-containing enzymes, including RIZ1, ESET, Eu-HMTase1, G9a, Suv39h1/h2, MLL1, and others, have been shown to transfer methyl groups to histones and to transcription factors; in particular, this has been shown at multiple lysine residues in histones, including H3-K4, K9, K27, K36, K79, H4-K20, and H1-K26, all of which have been reported in most cases to be mono-, di-, and trimethylated (reviewed in Martin and Zhang, 2005). It has been proposed that methyl groups may act as binding sites for a wide range of chromatin proteins, including the repressive heterochromatin protein 1 (HP1), which has been reported to bind methyl groups on histone H3 at lysine 9 (H3-K9; Nielsen et al.,2001), as well as the transcriptional activator WDR5 and the ATP-dependent chromatin-remodeling protein CDH1,which have been reported to bind methyl groups on H3 at lysine 4 (H3-K4; Wysocka et al., 2005; Dou et al.,2005; Flanagan et al., 2005).
Recently, a CoREST corepressor complex component (Tong et al., 1998; Andres et al., 1999; Ballas et al., 2001; Humphrey et al., 2001; You et al., 2001; Hakimi et al., 2002, 2003; Lunyak et al., 2002; Shi et al., 2003) known as LSD1/BHC110/KIAA0601/p110b was identified as the first histone lysine demethylase, which is involved in mediating neuron-restrictive silencing factor (NRSF)/REST-dependent repression of neuronal genes in non neuronal cells (Shi et al., 2004, 2005). In vitro, LSD1 specifically demethylates mono- and dimethylated H3-K4, which, in vivo, has been suggested to mediate gene repression by maintaining an unmethylated H3-K4 status on a specific set of regulated promoters (Shi et al., 2004, 2005). Interestingly, the demethylase activity of LSD1 has been shown to be modulated by other proteins, including CoREST and BHC80 (Shi et al., 2005; Lee et al., 2005), and by other histone marks displayed around the substrate (Forneris et al., 2005, 2006). Furthermore, LSD1 has been implicated in H3-K9 demethylation that is associated with PSA gene activation in an AR dependent manner (Metzger et al., 2005) and has been described as a component of the MLL1 coactivator complex (Nakamura et al., 2002). More recently, additional demethylase enzymes have been described and includeJHDM1, which specifically demethylates dimethyl histoneH3 at lysine 36 (H3-K36; Tsukada et al., 2006), JMJD1A/JHDM2A, which demethylates mono- and dimethyl H3-K9 (Yamane et al., 2006), and JMJD2A, which demethylates trimethyl H3-K9/K36 (Whetstine et al., 2006). These enzymes belong to a large family of proteins that contain a conserved JmjC domain, which has been shown to be critical for demethylation (Trewick et al., 2005; Tsukada et al., 2006). Together, these recent discoveries suggest an important but still mostly unknown role exerted by demethylation events in regulated gene transcription.
Here we report an unexpectedly widespread recruitment of the histone demethylase LSD1 to active promoters, including most ER α gene targets in MCF7 cells, and we find that LSD1 is needed for activation and is required to oppose the functions of three HMTs: RIZ1, ESET, and Eu-HMTase1. For gene transcription, these HMTs exert an inhibitory gatekeeper function, which is required to prevent recruitment of unliganded nuclear receptors and constitutive activation. Intriguingly, a similar molecular strategy is also employed for ER α gene targets that do not recruit LSD1 but utilize distinct combinations of HMTs and HDMs to provide promoter specificity to this HMT/HDM code. A similar code appears to exist also for a number of other signal-dependent transcription programs. In summary, we propose that a gatekeeper strategy imposed by a selective use of HMTs and HDMs is widely exploited in mammalian biology to determine the appropriate physiological amplitude of regulated gene transcription as orchestrated by nuclear receptors and other classes of DNA-binding transcription factors.
Results
A Broad Genome-Wide LSD1 Promoter-Binding Program Detected by ChIP-DSL
Based on our interest in the role of corepressor complexes in gene transcription regulation, we applied a new genome-wide chromatin immunoprecipitation (ChIP) assay based on DSL (DNA selection and ligation; details in Supplemental Experimental Procedures and in Kwon et al., 2007) to several CoREST corepressor complex components, including the specific histone lysine demethylase LSD1. Recruitment of LSD1 in MCF7 cells that had been treated with 17 β-estradiol (E2) was determined by the ChIP-DSL method using a specific α -LSD1 antibody (Figure S1) and an array containing 20,045 human-proximal promoters (Hu20K; Figure 1A). Statistical significance for promoter enrichment was based on the analysis of a genomic tiling component included on the array (Figure 1A, yellow dots). Based on data obtained from three independent biological replicates, we unexpectedly detected a total of 4212 LSD1-enriched (LSD1+) promoters in these cells (22% of the Hu20K array that showed reliable signals) even at a highly stringent selection criteria (p<0.0001), while 118 promoters were detected with an irrelevant antibody using the same criteria (Figures 1A and 1B, red dots). The number of LSD1+ promoters would increase to 5174 promoters (27% of the array) or 6570 promoters (34% of the array) if the selection criteria were relaxed to a more conventional stringency (p<0.001 and p<0.01, respectively; Figure 1C). These results were validated by conventional ChIP assay and quantitative PCR (qPCR) analysis on randomly selected LSD1+ promoters at p<0.0001 (Figure 1D).
Figure 1. Genome-Wide Promoter Analysis Reveals the Association of LSD1 with a Broad Gene Activation Program.
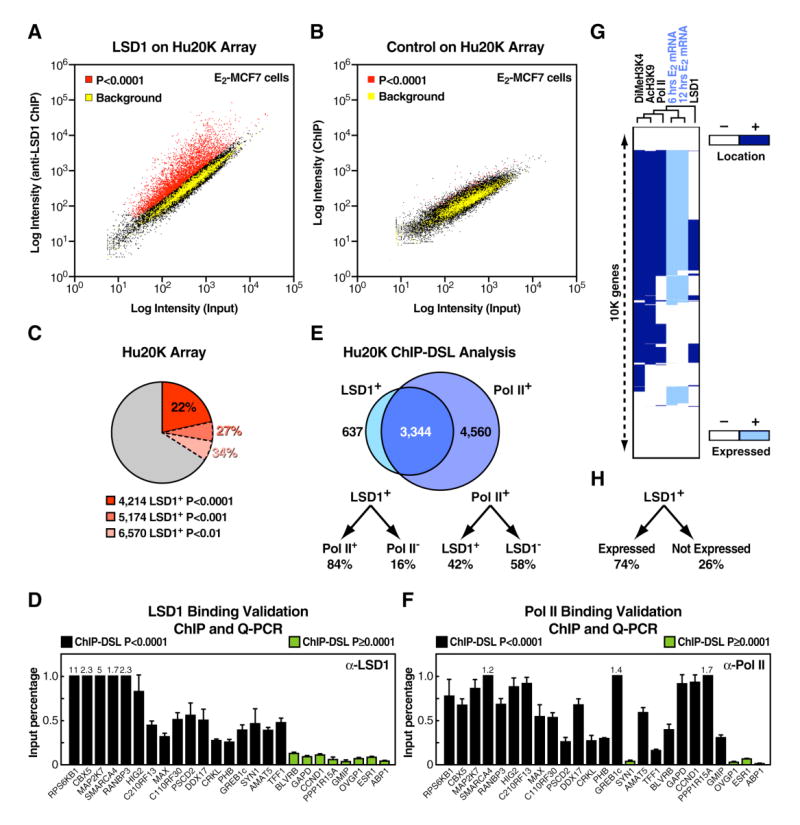
(A) Scatter plot of LSD1 recruitment to human gene promoters is shown. E2-induced MCF7 cells were profiled on the Hu20K array by ChIP-DSL. Three biological replicates were used to derive enriched promoters at p<0.0001 (red), shown in comparison with intergenic genomic sequences as negative controls (yellow). Weighted average is visualized on a single experiment scatter plot. (B) Scatter plot of a non relevant antibody profiled on the Hu20K array by ChIP-DSL is shown. Two biological replicates were used. (C) Number and percentage of LSD1+ promoters on the Hu20K array at different statistical cutoffs are shown. (D) ChIP/qPCR analysis of 17 LSD1+ and 8 LSD1− randomly selected promoters in E2-stimulated MCF7 cells is shown. The data are the average of three replicates, and error bars represent ± standard error mean. (E) Venn diagram of LSD1+ and Pol II+ promoters obtained by ChIP-DSL is shown. Only promoters with reliable signal intensities in both profiling experiments were included in the comparison. (F) ChIP/qPCR analysis of Pol II recruitment on selected LSD1+ and LSD1− promoters in E2-stimulated MCF7 cells is shown. The data are the average of three replicates, and error bars represent ± standard error mean. (G) Correlation of gene expression (light blue) with promoter occupancy (dark blue) is shown and includes histone modification marks (DiMeH3K4 and AcH3K9) as well as Pol II and LSD1, which were profiled in E2-induced MCF7 cells. Only genes included in both promoter- and expression-profiling arrays and reliably scored in all measurements were used to construct the binary map by unsupervised hierarchical clustering analysis. (H) LSD1+ genes classified by mRNA expression status are shown.
Consistent with the predictive value of this assay, virtually all known LSD1-target promoters represented on the Hu20K array were detected as LSD1+, including the repressed SYN1 (Hakimi et al., 2002) and SCN3A (Shi et al., 2004) promoters (Figure S2). The multiple experimentally confirmed or computationally predicted NRSF/REST-target genes found as LSD1+ (e.g., CX36, DNM1, PAX4, and SYT2; Andres et al., 1999; Ballas et al., 2001; Hakimi et al., 2002; Lunyak et al., 2002; Figure S3A) and the enrichment of NRSF/REST-predicted sites observed in LSD1+ neuronal-specific genes (Figure S3B) are consistent with the model that, as a component of the CoREST complex, LSD1 is functionally important in negatively regulating neuronal NRSF/REST-dependent genes in non neuronal cells (Andres et al., 1999; Ballas et al., 2001,2005; Humphrey et al., 2001; You et al., 2001; Hakimi et al., 2002; Lunyak et al., 2002; Shi et al., 2003, 2004; Bruce et al., 2004).
Broad LSD1 Promoter Binding Is Primarily Associated with Gene Activation Events
In addition to the role of LSD1 in transcriptional repression (Figure S2 and S3), ∼84% of LSD1+ promoters were Pol II+ and, quite unexpectedly, a full 42% of all Pol II+ promoters in MCF7 cells were LSD1+ (Figure 1E). These results were confirmed by conventional ChIP/qPCR assay (Figure 1F). Similarly, examination of histone marks was consistent with the preponderance of activated, rather than repressed, LSD1+ promoters. Indeed, dimethylation of histone H3 lysine 4 (diMeH3K4), a substrate for LSD1 demethylation activity associated with LSD1 repressive functions (Shi et al., 2004, 2005; Lee et al., 2005), and acetylation of histone H3 lysine 9 (acH3K9), another well established mark that correlates with gene activation, exhibited a considerable overlapping pattern with the presence of LSD1 on gene promoters (Figure 1G). These observations were corroborated by clustering ChIP-DSL and mRNA-profiling data sets obtained in MCF7 cells, thus observing ∼74% LSD1+ genes as expressed (Figures 1G and 1H). Taken together, our data suggest that the percentage of activated transcription units modulated by LSD1 (∼74%–84%) is substantially larger than the percentage of activated transcription units (∼16%–26%) included in the LSD1-repressed program (Figures 1E and 1H).
Recently, we identified 578 promoters that exhibit ERα binding in MCF7 cells (Kwon et al., 2007). Surprisingly, 58% of ER α -enriched (ERα+) promoters also exhibited LSD1 recruitment (319 ERα +/LSD1+ promoters); these represented ∼9% of all LSD1+ promoters (Figure 2A). In addition, genomic tiling analysis using ChIP-DSL assay of two well-established ERα-regulated target genes, TFF1/pS2 and GREB1, revealed LSD1- and ERα-corecruitment on presumed ERα-binding enhancer sites that were located ∼10 Kb upstream of TFF1/pS2 and ∼40Kb upstream of GREB1 gene start sites (Figure 2B). This ChIP-DSL analysis also showed that an ERα coactivator, CBP, and an “activation” histone mark, AcH3K9, exhibited similar profiles (Figure 2B). For all factors tested, including LSD1, the occupancy on both distal and proximal promoter cognate DNA sites was shown to be stimulated by E2 treatment as determined by ChIP/qPCR (Figure 2C). In addition, LSD1 recruitment on promoters of other LSD1+/ ERα+-target genes was also stimulated by E2 (Figure 2D). In agreement with these data, coimmunoprecipitation experiments in whole MCF7 cell extract using -LSD1 antibody demonstrated a specific E2-dependent interaction of LSD1 with endogenous ERα and CBP, and vice versa (Figures 2E and 2F).
Figure 2. LSD1 Associates with Most ERα-Promoter Targets in MCF7 Cells.
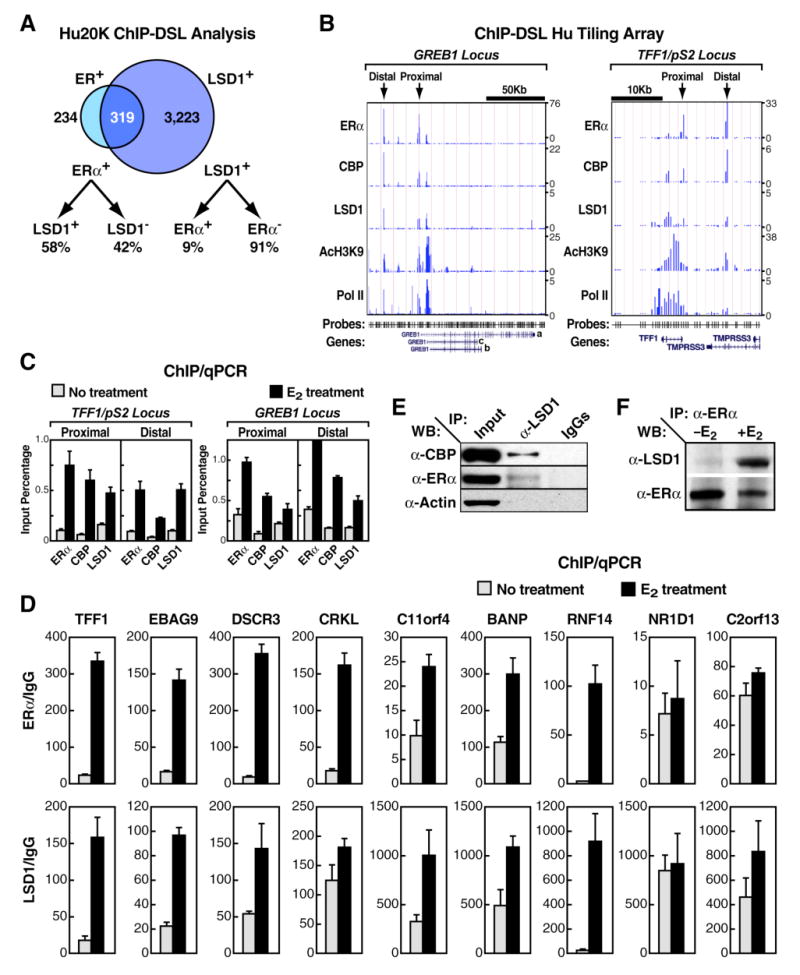
(A) Venn diagram of LSD1+ and ERα+ promoters as obtained by ChIP-DSL is shown. Only promoters with reliable signal intensities in both profiling experiments were included in the comparison. (B) ChIP-DSL tiling array analysis on GREB1 (left panel) and TFF1/pS2 (right panel) loci of ERα, CBP, LSD1, AcH3K9, and Pol II occupancy in E2-stimulated MCF7 cells is shown. Binding profiles represent ChIP versus input DNA intensity ratios (right). The most enriched ERα-binding sites (proximal and distal) are indicated by arrows. DSL probe location and RefSeq gene annotation are indicated in the bottom. (C) ChIP/qPCR recruitment analysis of ERα, CBP, and LSD1 on proximal and distal ERα-binding sites upon E2 treatment in MCF7 cells is shown. The data are the average of three replicates, and error bars represent ± standard error mean. (D) ChIP/qPCR analysis of LSD1 and ERα recruitment on additional LSD1+/ ERα+ promoters detected by ChIP-DSL assay upon E2 treatment in MCF7 cells is shown. The data are the average of three replicates, and error bars represent ± standard error mean. (E) Shows coimmunoprecipitation analysis of ERα and CBP by anti-LSD1 antibody in cell extracts obtained from E2-stimulated MCF7 cells. 5%input is shown. (F) Coimmunoprecipitation analysis of LSD1 by anti- ERα antibody in cell extracts obtained from unstimulated and E2-stimulated MCF7 cells is shown.
Ligand-Dependent Induction of ERα-Regulated Targets Requires Functionally Active LSD1
We next assessed the functional requirement of LSD1 in gene activation of ERα-regulated genes and found that E2-dependent induction of LSD1+/ERα+-target genes (pS2, GREB1, CTSD, and MYC) was essentially abolished in MCF7 cells in which LSD1 was knocked down by siRNA (Figures 3A, 3B, and S4A). By contrast, analysis on an LSD1−/ERα+-target gene, WISP2, revealed that depletion of LSD1 had no effect on its ligand-dependent activation (Figure 3C).
Figure 3. LSD1 Regulates E2-Dependent Gene Transcription While Both H3-K4 and H3-K9 Demethylation Events Are Observed.
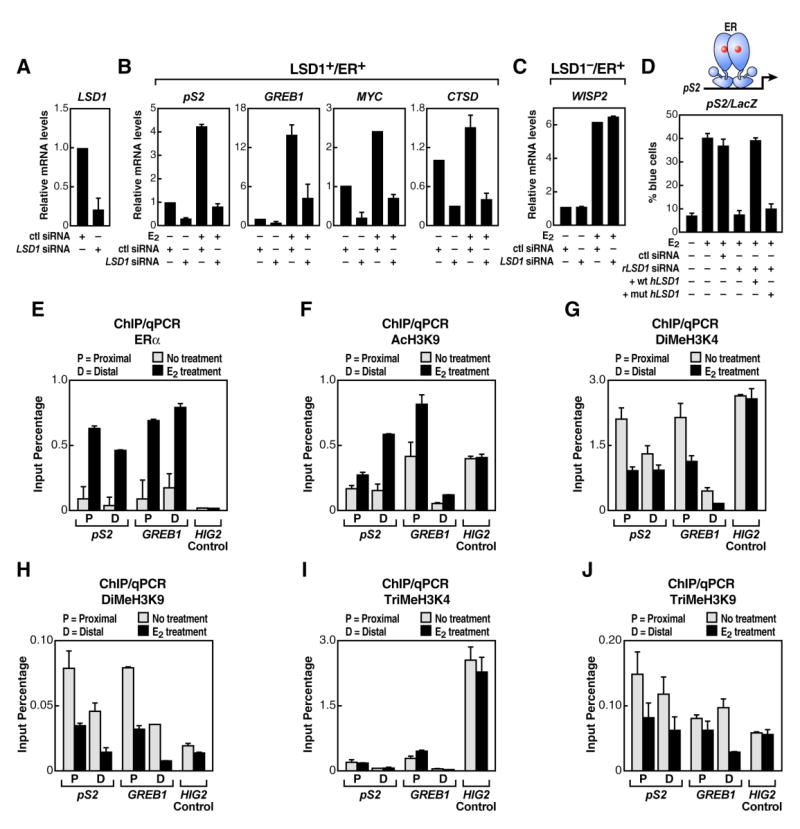
(A) Real-time qPCR (RT-qPCR) analysis is shown to document efficiency of LSD1 siRNA to diminish endogenous LSD1. (B) RT-qPCR analysis of several endogenous LSD1+/ ERα+-target genes upon LSD1 siRNA transfection is shown. (C) RT-qPCR analysis of an endogenous LSD1−/ ERα+-target gene upon LSD1 depletion by siRNA is shown. In (A)-(C) LSD1 siRNA was delivered by transient transfection in MCF7 cells, and β-actin mRNA expression levels as well as cell transfection efficiency were used for normalization. (D) Functional rescue analysis of the wild-type (wt) and the amine oxidase mutant (mut) human LSD1 form (hLSD1) in Rat-1 cells is shown. Endogenous rat LSD1 (rLSD1) expression was depleted by specific rat LSD1 siRNA, and ectopic wt hLSD1 and mut hLSD1 overexpression was accomplished by expression plasmids. Reporter plasmid and LSD1 siRNA were delivered by single-cell nuclear microinjection in Rat-1 cells. (E-J) Panels show ChIP/qPCR occupancy analysis of ERα (E), acH3-K9 (F), diMeH3-K4 (G), diMeH3-K9 (H), TriMeH3-K4 (I), and TriMeH3-K9 (J) on ERα-binding sites in vehicle- or E2-treated MCF7 cells. ERα proximal or distal binding sites on pS2 and GREB1 genomic loci were examined; HIG2 promoter was included as control. The data in (A)-(J) are the average of three replicates, and error bars represent ± standard error mean.
To test the role of the amine oxidase activity of LSD1 on regulation of LSD1+/ER+ targets, a rescue experiment was performed by single-cell nuclear microinjection assay; in this experiment, wild-type human LSD1 (wt hLSD1) or a predicted amine oxidase-inactive human form (mut hLSD1) was injected in Rat-1 cells that were simultaneously depleted of the endogenous rat LSD1 by microinjection of rLSD1 siRNA. While WT hLSD1 rescued the E2-induced activation of a pS2 promoter LacZ reporter, the mut hLSD1 was unable to rescue the reporter gene activity (Figure 3D). Similarly, transient transfection of wt hLSD1 but not mut hLSD1 caused an E2-dependent increase in the activity of an ERE-Luc reporter (Figure S5). Therefore, the amine oxidase activity, which was proven to be required for mediating LSD1-dependent H3-K4 and H3-K9 demethylation (Shi et al., 2004; Metzger et al., 2005), is also necessary for LSD1/ERα-dependent gene activation. We next addressed any H3-K4 and H3-K9 demethylation event that might occur upon E2 stimulation of LSD1/ER α -dependent gene targets. While ER α binding and acH3K9 were increased as a result of E2 treatment (Figures 3E and 3F), we observed, by quantitative ChIP/qPCR on promoter and distal pS2 and GREB1 ER α -target sites, a consistent decrease in both diMeH3K4 and diMeH3K9 (Figures 3G and 3H). Importantly, no simultaneous increases in trimethylation status were observed on the same sites (Figures 3I and 3J). In concert, these findings suggest that H3-K9 and H3-K4 methylation dynamics are key components of the gene activation program mediated by LSD1.
Mechanisms Underlying the LSD1-Dependent Gene Activation
To understand the mechanism underlying the functional requirement for LSD1 in E2-dependent induction, we examined the impact of removing various H3-K9 and H3-K4 HMTs (Martin and Zhang, 2005) by siRNA on E2-dependent induction. Depletion of known H3-K4 HMTs, including MLL1, had little effect on basal pS2 activity (data not shown). However, depletion of several specific H3-K9 HMTs, including RIZ1, ESET, and Eu-HMTase1,but not G9a or Suv39h1/h2, derepressed the pS2 promoter; this resulted in full activation in the absence of ligand (Figure 4A), while no significant effects were observed after depletion of these three enzymes in the presence of ligand (Figure 4B). Thus, in the absence of these specific H3-K9 HMTs, regulated activation of pS2 gene now became LSD1 independent (Figure 4C). Consistent with these observations, mRNA levels of endogenous pS2 and GREB1 genes were upregulated upon siRNA mediated RIZ1/ESET depletion even in the absence of ligand without any effect on response observed in the presence of ligand (Figures 4D, 4E, and S4B). To demonstrate the specificity in knockdown experiments, we observed at least a partial (∼70%) functional rescue of the repressive ESET activity in HeLa cells in which the endogenous human ESET (hESET) was depleted by specific hESET siRNA, and the mouse form of this HMT (mESET) was overexpressed (Figure 4F). In addition, the binding of ESET and RIZ1 on pS2 and GREB1 promoters was found to be decreased upon E2 stimulation as quantified by ChIP/qPCR (Figure 4G). Together, these data suggest that a critical function of LSD1 is to reverse the inhibitory effect of several H3-K9 HMTs: RIZ1, ESET, and Eu-HMTase1, the presence of which dictates LSD1 dependency for gene activation.
Figure 4. Specific H3-K9 HMTs Function as Inhibitory Gatekeepers and Dictate LSD1 Dependency for ERα-Regulated Gene Activation.
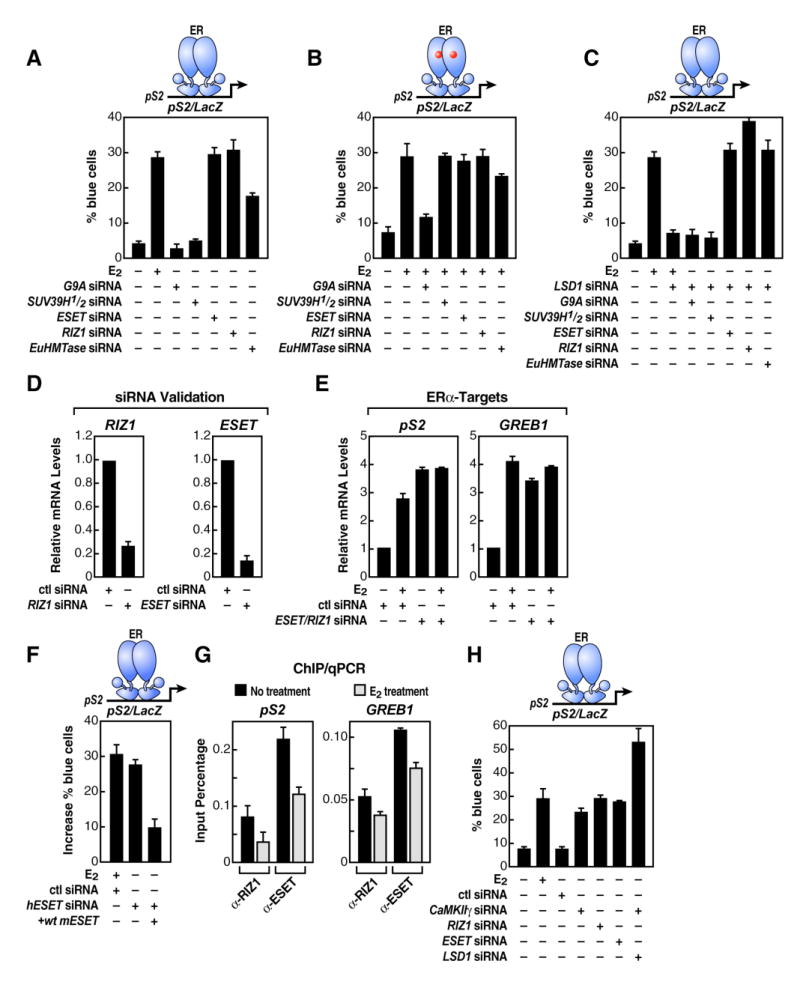
(A and B) Effect of different siRNAs to specific H3-K9 HMTs on pS2 promoter-LacZ reporter activity was analyzed by single-cell microinjection assay in the absence (A) and presence (B) of ligand (E2). (C) Functional LSD1 dependency of pS2 promoter-LacZ reporter activity was tested after depletion of specific H3-K9 HMTs. (D) RT-qPCR analysis was performed to document efficiency of H3-K9 HMTs siRNAs to diminish endogenous RIZ1 and ESET. (E) RT-qPCR analysis of endogenous ERα-target genes upon specific H3-K9 HMTs siRNA transfection is shown. (F) Functional rescue analysis of a mouse ESET form in HeLa cells is shown. Endogenous human ESET (hESET) expression was abolished by specific mouse ESET siRNA, and ectopic mouse ESET (mESET) overexpression was accomplished by expression plasmid. (G) Panel shows ChIP/qPCR recruitment analysis of RIZ1 and ESET on endogenous ERα-target promoters upon E2 stimulation in MCF7 cells. (H) Functional analysis of CaMKIIγ in absence of ligand (E2) on a pS2 promoter-LacZ reporter gene is shown. For experiments (A)-(C), (F), and (H) reporter plasmid and siRNAs were delivered by single-cell nuclear microinjection in MCF7 and HeLa (in F) cells. In (D) and (E) siRNAs were delivered by transient transfection in MCF7 cells, and β-actin expression levels and cell transfection efficiency were used for normalization. The data in (A)-(H) are the average of three replicates, and error bars represent ± standard error mean.
The requirement for more than one H3-K9 HMT implies that, at least for some genes, these enzymes are not fully functionally redundant; this has also been recently shown for similar non redundant HDM activities (Yamane et al., 2006). Interestingly, at least one of the specific H3-K9 HMTs, RIZ1, has been proposed to act as a tumor-suppressor gene that is silenced in many highly metastatic breast cancer cell lines (He et al., 1998; Duet al., 2001), and RIZ1 siRNA-depleted MCF7 cells showed increased levels of cell proliferation (Carling et al., 2004; Gazzerro et al., 2006). Indeed, cell proliferation and cell-cycle control were the top two ER+ gene ontology-enriched terms that were revealed by ChIP DSL analysis of ERα in MCF7 cells (Kwon et al., 2007), suggesting a global upregulation of ER+ gene targets in the absence of RIZ1.
Because of the connection between specific protein kinases, including CaMKs, and the actions of repressive HMTs (Ishitani et al., 2003; Zhang, et al., 2002; Kurahashi et al., 2005), we tested the possibility that CaMKII might modulate H3-K9 HMTs and, hence, LSD1- and E2-dependent gene activation events. Interestingly, specific CAMKIIγ siRNA resulted in full, LSD1-independent activation of the pS2-reporter gene (Figure 4H), which strongly suggests a link between specific signaling pathways and an HMT-dependent imposition of inhibitory histone marks.
A Balance between Histone Lysine Methylation and Demethylation Events Determines Ligand Dependency of ERα Targets
The observation of “ligand-independent” gene activation in the absence of specific H3-K9 HMTs raised the question of whether ERα is still critically required for activation in the absence of these enzymes and LSD1. To investigate this issue, siRNA depletion of ERα, along with RIZ1 or ESET, was performed in MCF7 cells and revealed that, in the absence of ERα, pS2 promoter activation was abolished (Figure 5A). These results, which are fully in concert with the observation that unliganded ERα can still bind to and even recycle at low levels on the pS2 promoter (Metivier et al., 2004; Perissi et al., 2004), were confirmed on endogenous ERα-target genes (Figures 5B and 5C).
Figure 5. H3-K9 HMTs Impose Ligand Dependency to the E2-Dependent Signaling Pathway.
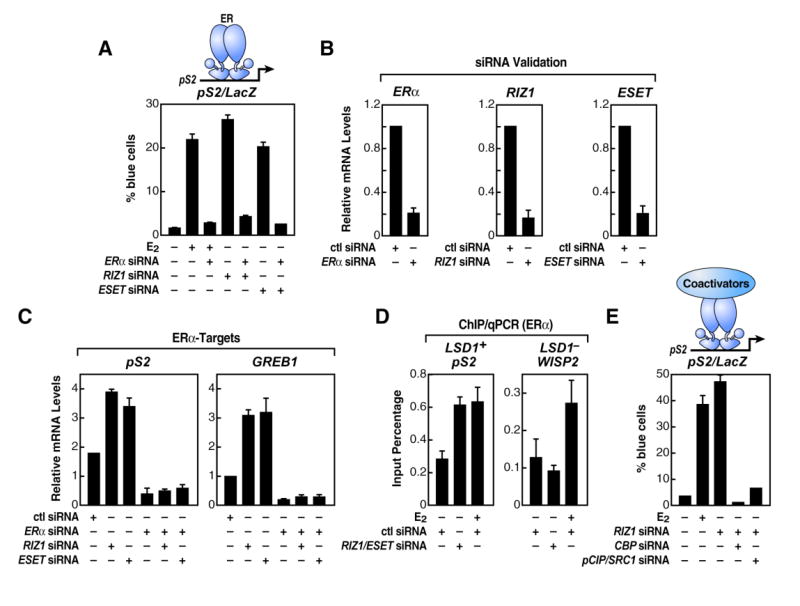
(A) ERα dependency of the pS2 promoter-LacZ reporter activity is analyzed in absence of specific H3-K9 HMTs. (B) RT-qPCR analysis was performed to document efficiency of ERα, RIZ1, and ESET siRNAs to diminish endogenous ERα, RIZ1, and ESET. (C) RT-qPCR analysis of endogenous ERα-target genes after removing H3-K9 HMTs and ERα by siRNA is shown. (D) ChIP/qPCR recruitment analysis of ERα in cells transfected with specific H3-K9 HMTs siRNAs in absence or presence of ligand (E2) is shown. (E) Analysis of pS2 promoter-LacZ reporter activity upon removing the ERα-associated coactivators CBP, pCIP, and SRC1 in cells depleted of the H3-K9 HMT RIZ1. For experiments (A) and (E), reporter plasmid and siRNAs were delivered by single-cell nuclear microinjection in MCF7 cells. In (B)-(D), siRNAs were delivered by transient transfection in MCF7 cells, and β-actin mRNA expression levels and cell transfection efficiency were used for normalization. The data in (A)-(E) are the average of three replicates, and error bars represent ± standard error mean.
ChIP analysis of RIZ1/ESET siRNA-depleted cells showed an increased recruitment of ERα, in absence of E2, to a level equivalent to that observed with E2, thus supporting the hypothesis that one critical function of H3-K9HMTs is to inhibit recruitment of unliganded ERα to its target genes (Figure 5D). Furthermore, well-established ERα coactivators, including CBP and SRC1-p/CIP, were required for this LSD1- and ligand-independent activation (Figure 5E). In contrast, the depletion of these same HMTs caused neither activation (data not shown) nor increased ERα binding on the LSD1-independent ERα target WISP2 promoter (Figure 5D), suggesting a role of other combinations of HMTs/HDMs in ERα regulation of LSD1-independent genes (see below).
Based on the broad recruitment of LSD1 on proximal promoters (Figure 1C), we determined the LSD1 dependency of several additional regulated gene activation programs. LSD1 siRNA microinjection experiments revealed a similar role of LSD1 in activation of AR, as previously shown (Metzger et al., 2005; Yamane et al., 2006). In addition, these experiments revealed a role of LSD1 in activation of NFκB-, AP-1-, and βRAR-regulated promoters, but not in the case of CREB-regulated gene targets (Figure S6). These data suggest that LSD1 is selectively involved in induction of a subset of regulated transcription units but is not universally required for gene activation. Analogous to ERα, RIZ1 and ESET appeared to exert a similar role in AR-dependent activation of the PSA gene (Figures 6A and S7A) as suggested by the partial derepression of PSA expression levels and the LSD1-independent activation of the same gene, which were again produced in the absence of specific H3-K9 HMTs. Similarly, depletion of RIZ1/ESET resulted in activation of NFκB-target genes in absence of signal, and this activation also exhibited dependency on the presence of NFκB factors (Figures S7B–S7D). Based on these data, we propose that a major biological role for the opposing actions of RIZ1/ESET/Eu-HMTase1 and LSD1 is to confer high amplitude of ligand response or signal dependency of gene activation on cohorts of regulated transcription units.
Figure 6. A Promoter-Specific HMT/HDM “Code” for Regulated Transcription Units.
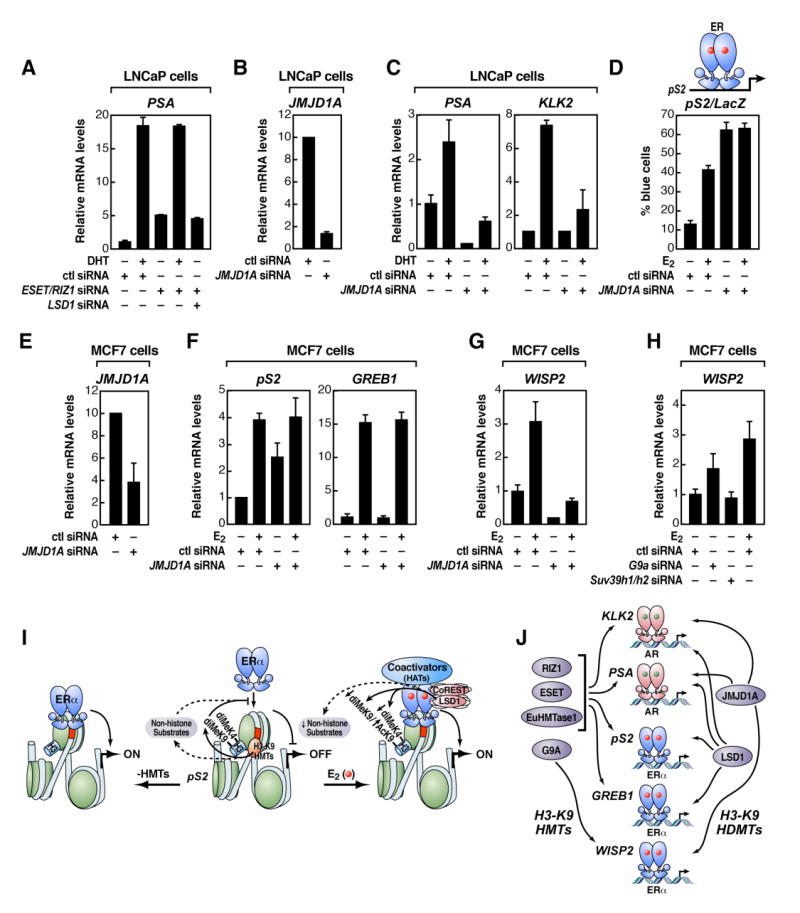
(A) RT-qPCR analysis of an endogenous LSD1+/AR+ gene target upon RIZ1/ESET and/or LSD1 depletion by siRNA in LNCaP cells is shown. (B) RT-qPCR analysis to document the efficiency of JMJD1A siRNA to diminish endogenous JMJD1A in LNCaP cells is shown. (C) RT-qPCR analysis of endogenous AR-target genes upon JMJD1A depletion by siRNA is shown. (D) Functional analysis of the pS2 promoter-LacZ reporter gene activity after removing JMJD1A by microinjection of siRNA in MCF7 cells is shown. (E) RT-qPCR analysis was performed to document efficiency of JMJD1A siRNA to diminish endogenous JMJD1A in MCF7 cells. (F) RT-qPCR gene expression analysis of endogenous LSD1+/ ERα+-target genes upon JMJD1A depletion by siRNA is shown. (G) RT-qPCR analysis of an endogenous LSD1−/ ERα+-target gene upon JMJD1A depletion by siRNA is shown. (H) RT-qPCR gene expression analysis of an endogenous LSD1−/ ERα+-target gene upon G9a or Suv39h1/h2 depletion by siRNA is shown. (I) Model of H3-K9 HMT requirement to inhibit constitutive ERα activation by blocking binding of the unliganded nuclear receptor to its cognate DNA sites is shown; HDMs, as LSD1, are required to demethylate H3-K9 HMTs substrates to permit activation by liganded ERα (see text for details). (J) Gene-specific use of HMT/HDMs to define regulated gene activation programs (see text for details) is shown. In all these experiments, siRNA was delivered by transient transfection in LNCaP (A)-(C) or MCF7 cells (D)-(H), and β-actin expression levels and cell transfection efficiency were used for normalization. The data in (A)-(H) are the average of three replicates ± standard error of the mean.
Promoter-Specific HMT/HDM “Code” for Regulated Transcription Units
Because pS2 and GREB1 require LSD1 for activation, while WISP2 does not, a key question is whether entirely distinct molecular strategies are used for LSD1+- and LSD1−-regulated transcription units or whether distinct HMTs and HDMs are required to mediate a similar regulatory mechanism. As a new AR-specific H3-K9 HDM, JMJD1A/JHDM2A, has recently been reported (Yamane et al., 2006), we examined whether this enzyme exerts a similar role on LSD1+/ERα-regulated targets and, alternatively, whether this factor might substitute for LSD1 on LSD1−/ERα+ gene targets. The specific depletion of JMJD1A by siRNA in LNCaP cells confirmed that this factor was required for ligand-dependent activation of AR target genes (Figures 6B and 6C), as previously shown (Yamane et al., 2006). Because LSD1 is also required (Figure 6A; Metzger et al., 2005), these AR-target genes demonstrate that at least two HDMs are required for full activation (Yamane et al., 2006). In contrast, ERα-dependent activation of pS2 and GREB1 gene targets depends on LSD1 but not on JMJD1A when similar experiments are performed in MCF7 cells (Figures 6D–6F). Interestingly, however, an unexpected activation of the pS2 promoter was observed in the absence of ligand (Figures 6D–6F). Based on these findings, we next explored whether an LSD1−/ERα+ gene target, WISP2, might use JMJD1A and HMTs other than ESET/RIZ1/EuHMTase to mediate a strategy of regulation similar to that utilized by LSD1 and these enzymes for pS2. Interestingly, while LSD1 siRNA did not inhibit ligand-dependent activation (Figure 3C), JMJD1A siRNA blocked E2-dependent stimulation of WISP2 (Figure 6G), and G9A but not SUV39H1/H2 siRNAs permitted a partial activation (∼50%) in the absence of ligand (Figure 6H). Together, these results provide compelling evidence for a widely used, promoter specific code in regulated transcription, with different cohorts of targets requiring distinct combinations of HMTs and HDMs. It is interesting to note that for some genes, loss of HMTs alone causes a level of activation similar to that observed in response to ligand, while for others, only a partial activation, with further enhancement when ligand is present, is observed.
Discussion
Here we report an unexpected mechanism that proves to be physiologically utilized to prevent constitutive gene activation by unliganded nuclear receptors and other classes of signal-dependent DNA-binding transcription factors. We have identified three specific H3-K9 HMTs that potentially act as gatekeepers for the ∼58% of ERα gene targets that are LSD1+ and that maintain a repressive status that precludes unliganded nuclear receptors, such as ERα, from effective binding and from functioning as ligand-independent, constitutive activators (Figure 6I). Indeed, transcriptional activation by unliganded nuclear receptors has been reported on removal of corepressors and on activation by specific signaling pathways (Culig et al., 1994; Nazareth and Weigel, 1996; Zwijsen et al., 1997; Rogatsky et al., 1999; Ueda et al., 2002; Ogawa et al., 2004; Kim et al., 2005).
Our data support the model that inhibitory HMTs prevent unregulated activation of these targets, at least in part, by blocking the binding of unliganded ERα to its cognate DNA sites. The binding of liganded ERα on these regulated promoters thereafter permits the recruitment of the LSD1 enzymatic activity that is required to dismiss the inhibitory marks and, potentially, methyl marks of other non histone substrates (I.G.-B. and M.G.R., unpublished results), thus permitting other ERα-recruited coactivators to initiate the regulated transcriptional response (Figure 6I). Thus, exchange of inhibitory histone lysine methyl marks based on the actions of specific, and not fully redundant, H3-K9 HMTs as well as the actions of critical HDMs are required to permit a ligand-dependent response. Therefore, while REST/CoREST-dependent genes clearly employ LSD1 in repression for at least a cohort of the REST-dependent program (Shi et al., 2004; Figures S2 and S3), our results have also revealed that LSD1, most likely associated with the MLL1 complex (Nakamura et al., 2002; Guenther et al., 2005), is required for a surprisingly broad range of regulated gene activation events. We further suggest that the HMT/HDM cycle provides a licensing mechanism for signal-dependent gene activation of other classes of regulated transcription factors that are required for normal development and homeostasis.
Gene-Specific Use of HMT/HDMs to Define Regulated Gene Activation Programs
Because LSD1 is recruited only to a subset of ERα-target genes, we were keen to initially examine the possibility that a similar inhibitory histone code and degree of HMT/HDM usage might act on genes even when LSD1 is not recruited to the promoter. Recently it has been reported that the mono- and dimethyl H3-K9 HDM, JMJD1A/JHDM2A, contributes with LSD1 to the hormone-induced H3-K9 demethylation of the PSA promoter/enhancer (Metzger et al., 2005; Yamane et al., 2006), and that an additional H3-K9demethylase, JMJD2A, displays specific trimethyl H3-K9/K36 activity (Whetstine et al., 2006). Therefore, analogous to histone deacetylase enzymes (HDACs), several H3-K9 HMTs and HDMs could be present in the same multiprotein complex (McKinsey et al., 2001; Yang and Seto, 2003; Ogawa et al., 2002; Shi et al., 2003) and might act to regulate gene expression in a non redundant fashion involving additional, distinct substrates.
Here, we have shown that distinct HMTs and HDMs are utilized in a promoter-specific fashion (Figure 6J). For example, the LSD1+/ERα+WISP2-target gene requires JMJD1A for ligand-dependent activation, in which G9a is selectively required to prevent ligand-independent constitutive activation. However, LSD1 but not JMJD1A is necessary for E2-dependent activation of the ERα+-target genes pS2 and GREB1, while both LSD1 and JMJD1A are required for DHT activation of the KLK2 and PSA genes. It is therefore tempting to speculate that there will be multiple cohorts of target genes for each nuclear receptor that depend on a similar strategy to prevent constitutive activation, but each will utilize unique combinations of HMT sand HDMs to license ligand-regulated expression.
While we would predict the existence of additional substrates other than H3-K9, for H3-K9 HMTs, and H3-K4/K9, for LSD1, we propose that ultimately the inhibitory gatekeeper strategy that is imposed by different HMTs blocks ligand-independent receptor binding and constitutive gene activation. This gatekeeper function is biologically overcome on binding of liganded receptors and specific H3-K9 HDMs. The requirement for three different HMTs, RIZ1/PRDM5 (Steele-Perkins et al., 2001), ESET/SETDB1 (Yang et al., 2002; Schultz et al., 2002; Wang et al., 2003), and Eu-HMTase1/GLP (Ogawa et al., 2002), for at least some ERα-target genes, suggests that these SET-domain enzymes are not fully functionally redundant (Schultz et al., 2002; Wang et al., 2003), as seems to similarly occur for at least two HDMs, LSD1 and JMJD1A/JHDM2A, for the regulation of some AR-target genes (Metzger et al.,2005; Yamane et al., 2006). As this represents an analogous strategy involving different HMTs and HDMs, the link between histone marks in preventing constitutive activation of physiologically regulated genes appears to be a general strategy.
H3-K4 and H3-K9 Demethylation in E2-Dependent Gene Activation
At least two different dimethylated lysines have been proposed to be demethylated by LSD1: the well-documented H3-K4 (Shi et al., 2004, 2005; Lee et al., 2005) and H3-K9 (Metzger et al., 2005). LSD1 was found by ChIP-DSL to be mainly associated with the presence of dimethyl H3-K4 on the genome (Figure 1G). Indeed, although a certain degree of H3-K4 demethylase activity was observed upon activation of ERα targets, high remaining levels of this mark were still observed after LSD1 binding and activation, which would agree with the high overlapping pattern detected (Figure 3G). In contrast, it is proposed that this mark is fully removed by LSD1 on targets that are repressed by NRSF, which is supposed to confer gene repression (Shi et al., 2004). Thus, LSD1 might switch activity depending on the promoter, which is consistent with the ability of CoREST and BHC80 to modify LSD1 enzymatic activity in vitro (Shi et al., 2005; Lee et al., 2005), therefore displaying H3-K4 demethylase on NRSF targets and H3-K9 demethylase on AR, ERα, and other factors' target promoters. Also, one of the two reported LSD1 activities might be selectively inactivated by the surrounding histone marks present around the promoter. Indeed, specific histone marks, such as AcH3-K9, inhibit H3-K4 demethylation in vitro (Forneris et al., 2005, 2006). Of course, both activities could be also equally utilized on all promoters, with the final levels of histone methylation being indeed determined by the predemethylation levels.
The regulatory roles of these two histone methylation marks on gene activation of euchromatic promoters have been the subject of intense investigation during the past few years. For example, methylation of H3-K9 has been described to decrease on a subset of inflammatory genes upon LPS stimulation (Saccani and Natoli, 2002).Similarly, an inverse relationship between H3-K9 methylation and NFκB p65 binding on the IL-1β promoter has been observed upon LPS stimulation in THP-1 cells (Chan et al., 2005). Dimethylation of H3-K9 but also H3-K4 has been described to decrease when T3R shifted from an unliganded state to a liganded, active state (Liet al., 2002), and a dramatic decrease in H3-K4 occurred on a tandem array of the mouse mammary tumor virus (MMTV) promoter upon induction by glucocorticoids (Ma et al., 2001). Finally, a decline in both dimethyl H3-K4 (Kim et al., 2003) and dimethyl H3-K9 (Metzger et al., 2005) has been reported on the PSA promoter upon DHT induction, and here we show that both histone marks are simultaneously decreased upon ERα/E2-dependentactivation of several promoters. Thus, dynamic changes of H3-K4 and H3-K9 methylation seem to occur in a diversity of programs.
In conclusion, an important aspect of the serial posttranslational methylation modifications of histones and transcription factors, which constitute a component of the hypothesized “histone code,” is to impose signal/ligand dependency to regulated DNA-binding transcription factors by modulatory binding of unliganded receptors and other classes of transcription factors, thereby preventing constitutive activation of cohorts of target genes.
Experimental Procedures
Materials and Reagents
Anti-LSD1 antibody was generated in guinea pigs against bacterially expressed N'-terminal region (13–219 aa) of the human LSD1. Anti-CoREST antibody was a generous gift from Dr. Gail Mandel. Commercially available antibodies and siRNAs are documented in Supplemental Data.
ChIP Assay
MCF7 breast cancer cells were hormone deprived for 4 days and then treated for 1 hr with 100 nM 17b-estradiol (E2). ChIP assay was conducted as previously described (Zhu et al., 2006). Further details and list of primers used for PCR validation are available in Supplemental Data.
ChIP-DSL Assay
Genome-wide promoter location analysis by ChIP-DSL on Hu20K and HuTiling arrays was performed and analyzed as described in Supplemental Experimental Procedures and in Kwon et al., 2007. Biological triplicates were performed in each case.
Single-Cell Nuclear Microinjection Assay
The small interfering RNAs (siRNAs) were delivered into cells by single cell microinjection assay as previously described (Kamei et al., 1996; Heinzel et al., 1997). Details and siRNA information are available in Supplemental Experimental Procedures.
Transient Transfection Assay
Transient transfection assay in MCF7 cells was performed using Lipofectamin 2000, following manufacturer's protocol (Invitrogen). RNA Isolation, Real-Time PCR, and RNA
Profiling Analysis
RNA was extracted by RNeasy Assay (Qiagen), and cDNA was obtained by SuperScript First-Strand Synthesis System (Invitrogen) following both manufacturers' protocols. RNA profiling was performed as described by Kwon et al., 2007. Further details and list of primers used for PCR amplification are available in Supplemental Experimental Procedures.
Immunoprecipitation
Immunoprecipitation assay was conducted as previously described (Bannister and Kouzarides., 1996). Details are available in Supplemental Experimental Procedures.
Real-Time qPCR
Semi-qPCR was carried out according to the Mx3000P Real-Time PCR Systems manual and the Brilliant qPCR reagent kit (Stratagene). Details are available in Supplemental Experimental Procedures.
Supplementary Material
Supplemental Data include seven figures, Supplemental Experimental Procedures, and Supplemental References and can be found with this article online at http://www.cell.com/cgi/content/full/128/3/505/DC1/.
Acknowledgments
We thank colleagues in our laboratories, especially C.A. Nelson and A. Krones for technical assistance. We also thank Dr. V. Perissi for critical reading of the manuscript and Dr. A. Almenar-Queralt for generous help; Dr. A. Aggarwal for advice on prediction of essential enzymatic LSD1 residues; Dr. G. Mandel for kindly providing CoREST antibodies; A. Gonzalez (Santa Cruz Technologies) for advice on reagents; and J. Hightower and M. Fisher for figure and manuscript preparation. We especially acknowledge Ron Margolis and Phil Smith (NIDDK) and Laura Mamounas (NINDS) for their critical support in permitting development of this technology. M.G.R. is an investigator with Howard Hughes Medical Institute. This work is supported by CA97134, DK39949, and NS34934 NIH grants, Vitamin Cases Consumer Settlement Fund, and Army Concept Award BC0464 to M.G.R.; DK063491 and CA52599 NIH grants to C.K.G.; and CA114184 and HG003119 NIH grants to X.-D.F; and Postdoctoral Traineeship Award (PC040247, CDMRP-USAMRAA, DoD) for I.G-B.
References
- Andres ME, Burger C, Peral-Rubio MJ, Battaglioli E, Anderson ME, Grimes J, Dallman J, Ballas N, Mandel G. CoREST: a functional corepressor required for regulation of neuralspecific gene expression. Proc Natl Acad Sci USA. 1999;96:9873–9878. doi: 10.1073/pnas.96.17.9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas N, Battaglioli E, Atouf F, Andres ME, Chenoweth J, Anderson ME, Burger C, Moniwa M, Davie JR, Bowers WJ, et al. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31:353–365. doi: 10.1016/s0896-6273(01)00371-3. [DOI] [PubMed] [Google Scholar]
- Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–657. doi: 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384:641–643. doi: 10.1038/384641a0. [DOI] [PubMed] [Google Scholar]
- Bruce AW, Donaldson IJ, Wood IC, Yerbury SA, Sadowski MI, Chapman M, Gottgens B, Buckley NJ. Genome wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc Natl Acad Sci USA. 2004;101:10458–10463. doi: 10.1073/pnas.0401827101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling T, Kim KC, Yang XH, Gu J, Zhang XK, Huang S. A histone methyltransferase is required for maximal response to female sex hormones. Mol Cell Biol. 2004;24:7032–7042. doi: 10.1128/MCB.24.16.7032-7042.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C, Li L, McCall CE, Yoza BK. Endotoxin tolerance disrupts chromatin remodeling and NF-κB transactivation at the IL-1beta promoter. J Immunol. 2005;175:461–468. doi: 10.4049/jimmunol.175.1.461. [DOI] [PubMed] [Google Scholar]
- Chen JD, Evans RM. A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature. 1995;377:454–457. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, Bartsch G, Klocker H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Res. 1994;54:5474–5478. [PubMed] [Google Scholar]
- Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J, Allis CD, Chait BT, Hess JL, Roeder RG. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121:873–885. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Du Y, Carling T, Fang W, Piao Z, Sheu JC, Huang S. Hypermethylation in human cancers of the RIZ1 tumor suppressor gene, a member of a histone/protein methyltransferase superfamily. Cancer Res. 2001;61:8094–8099. [PubMed] [Google Scholar]
- Fischle W, Wang Y, Allis CD. Binary switches and modification cassettes in histone biology and beyond. Nature. 2003;425:475–479. doi: 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]
- Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim Y, Minor W, Rastinejad F, Khorasanizadeh S. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- Forneris F, Binda C, Vanoni MA, Battaglioli E, Mattevi A. Human histone demethylase LSD1 reads the histone code. J Biol Chem. 2005;280:41360–41365. doi: 10.1074/jbc.M509549200. [DOI] [PubMed] [Google Scholar]
- Forneris F, Binda C, Dall'Aglio A, Fraaije MW, Battaglioli E, Mattevi A. A highly specific mechanism of histone H3-K4 recognition by histone demethylase LSD1. J Biol Chem. 2006;46:35289–35295. doi: 10.1074/jbc.M607411200. [DOI] [PubMed] [Google Scholar]
- Gazzerro P, Abbondanza C, D'Arcangelo A, Rossi M, Medici N, Moncharmont B, Puca GA. Modulation of RIZ gene expression is associated to estradiol control of MCF-7 breast cancer cell proliferation. Exp Cell Res. 2006;312:340–349. doi: 10.1016/j.yexcr.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Guenther MG, Jenner RG, Chevalier B, Nakamura T, Croce CM, Canaani E, Young RA. Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci USA. 2005;102:8603–8608. doi: 10.1073/pnas.0503072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi MA, Bochar DA, Chenoweth J, Lane WS, Mandel G, Shiekhattar R. A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc Natl Acad Sci USA. 2002;99:7420–7425. doi: 10.1073/pnas.112008599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi MA, Dong Y, Lane WS, Speicher DW, Shiekhattar R. A candidate X-linked mental retardation gene is a component of a new family of histone deacetylase-containing complexes. J Biol Chem. 2003;278:7234–7239. doi: 10.1074/jbc.M208992200. [DOI] [PubMed] [Google Scholar]
- He L, Yu JX, Liu L, Buyse IM, Wang MS, Yang QC, Nakagawara A, Brodeur GM, Shi YE, Huang S. RIZ1, but not the alternative RIZ2 product of the same gene, is underexpressed in breast cancer, and forced RIZ1 expression causes G2-M cell cycle arrest and/or apoptosis. Cancer Res. 1998;58:4238–4244. [PubMed] [Google Scholar]
- Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, Laherty CD, Torchia J, Yang WM, Brard G, Ngo SD, Davie JR, et al. A complex containing N-CoR, mSin3 and histone deacetylase mediates transcriptional repression. Nature. 1997;387:43–48. doi: 10.1038/387043a0. [DOI] [PubMed] [Google Scholar]
- Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass CK, et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- Humphrey GW, Wang Y, Russanova VR, Hirai T, Qin J, Nakatani Y, Howard BH. Stable histone deacetylase complexes distinguished by the presence of SANT domain proteins CoREST/kiaa0071 and Mta-L1. J Biol Chem. 2001;276:6817–6824. doi: 10.1074/jbc.M007372200. [DOI] [PubMed] [Google Scholar]
- Ishitani T, Kishida S, Hyodo-Miura J, Ueno N, Yasuda J, Waterman M, Shibuya H, Moon RT, Ninomiya-Tsuji J, Matsumoto K. The TAK1-NLK mitogen-activated protein kinase cascade functions in the Wnt-5a/Ca(2+) pathway to antagonize Wnt/beta-catenin signaling. Mol Cell Biol. 2003;23:131–139. doi: 10.1128/MCB.23.1.131-139.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B, Lin SC, Heyman RA, Rose DW, Glass CK, Rosenfeld MG. A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell. 1996;85:403–414. doi: 10.1016/s0092-8674(00)81118-6. [DOI] [PubMed] [Google Scholar]
- Kim J, Jia L, Tilley WD, Coetzee GA. Dynamic methylation of histone H3 at lysine 4 in transcriptional regulation by the androgen receptor. Nucleic Acids Res. 2003;31:6741–6747. doi: 10.1093/nar/gkg909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jia L, Stallcup MR, Coetzee GA. The role of protein kinase A and cAMP response element-binding protein in androgen receptor-mediated transcription at the prostate-specific antigen locus. J Mol Endocrinol. 2005;34:107–118. doi: 10.1677/jme.1.01701. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Histone methylation in transcriptional control. Curr Opin Genet Dev. 2002;12:198–209. doi: 10.1016/s0959-437x(02)00287-3. [DOI] [PubMed] [Google Scholar]
- Kurahashi T, Nomura T, Kanei-Ishii C, Shinkai Y, Ishii S. The Wnt-NLK signaling pathway inhibits A-Myb activity by inhibiting the association with coactivator CBP and methylating histone H3. Mol Biol Cell. 2005;16:4705–4713. doi: 10.1091/mbc.E05-05-0470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YS, Garcia-Bassets I, Hutt KR, Cheng CS, Jin M, Liu D, Benner C, Wang D, Ye Z, Bibikova M, Fan JB, Duan L, Glass CK, Rosenfeld MG, Fu XD. Sensitive ChIP DSL technology reveals an extensive ERa binding program on human gene promoters. Proc Natl Acad Sci USA. 2007 doi: 10.1073/PNAS 0700715104. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachner M, Jenuwein T. The many faces of histone lysine methylation. Curr Opin Cell Biol. 2002;14:286–298. doi: 10.1016/s0955-0674(02)00335-6. [DOI] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437:432–435. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- Li J, Lin Q, Yoon HG, Huang ZQ, Strahl BD, Allis CD, Wong J. Involvement of histone methylation and phosphorylation in regulation of transcription by thyroid hormone receptor. Mol Cell Biol. 2002;22:5688–5697. doi: 10.1128/MCB.22.16.5688-5697.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunyak VV, Burgess R, Prefontaine GG, Nelson C, Sze SH, Chenoweth J, Schwartz P, Pevzner PA, Glass C, Mandel G, Rosenfeld MG. Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science. 2002;298:1747–1752. doi: 10.1126/science.1076469. [DOI] [PubMed] [Google Scholar]
- Ma H, Baumann CT, Li H, Strahl BD, Rice R, Jelinek MA, Aswad DW, Allis CD, Hager GL, Stallcup MR. Hormone-dependent, CARM1-directed, arginine-specific methylation of histoneH3ona steroid-regulatedpromoter. Curr Biol. 2001;11:1981–1985. doi: 10.1016/s0960-9822(01)00600-5. [DOI] [PubMed] [Google Scholar]
- Margueron R, Trojer P, Reinberg D. The key to development: interpreting the histone code? Curr Opin Genet Dev. 2005;15:163–176. doi: 10.1016/j.gde.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Martin C, Zhang Y. The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol. 2005;6:838–849. doi: 10.1038/nrm1761. [DOI] [PubMed] [Google Scholar]
- McKenna NJ, O'Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108:465–474. doi: 10.1016/s0092-8674(02)00641-4. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN. Control of muscle development by dueling HATs and HDACs. Curr Opin Genet Dev. 2001;11:497–504. doi: 10.1016/s0959-437x(00)00224-0. [DOI] [PubMed] [Google Scholar]
- Metivier R, Penot G, Carmouche RP, Hubner MR, Reid G, Denger S, Manu D, Brand H, Kos M, Benes V, Gannon F. Transcriptional complexes engaged by apo-estrogen receptor-alpha isoforms have divergent outcomes. EMBO J. 2004;23:3653–3666. doi: 10.1038/sj.emboj.7600377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger E, Wissmann M, Yin N, Muller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature. 2005;437:436–439. doi: 10.1038/nature04020. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Mori T, Tada S, Krajewski W, Rozovskaia T, Wassell R, Dubois G, Mazo A, Croce CM, Canaani E. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. 2002;10:1119–1128. doi: 10.1016/s1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- Narlikar GJ, Fan HY, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108:475–487. doi: 10.1016/s0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- Nazareth LV, Weigel NL. Activation of the human androgen receptor through a protein kinase A signaling pathway. J Biol Chem. 1996;271:19900–19907. doi: 10.1074/jbc.271.33.19900. [DOI] [PubMed] [Google Scholar]
- Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O'Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE, Kouzarides T. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412:561–565. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- Ogawa H, Ishiguro K, Gaubatz S, Livingston DM, Nakatani Y. A complex with chromatin modifiers that occupies E2F and Myc-responsive genes in G0 cells. Science. 2002;296:1132–1136. doi: 10.1126/science.1069861. [DOI] [PubMed] [Google Scholar]
- Ogawa S, Lozach J, Jepsen K, Sawka-Verheller D, Perissi V, Sasik R, Rose DW, Johnson RS, Rosenfeld MG, Glass CK. A nuclear receptor corepressor transcriptional checkpoint controlling activator protein 1-dependent gene networks required for macrophage activation. Proc Natl Acad Sci USA. 2004;101:14461–14466. doi: 10.1073/pnas.0405786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perissi V, Aggarwal A, Glass CK, Rose DW, Rosenfeld MG. A corepressor/coactivator exchange complex required for transcriptional activation by nuclear receptors and other regulated transcription factors. Cell. 2004;116:511–526. doi: 10.1016/s0092-8674(04)00133-3. [DOI] [PubMed] [Google Scholar]
- Peterson CL, Laniel MA. Histones and histone modifications. Curr Biol. 2004;14:R546–R551. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Privalsky ML. The role of corepressors in transcriptional regulation by nuclear hormone receptors. Annu Rev Physiol. 2004;66:315–360. doi: 10.1146/annurev.physiol.66.032802.155556. [DOI] [PubMed] [Google Scholar]
- Rogatsky I, Trowbridge JM, Garabedian MJ. Potentiation of human estrogen receptor alpha transcriptional activation through phosphorylation of serines 104 and 106 by the cyclin ACDK2 complex. J Biol Chem. 1999;274:22296–22302. doi: 10.1074/jbc.274.32.22296. [DOI] [PubMed] [Google Scholar]
- Rosenfeld MG, Lunyak VV, Glass CK. Sensors and signals: a coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2006;20:1405–1428. doi: 10.1101/gad.1424806. [DOI] [PubMed] [Google Scholar]
- Saccani S, Natoli G. Dynamic changes in histone H3 Lys 9 methylation occurring at tightly regulated inducible inflammatory genes. Genes Dev. 2002;16:2219–2224. doi: 10.1101/gad.232502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ., 3rd SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002;16:919–932. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Sawada J, Sui G, Affar el B, Whetstine JR, Lan F, Ogawa H, Luke MP, Nakatani Y, Shi Y. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell. 2005;19:857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Steele-Perkins G, Fang W, Yang XH, Van Gele M, Carling T, Gu J, Buyse IM, Fletcher JA, Liu J, Bronson R, et al. Tumor formation and inactivation of RIZ1, an Rb-binding member of a nuclear protein-methyltransferase superfamily. Genes Dev. 2001;15:2250–2262. doi: 10.1101/gad.870101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong JK, Hassig CA, Schnitzler GR, Kingston RE, Schreiber SL. Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature. 1998;395:917–921. doi: 10.1038/27699. [DOI] [PubMed] [Google Scholar]
- Trewick SC, McLaughlin PJ, Allshire RC. Methylation: lost in hydroxylation? EMBO Rep. 2005;6:315–320. doi: 10.1038/sj.embor.7400379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439:811–816. doi: 10.1038/nature04433. [DOI] [PubMed] [Google Scholar]
- Ueda T, Mawji NR, Bruchovsky N, Sadar MD. Ligand independent activation of the androgen receptor by IL-6 and the role of the coactivator SRC-1 in prostate cancer cells. J Biol Chem. 2002;277:38087–38094. doi: 10.1074/jbc.M203313200. [DOI] [PubMed] [Google Scholar]
- Wang H, An W, Cao R, Xia L, Erdjument-Bromage H, Chatton B, Tempst P, Roeder RG, Zhang Y. mAM facilitates conversion by ESET of dimethyl to trimethyl lysine 9 of histone H3 to cause transcriptional repression. Mol Cell. 2003;12:475–487. doi: 10.1016/j.molcel.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylase. Cell. 2006;125:467–481. doi: 10.1016/j.cell.2006.03.028. [DOI] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, Roeder RG, Brivanlou AH, Allis CD. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell. 2005;121:859–872. doi: 10.1016/j.cell.2005.03.036. [DOI] [PubMed] [Google Scholar]
- Yamane K, Toumazou C, Tsukada Y, Erdjument-Bromage H, Tempst P, Wong J, Zhang Y. JHDM2A, a JmjC-containing H3K9 demethylase, facilitates transcription activation by androgen receptor. Cell. 2006;125:483–495. doi: 10.1016/j.cell.2006.03.027. [DOI] [PubMed] [Google Scholar]
- Yang L, Xia L, Wu DY, Wang H, Chansky HA, Schubach WH, Hickstein DD, Zhang Y. Molecular cloning of ESET, a novel histone H3-specific methyltransferase that interacts with ERG transcription factor. Oncogene. 2002;21:148–152. doi: 10.1038/sj.onc.1204998. [DOI] [PubMed] [Google Scholar]
- Yang XJ, Seto E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr Opin Genet Dev. 2003;13:143–153. doi: 10.1016/s0959-437x(03)00015-7. [DOI] [PubMed] [Google Scholar]
- You A, Tong JK, Grozinger CM, Schreiber SL. CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc Natl Acad Sci USA. 2001;98:1454–1458. doi: 10.1073/pnas.98.4.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, McKinsey TA, Olson EN. Association of class II histone deacetylases with heterochromatin protein 1: potential role for histone methylation in control of muscle differentiation. Mol Cell Biol. 2002;22:7302–7312. doi: 10.1128/MCB.22.20.7302-7312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Baek SH, Bourk EM, Ohgi KA, Garcia-Bassets I, Sanjo H, Akira S, Kotol PF, Glass CK, Rosenfeld MG, Rose DW. Macrophage/cancer cell interactions mediate hormone resistance by a nuclear receptor derepression pathway. Cell. 2006;124:615–629. doi: 10.1016/j.cell.2005.12.032. [DOI] [PubMed] [Google Scholar]
- Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. CDK-independent activation of estrogen receptor by cyclin D1. Cell. 1997;88:405–415. doi: 10.1016/s0092-8674(00)81879-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data include seven figures, Supplemental Experimental Procedures, and Supplemental References and can be found with this article online at http://www.cell.com/cgi/content/full/128/3/505/DC1/.