

Abstract
Previous work has established that stably-transfected human MCF7 cells over-expressing high mobility group A1 proteins (HMGA1) are deficient in global genomic repair (GGR) following exposure to either UV light or cisplatin. To investigate whether HMGA1 over-expression also interferes with gene-specific repair, we employed a rapid and convenient quantitative polymerase chain reaction assay for measuring repair in unique DNA sequences. Efficiency of UV-induced lesion removal was assessed for two genes in MCF7 cells either induced, or not, to over-express transgenic HMGA1 proteins: the constitutively active HPRT gene and the transcriptionally silent β-globin gene. As controls, similar experiments were also performed in non-transgenic MCF7 cells that do not express detectable levels of HMGA1 and in normal human embryonic fibroblasts that naturally over-express HMGA1 proteins. Our results indicate that exposure of cells to a UV dose of 20 J/m2 produced an average of 0.21 ± 0.03 lesions/kb and 0.19 ± 0.02 lesions/kb in the HPRT and β-globin genes, respectively, with no significant difference between HMGA1 over-expressing cells and non-expressing cells. On the other hand, analysis of repair following UV exposure revealed that, compared to controls, HMGA1 over-expressing cells take considerably longer to repair photo-lesions in both the active HPRT and the silent β-globin loci, with non-expressing cells repairing 50% of lesions in HPRT 3-4 hours faster than HMGA1 over-expressing cells. Interestingly, the delay in repair is even more prolonged in the silent β-globin locus in HMGA1 over-expressing cells compared to control cells. To our knowledge, this is the first report of HMGA1 proteins inhibiting NER within specific genes located in either transcriptionally active “open”, or inactive “closed”, chromatin domains. Furthermore, taken together with previous findings, these results suggest that HMGA1 over-expression interferes with repair processes common to both the GGR and transcription coupled repair pathways.
Keywords: High Mobility group A1, Nucleotide excision repair, (NER), Transcription-coupled repair (TCR), DNA repair, Cyclobutane pyrimidine dimmer (CPD), Quantitative Polymerase Chain Reaction (QPCR)
1. Introduction
Heterogeneity of nucleotide excision repair (NER) in different regions of the genome has been well established, and prescribes that DNA damage occurring in transcriptionally active regions will be preferentially removed as compared to damage in non-transcribed, inactive regions of the genome [1]. Thus, mammalian cells possess two specialized sub-pathways of NER, one that removes lesions from active genes quickly and efficiently (transcription coupled repair or TCR), and one that removes lesions from the inactive, “silent” majority of the genome (global genomic repair or GGR), albeit more slowly and inefficiently than TCR [2, 3].
Previously we demonstrated that human breast epithelial cells over-expressing high mobility group A1 (HMGA1) proteins are compromised in their ability to carry out GGR of UV-induced DNA damage [4]. Additionally, work by others has shown that HMGA1 over-expressing human breast epithelial cells are more sensitive to cisplatin treatment, an indication that HMGA1 inhibits GGR removal of these types of chemical adducts as well [5]. Since HMGA1 proteins have been implicated in a variety of intracellular processes and harbor both DNA binding and chromatin remodeling ability (reviewed in: [6]), we wanted to determine if, as in the case of GGR, increased expression of HMGA1 also inhibited repair of unique sequence genes that are either transcriptionally active or inactive and, thus, assess whether the affect of HMGA1 on repair process was influenced by chromatin structure. Additionally, since little is known regarding the means by which HMGA1 proteins interfere with normal NER processing in over-expressing cells, we reasoned that comparing their effects on the efficiency of GGR with their effects on repair of individual genes that are transcriptionally active, or not, could lend insight into potential molecular mechanisms of NER inhibition by HMGA1 proteins.
Utilizing a recently developed quantitative long-range PCR-based assay for measuring gene-specific removal of UV-induced lesions [7], we assessed the efficiency of removal of UV-induced DNA damage in two different human genes: the hypoxanthine phosphoribosyl transferase (HPRT) gene, an allele of which is constitutively expressed in all normal somatic cells [8-10]), and the β-globin gene, which is transcriptionally silent in all cells but those of erythroid lineage [11]. These genes were chosen because microarray and other analyses indicated that HMGA1 proteins do not regulate the transcription of either gene in the cell lines being investigated (unpublished data), an important consideration given that these proteins can either positively or negatively regulate the transcription of a large number of genes in vivo (reviewed in: [12]). The cell lines used in this study include stable transgenic MCF7 human breast cancer epithelial cell lines that contain a tetracycline-regulated HMGA1a transgene which can be experimentally induced (or not) to over-express HMGA1 protein, as previously described [13]. For comparison, we also assayed the efficiency of UV-induced lesion removal in non-transgenic MCF7 cells that express little, or no, detectable endogenous HMGA1 and in human embryonic fibroblasts (IMR90 cells), which naturally over-expresses high levels of these proteins.
Here, we demonstrate that NER efficiency associated with both the transcriptionally active HPRT gene as well as the transcriptionally inactive β-globin gene is significantly compromised in HMGA1 over-expressing cells. These findings, coupled with previously published observations, demonstrate that HMGA1 over-expression (either naturally occurring or artificially induced) negatively impacts both GGR and the repair of transcriptionally active genes in living cells, suggesting that abnormally high levels of these proteins may inhibit one or more steps common to both NER pathways. Regardless of molecular mechanism(s) involved, however, these results support the emerging role for HMGA1 in contributing to the accumulation of mutations and genetic instabilities associated with naturally occurring cancer cells that, almost universally, over-express these nonhistone proteins.
2. Materials and methods
2.1 Cell culture
Human MCF7 cells, a line of breast adenocarcinoma cells that retains many characteristics of differentiated mammary epithelium (ATCC, Manassas, VA; Cat. No. HTB-22), were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 10mM HEPES, 100 units/mL penicillin G sodium and 100μg/mL streptomycin sulfate. For routine maintenance, almost confluent monolayer cultures of cells were digested to single cell suspensions by gentle trypsinization, washed with phosphate buffered saline (PBS), counted and seeded into 100 mm culture dishes, as previously described [13]. Clonal cell lines MCF7-7C-Cs8 and MCF7-7C-Cs9 are stably transfected derivatives of MCF7/Tet-“OFF” cells containing a tetracycline-regulated pTRE vector encoding Hemagglutinin (HA)-tagged HMGA1a cDNA, maintained in the presence of 100 μg/ml of hygromycin [13]. These different transgenic cell lines express high, but variable levels of transgenic HA-tagged HMGA1a protein when grown in medium lacking tetracycline (these are referred to as HMGA1 “ON” cells). To prevent expression of HMGA1 transgenes, MCF7-7C-Cs cells were cultured in media containing 2μg/ml tetracycline (and are referred to as HMGA1 “OFF” cells). Diploid human embryonic IMR90 fibroblast cells (Coriell Cell Repositories, Camden, NJ; repository # I9078) were cultured in DMEM containing 5% FBS and 5% newborn calf serum (NCS), supplemented with 2 mM L-glutamine, 10mM HEPES, 100 units/mL penicillin G sodium and 100μg/mL streptomycin sulfate.
2.2 Western blot analysis
Levels of intracellular HMGA1 proteins were determined by Western blot analysis of cell extracts prepared with TRIzol (Invitrogen, Carlsbad, CA), according to the manufacturer’s instructions. Ten μg total protein from cell isolates was loaded onto a 12% polyacrylamide gel in the presence of sodium dodecyl sulfate (SDS). Proteins were separated electrophoretically at 100V for 2.5 hours before transferring to Immobilon-P membrane (Millipore, Billerica, MA) via tank transfer in buffer containing 25mM Tris, pH 7.5, 0.2M glycine and 20% methanol at 100V for 1 hour. Prior to addition of primary antibodies, membranes were blocked in Tris-buffered saline (TBS) containing 5% nonfat dry milk. Membranes were probed for HMGA1 and total actin (as a loading control) using either a specific polyclonal antibody against HMGA1 proteins (1:1000 dilution) [14], or polyclonal anti-actin rabbit antibody (1:5000; Sigma, Co., St. Louis, MO). Secondary antibody was horseradish peroxidase (HRP)-conjugated goat-anti-rabbit (1:5000; Santa Cruz). Blots were developed using SuperSignal chemiluminescent substrate (Pierce, Rockford, IL). Films were scanned and bands quantified using a densitometer and employing ImageQuant® software to determine fold-differences in band intensities.
2.3 UV Irradiation and DNA extraction
For UV-induction, cells were cultured as described above and, after the last passage, grown until confluent. To suppress DNA synthesis, which could mimic repair, 2mM hydroxyurea (HU) was added to plates one hour prior to irradiation [15]. For UV exposure, media was removed from culture plates, cells were washed with phosphate buffered saline (PBS) and then irradiated using low pressure Hg lamps (Sylvania, Model G30T8, Danvers, MA) at a dose of 20 J/m2 (measured with a Spectroline DM-254N short wave UV meter) (Spectronic Corp., Westbury, NY), as previously described [16]. For time course experiments, cells were either harvested immediately after UV exposure (0 hour samples), or the medium was replaced and cells harvested at different times after irradiation and then immediately frozen. Control (UV -) cells were handled identically except for UV treatment. All cells were stored as pellets at -70°C until DNA extraction could be performed.
High molecular weight DNA was extracted from frozen cell pellets using the Qiagen Blood and Cell Culture DNA Mini Kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol. To determine the concentration and purity of extracted high molecular weight DNA, samples were diluted 1:2 in Tris-EDTA (TE) buffer and the absorbance determined spectrophotometrically at 260 nm and 280 nm. To verify DNA concentration, sample stocks were diluted to 10 ng/μl (the approximate working concentration for quantitative PCR) and the absorbance re-determined spectrophotometrically.
2.4 Gene-specific quantitative PCR (QPCR)
Fifty μl amplification reactions were performed in 0.5 ml thin-wall PCR tubes (Phenix, Hayward, CA) on a Techne Genius thermal cycler (Model FGEN05TP, Techne, Cambridge, UK). A 10.4 kb fragment of the HPRT gene (GenBank Accession # J00205) containing exons 2-5 was amplified using previously published PCR primers 14577 (forward) and 24997 (reverse) [7]. A 13.5kb fragment of the β-globin gene locus (GenBank Accession # J00179) containing the 5’flanking region of the gene was also amplified using primers 48510 (forward) and 62007 (reverse) reported by Santos et al., [7]. PCR conditions for both sets of reactions were as follows: 20 ng total genomic DNA, 1 X Buffer XL II (Applied Biosystems, Foster City, CA), 100 ng/μL bovine serum albumin (BSA), 200 μM (each) of the four deoxynucleotides, 20 pmol (each) forward and reverse primers, 0.7 mM Mg(OAc)2, and 1 unit rTth DNA polymerase XL (Applied Biosystems, Foster City, CA). For radioactive labeling to verify ethidium bromide-based repair quantitation, 2 μCi [P32]-α-dATP was added to each reaction. As controls, following the recommendations of Santos et al. [7], both a small fragment amplification reaction and a half-template (50%) reaction were run for each set of experiments. Small fragment amplification reactions utilized primers HPRT 1 and HPRT 2 (Clonetech, Palo Alto, CA) to amplify a 250bp fragment of the HPRT gene. 50% control reactions contained the same long-fragment HPRT and β-globin gene primers as above, but one half the concentration (i.e., 10 ng).of template genomic DNA.
Amplification parameters included a PCR ‘hot start’, in which samples were heated to 75°C for 5 minutes before adding rTth DNA polymerase to each reaction. Genomic samples were then denatured by heating at 94°C for 1 minute, followed by 27 cycles of denaturation at 94°C for 15 seconds and annealing and extension at 64°C for 12 minutes. Samples were then heated to 72°C for 10 minutes for final elongation and stored at 4°C until electrophoretic analysis could be completed.
2.5 DNA repair analysis
Eight μL of each PCR reaction in loading buffer (2.5% glycerol, 10 mM EDTA, pH 8.0 plus 100 ug/ml each of bromephenol blue and xylene cyanol) were loaded onto a 0.8% agarose gel containing 2 μM ethidium bromide and amplified DNA products separated by electrophoresis at 60V for approximately 2.5 hours. Stained DNA bands were visualized and digitally captured using a GelDoc™EQ Imager (BioRad, Richmond, CA) and band intensities quantified using Quantity One software (BioRad). Band intensities were first corrected for gel background (average above and below each band) and then normalized to both 100% and 50% (i.e., half the original template concentration) control reactions to measure variances observed in repair samples and also to insure that DNA amplification is measured in the linear range of the PCR reactions, respectively [17]. During initial optimization procedures, small fragments of the HPRT gene were PCR amplified in independent reactions as a measure of template concentration per sample, as recommended by Santos et al. [17]. The results of these small fragment (250 bp) controls demonstrated that variances in DNA concentration among all samples was minimal and did not impact the results obtained in the repair assays themselves (data not shown). The normalized value of each UV-irradiated sample was then divided by the normalized band intensity of the non-UV-irradiated sample. This provides the fraction of non-damaged templates at a given UV dose, as described by Van Houten and coworkers [17, 18]. Assuming a Poisson distribution of UV-induced lesions between amplified strands (e-S, where S = average lesion frequency), the lesion frequency per strand was calculated using the equation S = -ln(AD/A0), where A0 = amount of normalized full length (uncut) band intensity from non-UV-irradiated DNA and AD = the amount of normalized full length band intensity from a UV irradiated sample at a given time after DNA damage. Repair efficiencies at each time point were measured from the difference in average lesion frequency between that repair time and un-repaired DNA (0 hours).
The mean value and standard deviation for lesion incidence and efficiency of UV-induced lesion repair were calculated for at least 3 independent experiments for each gene amplification in each of the different cell lines. Best fit curve plots of the data were derived employing the PivotChart Wizard function of Microsoft Excel®.
3. Results
Previous work from our laboratory demonstrated that global genomic repair of UV-induced lesions is compromised in stably transfected MCF7 cells induced to over-express HMGA1 as well as in primary tumor cells that naturally over-express these proteins [4]. To assess whether a similar inhibition of repair of UV-induced damage also occurs in unique DNA sequences, we employed a long-range, gene-specific quantitative PCR (QPCR)-based repair assay [19] to monitor the time course of NER following irradiation of cells over-expressing (or not) HMGA1 proteins. This repair assay relies on the inability of the DNA polymerase employed in the PCR reaction to synthesize new DNA past bulky, helix-distorting lesions such as UV-induced CPD or (6-4) photoproducts. Thus, in the QPCR assay, the more lesions present in the DNA template, the less overall amplification of a full-length product. An additional question of biological importance addressed by these same experiments is whether or not there is a differential effect of HMGA1 over-expression on the efficiency of NER of lesions located in an “active” gene that is continuously transcribed and has a relatively “open” chromatin structure compared to the efficiency of repair in a constitutively silent, inactive gene that has a more condensed chromatin structure (reviewed in: [20]).
3.1 HMGA1 protein levels in “OFF” and “ON” cells
Two different human gene loci, neither of which is regulated by HMGA1 proteins in the cell types being investigated (unpublished data), were analyzed for repair efficiency: the HPRT gene locus, which has previously been demonstrated to be constitutively expressed in normal somatic cells [10] and the β-globin gene locus, which is transcriptionally silent in all cells except those of erythroid lineage 11]. The cell lines used to assess repair efficiency in these two gene loci were the previously described transgenic HMGA1 “OFF” and HMGA1 “ON” cells[4], as well as non-transgenic MCF7 human breast epithelial cells and normal human embryonic fibroblasts (strain IMR90). As shown by the western blot in Figure 1A, MCF7 and HMGA1 “OFF” cells express little to no detectable HMGA1, while transgenic HMGA1 “ON” cells express up to 45-fold more HMGA1 than their non-expressing counterparts. The embryonic IMR90 cells naturally express HMGA1 proteins by as much as 12-fold over MCF7 and HMGA1 “OFF” cells (Fig. 1B).
Figure 1.
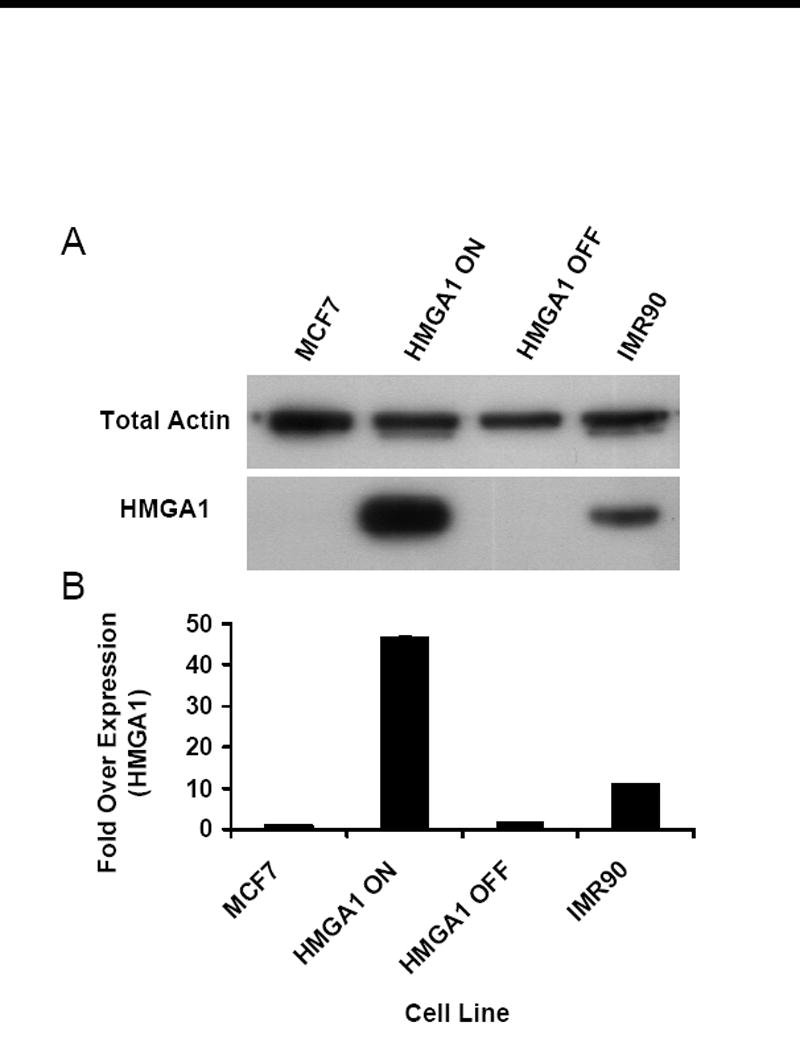
Characterization of HMGA1 expression levels. (A) Western blot of HMGA1 proteins (bottom panel) in transgenic HMGA1 OFF and ON cell lines as well as parental MCF7 human breast epithelial cells and IMR90 human embryonic fibroblast cells. Total actin was used as loading control in these studies (top panel). (B) Graphical representation of densitometric analyses performed on films from HMGA1 western blots. Each bar represents the mean density observed for three independent whole-cell lysate analyses normalized to a total actin loading control. Error bars represent ± 1 standard deviation (SD) from the mean density.
3.2 Long-range QPCR analyses of NER in the active HPRT and inactive β-globin loci
Figure 2 shows the results of representative long-range QPCR-based repair time course experiments for HMGA1 “ON” and HMGA1 “OFF” cells for both the HPRT gene (panel A; 10.4 kb DNA fragment) and the β-globin gene (panel B; 13.5 kb DNA fragment) amplification products. Lanes 2 through 7 show the relative amplifications of 20 ng template DNA from non-irradiated (lane 2) and UV-irradiated (20 J/m2) (lanes 3-7) cells at various time points after irradiation (0 to 24 hours). Lane 1 shows the amplification of 50% (10 ng) of the initial non-irradiated template DNA, a control to demonstrate that the PCR amplification reaction is in the linear (and therefore relevant) range. The number of hours cells were allowed to repair UV-induced damage is indicated above the lanes. As shown in both Panels A and B, by 24 hours DNA bands are clearly present in all samples, indicating that some amplification (or repair) has occurred within template sequences. Also, as previously demonstrated by Van Houten et al.[19], repair of the β-globin sequences occurs much more slowly than does repair of HPRT sequences, as indicated by band intensities between 2 and 24 hours (compare panels A and B, lanes 2-7).
Figure 2.
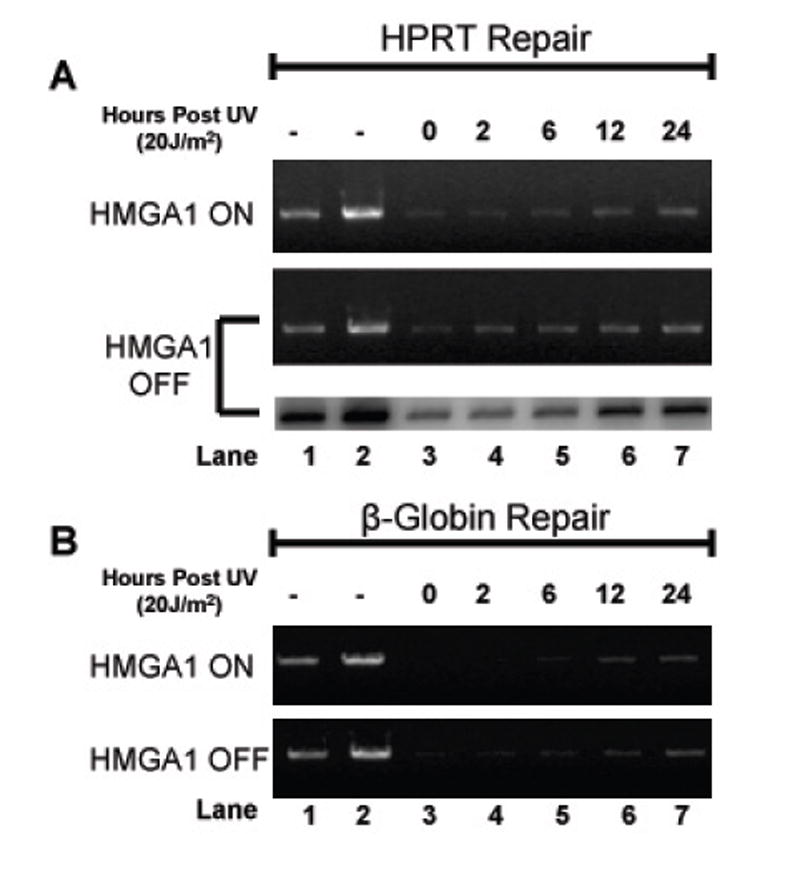
Long-range quantitative PCR analyses of UV-induced lesion removal in HPRT and β-globin genes. (A) Long-range QPCR analysis of lesion removal in the HPRT gene locus for both HMGA1 ON (top panel) and HMGA1 OFF cells (middle and bottom panels). Both ethidium bromide-stained agarose gel and phosphorimage analysis are included for the HMGA1 OFF cell line. Briefly, long-range QPCR products were electrophoresed in 0.8% agarose and either visualized using ethidium bromide (top and middle panel), or, in the bottom panel, dried and exposed to a phosphor-storage screen for detection of [32P]-α-dATP incorporated into the amplified DNA fragments. Lanes 1 and 2 contain, respectively, 50% (10 ng) and 100% (20 ng) of non-UV-treated genomic DNA as controls. (B) Long-range QPCR analysis of lesion removal in the β-globin gene locus for both HMAG1 ON (top panel) and HMGA1 OFF cells (bottom panel). Both panels are representative of ethidium bromide-stained agarose gels. As in panel A, lanes 1 and 2 contain 50% and 100% non-UV-treated genomic DNA as a control.
As a further control for quantification, radioactive QPCR reactions were performed (Fig. 2A, lower panel of HMGA1 “OFF” sample set) incorporating [32P]-α-dATP into amplified fragments. Samples were then electrophoretically separated as described in the Materials and Methods section and agarose gels were dried and exposed overnight to a phosphor storage screen. As shown in Fig. 2A, changes in the radioactive DNA band intensities in these repair experiments paralleled the changes seen with stained DNA and further quantification revealed no statistically significant differences between repair efficiencies calculated by ethidium bromide-stained digital gel analysis and phosphorimaging analysis (data not shown).
3.3 Levels of UV-induced damage are equivalent in HMGA1 “OFF” and “ON’ cells
Tables 1 and 2 represent data quantified from at least three independent experiments for each of the different cell lines for the HPRT and β-globin genes, respectively. As indicated by the number of UV-induced lesions per kb of DNA, there were no significant differences in the amount of DNA damage induced in the four independent cell lines immediately following a 20 J/m2 UV exposure, demonstrating that HMGA1 over-expression does not appreciably alter the amount of DNA damage induced in these cells. On the other hand, comparison of the % of lesions repaired at 12 hours post-UV exposure between Table 1 (for the active HPRT gene) and Table 2 (for the silent β-globin gene) indicates that repair of the transcriptionally inactive β-globin gene is significantly retarded in all four cell lines compared to amount of repair occurring in the active HPRT gene in these same cells. These results are consistent with the widespread observation that removal of DNA damage from transcriptionally active genes frequently occurs more rapidly than removal of lesions from non-transcribed genes [21]. Most importantly, however, at 12 hours post-UV exposure it is apparent that there is a substantial difference in the amount of repair between cell lines with little or no detectable HMGA1 (i.e., MCF7 and HMGA1 “OFF”), and those that are either induced to over-express (HMGA1 ON), or naturally over-express (IMR90), HMGA1 proteins. Tables 1 and 2 demonstrate that gene-specific repair of UV-induced lesions in both the HPRT and β-globin loci are significantly lower in HMGA1 over-expressing cells than in non-HMGA1-expressing cells.
Table 1.
HPRT Gene Locus Repair Statistics
| Cell Line | Lesions/kb | % Lesions Repaired at 12 hours |
|---|---|---|
| MCF7 | 0.21 ± 0.03 | 43.8 ± 2.9 |
| HMGA1 OFF | 0.24 ± 0.02 | 55.7 ± 0.1 |
| HMGA1 ON | 0.21 ± 0.04 | 35.0 ± 1.7 |
| IMR90 | 0.25 ± 0.02 | 32.8 ± 1.7 |
Table 2.
β-Globin Gene Locus Repair Statistics
| Cell Line | Lesions/kb | % Lesions Repaired at 12 hours |
|---|---|---|
| MCF7 | 0.18 ± 0.02 | 23.9 ± 0.7 |
| HMGA1 OFF | 0.20 ± 0.03 | 34.7 ± 4.0 |
| HMGA1 ON | 0.17 ± 0.05 | 3.1 ± 1.5 |
| IMR90 | 0.21 ± 0.02 | 6.8 ± 2.4 |
3.4 Rate of NER is significantly delayed in the HPRT and β-globin loci in HMGA1 over-expressing cells
To further characterize the decreased repair in HMGA1 over-expressing cells, the time course of repair over a 24 hour period was determined for both the HPRT and β-globin genes in the parental MCF7, HMGA1 “OFF” and HMGA1 “ON” cell lines. As shown by the results in Figure 3, repair of DNA lesions in both the HPRT and β-globin genes commences (albeit, at very different rates) soon after exposure of either the parental MCF7 or the HMGA1 “OFF” cells to UV light and continues for the next 24 hours. In marked contrast, in HMGA1 “ON” cells, repair of lesions in the inactive β-globin gene is almost totally absent for up to 8-10 hours (Fig. 3B) following UV exposure and is also significantly delayed by about 2-4 hours in the active HPRT gene (Fig. 3A).
Figure 3.
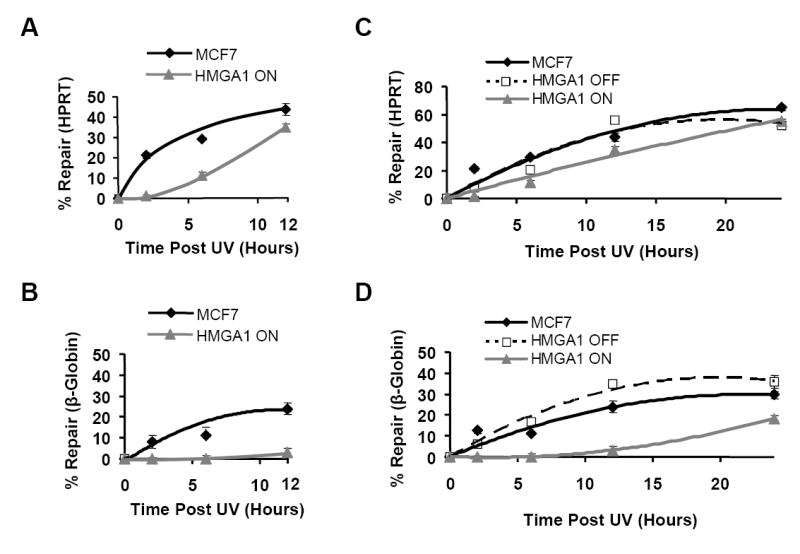
Gene-specific repair profiling reveals that HMGA1-overexpressing ON cells are less efficient at removing UV-induced lesions compared to non-expressing HMGA1 OFF and parental MCF7 cells. Best fit repair curves for removal of UV-induced lesions in both the transcriptionally active HPRT gene locus (A) and the silent β-globin locus (B) in HMGA1 ON (Δ) and non-expressing MCF7 (♦) cells during the first 12 hours following irradiation. Each time point represents the mean ± 1 SD of the repair efficiency observed in at least three independent experiments with each cell line. (C) Repair curves for UV-induced lesion removal in the active HPRT locus in HMGA1 ON (△), HMGA1 OFF (■) and MCF7 (♦) cells over a 24 hour period following irradiation. Each point represents the mean of at least three independent experiments. (D) Repair curves for UV-induced lesion removal in the silent β-globin gene locus in HMGA1 ON (△), HMGA1 OFF (■) and MCF7 (♦) cells over a 24 hour period following irradiation. Each point represents the mean ± 1 SD of at least three independent experiments
Nevertheless, even though lesion repair in the HPRT gene is initially impaired for several hours in “ON” cells, by 12 hours post-irradiation the extent of repair in ON cells approaches the level found in non-expressing control cells (Fig. 3A; Table 1) and by 24 hours post-UV exposure repair is nearly identical to that observed in both the parental MCF7 and “OFF” cells (Fig. 3C). On the other hand, compared to parental and HMGA1 “OFF” cells, repair of the β-globin gene is severely compromised throughout the entire 24 hour time course in “ON” cells (Figs. 3B & D; Table 2). For example, in HMGA1 “ON” cells at 12 hours post-irradiation only ~3% of lesions have been removed from the β-globin gene, compared to MCF7 cells and HMGA1 “OFF” cells which have removed between 7-10 times as many lesions (Fig. 3B & Table 2). Significantly, even after 24 hours of repair the silent β-globin locus still contains nearly twice as many lesions in the ON cells compared to the control MCF7 and HMGA1 OFF cells (Fig. 3D). It is also important to note that similar repair profiles were obtained from time course studies examining non-HMGA1-expressing IMR90 embryonic fibroblast cell line, which naturally over-expresses HMGA1 proteins (data not shown).
4. Discussion
In this study, we demonstrate that transgenic MCF7 cells induced to over-express HMGA1 proteins (i.e., “ON” cells) exhibit compromised nucleotide excision repair of UV-induced photoproducts both in a constitutively active HPRT gene allele (which exists in a more “open” or accessible chromatin structure; [8]) and the silent β-globin gene (which exists in a “closed”, or compacted, chromatin structure; [11]) compared to non-HMGA1-expressing “OFF” cells. While repair of both gene loci was significantly inhibited in the presence of HMGA1 proteins, this effect was much more pronounced in the transcriptionally silent β-globin gene. These results were confirmed in two additional human cell lines, the parental (non-transgenic) MCF7 breast epithelial cell line, which expresses little detectable HMGA1, and the IMR90 embryonic lung fibroblast cell strain, which naturally expresses as much as 12-fold more HMGA1 than either MCF7 or HMGA1 “OFF” cells (Fig. 1B). The confirmatory results obtained with the IMR90 cells are of particular note since they rule out the possibility that the reduction of NER efficiency observed in the HMGA1 over-expressing “ON” cells is an experimental artifact due to the use of artificially created transgenic cells. Additionally, the results of these control experiments (Tables 1 and 2) also indicated that there were no significant differences in the amount of DNA damage induced in cells exposed to a 20 J/m2 dose of UV. This was the case regardless of whether HMGA1 proteins are present in cells, or whether the genomic regions being analyzed were transcriptionally active, thus excluding the possibility that variations in overall DNA damage levels contributed to the inhibition of NER observed in over-expressing cells.
Strikingly, repair of the transcriptionally active HPRT gene in HMGA1 “ON” cells, while significantly delayed immediately following irradiation (Fig. 3A), reaches similar levels of repair to that of both the MCF7 and HMGA1 “OFF” cells by 24 hours post-UV exposure (Fig. 3C). Interestingly, this repair profile for the UV-damaged HPRT gene in living cells is reminiscent of the results of our previously reported in vitro analyses of the repair of HMGA1-bound UV-damaged DNA fragments by Xenopus oocyte nuclear extracts [4]. In those studies we demonstrated that while removal of damage from the HMGA1-bound DNA fragments was markedly inhibited early during incubation in the repair extracts (~60 min), by ~190 min of incubation nearly all of the lesions were removed from the DNA. These results clearly established that binding of HMGA1 to UV-induced lesions inhibits their removal from DNA by repair extracts but they also showed that over an extended period of time nearly complete repair is possible, most likely as a consequence of the known rapid and reversible association of HMGA1 proteins with DNA substrates in vitro [4]. Similar considerations may also be at least partially applicable in helping to explaining the in vivo repair profile seen for the HPRT gene in HMGA1 “ON” cells (Figs. 3A & C). Recent fluorescence recovery after photobleaching (FRAP) studies have demonstrated that in the nuclei of living cells HMGA1 proteins are highly mobile, rapidly and reversibly binding to DNA and chromatin substrates on a time scale of seconds [22]. Such in vivo mobility potentially allows for “windows of opportunity” to occur when HMGA1 proteins are not bound to damaged DNA, thus allowing for lesion repair even in over-expressing cells. Additional support for this idea is provided by the results of FRAP experiments demonstrating that the reversible binding of HMGA1 proteins is considerably more rapid in regions of active “euchromatin” than in regions of inactive heterchromatin [23]. These findings are in agreement with the present observations showing that repair of UV-induced lesions, although inhibited in HMGA1 over-expressing “ON” cells, still occurs and is much more rapid in the transcriptionally active HPRT gene than in the silent β-globin locus (Fig. 3).
HMGA1 “OFF” cells display a ‘typical’ repair time course profile for lesion removal in both the HPRT and β-globin genes, similar (in shape) to that of previously published “normal” cells [24, 25], albeit with overall repair being slower in the β-globin gene compared to the active HPRT gene (Fig. 3; compare HMGA1 “OFF” cell line shapes between panels C and D). Repair profiles for both genes are also similar in HMGA1 “ON” cells, but differ markedly in shape from that of HMGA1 “OFF” cells, and also from previously published normal cell repair data [24]. These results suggest that common mechanisms may be operating by which HMGA1 interferes with normal NER processes in both the HPRT and β-globin genes, the effects of which are exaggerated in more compacted, transcriptionally inactive regions of the genome. One of these mechanisms might well be the reversible binding of HMGA1 proteins directly to UV-induced DNA lesions, as discussed above. But this is unlikely to be the only way in which HMGA1 over-expression reduces the efficiency of NER in cells.
Previous work from our laboratory demonstrated that GGR of UV-induced lesions is inhibited in both transgenic MCF7 cells that have been induced to over-express HMGA1 as well as in non-transgenic cancer cells that naturally over-express this protein [4]. The research presented here extends these findings by demonstrating that HMGA1 over-expression also significantly reduces the efficiency of NER in both transcriptionally active (HPRT) and inactive (β-globin) unique sequence genes during the first 8-10 hours following UV-exposure, with the silent β-globin locus showing the most dramatic effect with an almost complete inhibition of repair during this period (Figs. 2 and 3). It has long been known that transcriptionally active mammalian genes are often repaired more efficiently than inactive genes [1]. Therefore, the present finding that lesion repair of the active HPRT gene is significantly reduced for several hours immediately following UV irradiation suggests that in over-expressing cells HMGA1 may be interfering with the process of transcription coupled repair (TCR). A number of potential caveats to this idea should be considered, however. One, for example, is the fact that the human HPRT gene is located on the X chromosome and since both the MCF7 and IMR90 cells employed in this study were derived from females, only one of the two HPRT gene copies present in the cells is transcriptionally active. Thus the delay in repair of the HPRT gene observed in HMGA1 over-expressing cells (Figs. 3A and C) could well be an underestimate of the actual rate of repair going on in the bona fide transcriptionally active HPRT gene, given that our assay monitors repair in both alleles. Therefore, similar types of repair studies should be performed on transcriptionally active autosomal genes to establish if our current findings are indeed a universal effect. Another caveat is the fact that the long-range QPCR technique employed to assess repair efficiency in these experiments monitors both the transcribed and non-transcribed strands of the HPRT gene and thus can not distinguish between the contributions of stand-specific TCR and GGR at this locus. Additional analyses of the transcribed and non-transcribed strands of both a transcriptionally active and an inactive gene are, therefore, required to determine the precise contribution of HMGA1 over-expression to possible suppression of strand-specific repair. Another caveat is the fact that the long-range QPCR technique employed to assess repair efficiency in these experiments monitors both the transcribed and non-transcribed strands of the HPRT gene and thus can not distinguish between the contributions of stand-specific TCR and GGR at this locus. Additional analyses of the transcribed and non-transcribed strands of both a transcriptionally active and an inactive gene are, therefore, required to determine the precise contribution of HMGA1 over-expression to possible suppression of strand-specific repair. It is noteworthy, nevertheless, that TCR was first recognized in an active gene (DHFR) through the use of double-stranded DNA probes, initially utilizing DNA blot hybridization techniques [26-28] and later through the use of long-range QPCR repair assays [29] and that both techniques have also been successfully applied to investigations of excision repair of lesions in the human HPRT gene [30, 31].
Nevertheless, the several hour delay in the onset of lesion removal following UV exposure that is apparent in the repair profiles for both the HPRT and β-globin genes in “ON” cells (Figs. 3A & B) suggests that HMGA1 over-expression might be affecting a step(s) common to both the GGR and TCR sub-pathways rather than just being confined to an inhibition of GGR. In this regard, we have demonstrated, using microarray analyses [32], that XPA transcripts are down-regulated in HMGA1 over-expressing cells. Thus a second possible mechanism by which HMGA1 may be inhibiting NER (and one that could operate in conjunction with direct lesion ‘masking’) is by interference with XPA gene expression in over-expressing cells. Indeed, in direct support of this possibility, we have recently obtained evidence linking repression of XPA transcription (and a reduction in cellular XPA protein levels) to binding of HMGA1 to a negative regulatory element in the endogenous XPA gene promoter in over-expressing cells (unpublished data).
The XPA gene product, while exhibiting no known catalytic function within the NER complex, is absolutely required for both TCR and GGR [33]. Furthermore, previous work has shown that decreased amounts of functional intracellular XPA protein decreases the efficiency of removal of both CPDs and UV- induced (6-4) photoproducts, albeit to different degrees [34, 35]). And, of particular note with regard to the current findings, other researchers have demonstrated that the efficiency of TCR in cells is less affected by decreases in XPA protein concentration than is the efficiency of GGR [36]. Experiments are, therefore, currently underway to examine more closely the possible involvement of HMGA1 in inhibiting XPA gene transcription and, thereby, reducing the overall efficiency of both GGR and TCR in over-expressing cells.
The data presented here are the first, to our knowledge, to link transcriptional activity and chromatin context with observed HMGA1-mediated defects in NER, further supporting a role for abnormal expression of these proteins with an increasing likelihood of accumulating genetic damage over time. It also seems reasonable to speculate that the influence of HMGA1 proteins on the efficiency of DNA repair could play an important role in the increasing frequency of chromosomal instabilities displayed by cancer cells, which, almost universally, over-express HMGA proteins. In this connection, HMGA1 over-expression has recently been linked to both chromosomal rearrangements in prostate cancer cells [37] and to aneuploidy in colon carcinomas [38].
On the other hand, based on the present data it also seems reasonable to suspect that tumor cells over-expressing HMGA1 proteins would exhibit a greater sensitivity to DNA damaging agents than normal cells, and that such over-expression might contribute to more positive therapeutic outcomes. A number of observations are indeed consistent with this suggestion. These include the fact that induced HMGA over-expression has been reported to increase cell sensitivity to killing by genotoxic agents such as UV light [4], cisplatin and bleomycin [5], doxorubicin (a topoisomerase II inhibitor) and X-ray irradiation [39]. Additionally, cells that naturally over-express HMGA proteins [4, 5, 39-42] have been demonstrated to not only be deficient in NER [4, 43] but also to exhibit increased sensitivity to DNA damage induced by both chemical agents, such as cisplatin or bleomycin [5, 39], and UV light [4, 43]. Intriguingly, it has been speculated that the deficiency in NER and increased sensitivity to UV photolesions observed in mouse ES cells that express high levels of HMGA proteins [42] are likely mechanisms to avoid the accumulation of mutated cells during embryogenesis [43].
Acknowledgments
This work was supported by NIH grant No. GM071760 (to R.R. and M.J.S.) and NIH grant No. T32 GM008336 for predoctoral training in biotechnology (to J.E.A.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2. ASM Press; Washington D.C: 2006. [Google Scholar]
- 2.Pfeiffer P, Gottlich B, Reichenberger S, Feldmann E, Daza P, Ward JF, Milligan JR, Mullenders LH, Natarajan AT. DNA lesions and repair. Mutat Res. 1996;366:69–80. doi: 10.1016/s0165-1110(96)90029-9. [DOI] [PubMed] [Google Scholar]
- 3.Sancar GB, Siede W, van Zeeland AA. Repair and processing of DNA damage: a summary of recent progress. Mutat Res. 1996;362:127–46. doi: 10.1016/0921-8777(95)00029-1. [DOI] [PubMed] [Google Scholar]
- 4.Adair JE, Kwon Y, Dement GA, Smerdon MJ, Reeves R. Inhibition of nucleotide excision repair by high mobility group protein HMGA1. J Biol Chem. 2005;280:32184–32192. doi: 10.1074/jbc.M505600200. [DOI] [PubMed] [Google Scholar]
- 5.Baldassarre G, Belletti B, Battista S, Nicoloso MS, Pentimalli F, Fedele M, Croce CM, Fusco A. HMGA1 protein expression sensitizes cells to cisplatin-induced cell death. Oncogene. 2005;24:6809–6819. doi: 10.1038/sj.onc.1208831. [DOI] [PubMed] [Google Scholar]
- 6.Reeves R. Structure and function of the HMGI(Y) family of architectural transcription factors. Environ Health Perspect. 2000;108(Suppl 5):803–809. doi: 10.1289/ehp.00108s5803. [DOI] [PubMed] [Google Scholar]
- 7.Santos JH, Meyer JN, Mandavilli BS, Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol. 2006;314:183–199. doi: 10.1385/1-59259-973-7:183. [DOI] [PubMed] [Google Scholar]
- 8.Lin D, Chinault AC. Comparative study of DNase I sensitivity at the X-linked human HPRT locus. Somat Cell Mol Genet. 1998;14:261–272. doi: 10.1007/BF01534587. [DOI] [PubMed] [Google Scholar]
- 9.Kang SH, Kiefer MC, Yang TP. Role of the promoter in maintaining transcriptionally active chromatin structure and DNA methylation patterns in vivo. Mol Cell Biol. 2003;23:4150–4161. doi: 10.1128/MCB.23.12.4150-4161.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koyama Y, Banzai T, Sonezaki S, Kusano K. Stable expression of a heterologous gene introduced via gene targeting into the HPRT locus of human fibrosarcoma cells. Biotechnol Bioeng. 2006;95:1052–1060. doi: 10.1002/bit.21058. [DOI] [PubMed] [Google Scholar]
- 11.Stamatoyannopoulos G. Control of globin gene expression during development and erythroid differentiation. Exp, Hematol. 2005;33:259–271. doi: 10.1016/j.exphem.2004.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reeves R, Beckerbauer L. HMGI/Y proteins: flexible regulators of transcription and chromatin structure. Biochim Biophys Acta. 2001;1519:13–19. doi: 10.1016/s0167-4781(01)00215-9. [DOI] [PubMed] [Google Scholar]
- 13.Reeves R, Edberg D, Li Y. The architectural transcription factor HMGI(Y) promotes tumor progression and mesenchymal transition of human epithelial cells. Molec Cell Biol. 2001;21:575–594. doi: 10.1128/MCB.21.2.575-594.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reeves R, Nissen MS. Purification and assays for high mobility group HMG-I(Y) protein function. Meths Enzymol. 1999;304:155–188. doi: 10.1016/s0076-6879(99)04011-2. [DOI] [PubMed] [Google Scholar]
- 15.Baxter BK, Smerdon MJ. Nucleosome unfolding during DNA repair in normal and xeroderma pigmentosum (group C) human cells. J Biol Chem. 1998;273:17517–17524. doi: 10.1074/jbc.273.28.17517. [DOI] [PubMed] [Google Scholar]
- 16.Smerdon MJ, Kastan MB, Lieberman MW. Distribution of repair-incorporated nucleotides and nucleosome rearrangement in the chromatin of normal and xeroderma pigmentosum human fibroblasts. Biochem. 1979;18:3732–3739. doi: 10.1021/bi00584a014. [DOI] [PubMed] [Google Scholar]
- 17.Santos JH, Mandavilli BS, Van Houten B. Measuring oxidative mtDNA damage and repair using quantitative PCR. Meths Mol Biol. 2002;197:159–176. doi: 10.1385/1-59259-284-8:159. [DOI] [PubMed] [Google Scholar]
- 18.Wang YC, Lee PJ, Shih CM, Chen HY, Lee CC, Chang YY, Hsu YT, Liang YJ, Wang LY, Han WH, Wang YC. Damage formation and repair efficiency in the p53 gene of cell lines and blood lymphocytes assayed by multiplex long quantitative polymerase chain reaction. Anal Biochem. 2003;319:206–215. doi: 10.1016/s0003-2697(03)00330-0. [DOI] [PubMed] [Google Scholar]
- 19.Van Houten B, Cheng S, Chen Y. Measuring gene-specific nucleotide excision repair in human cells using quantitative amplification of long targets from nanogram quantities of DNA. Mutat Res. 2000;460:81–94. doi: 10.1016/s0921-8777(00)00018-5. [DOI] [PubMed] [Google Scholar]
- 20.Wolffe A. Chromatin: Structure and Function. Academic Press; San Diego, CA: 1998. [Google Scholar]
- 21.Balajee AS, Bohr VA. Genomic heterogeneity of nucleotide excision repair. Gene. 2000;250:15–30. doi: 10.1016/s0378-1119(00)00172-4. [DOI] [PubMed] [Google Scholar]
- 22.Catez F, Yang H, Tracey KJ, Reeves R, Misteli T, Bustin M. Network of dynamic interactions between histone H1 and high-mobility group proteins in chromatin. Molec Cell Biol. 2004;24:4321–4328. doi: 10.1128/MCB.24.10.4321-4328.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harrer H, Luhrs H, Bustin M, Scheer U, Hock R. Dynamic interaction of HMGA1a protein with chromatin. J Cell Sci. 2004;117:3459–3471. doi: 10.1242/jcs.01160. [DOI] [PubMed] [Google Scholar]
- 24.Vreeswijk MPG, van Hoffen A, Westland BE, Vrieling H, van Zeeland AA, Mullenders LH. Analysis of repair of cylcobutane pyrimidine dimmers and pyrimidine-6-4-pyrimidone photoproducts in transcriptionally active and inactive genes in Chinese hamster cells. J Biol Chem. 1994;269:31858–31863. [PubMed] [Google Scholar]
- 25.Tung BS, McGregor WG, Wang YC, Maher VM, McCormick JJ. Comparison of the rate of excision of major ultraviolet photoproducts in the strands of the HPRT gene of normal and xeroderma pigmentosum variant cells. Mutat Res. 1996;362:65–74. doi: 10.1016/0921-8777(95)00034-8. [DOI] [PubMed] [Google Scholar]
- 26.Bohr VA, Smith CA, Okumoto DS, Hanawalt PC. DNA repair in an active gene: removal of pyrimidine dimmers from the DHFR gene of CHO cells is much more efficient than in the gnome overall. Cell. 1985;40:359–369. doi: 10.1016/0092-8674(85)90150-3. [DOI] [PubMed] [Google Scholar]
- 27.Mellon I, Bohr VA, Smith CA, Hanawalt PC. Preferential DNA repair of an active gene in human cells. Proc Natl Acad Sci USA. 1986;83:8878–8882. doi: 10.1073/pnas.83.23.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bohr VA, Okumoto DS, Ho L, Hanawalt PC. Characterization of a DNA repair domain containing the dihydrofolate reductase gene in Chinese hamster ovary cells. J Biol Chem. 1986;261:16666–16672. [PubMed] [Google Scholar]
- 29.McCarthy MJ, Rosenblatt JI, Lloyd RS. A modified quantitative polymerase chain reaction assay for measuring gene-specific repair of UV photoproducts in human cells. Mutat Res. 1996;363:57–66. doi: 10.1016/0921-8777(95)00061-5. [DOI] [PubMed] [Google Scholar]
- 30.Chen RH, Maher VM, Brouwer J, Van de Putte P, McCormick JJ. Preferential repair and strand-specific repair of benzo[a]pyrene dio epoxide adducts in the HPRT gene of diploid human fibroblasts. Proc Natl Acad Sci USA. 1992;89:5413–5417. doi: 10.1073/pnas.89.12.5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei D, Maher VM, McCormick JJ. Site-specific rates of excision repair of benzo[a]pyrene dio epoxide adducts in the hypoxanthine phosphoribosyltransferase gene of human fibroblasts: correlation with mutation spectra. Proc Natl Acad Sci USA. 1995;92:2204–2208. doi: 10.1073/pnas.92.6.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reeves R, Adair JE. Role of high mobility group (HMG) chromatin proteins in DNA repair. DNA Repair (Amst) 2005;4:926–938. doi: 10.1016/j.dnarep.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 33.Kobayashi T, Takeuchi S, Saijo M, Nakatsu Y, Morioka H, Otsuka E, Wakasugi M, Nikaido O, Tanaka K. Mutational analysis of a function of xeroderma pigmentosum group A (XPA) protein in strand-specific DNA repair. Nucl Acids Res. 1998;26:4662–4668. doi: 10.1093/nar/26.20.4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cleaver JE, Charles WC, McDowell ML, Sadinski WJ, Mitchell DL. Overexpression of the XPA repair gene increases resistance to ultraviolet radiation in human cells by selective repair of DNA damage. Cancer Res. 1995;55:6152–60. [PubMed] [Google Scholar]
- 35.Koberle B, Roginskaya V, Wood RD. XPA protein as a limiting factor nucleotide excision repair and UV sensitivity in human cells. DNA Repair (Amst) 2006;5:641–648. doi: 10.1016/j.dnarep.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 36.Bohr VA. DNA Repair fine structure and its relations to genomic instability. Carcinogen. 1995;16:2885–2892. doi: 10.1093/carcin/16.12.2885. [DOI] [PubMed] [Google Scholar]
- 37.Takaha N, Hawkins AL, Griffin CA, Isaacs WB, Coffey DS. High mobility group protein I(Y): a candidate architectural protein for chromosomal rearrangements in prostate cancer cells. Cancer Res. 2002;62:647–651. [PubMed] [Google Scholar]
- 38.Grade M, Hormann P, Becker S, et al. Gene expression profiling reveals a massive, aneuploidy-dependent transcriptional deregulation and distinct differences between lymph node-negative and lymph node-positive colon carcinomas. Cancer Res. 2007;67:41–56. doi: 10.1158/0008-5472.CAN-06-1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boo LM, Lin HH, Chung V, Zhou B, et al. High mobility group A2 potentiates genotoxic stress in part through the modulation of basal and DNA-damage-dependent phosphatidylinositol 3-kinase-related protein kinase activation. Cancer Res. 2005;65:6622–6630. doi: 10.1158/0008-5472.CAN-05-0086. [DOI] [PubMed] [Google Scholar]
- 40.Richards M, Tan S-P, Tan J-H, Chan W-K, Bongso A. The transcriptome profile of human embryonic stem cells defined by SAGE. Stem Cells. 2004;22:51–64. doi: 10.1634/stemcells.22-1-51. [DOI] [PubMed] [Google Scholar]
- 41.Li O, Vasudenvan D, Davey CA, Droge P. High-level expression of DNA architectural factor HMGA2 and its association with nucleosomes in human embryonic stem cells. Genesis. 2006;44:523–529. doi: 10.1002/dvg.20242. [DOI] [PubMed] [Google Scholar]
- 42.Zhou X, Chada K. HMGI family proteins: architectural transcription factors in mammalian development and cancer. Keio J Med. 1998;47:73–77. doi: 10.2302/kjm.47.73. [DOI] [PubMed] [Google Scholar]
- 43.Van Sloun PP, Jansen JG, Weeda G, Mullenders LH, et al. The role of nucleotide excision repair in protecting embryonic stem cells from genotoxic effects of UV-induced DNA damage. Nucl Acids Res. 1999;27:3276–3282. doi: 10.1093/nar/27.16.3276. [DOI] [PMC free article] [PubMed] [Google Scholar]