

Abstract
Cysteine sulfenic acid (Cys-SOH) is an elusive intermediate in reactive oxygen species-induced oxidation reactions of many proteins such as peroxiredoxins and tyrosine phosphatases. Cys-SOH is proposed to play a vital role in catalytic and signaling functions. The formation of cysteine sulfinic acid (Cys-SO2H) and cysteine sulfonic acid (Cys-SO3H) has been implicated in the activation of matrix metalloproteinase-7 (MMP-7) and oxidation of thiol to cysteine sulfinic acid has been associated with the autolytic cleavage of MMP-7. We have examined the formation of cysteine sulfenic acid in a synthetic peptide PRCGVPDVA which is a cysteine switch domain of MMP-7 and other matrix metalloproteases. We have prepared the cysteine sulfenic acid containing peptide, PRC(SOH)GVPDVA, by reaction with hydroxyl radicals generated by the Fenton reaction (Fe+2/H2O2). We characterized this modified peptide by tandem mass spectrometry and accurate mass measurement experiments. In addition, we used 7-chloro-4-nitrobenzo-2-oxa-1,3-diazol (NBD-Cl) reagent to form an adduct with PRC(SOH)GVPDVA to provide additional evidence for the viability of PRC(SOH)GVPDVA in solution. We also characterized an intramolecular cysteine sulfinamide cross-link product PRC[S(O)N]GVPDVA based on tandem mass spectrometry and accurate mass measurement experiments. These results contribute to the understanding of a proteolytic cleavage mechanism that is traditionally associated with MMP activation.
Introduction
Cysteine sulfenic acid (Cys-SOH) formation plays a vital role in regulating the catalysis, function and redox signaling of several proteins such as peroxiredoxins, phosphatases, transcription factors, and members of the NAD(P)H/disulfide reductase family [1–5]. Cysteine sulfenic acids in proteins are also widespread posttranslational modifications that can result from oxidative stress conditions [6]. Cysteine residues that possess a low pKa in proteins can be selectively oxidized by reactive oxygen species (H2O2, alkyl hydroperoxides, peroxy nitrite, hypochlorous acid) to cysteine sulfenic acid via a cysteine thiolate anion (Cys-S−) intermediate [7–10]. Since cysteine sulfenic acid is a highly reactive intermediate, it further reacts with any accessible thiol to form a disulfide or rapidly undergoes further oxidation with oxidants such as H2O2 to give cysteine sulfinic acid (Cys-SO2H) and cysteine sulfonic acid (Cys-SO3H) [2].
Cysteine sulfenic acid has been identified by x-ray crystallography and 13C-NMR techniques in the active site of the NADH peroxidase protein from Enterococcus faecalis.[11–12]. However, cysteine sulfenic acid was not observed in solution in the case of PTP1-B [3] although it was characterized in the crystal form of this protein [13]. Because of the high reactivity of cysteine sulfenic acid due to unfavorable solvent interactions and local environment of most proteins [2], its definitive identification became possible only after trapping with chemical reagents such as dimedone [14], a fluorophore-containing analog of dimedone [15], NBD-Cl [16], and recent labeling experiments with biotin-maleimide after arsenite specific reduction [6] followed by mass spectrometry. Fuangthong and Helmann [17] observed a mass increase consistent with cysteine sulfenic acid in intact OhrR repressor protein by ESI-MS using NBD-Cl trapping experiments. However, to the best of our knowledge, there is no report in the literature describing the characterization of underivatized cysteine sulfenic acid at the peptide level by tandem mass spectrometry experiments, which are very important for ruling out possible oxidative modifications of other amino acids such as methionine and phenylalanine.
Macrophage-derived matrix metalloproteinases (MMPs) are a family of zinc and calcium dependent enzymes implicated in regulating matrix degradation in inflammation, arthritis, cancer, cardiovascular and neurodegenerative diseases [18–23]. A highly conserved domain called the cysteine switch (amino acid sequence PRCXXPD) is proposed to regulate the activity of all MMPs except for MMP-23 [19,22,23]. Likewise, MMP-7 also contains another cysteine switch sequence, PRCGVPDVA, in its pro-domain. Fu et al. demonstrated that pro-MMP-7 can be activated by HOCl oxidation, but not H2O2 oxidation, converting cysteine to cysteine sulfinic acid and cysteine sulfonic acid [24]. Thus oxygenation of thiol residue in the cysteine switch peptide PRCGVPDVA was found to be a key event in the autolytic cleavage of pro-MMP-7. However, these authors did not investigate the formation of a cysteine sulfenic acid intermediate that could be involved in the activation mechanism of MMP-7. This prompted us to investigate cysteine sulfenic acid formation in the cysteine switch sequence PRCGVPDVA of MMPs.
In this study, we have used Fenton reagents (Fe+2/H2O2) to produce highly reactive hydroxyl radicals (OH.) that can react with sulfhydryl group in cysteine to give a variety of oxidation products. The formation of hydroxyl radicals in the Fenton reaction has been shown to be potentially more damaging to cells than other reactive oxidants [25]. In fact, recently we have prepared cysteine sulfinic acid and cysteine sulfonic acid in PRCGVPDVA by hydroxyl radicals generated in the Fenton reaction and studied their fragmentation mechanism by ion trap tandem mass spectrometry [26]. Here we report the preparation and characterization by HPLC, tandem mass spectrometry and accurate mass measurement of a stable cysteine sulfenic acid in a synthetic peptide, PRC(SOH)GVPDVA that mimics the cysteine switch sequence of MMPs.
Experimental
The peptide PRCGVPDVA was a gift by Prof. Yinsheng Wang, University of California at Riverside, Riverside, CA. NBD-Cl was purchased from Aldrich. PRCGVPDVA was oxidized using Fenton reagents (Fe+2/H2O2) as reported previously [26]. Briefly, 1mM peptide solution was incubated at 37°C in the presence of 0.5 mM FeSO4 and 0.2 mM H2O2 for approximately 30 min. The reaction was terminated by adding aliquots of methionine solution until its concentration reached 1 mM. Based on the number of counts in the extracted ion chromatograms (Figure 1), cysteine sulfonic acid was found to be the major product followed by cysteine sulfinic acid, cysteine sulfenic acid and cysteine sulfinamide (-C[S(O)N]-) products. Adducts of PRCGVPDVA and cysteine sulfenic acid with NBD-Cl were synthesized according to Ellis et al. [16] except that we carried out the reaction under aerobic conditions. We did not rigorously optimize the reaction conditions to increase the yield of cysteine sulfenic acid and cysteine sulfinamide products or the adducts of PRC(S-NBD)GVPDVA and PRC[S(O)-NBD]GVPDVA.
Figure 1.

Extracted Ion Chromatograms of major cysteine oxidation products of the cysteine switch domain peptide PRCGVPDVA from MMP-7
Data-dependent nano LC-MS/MS experiments were carried out using a Waters CapLC (Milford, MA) coupled directly to a Waters Q-TOF Micro mass spectrometer (Milford, MA) through a Picotip needle (New Objective) and a home-made pre-column flow splitter. The analytical column was a 75 μm I.D. X 15 cm PepMap C18 (LC Packings). Mobile phase A was 2% acetonitrile/0.1% formic acid, and mobile phase B was 90% acetonitrile/0.1% formic acid in water. The gradient was 5%–90% B over 45 min with a flow rate of 400 nL/min. Automatic switching between MS and MS/MS modes was controlled by MassLynx 4.0 (Micromass) software, dependent on both signal intensity and charge states from MS to MS/MS and on either signal intensity or time from MS/MS to MS. In the case of direct infusion MS/MS experiments, 50% acetonitrile in 0.1% formic acid was used as the carrier and electrospray solvent at a flow rate of 500 nL/min. A 1 μL aliquot of 2 μM sample solution was injected via a divert valve in each run under following source conditions; spray voltage 1500 V to 1800 V, cone voltage 40 V, source temperature 80°C. Argon was used as a collision gas for all MS/MS experiments. The quadrupole mass filter before the TOF analyzer was set with LM and HM resolution of 14.0 (arbitrary units), which is equivalent to a 1.0-Da mass window for transmission of precursor ions. Each spectrum was obtained by averaging approximately 25 scans, and the scan time was 1 sec/scan. The Q-TOF Micro instrument was calibrated with [Glu1]-fibrinopeptide B and the data in both MS and MS/MS modes were acquired at 5000 resolution. In the case of accurate mass measurement experiments, the precursor ion was used as a lock mass to calibrate the masses of the fragment ions. To assess stability of the oxidized peptide forms, 5 μL of a single sample stored at 4° C was injected every two hours after quenching the Fenton reaction onto a Symmetry 5Im particle, 180 μm × 20 mm C18 precolumn (Waters, Milford, MA), then washed 5 min with 1% acetonitrile in 0.1% formic acid at a flow rate of 20 μL/min. After washing, peptides were eluted and passed through an Atlantis 3 μm particle, 75 μm × 100 mm C18 analytical column (Waters, Milford, MA) with a gradient of 1–80% Acetonitrile in 0.1% formic acid. The gradient was delivered over 60 min by a nanoACQUITY UPLC (Waters, Milford, MA) at a flow rate of 250 nL/min to a fused silica distal end-coated tip nano-electrospray needle (New Objective, Woburn, MA). MS survey scans were collected on a Q-TOF Premiere (Waters/Micromass, Milford, MA). Extracted ion chromatograms were generated for the m/z of interest (927.4, 929.4, 945.4, 961.4) and an equal number of scans were summed across the signal peak for each corresponding time point. Normalized intensities were calculated by dividing reaction products ion counts by those of an internal standard ([Glu1]-fibrinopeptide B, +2 m/z 785.8). Values for two replicates experiments were averaged and standard deviations calculated.
Results and Discussion
Our goal was to separate and identify by nanoLC-MS/MS all the cysteine oxidation products formed in the hydroxyl radical reaction. The oxidation reaction mixture including the PRCGVPDVA peptide was analyzed in a data-dependent nanoLC-MS/MS experiment with the emphasis on the identification of the cysteine sulfenic acid product. Figure 1 shows the extracted ion chromatograms (XIC) of possible products that resulted from the Fenton oxidation reaction and their masses are shown in their respective mass spectra (Figure 2). The products at m/z 927.4, m/z 929.4, m/z 945.4, m/z 961.4 in Figure 2 differed by 14 Da, 16 Da, 32 Da and 48 Da, respectively, from the unmodified precursor peptide PRCGVPDVA (m/z 913.5). The latter two products, m/z 945.5 (+36 Da) and m/z 961.4 (+48 Da) were characterized as cysteine sulfinic acid and cysteine sulfonic acid, respectively, as reported previously by us [26]. However, we could not obtain sequence information for the products of m/z 929.5(+1)/m/z 465.2 (+2) or m/z 927.5(+1)/m/z 464.2 (+2) in our on-line LC-MS/MS experiments. Therefore, we carried out direct infusion MS/MS experiments for the singly charged ions of all modified peptides including the ion of m/z 929.5, for which a precursor selection window of only 1 Da was used to avoid fragmenting impurities from adjacent peaks.
Figure 2.

Partial mass spectra of major cysteine oxidation products of the PRCGVPDVA peptide
Characterization of cysteine sulfenic acid [PRC(SOH)GVPDVA]
The MS/MS spectra of unmodified PRCGVPDVA (m/z 913.5) and the modified product at m/z 929.5 are showed in Figure 3. In Figure 3b, the formation of the y4 (m/z 401) and b2 (m/z 254) ions rules out the possibility of modification of either of the two prolines or the arginine in PRCGVPDVA. However, the formation of the ion at m/z 741 (b7 +16 Da) indicates that cysteine is most likely modified. In addition to this, the formation of the diagnostic ion at m/z 879 (-H2SO) and the loss of neutral H2SO from b3, b4, and b7 ions confirm that the cysteine is modified with an oxygen atom to cysteine sulfenic acid, PRC(SOH)GVPDVA. This m/z 879 ion and an ion at m/z 691, both due to loss of H2S from the unmodified peptide, are also formed in the MS/MS spectrum of PRCGVPDVA (Figure 3a). Furthermore, we observed that the MS/MS spectra of higher oxidation products, cysteine sulfinic acid and cysteine sulfonic acid also exhibit similar neutral losses from precursor ions as well as fragment ions (data not shown). In fact, the facile loss of neutral H2SO3 from both parent and fragment ions are well documented in the literature [26, 27–29]. The structural assignments for some of the key fragment ions formed in the MS/MS spectra of m/z 929.4 and m/z 913.4 are further supported by the results of high resolution MS/MS experiments (Tables 1, 2 and 4). We have observed that the formation of strong b3 and y6 ions due to preferential cleavage c-terminal to the oxidized cysteine occurs only in the cysteine sulfinic acid but not other oxidized forms. This is due to the formation of stable cyclic CID fragmentation products from the cysteine sulfinic acid form in an energetically favorable cleavage pathway similar to those formed c-terminal to aspartic acid residues (Figure 3 and reference 26). In cases where observed m/z values differ by more than 5 ppm from predicted values, this was due to low intensity ion signals for the CID fragment ions derived from the low abundance oxidation products.
Figure 3.

MS/MS spectra of the precursor peptides shown in Figure 2: a) unmodified cysteine switch peptide PRCGVPDVA (m/z 913.4) and b) cysteine sulfenic acid product PRC(SOH)GVPDVA (m/z 929.4)
Table 1.
High resolution data for the (M+H)+ ions of PRCGVPDVA, PRC[S(O)N]GVPDVA and PRC(SOH)GVPDVA
| Parent Ion (M+H)+ | Measd Mass | Calc Mass | ΔPPM | Formula |
|---|---|---|---|---|
| PRCGVPDVA | 913.4565 | 913.4566 | −0.1 | C38 H65 N12 O12 S |
| PRC[S(O)N]GVPDVA | 927.4390 | 927.4358 | 3.5 | C38 H63 N12 O13 S |
| PRC(SOH)GVPDVA | 929.4442 | 929.4515 | −7.8 | C38 H65 N12 O13 S |
Table 2.
High resolution data for fragment ions in the MS/MS spectrum of (M+H)+ ions of PRCGVPDVA
| Ion | Measd Mass | Calc Mass | ΔPPM | Formula |
|---|---|---|---|---|
| (M+H)+ | 913.4565 | 913.4565 | 0.0 | C38 H65 N12 O12 S |
| -H2S | 879.4518 | 879.4688 | −19.4 | C38 H63 N12 O12 |
| b7 | 725.3406 | 725.3405 | 0.2 | C30 H49 N10 O9 S |
| b5-NH3 | 496.2332 | 496.2342 | −2.0 | C21 H34 N7 O5 S |
| a5 | 485.2651 | 485.2658 | −1.5 | C20 H37 N8 O4 S |
| a5-NH3 | 468.2420 | 468.2393 | 5.8 | C20 H34 N7 O4 S |
| b3-NH3 | 340.1415 | 340.1443 | −8.3 | C14 H22 N5 O3 S |
| a3-NH3 | 312.1568 | 312.1494 | 23.6 | C13 H22 N5 O2 S |
Table 4.
High resolution data for fragment ions in the MS/MS spectrum of (M+H)+ ions of PRC(SOH)GVPDVA
| Ion | Measd Mass | Calc Mass | ΔPPM | Formula |
|---|---|---|---|---|
| (M+H)+ | 929.4515 | 929.4515 | 0.0 | C38 H65 N12 O13 S |
| -H2O | 911.4355 | 911.4409 | −5.9 | C38 H63 N12 O12 S |
| -H2SO | 879.4837 | 879.4688 | 16.9 | C38 H63 N12 O12 |
| b7 | 741.3430 | 741.3354 | 10.3 | C30 H49 N10 O10 S |
| b7-H2SO | 691.3450 | 691.3527 | −11.2 | C30 H47 N10 O9 |
| y4 | 401.2066 | 401.2036 | 7.4 | C17 H29 N4 O7 |
| b4-H2SO | 380.2084 | 380.2046 | 9.9 | C16 H26 N7 O4 |
In addition to the above experimental evidence demonstrating that a stable cysteine sulfenic acid [PRC(SOH)GVPDVA] was produced in solution, we further supported this finding by forming the adduct of cysteine sulfenic acid with NBD-Cl [16]. The MS/MS spectra of NBD adducts of PRCGVPDVA and PRC(SOH)GVPDVA are shown in Figure 4. The spectrum in Figure 4b clearly showed a mass shift for the b7 (m/z 904) ion due to the cysteine sulfenic acid adduct with NBD. In addition, a diagnostic ion at m/z 879 [-NBD-S(O)H] indicated the formation of a PRC[S(O)-NBD]GVPDVA adduct and confirmed the viability of PRC(SOH)GVPDVA in solution. Thus both PRC(SOH)GVPDVA and PRC[S(O)-NBD]GVPDVA underwent very similar fragmentation pathways under these experimental conditions.
Figure 4.

MS/MS spectra of a) PRC(S-NBD)GVPDVA (m/z 1076.5) and b) PRC[S(O)-NBD]VPDVA (m/z 1092.5)
The formation of stable cysteine sulfenic acid in PRC(SOH)GVPDVA is also in agreement with the reported reactivity order of all 20 amino acids towards hydroxyl radicals generated in radiolysis experiments [30] where cysteine was shown to be the most highly reactive amino acid residue. According to rules for the stabilization of cysteine sulfenic acid in solution[2], hydrogen bonding is one of the main factors that can influence the stability of cysteine sulfenic acid in proteins in addition to the absence of proximal Cys-SH and limited nonpolar solvent access. We propose a hydrogen bonding effect in the formation of stable cysteine sulfenic acid in PRC(SOH)GVPDVA due to the formation of a strong intramolecular hydrogen bond between the cysteine-SOH group and the guanidine group of arginine (Scheme 1).
Scheme 1.
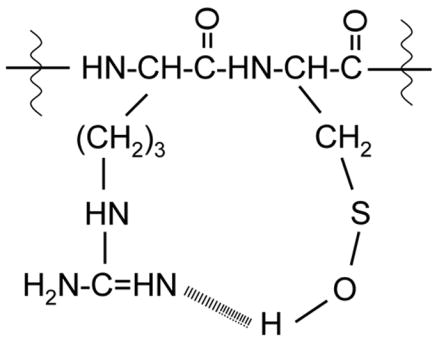
We performed a time course experiment to demonstrate the stability of the cysteine sulfenic acid peptide after quenching the Fenton reaction. Within experimental error, we found no detectable loss of the sulfenic acid form after 25 hours (Figure 5).
Figure 5.

The stability of reaction products at m/z 927.4 (█), 929.4 (□), 945.4 (
 ), and 961.4 (
), and 961.4 (
 ) was monitored over a period of 25 hours. Samples were analyzed via LC-MS approximately every two hours after quenching the Fenton reaction. The relative ion count for each product was calculated according to accurate mass and elution time, normalized to an internal control ([Glu1]-fibrinopeptide B doubly charged peptide, m/z 785.8), and values from two experiments were averaged. The thirty minute time point (*) was acquired once. The inset shows an expanded scale of the key products cysteine sulfinamide cross-link (927.4), and cysteine sulfenic acid (929.4) peptides.
) was monitored over a period of 25 hours. Samples were analyzed via LC-MS approximately every two hours after quenching the Fenton reaction. The relative ion count for each product was calculated according to accurate mass and elution time, normalized to an internal control ([Glu1]-fibrinopeptide B doubly charged peptide, m/z 785.8), and values from two experiments were averaged. The thirty minute time point (*) was acquired once. The inset shows an expanded scale of the key products cysteine sulfinamide cross-link (927.4), and cysteine sulfenic acid (929.4) peptides.
Characterization of cysteine sulfinamide cross-link (PRC[S(O)N]GVPDVA)
To characterize the structure of the modified product at m/z 927.5 (Figure 1d), its MS/MS spectrum was acquired as shown in Figure 6. The formation of the ion at m/z 401.5 (y4) eliminates the possibility of modification at the C-terminal P,D,V,A residues. The formation of ions at m/z 739.4 (b7+14 Da) and m/z 482.2 (a5-NH3, +14 Da) suggested that the modification could be on the C, G or V residues. However, the presence of low abundance diagnostic ions at m/z 879.5 (-SO) and m/z 865.5 (-CH2SO), suggested that cysteine is modified by gain of one oxygen and loss of two hydrogens. Thus, in addition to the cysteine, R or G may also be involved in the modification to account for the 14 Da increment from the unmodified PRCGVPDVA peptide. These observations strongly suggest that the cysteine residue of PRCGVPDVA is oxidized to form an intramolecular cysteine sulfinamide cross-link (PRC[S(O)N]GVPDVA) and that the cross-link involves the guanidine group of the arginine residue as reported previously in PFRCG and PFKCG peptides [31]. The high resolution data obtained for some of these informative fragment ions (Table 3) further supported the formation of a cysteine sulfinamide cross-link product. The ion at m/z 254 may be formed by a rearrangement reaction. This type of rearrangement ion (m/z 373, b3) was observed in the MS/MS spectra of cysteine sulfonamide, cysteine sulfinamide and cysteine sulfenamide products that were formed in the oxidation of PFKCG by HOCl [31].
Figure 6.

MS/MS spectrum of PRC[S(O)NH]GVPDVA (m/z 927.4)
Table 3.
High resolution data for fragment ions in the MS/MS spectrum of (M+H)+ ions of PRC[S(O)N]GVPDVA
| Ion | Measd Mass | Calc Mass | ΔPPM | Formula |
|---|---|---|---|---|
| (M+H)+ | 927.4358 | 927.4358 | 0.0 | C38 H63 N12 O13 S |
| -SO | 879.4617 | 879.4688 | −8.1 | C38 H63 N12 O12 |
| -CH2SO | 865.4400 | 865.4532 | −15.2 | C37 H61 N12 O12 |
| b7 | 739.3200 | 739.3197 | 0.4 | C30 H47 N10 O10 S |
| a5-NH3 | 482.2100 | 482.2186 | −17.8 | C20 H32 N7 O5 S |
| y4 | 401.2066 | 401.2036 | −9.0 | C17 H29 N4 O7 |
| b3-SO | 323.1865 | 323.1832 | 10.3 | C14 H23 N6 O3 |
Possible role for cysteine sulfenic acid formation in MMP activation
The activation mechanism of MMPs is believed to involve the disruption of a bond between the thiol residue of the pro-domain and a catalytic Zn atom [22]. For example, in MMP-7 the oxygenation of thiol by HOCl disrupts the thiol-Zinc bond and leads to conformational changes that allow autolytic cleavage and access to the enzyme active site. Cysteine sulfenic acid is a central reactive intermediate in many cellular oxidative reaction pathways including the pathway in which cysteine sulfinic acid and cysteine sulfonic acid can be produced [1–5]. The cysteine sulfinamide characterized in this study and as identified previously in the synthetic peptides PFKCG and PFRCG [31] and S100A8 protein [34], seems to be a common product in the oxidation reactions of cysteine containing peptides and proteins. Interestingly, in the case of S100A8 protein [34], a cysteine sulfenic acid intermediate was proposed to be involved in the mechanism of cysteine sulfinamide formation. Since we also identified both cysteine sulfenic acid and cysteine sulfinamide cross link products in the cysteine switch peptide PRCGVPDVA, we believe that PRC(SOH)GVPDVA is a key intermediate in the oxidation reactions of MMP-7 that could further react with reactive oxygen species to form cysteine sulfinic acid and cysteine sulfonic acid [35] or gives rise to novel oxidation products like cysteine sulfinamide cross-link with the loss of a hydrogen molecule [34].
Conclusions
We used an oxidation reaction by hydroxyl radicals generated in the Fenton reaction to generate for the first time a stable cysteine sulfenic acid, [PRC(SOH)GVPDVA], in a synthetic cysteine switch peptide of the matrix metalloproteinase MMP-7. We used tandem mass spectrometry to characterize the structure of the cysteine sulfenic acid as well as an intramolecular cysteine sulfinamide cross-link product, PRC[S(O)N]GVPDVA, in solution. Our results suggest that cysteine sulfenic acid may be involved in regulating the activation of MMPs that contain the cysteine switch sequence.
Acknowledgments
We gratefully acknowledge support from NIH grants S10 RR017990 and 2P30CA016087.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 2.Philip E. Protein thiol oxidation in health and disease: Techniques for measuring disulfides and related modifications in complex protein mixtures. Free Radical Biology & Medicine. 2006;40:1889–1899. doi: 10.1016/j.freeradbiomed.2005.12.037. [DOI] [PubMed] [Google Scholar]
- 3.Salmeen A, Andersen JN, Myers MP, Meng TC, Hinks JA, Tonks NK, Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature. 2003;423:769–773. doi: 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- 4.Claiborne A, Mallett TC, Yeh JI, Luba J, Parsonage D. Structural, Redox, and Mechanistic Parameters for Cysteine-Sulfenic Acid Function in Catalysis and Regulation. Adv Protein Chem. 2001;58:215–276. doi: 10.1016/s0065-3233(01)58006-7. [DOI] [PubMed] [Google Scholar]
- 5.Paget MSB, Buttner MJ. Thiol-based regulatory switches. Annu Rev Genet. 2003;37:91–121. doi: 10.1146/annurev.genet.37.110801.142538. [DOI] [PubMed] [Google Scholar]
- 6.Saurin AT, Neubert H, Brennan JP, Eaton P. Widespread sulfenic acid formation in tissues in response to hydrogen peroxide. Proc Natl Acad Sci USA. 2004;101:17982–17987. doi: 10.1073/pnas.0404762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bryk R, Griffin P, Nathan C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature. 2000;407:211–215. doi: 10.1038/35025109. [DOI] [PubMed] [Google Scholar]
- 8.Raftery MJ, Yang Z, Valenzuela SM, Geczy CL. Novel intra and intermolecular sulfinamide bonds in S100A8 produced by hypochlorite oxidation. J Biol Chem. 2001;276:33393–401. doi: 10.1074/jbc.M101566200. [DOI] [PubMed] [Google Scholar]
- 9.Kim JR, Yoon HW, Kwon KS, Lee SR, Rhee SG. Identification of Proteins Containing Cysteine Residues that are Sensitive to Oxidation by Hydrogen Peroxide at Neutral pH. Anal Biochem. 2000;283:214–221. doi: 10.1006/abio.2000.4623. [DOI] [PubMed] [Google Scholar]
- 10.Allison WS. Formation and reactions of sulfenic acids in proteins. Acc Chem Res. 1976;9:293–299. [Google Scholar]
- 11.Yeh JI, Claiborne A, Hol WGJ. Structure of the native cysteine-sulfenic acid redox center of enterococcal NADH peroxidase refined at 2.8 A resolution. Biochemistry. 1996;35:9951–9957. doi: 10.1021/bi961037s. [DOI] [PubMed] [Google Scholar]
- 12.Crane EJ, III, Vervoort J, Claiborne A. 13C NMR analysis of the cysteine-sulfenic acid redox center of enterococcal NADH peroxidase. Biochemistry. 1997;36:8611–8618. doi: 10.1021/bi9707990. [DOI] [PubMed] [Google Scholar]
- 13.van Montfort RLM, Congreve M, Tisi D, Carr R, Jhoti H. Oxidation state of the active-site cysteine in protein tyrosine phosphatase 1B. Nature. 2003;423:773–777. doi: 10.1038/nature01681. [DOI] [PubMed] [Google Scholar]
- 14.Willett WS, Copley SD. Identification and localization of a stable sulfenic acid in peroxide-treated tetrachlorohydroquinone dehalogenase using electrospray mass spectrometry. Chem Biol. 1996;3:851–857. doi: 10.1016/s1074-5521(96)90071-x. [DOI] [PubMed] [Google Scholar]
- 15.Poole LB, Zeng B, Knaggs SA, Yakubu M, king SB. Synthesis of Chemical Probes to Map Sulfenic Acid Modifications on Proteins. Bioconjugate Chem. 2005;16:1624–1628. doi: 10.1021/bc050257s. [DOI] [PubMed] [Google Scholar]
- 16.Ellis HR, Poole LB. Novel application of 7-chloro-4-nitrobenzo-2-oxa-1.3-diazole to identify cysteine sulfenic acid in the AhpC component of alkyl hydroperoxide reductase. Biochemistry. 1997;36:15013–15018. doi: 10.1021/bi972191x. [DOI] [PubMed] [Google Scholar]
- 17.Fuangthong M, Helmann JD. The OhrR repressor senses organic hydroperoxides by reversible formation of a cysteine-sulfenic acid derivative. Proc Natl Acad Sci USA. 2002;99:6690–6695. doi: 10.1073/pnas.102483199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sillanpaa SM, Anttila MA, Voutilainen KA, Ropponen KM, Sironen RK, Saarikoski SV, Kosma VM. Prognostic significance of matrix metalloproteinase-7 in epithelial ovarian cancer its relation to beta-catenin expression. Int J Cancer. 2006;119:1792–1799. doi: 10.1002/ijc.22067. [DOI] [PubMed] [Google Scholar]
- 19.Woessner JF, Nagase H. Matrix Metalloproteinases and TIMPs. Oxford University Press; New York, NY: 2000. [Google Scholar]
- 20.Marchenko GN, Ratnikov BI, Rozanov DV, Godzik A, Deryugina EI, Strongin AY. Characterization of matrix metalloproteinase-26, a novel metalloproteinase widely expressed in cancer cells of epithelial origin. Biochem J. 2001;356:705–718. doi: 10.1042/0264-6021:3560705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wielockx B, Libert C, Wilson C. Matrilysin (matrix metalloproteinase- 7): a new promising drug target in cancer inXammation. Cytokine Growth Factor Rev. 2004;15:111–115. doi: 10.1016/j.cytogfr.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 22.Van Wart HE, Birkedal-Hansen H. The cysteine switch: a principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc Natl Acad Sci USA. 1990;87:5578–5582. doi: 10.1073/pnas.87.14.5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siwik DA, Pagano PJ, Colucci WS. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Physiol Cell Physiol. 2001;280:C53–C60. doi: 10.1152/ajpcell.2001.280.1.C53. [DOI] [PubMed] [Google Scholar]
- 24.Fu X, Kassim SY, Parks WC, Heinecke JW. Hypochlorous Acid Oxygenates the Cysteine Switch Domain of Pro-matrilysin (MMP-7): A mechanism for matrix metalloproteinase activation and atherosclerotic plaque rupture by myeloperoxidase. 2001;276:41279–41287. doi: 10.1074/jbc.M106958200. [DOI] [PubMed] [Google Scholar]
- 25.Dalle-Donne I, Scaloni A, Giustarini D, Cavarra E, Tell G, Lungarella G, Colombo R, Rossi R, Milzani A. Proteins as biomarkers of oxidative/nitrosative stress in diseases: The contribution of redox proteomics. Mass Spectrometry Reviews. 2005;24:55–99. doi: 10.1002/mas.20006. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Vivekananda S, Men L, Zhang Q. Fragmentation of Protonated Ions of Peptides Containing Cysteine Cysteine Sulfinic Acid Cysteine, Sulfonic Acid. J Am Soc Mass Spectrom. 2004;15:697–702. doi: 10.1016/j.jasms.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 27.Men L, Wang Y. Further studies on the fragmentation of protonated ions of peptides containing aspartic acid glutamic acid cysteine sulfinic acid and cysteine sulfonic acid. Rapid Commun Mass Spectrom. 2005;19:23–30. doi: 10.1002/rcm.1748. [DOI] [PubMed] [Google Scholar]
- 28.Tsaprailis G, Nair H, Somogyi A, Wysocki VH, Zhong W, Futrell JH, Summerfield SG, Gaskell SJ. Influence of Secondary Structure on the Fragmentation of Protonated Peptides. J Am Chem Soc. 1999;121:5142–5154. [Google Scholar]
- 29.Summerfield SG, Cox KA, Gaskell SJ. The Promotion of d-Type Ions During the Low Energy Collision-Induced Dissociation of Some Cysteic Acid-Containing Peptides. J Am Soc Mass Spectrom. 1997;8:25–31. [Google Scholar]
- 30.Xu G, Chance MR. Radiolytic Modification and Reactivity of Amino Acid Residues Serving as Structural Probes for Protein Footprinting. Anal Chem. 2005;7:4549–4555. doi: 10.1021/ac050299+. [DOI] [PubMed] [Google Scholar]
- 31.Fu X, Mueller DM, Heinecke JW. Generation of Intramolecular and Intermolecular Sulfenamides, Sulfinamides, and Sulfonamides by Hypochlorous Acid: A Potential Pathway for Oxidative Cross-Linking of Low-Density Lipoprotein by Myeloperoxidase. Biochemistry. 2002;41:1293–1301. doi: 10.1021/bi015777z. [DOI] [PubMed] [Google Scholar]
- 32.Park AJ, Matrisian LM, Kells AF, Pearson R, Yuan ZY, Navre M. Mutational analysis of the transin (rat stromelysin) autoinhibitor region demonstrates a role for residues surrounding the “cysteine switch”. J Biol Chem. 1991;266:1584–90. [PubMed] [Google Scholar]
- 33.Gu Z, Kaul M, Yan Boxu, Kridel SJ, Cui Jiankun, Strongin Alex, Smith Jeffrey W, Liddington RC, Lipton SA. S-Nitrosylation of Matrix Metalloproteinases: Signaling Pathway to Neuronal cell Death. Science. 2002;297:1186–1190. doi: 10.1126/science.1073634. [DOI] [PubMed] [Google Scholar]
- 34.Raftery MJ, Zheng Yang, Valenzuela SM, Geczy CL. Novel Intra-Intermolecular, Sulfinamide Bonds in S100A8 Produced by Hypochlorite Oxidation. J Biol Chem. 2001;276:33393–33401. doi: 10.1074/jbc.M101566200. [DOI] [PubMed] [Google Scholar]
- 35.Hamann M, Zhang T, Hendrich S, Thomas JA. Quantitation of Protein Sulfinic and Sulfonic Acid, Irreversibly Oxidized Protein Cysteine Sites in Cellular Proteins. Methods Enzymol. 2002;348:146–156. doi: 10.1016/s0076-6879(02)48634-x. [DOI] [PubMed] [Google Scholar]