

Abstract
Histone deacetylase inhibitors (HDACi) induce growth arrest and differentiation, particularly in the colon where they are potential chemotherapeutic agents. A key mediator of HDACi action is the cyclin dependent kinase (CDK) inhibitor p21waf1. HDACi treatment of colonic cells promotes the formation of an ATM/ZBP-89/p300 complex on p21waf1 proximal promoter, and this multi-molecular complex plays an important role in HDACi induction of p21waf1 expression in vitro and mucosal protection in vivo. Here we found that ZBP-89 is phosphorylated by ATM kinase in vitro and in vivo. Disruption of the ATM phosphorylation motif 202SQ within the zinc finger domain of ZBP-89 attenuated its ability to enhance p21waf1 activation by butyrate. Moreover, disruption of the ATM phosphorylation site abrogated the ability of ZBP-89 to potentiate butyrate induction of endogenous p21waf1 expression. These results demonstrate that ATM phosphorylation of ZBP-89 contributes to HDACi induction of p21waf1 gene expression.
Keywords: histone deacetylase inhibitor, trichostatin A, acetylation, phosphorylation, ZNF148
Introduction
Inhibition of histone deacetylases (HDACs) by trichostatin A (TSA) has been shown by Kastan and coworkers to activate ataxia-telangiectasia mutated (ATM) kinase demonstrating that hyperacetylation and subsequently chromatin remodeling alone is sufficient to activate ATM in the absence of DNA damage [1]. Butyrate is a short chain fatty acid produced in millimolar concentrations by bacterial fermentation of fiber in the colon and is also a potent histone deacetylase inhibitor (HDACi) [2-7]. Numerous studies have shown that butyrate induces cell growth arrest and stimulates apoptosis and differentiation in a variety of cancer cells in vitro [3]. In animal models, butyrate inhibits the growth of carcinogen-induced colonic tumors [8-10].
Chromatin becomes more accessible to transcription factors with HDAC inhibition resulting in the activation of ∼2% of all genes and subsequently cell growth arrest [11]. The GC-rich elements within these genes are typically the DNA sequences responsive to chromatin remodeling [7, 12, 13]. p21waf1 is a CDK inhibitor that plays a critical role in cell growth arrest [14, 15]. Morever, the p21waf1 promoter is GC-rich. Previous studies have shown that the GC-rich DNA elements of the proximal promoter are critical for HDACi-induced expression of p21waf1 [16-18] and this induction is enhanced by the transcription factor ZBP-89 (ZNF148, Zfp148, BFCOL1) [18]. ZBP-89 is a DNA binding protein that regulates a variety of genes through direct binding to GC-rich elements that are also bound by Sp1/Sp3 [19]. ZBP-89 is required for p53 phosphorylation at Ser15, the same site targeted by the ATM kinase [20]. Our most recent studies have shown that HDACi promotes the formation of an ATM/ZBP-89/p300 multimolecular complex on the p21waf1 proximal promoter and that this complex plays an important role in butyrate induction of p21waf1 expression in vitro and in mucosal protection in vivo [21]. Since HDACi activation of p21waf1 gene expression requires ATM [22] and ATM has no identified DNA binding domain, we hypothesized that ATM contributes to HDACi induction of p21waf1 expression by modulating ZBP-89 transcriptional activity. Here we show that ATM phosphorylates ZBP-89 in vitro and in vivo. Mutation of the ATM phosphorylation motif in ZBP-89 attenuated butyrate induction of p21waf1 expression.
Materials and Methods
Reagents
Generation of rabbit anti-ZBP-89 has been described previously [23]. Polyclonal antibodies against Ac-H2A and monoclonal antibodies against phospho-p53Ser15 were obtained from Cell Signaling (Beverly, MA). Monoclonal antibodies against p21waf1, and rabbit polyclonal antibodies against phospho-p53 Ser15 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal Flag M2 antibody, sodium butyrate and trichostatin A were purchased from Sigma (St Louis, MO). Lambda protein phosphatase was purchased from New England Biolabs (Beverly, MA).
Cell Culture
HCT116 cells (ATCC) were cultured in McCoy's 5A medium with 10% FBS. GM00637I and GM05849D, human fibroblast cell lines, were purchased from Coriell Cell Repository (Camden, NJ) and cultured in modified Eagle's medium containing 10% fetal bovine serum. To generate stable cell lines that express full-length or mutant forms of ZBP-89, HCT116 cells were transfected with the pcDNA3 vector or different forms of ZBP-89 expressing vectors and selected with G418 at 1 mg/ml for one to two weeks. The pools of G418-resistent cells that expressed the different forms of ZBP-89 were used for further analyses.
Plasmids
Expression vectors for ZBP-89/FL, ZBP-89/ΔZnF and ZBP-89/ΔNter, and expression vectors for GST-ZBP-89(1-300), GST-ZBP-89(150-300) and GST-ZBP-89(300-794) were described before [24]. The pcDNA3-ATM (WT) and pcDNA3-ATM (KD) plasmids and p21waf1 0-Luc plasmid, which contains 2.4 kb of the human p21waf1 promoter, were kindly provided by Dr. Michael Kastan (St Jude Children's Research Hospital) and Dr. Bert Vogelstein (John Hopkins University), respectively.
Site-directed mutagenesis of ZBP-89
Site-directed mutagenesis kit (Stratagene) was used to introduce Ser202→Gly202 mutation in ZBP-89 using pcDNA3-Flag-ZBP-89 as a template. The following primers were used:
forward: 5′-GCGAAAAACCGTTTCAATGTGGTCAGTGTGACATGCGTTTC-3′
backward: 5′-GAAACGCATGTCACACTGACCACATTGAAACGGTTTTTCGC-3′.
The underlined nucleotide is the point mutation that was introduced into the ZBP-89 cDNA. The mutation was verified by DNA sequencing. The expression of Flag-ZBP-89/S202G was verified by immunoblotting.
Transient transfection and luciferase assay
Transient transfection and luciferase reporter assays were performed as described before [18]. Briefly, cells cultured in 48-well plates were transfected with FUGENE 6 (Roche). The cells were treated with 2.5 mM sodium butyrate for 18 h prior to performing luciferase reporter assays that were normalized to protein [18].
In vitro kinase assay
GST fusion proteins of ZBP-89 were expressed and purified as described before [24]. HCT116 cells were transfected with 10 μg of plasmids encoding Flag-tagged wild-type ATM expression vector. Two days later, cells were collected and lysed in lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Tween 20, 0.3% NP-40, 1 mM NaF, 1 mM Na3VO4, 1 mM PMSF with α-complete protease inhibitors (Roche)). Cleared supernatants were immunoprecipitated with protein-A-agarose conjugated anti-Flag M2 antibody [25]. The beads were washed twice with lysis buffer and twice with kinase buffer (20 mM HEPES pH 7.5, 50 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol (DTT), and 10 mM MnCl2). Finally, the immunoprecipitate was resuspended in 50 μl of kinase buffer containing 10 μCi of [γ- 32P] ATP and GST fusion proteins of ZBP-89. The kinase reaction was conducted at 30 °C for 30 min. Proteins were separated on a 4-12% Bis-Tris gel, stained with Coomassie brilliant blue G250 and dried for autoradiography.
In vivo labeling with [32P] orthophosphate
HCT116 cells were transfected with 5 μg of plasmids encoding Flag-tagged ZBP-89, wild-type or kinase dead ATM. Two days after transfection, the cells were washed with phosphate-free DMEM and incubated for 3 h with the same medium containing [32P] orthophosphate (Amersham) at 0.2 mCi/ml. Cells were lysed in lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Tween 20, 0.3% NP-40, 1 mM NaF, 1 mM Na3VO4, 1 mM PMSF with α-complete protease inhibitors (Roche)). ZBP-89 was immunoprecipitated from cell extracts with ZBP-89 antibody coupled to protein A-sepharose (Santa Cruz). Immune complexes were washed five times with lysis buffer containing 0.5 M NaCl. The beads were mixed with Laemmli buffer, boiled, and subjected to SDS-PAGE and autoradiography.
In vitro dephosphorylation and Western blots
Whole cell extracts were dephosphorylated with lambda protein phosphatase (New England Biolabs) following the manufacturer's protocol. For the detection of p53, proteins were separated on the Novex 4-12% Bis-Tris gels (Invitrogen). For the detection of ZBP-89, protein was separated on the Novex 3-8% Tris-Acetate gels (Invitrogen); and the gels were run at 120 V for 8-10 h in the cold room. Proteins were transferred to the PVDF membrane, and Western blots were performed using the corresponding antibodies.
Results and Discussion
ATM phosphorylates ZBP-89 in vivo
In our recent studies, we have shown that the ATM kinase is activated during HDACi-induced chromatin remodelling in human colon cancer cell lines [21]. In addition, HDACi promotes the formation of the ATM/ZBP-89/p300 complex on the p21waf1 promoter, suggesting that there is interplay between these two molecules [21]. To determine whether the expression or protein mobility of ZBP-89 was affected by HDACi-induced chromatin remodelling and ATM kinase activation, HCT116 cells were treated with 1 μM TSA for 6 h and ZBP-89 protein was analyzed by Western blot. Both isoforms of ZBP-89 (∼110-120 kD) showed slower migration upon TSA treatment (Fig 1A). The altered mobility was reversed by treatment with lambda protein phosphatase (PPase), indicating that ZBP-89 is modified by phosphorylation after TSA treatment. TSA treatment increased the level of p-p53Ser15, confirming the presence of ATM kinase activity [26]. Treatment with PPase completely removed the phosphate at Ser15 (Fig 1A), which validated the effectiveness of the PPase dephosphorylation treatment. To further show that ATM has an impact on ZBP-89 phosphorylation, whole cell extracts were prepared from normal human fibroblasts and A-T fibroblasts that are null for ATM (A-T) and separated the proteins by SDS-PAGE (Fig 1B). As shown in Fig 1B, both ZBP-89 and p53 proteins from the A-T cells migrated faster in the gel compared to ZBP-89 from normal fibroblasts. In contrast, there was no significant difference in the electrophoretic mobility of GAPDH protein from the normal and A-T fibroblasts (Fig 1B). Moreover, the levels of p-p53Ser15 and Ac-p53K373 were also significantly reduced in the A-T cells (Fig 1B). Protein phosphatase treatment increased the electrophoretic mobility of ZBP-89 from the normal fibroblasts but not that from the A-T cells, demonstrating that phosphorylation is responsible for the shift in electrophoretic mobility of ZBP-89 (Fig 1C).
Figure 1. TSA treatment induces the phosphorylation of ZBP-89 and p53.

(A) HCT116 cells were treated with 1 μM trichostatin A (TSA) for 6 h. Whole cell extracts were treated with or without lambda protein phosphatase (PPase) before separation by SDS-PAGE and detection by Western blot. (B) Whole cell extracts were prepared from GM0637I (normal fibroblasts) and GM05849D (A-T fibroblasts) then analyzed by Western blot. (C) Whole cell extracts from GM0637I (normal) and GM05849D (A-T) were treated with or without PPase before analysis by Western blot. IB= immunoblot antibody used
To determine whether ZBP-89 is a substrate of ATM kinase, a ZBP-89 expression vector was co-transfected with a wild-type (WT) or a kinase dead (KD) ATM expression vector into HCT116 cells. Following transfection, cells were labelled with [32P] orthophosphate, and ZBP-89 was immunoprecipitated from cell extracts with its specific antibody. As shown in Fig 2A, ZBP-89 protein was readily phosphorylated by catalytically active (WT), but not inactive (KD), ATM protein in vivo.
Figure 2. ATM phosphorylates ZBP-89 in vitro and in vivo.

(A) HCT116 cells were transfected with expression vectors encoding a Flag-tagged ZBP-89, and wild-type (WT) or kinase-dead (KD) full-length ATM protein. Two days after transfection, the cells were washed with phosphate-free DMEM and incubated for 3 h with the same medium containing [32P] orthophosphate. ZBP-89 was immunoprecipitated from whole cell extracts with anti-ZBP-89 antibody, separated on a 4-12% Bis-Tris gel, dried the analyzed by autoradiography. (B) HCT116 cells were transfected with wild-type ATM expression vector. Two days later, ATM was immunoprecipitated from whole cell lysis and used for in vitro kinase assays. Top panel: GST fusion protein levels determined by Coomassie brilliant blue G250 staining. The asterisk indicates a nonsepecific protein band. The arrows indicate GST-ZBP-89 fusion proteins. Bottom panel: extent of 32P incorporation into each fusion protein. The same amount of WT ATM was used in each assay. The asterisk indicates nonsepecific signals. The arrows indicate phosphorylated GST-ZBP-89 fusion proteins. (C) S/TQ motif in human ZBP-89 proteins. The 202SQ motif and its flanking sequence are highly conserved among human, rat and mouse ZBP-89 proteins. ZnF: zinc finger DNA binding domain. The arrowheads indicate all S/TQ motifs in the ZBP-89 protein.
To identify the site in ZBP-89 that is phosphorylated by ATM, we performed in vitro kinase assays using glutathione S-transferase (GST) fusion proteins of ZBP-89. Our in vitro kinase assay demonstrated that the N terminus, but not the C terminus, of ZBP-89 is phosphorylated by ATM (Fig 2B). Sequence annotation revealed that, within the N terminus of ZBP-89, there was only one ATM phosphorylation motif at 202SQ, which is highly conserved among human, rat and mouse proteins (Fig 2C). Therefore, Serine 202 appears to be the ZBP-89 phosphorylation site for ATM.
ATM phosphorylation of ZBP-89 at Ser202 contributes to butyrate activation of p21waf1
Since ZBP-89 plays an important role in HDACi-induced p21waf1 expression [18, 19, 21], we set out to determine whether ATM phosphorylation of ZBP-89 at Ser202 has any effect on ZBP-89 regulated p21waf1 expression. To this end, site-directed mutagenesis was employed to mutate Ser202 to the neutral amino acid glycine (Fig 3A). Since HDAC inhibitors (e.g., butyrate and TSA) activate ATM signaling in HCT116 cells [21] and phosphorylation of endogenous ZBP-89 (Fig 1A), we determined whether disruption of the ATM phosphorylation site at Ser202 had any effect on butyrate-induced phosphorylation of ZBP-89. As shown in Fig 3B, Flag-tagged wild-type ZBP-89 migrated more slowly in butyrate-treated cells compared to that from untreated cells (Fig 3B). Dephosphorylation with protein phosphatase reversed the mobility shift (Fig 3B), indicating that phosphorylation is responsible for the slower migration of ZBP-89 in butyrate-treated cells. Conversely, a less significant shift in the electrophoretic mobility of Flag-tagged ZBP-89/S202G was observed with butyrate treatment (Fig 3B), further confirming that Ser202 is a major phosphorylation site on ZBP-89 upon butyrate treatment. Elevated levels of exogenous Flagged-ZBP-89 proteins (wild-type and S202G) were observed in the presence of butyrate because the cytomegaviral promoter of pcDNA3 is GC-rich and butyrate inducible (Fig 3B) [21].
Figure 3. Butyrate induces ZBP-89 phosphorylation at Ser202.

(A) Schematic structure of the full length (FL) and Ser202 mutant (S202G) ZBP-89 proteins. (B) HCT116 cells were transfected with pcDNA3-ZBP-89/WT and pcDNA3-ZBP-89/S202G expression vectors. One day later, cells were treated with 2.5 mM sodium butyrate (NaBu) for 16 h before collected. Whole cell extracts were prepared and treated with or without protein phosphatase. Proteins were separated on a 3-8% Tris-Acetate gel. Elevated levels of exogenous ZBP-89 proteins (WT and S202G) were observed in the presence of butyrate because the cytomegaviral promoter of pcDNA3 is GC-rich and butyrate inducible. IB= immunoblot antibody used.
To determine the effect of Ser202 phosphorylation on ZBP-89 mediated butyrate induction of p21waf1 expression, either wild-type or the S202G ZBP-89 mutant was cotransfected with a p21waf1 reporter followed by butyrate treatment (Fig 4A). Consistent with our previous observation [26], wild-type ZBP-89 significantly enhanced butyrate-induced p21waf1 promoter activity (Fig 4A). However, the disruption of the ATM phosphorylation site at the 202SQ motif (ZBP-89/S202G) reduced butyrate-mediated p21waf1 transcriptional activation by 31% (Fig 4B). Complete inhibition of the induction was likely not observed due to the presence of WT ZBP-89 in the HCT116 cells. Unfortunately, ZBP-89 null cells do not exist. Thus, phosphorylation at Ser202 in ZBP-89 contributes to transcriptional activation of the p21waf1 promoter by butyrate.
Figure 4. Mutation of Ser202 in ZBP-89 attenuates the transcriptional activity of ZBP-89 in butyrate activation of p21waf1 expression.
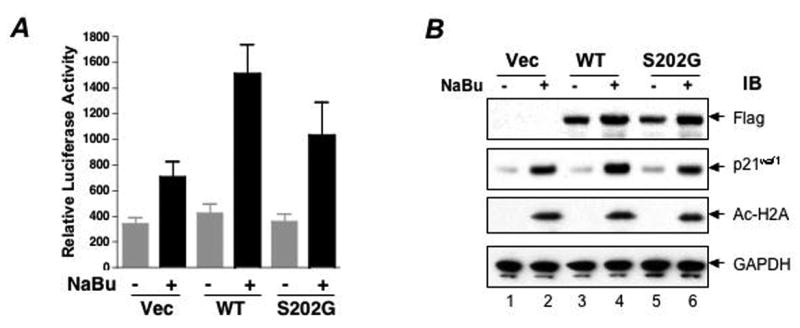
(A) HCT116 cells cultured in 48-well plates were transfected with 25 ng of the p0-Luc p21 waf1 reporter plasmid and 10 ng of different ZBP-89 expression vectors. Twenty-four hours after transfection, the cells were treated with 2.5 mM sodium butyrate (NaBu) for 18h before lysing and assaying for luciferase activity. Luciferase activity was normalized to protein. The data shown are the mean ±SEM for six individual experiments performed in triplicate. (B) HCT116 cells were transfected with pcDNA3, pcDNA3-ZBP-89/WT and pcDNA3-ZBP-89/S202G expression vectors and were selected with G418 at 1 mg/ml for two weeks. Cells from G418-resistant pools were treated with 2.5 mM sodium butyrate for 8 h before they were harvested for immunoblotting. IB= immunoblot antibody used
To further demonstrate that disruption of phosphorylation at the Ser202 site inhibited endogenous p21waf1 expression, we overexpressed ZBP-89/S202G in HCT116 cells. As shown in Fig 4B and consistent with our previous observation [18, 21], overexpression of wild-type ZBP-89 significantly enhanced butyrate induction of endogenous p21waf1 protein without affecting the acetylation of histone 2A, whereas ZBP-89/S202G failed to enhance butyrate induction of endogenous p21waf1 (Fig 4B), suggesting a critical role for ATM phosphorylation of ZBP-89 in butyrate induction of p21waf1 expression.
ATM is a protein kinase that is activated upon DNA damage-induced strand breaks and HDACi-induced chromatin remodeling [1, 26]. It phosphorylates serine or threonine within the S/TQ motif, and its substrates include p53, p95/NBS, BRCA1, Chk2 and SMC1 [26]. The phosphorylation and activation of its substrates promote cell cycle arrest and DNA repair. We have shown recently that HDACi activates ATM signaling in human colon cancer cell lines and promotes the formation of an ATM/ZBP-89/p300 complex on p21waf1 promoter [21]. Our results here demonstrate that ATM phosphorylates ZBP-89 in vitro and in vivo and that the phosphorylation of ZBP-89 at Ser202 contributes to the transcriptional activity of ZBP-89 in butyrate induction of p21waf1 expression.
The CDK inhibitor p21waf1 is a key mediator of HDACi induced cell growth arrest [15, 27]. Understanding the molecular mechanism by which butyrate and other HDACis control cell growth has broad implications in cancer biology. Given the potential importance of HDAC inhibitors as chemotherapeutic agents, these studies will further our understanding of this pathway and possibly reveal new targets for intervention for cancer and suppress of inflammation [21].
Acknowledgments
This work was supported by the NIH grant R01-DK55732 (JLM).
Abbreviations
- ZBP-89
zinc finger binding protein 89 kDa
- ATM
ataxia-telangiectasia mutated
- HDACi
histone deacetylase inhibitory
- TSA
trichostatin A
- PPase
lambda protein phosphatase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 2.Boffa LC, Vidali G, Mann RS, Allfrey VG. Suppression of histone deacetylation in vivo and in vitro by sodium butyrate. J Biol Chem. 1978;253:3364–3366. [PubMed] [Google Scholar]
- 3.Candido EP, Reeves R, Davie JR. Sodium butyrate inhibits histone deacetylation in cultured cells. Cell. 1978;14:105–113. doi: 10.1016/0092-8674(78)90305-7. [DOI] [PubMed] [Google Scholar]
- 4.Sealy L, Chalkley R. The effect of sodium butyrate on histone modification. Cell. 1978;14:115–121. doi: 10.1016/0092-8674(78)90306-9. [DOI] [PubMed] [Google Scholar]
- 5.Kruh J. Effects of sodium butyrate, a new pharmacological agent, on cells in culture. Mol Cell Biochem. 1982;42:65–82. doi: 10.1007/BF00222695. [DOI] [PubMed] [Google Scholar]
- 6.Hassig CA, Tong JK, Schreiber SL. Fiber-derived butyrate and the prevention of colon cancer. Chem Biol. 1997;4:783–789. doi: 10.1016/s1074-5521(97)90111-3. [DOI] [PubMed] [Google Scholar]
- 7.Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr. 2003;133:2485S–2493S. doi: 10.1093/jn/133.7.2485S. [DOI] [PubMed] [Google Scholar]
- 8.G P, McIntyre A, Young GP. Butyrate production from dietary fibre and protection against large bowel cancer in a rat model. Gut. 1993;34:386–391. doi: 10.1136/gut.34.3.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.A J, Medina V, Alvarez-Arguelles H, Hernandez C, Gonzalez F. Sodium butyrate inhibits carcinoma development in a 1,2-dimethylhydrazine-induced rat colon cancer. JPEN J Parenter Enteral Nutr. 1998;22:14–17. doi: 10.1177/014860719802200114. [DOI] [PubMed] [Google Scholar]
- 10.T T, Kameue C, Yamada K, Koyama H, Iwasaki Y, Nakayama K, Ushida K. Dietary sodium gluconate protects rats from large bowel cancer by stimulating butyrate production. J Nutr. 2004;134:940–944. doi: 10.1093/jn/134.4.940. [DOI] [PubMed] [Google Scholar]
- 11.Van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr. 1996;5:245–253. [PMC free article] [PubMed] [Google Scholar]
- 12.Park C, Chamberlin ME, Pan CJ, Chou JY. Differential expression and butyrate response of human alkaline phosphatase genes are mediated by upstream DNA elements. Biochemistry. 1996;35:9807–9814. doi: 10.1021/bi9602223. [DOI] [PubMed] [Google Scholar]
- 13.Koyama N, Hoelzer D, Ottmann OG. Regulation of human IL-18 gene expression: interaction of PU.1 with GC-box binding protein is involved in human IL-18 expression in myeloid cells. Eur J Immunol. 2004;34:817–826. doi: 10.1002/eji.200324420. [DOI] [PubMed] [Google Scholar]
- 14.Boulaire J, Fotedar A, Fotedar R. The functions of the cdk-cyclin kinase inhibitor p21WAF1. Pathol Biol (Paris) 2000;48:190–202. [PubMed] [Google Scholar]
- 15.Ocker M, Schneider-Stock R. Histone deacetylase inhibitors: Signalling towards p21(cip1/waf1) Int J Biochem Cell Biol. 2007 doi: 10.1016/j.biocel.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 16.Huang L, Sowa Y, Sakai T, Pardee AB. Activation of the p21WAF1/CIP1 promoter independent of p53 by the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) through the Sp1 sites. Oncogene. 2000;19:5712–5719. doi: 10.1038/sj.onc.1203963. [DOI] [PubMed] [Google Scholar]
- 17.Xiao H, Hasegawa T, Isobe K. p300 collaborates with Sp1 and Sp3 in p21(waf1/cip1) promoter activation induced by histone deacetylase inhibitor. J Biol Chem. 2000;275:1371–1376. doi: 10.1074/jbc.275.2.1371. [DOI] [PubMed] [Google Scholar]
- 18.Bai L, Merchant JL. Transcription factor ZBP-89 cooperates with histone acetyltransferase p300 during butyrate activation of p21waf1 transcription in human cells. J Biol Chem. 2000;275:30725–30733. doi: 10.1074/jbc.M004249200. [DOI] [PubMed] [Google Scholar]
- 19.Merchant JL, Bai L, Okada M. ZBP-89 mediates butyrate regulation of gene expression. J Nutr. 2003;133:2456S–2460S. doi: 10.1093/jn/133.7.2456S. [DOI] [PubMed] [Google Scholar]
- 20.Takeuchi A, Mishina Y, Miyaishi O, Kojima E, Hasegawa T, Isobe KI. Heterozygosity with respect to Zfp148 causes complete loss of fetal germ cells during mouse embryogenesis. Nat Genet. 2003;33:172–176. doi: 10.1038/ng1072. [DOI] [PubMed] [Google Scholar]
- 21.Bai L, Kao JY, Law DJ, Merchant JL. Recruitment of ataxia-telangiectasia mutated to the p21(waf1) promoter by ZBP-89 plays a role in mucosal protection. Gastroenterology. 2006;131:841–852. doi: 10.1053/j.gastro.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 22.Ju R, Muller MT. Histone deacetylase inhibitors activate p21(WAF1) expression via ATM. Cancer Res. 2003;63:2891–2897. [PubMed] [Google Scholar]
- 23.Taniuchi T, Mortensen ER, Ferguson A, Greenson J, Merchant JL. Overexpression of ZBP-89, a zinc finger DNA binding protein, in gastric cancer. Biochem Biophys Res Commun. 1997;233:154–160. doi: 10.1006/bbrc.1997.6310. [DOI] [PubMed] [Google Scholar]
- 24.Bai L, Merchant JL. ZBP-89 promotes growth arrest through stabilization of p53. Mol Cell Biol. 2001;21:4670–4683. doi: 10.1128/MCB.21.14.4670-4683.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuwabara MD, Sigman DS. Footprinting DNA-protein complexes in situ following gel retardation assays using 1,10-phenanthroline-copper ion: Escherichia coli RNA polymerase-lac promoter complexes. Biochemistry. 1987;26:7234–7238. doi: 10.1021/bi00397a006. [DOI] [PubMed] [Google Scholar]
- 26.L D, Kastan MB. The many substrates and functions of ATM. Nat Rev Mol Cell Biol. 2000;1:179–186. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- 27.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Panini SR, Gupta A, Sexton RC, Parish EJ, Rudney H. Regulation of sterol biosynthesis and of 3-hydroxy-3-methylglutaryl-coenzyme A reductase activity in cultured cells by progesterone. J Biol Chem. 1987;262:14435–14440. [PubMed] [Google Scholar]