

Abstract
Despite advances in diagnostic imaging and drug discovery, primary malignant brain tumors remain fatal. Median survival for patients with the most severe forms is rarely past eight months. The severity of the disease and the lack of substantial improvement in patient survival demand that new approaches be explored in drug delivery to brain tumors. Recently, local delivery of chemotherapy to brain tumors has provided a way to circumvent the blood-brain barrier, allowing delivery of chemotherapy drugs directly to malignant cells in the brain. Two methods of local delivery have been developed: polymeric-controlled release and convection-enhanced delivery. Controlled release utilizes degradable or non-degradable polymers as carriers of chemotherapy; polymer implants or microparticles are implanted locally to introduce a sustained source of drug for periods of days or months. Convection-enhanced delivery employs the bulk flow of drugs dissolved in fluid, which is introduced intracranially using a catheter and pump. The convective fluid flow is capable of delivering drugs great distances within the brain, potentially treating invasive cells at a distance from the catheter infusion site. These two new delivery strategies are capable of delivering both standard chemotherapeutic drugs and new methods of anti-cancer therapy. Taken individually, or used in tandem, they represent a potential revolution in brain cancer treatment.
Introduction
Each year, approximately 14,000 people are stricken with brain cancer. The disease occurs across both social and economic lines, with incidence rates peaking both in childhood and later in old age. Despite advances in imaging technology — which has led to earlier diagnosis of many tumors — the ability to treat the most aggressive form of brain cancer, glioblastoma multiforme (GBM), has not improved since 1980. The one-year survival rate for invasive central nervous system (CNS) cancer was 57.9 percent in 2002, and survival for GBM in particular is even lower [1].
Gliomas are primary CNS tumors arising from the glial cells. Malignant gliomas have a characteristic ability to infiltrate healthy brain tissue and form satellite tumors. This capacity for migration makes them exceedingly difficult to treat and invariably fatal. Even after resection, invasive cells can give rise to tumors within centimeters of the resection site [2]. Untreated malignant tumors can eventually spread to the contralateral hemisphere [3]. Many forms of systemic chemotherapy are excluded from the CNS by the blood-brain barrier (BBB) [4]. A few compounds — such as the class of antiproliferative drugs called nitrosoureas (including carmustine and lomustine) or other alkylating agents (temozolomide) — have some ability to cross the BBB and have been used clinically [5]. Unfortunately, systemic delivery of these agents appears to offer modest benefit as a supplement to radiotherapy [6,7].
Over the past two decades, a variety of approaches to enhance the activity of systemically delivered chemotherapy drugs have been tested. Hyperosmolar BBB disruption has been used to enhance BBB transfer of chemotherapy agents, with mixed results. One study, using PET imaging to evaluate a combination of methotrexate and hyperosmolar BBB disruption, indicated a negligible effect in brain tumors, which is echoed by the marginal findings in clinical trials [8,9]. A variety of approaches have been tested for enhancing BBB permeability of systemically administered drugs — by modification with hydrophobic side groups, conjugation to ligands with known BBB carriers, such as transferrin, or encapsulation in liposomes or nanoparticles — but none of these approaches have impacted clinical treatment of glioma [10].
The failure of conventional systemic drug delivery for glioma has motivated more direct approaches to drug delivery. Direct intracranial drug delivery would eliminate the need for a chemotherapeutic agent to cross the BBB. The ability to bypass the BBB would enable a wider range of agents — such as paclitaxel, doxorubicin, immunotoxins, and even gene therapy vectors — to be evaluated for brain cancer treatment. This review describes two of the most promising approaches for direct delivery of agents to intracranial tumors: polymeric-controlled release and convection-enhanced delivery.
Polymeric-controlled release
Polymeric-controlled release has long been used for drug delivery. Some early systems — the five-year subcutaneous Norplant® contraceptive and the conjuctival Ocusert® system for glaucoma — have proven the effectiveness of this approach for both systemic and local therapy [11]. The controlled release of drug also protects it from elimination [12].
Controlled release systems can be designed from both degradable and nondegradable polymers. When constructed from nondegradable polymers, drug release is usually governed by diffusion of the drug through the matrix. In contrast, release from degradable polymers is governed by a combination of drug diffusion through the polymer and erosion of the polymer matrix. Polymers can be combined into copolymers to tune degradation and release characteristics. Correctly designed, polymer and drug systems can provide reliable sustained release for periods of days or many years (see review in [12]).
Non-degradable polymer systems
Delivery systems constructed from nondegradable polymers can be employed when a removable system is required. The most common nondegradable polymer system is a copolymer of ethylene and vinyl acetate. Polyethylene-co-vinyl acetate (EVAc) materials exhibit excellent biocompatibility and are typically implanted chronically. EVAc have been developed for the controlled release of DNA, antibodies, as well as chemotherapeutics [13,14]. Two studies that examined the delivery of amsacrine and mitoxantrone in rat glioma models observed potent anti-tumor effects [15,16]. While the engineering of these systems is highly developed — so that delivery systems of virtually any size, shape, rate, and duration of release can be produced — the persistence of the polymer after delivery limits their clinical use.
Degradable polymer systems
Controlled release systems using biodegradable polymers are more common than non-degradable systems. The biodegradable systems offer the same advantages as the persistent wafers, but they completely erode during delivery and are cleared from the body. This disappearance attenuates the response to the implant and makes it clinically attractive. Most systems are diffusion regulated and the kinetics are well-characterized [17]. Many degradable systems are designed to erode via hydrolysis, taking advantage of the prevalence of water in the human body. For example, a variety of controlled release systems have been designed around the hydrolysable anhydride bond. Copolymers are typically coupled with anhydride bonds to control the rate of hydrolysis [12]. The addition of a copolymer affects the degradation of the polymer device. The correct copolymer system can achieve ordered erosion, which provides a consistent release of drug. The most common copolymer systems used intracranially is polybis(p-carboxyphenoxy)propane-sebacic acid (p(CPP-SA).
p(CPP-SA)-based delivery systems have been characterized for a variety of drugs and already are used clinically. Many controlled release systems are based on an implantable wafer, which has been studied with the drugs mitoxantrone, carmustine (BCNU), 4-hydroperoxycyclophosphamide (4-HC), paclitaxel, carboplatin, and adriamycin [18-21]. The characteristics of drug release from wafers depend on the polymer and drug used. A typical controlled release curve for BCNU is shown in [Figure 1] [22]. The linear region in Figure 1b indicates that the drug release is diffusion controlled [12]. In drug-polymer systems with a high degree of surface associated drug, a burst release phase is observed. p(CPP-SA) wafers of BCNU and 4-HC have demonstrated release for 50 days. The release of paclitaxel proceeded at a much lower rate, less than 0.01 mg/day, and for a much longer period of time (160 days). The slower release rate can be attributed to the hydrophobicity of the drug that causes a strong affinity for the polymer. The controlled release of drugs from these wafers has demonstrated performance in many in vitro and in vivo glioma models. Studies have demonstrated improved performance of these wafers when compared to free drug administration in a rat model challenged with an intracranial 9L tumor [23]. Small cylindrical wafers have been studied in the same way [18].
Figure 1.
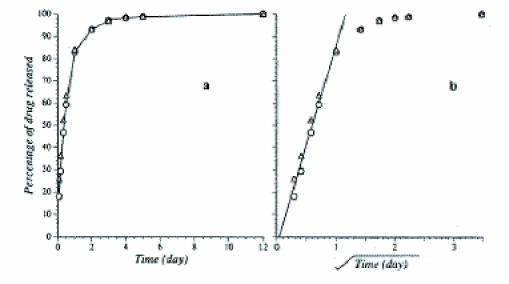
The cumulative release of BCNU into a well-stirred reservoir as a function of time (a) and the square root of time (b). The linear portion indicated in b is indicative of diffusion controlled release from a planar geometry. Reprinted with permission from Springer Science and Business Media: Fung, L. K. et al. Chemotherapeutic drugs released from polymers: distribution of 1,3-bis(2-chloroethyl)-1-nitrosourea in the rat brain. Figure 2. Pharmaceutical Research. 1996;13:671-82.
The p(CPP-SA) wafer loaded with BCNU has undergone further characterization and is available clinically as Gliadel® [24]. The Gliadel® wafer is 14 mm in diameter and 1 mm thick. It is loaded with 7.7 mg of the drug carmustine and is implanted intracranially after surgical debulking of the tumor [Figure 2]. Gliadel® has been studied for clinical use in initial treatment, treatment of recurrences, and in conjunction with radiotherapy for malignant gliomas [25-27]. In all cases, therapy with Gliadel® was well tolerated with no significant increase in toxicity, infection, or inflammation. The majority of studies indicated a modest improvement in survival for patients with malignant gliomas that received Gliadel® [28,29]. Further research is directed at improving the Gliadel® wafer by examining dosing and combination therapy [30].
Figure 2.
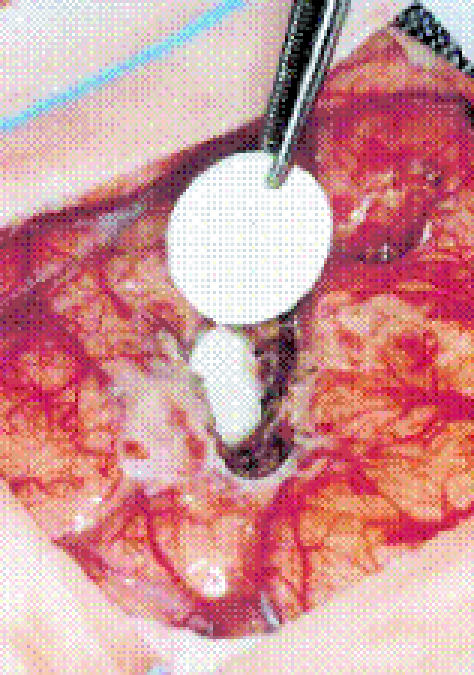
Intracranial placement of Gliadel® wafer. The wafer is implanted in the cavity left after surgical resection of the tumor. Wafers are dime-sized and impregnated with carmustine. Reprinted with permission from Fleming. Pharmacokinetics of the carmustine implant. Clinical Pharmacokinetics. 2002;4(6):403-419 (Figure 1, page 405).
Fatty acid dimer copolymers use the same polyanhydride linkages, but they offer a distinct advantage over carboxyphenoxy propane polymer systems. FAD-SA is typically formed into a disk shape and then implanted intracranially. The primary use of FAD-SA is the controlled delivery of the drug 4-hydroperoxycyclophosphamide (4HC) [31]. FAD-SA is used for delivery of this drug because 4-HC is hydrolytically unstable in the p(CPP-SA) wafer [32]. The ability to tailor the chemical and physical properties of the vehicle to the drug is one of the advantages of polymeric delivery.
Other degradable polymers are also useful for drug delivery. Biodegradable polymer matrices based on polymers of lactide and glycolide are perhaps the most popular platform for local drug delivery. Polylactic acid (PLA), polyglycolic acid (PGA), and their copolymer polylactic-co-glycolic acid (PLGA) have a long history of use as biomaterials, beginning in the 1970s with biodegradable sutures. Matrices made from these polymers are biocompatible, hydrophobic, and degrade to the naturally occurring monomers via ester hydrolysis. Polymer nanospheres, microspheres, and wafers of varying sizes are made via emulsion/solvent evaporation, salt leaching, and other methods [33]. When making spherical particles using the emulsion process, the solvent, surfactant, and polymer properties can be modified to tune size and release characteristics [34,35].
Implantable PLGA matrices loaded with chemotherapeutics are currently under development. The implants can be in the form of electrospun scaffolds, thin films, or even wafers. The process of electrospinning uses a voltage differential to produce a non-woven mesh. Paclitaxel has been mixed with the polymer solution before electrospinning to create a mesh with drug-delivery capability [36]. The drug-loaded mesh has controlled release properties and was effective in vitro against C6 glioma cells. A mesh delivery system might have advantages over a wafer, because the mesh can potentially conform to the shape of the resection cavity. The C6 glioma line was also used to evaluate the controlled release of radioiododeoxyuridine from a thin film of PLGA [37]. Biodegradable PLGA wafers containing BCNU can be used in an analogous method to Gliadel®. The release profile of BCNU from the PLGA wafer shows a similar profile and duration when compared to the p(CPP-SA) wafer [38]. A reformulated wafer made of compressed BCNU containing microparticles yielded a longer in vitro release profile, 70 days compared to seven [39].
PLGA nanoparticles and microparticles can be fabricated using the single emulsion method to encapsulate hydrophobic compounds and the double emulsion method to encapsulate hydrophilic compounds [Figure 3] [41,41]. The advantages of polymer particles lie in the route of administration. Polymer nanoparticles may be administered using a burr hold and catheter, which is significantly less invasive than the implantation of polymer wafers.
Figure 3.

SEM image of PLGA microparticles loaded with BCNU. The particles were formed using the solvent extraction/evaporation method. Average sphere diameter is 30 mm. Reprinted with permission from Elsevier. Painbeni T, et al., Internal morphology of poly(D,L-lactide-co-glycolide) BCNU-loaded microspheres. Influence on drug stability. European Journal of Pharmaceutics and Biopharmaceutics. 1998;45:31-9.
Polymeric-controlled release is able to administer drugs intracranially, which would be excluded for systemic use or as a new route of administration. This is the case with doxorubicin (adriamycin), which exhibits dose-limiting cardiotoxicity [42]. The drug temozolomide (TM) has demonstrated clinical efficacy when given systemically [5]. A controlled release form of TM using PLGA microparticles gave the characteristic biphasic release profile, burst release followed by a linear period, with a 35-day release period [43]. The TM microparticles demonstrated cytotoxicity in culture with C6 cells and could be administered intracranially as part of the same regimens currently evaluating systemic TM.
Polymeric-controlled release systems can be engineered for drugs, such as paclitaxel, that are difficult to administer with conventional methods. Two drug properties that can limit traditional use are hydrophobicity and toxicity. Paclitaxel is hydrophobic and sparingly soluble in water; this restricts its use in injection buffers. Intravenous chemotherapy with paclitaxel utilizes an adjuvant called Cremophor EL to disperse the drug, which can cause serious side effects. PLGA is naturally hydrophobic and encapsulates hydrophobic drugs efficiently. The hydrophobicity of a drug also affects its release profile. Paclitaxel preferentially partitions inside PLGA and exhibits a lengthy release curve. A study with PLGA microparticles showed a sustained release of paclitaxel for more than 60 days without a burst release period [44]. The controlled release of paclitaxel also limits the systemic toxicity. The release is so slow that a variety of coencapsulants have been evaluated to quicken the release of paclitaxel from PLGA matrices. Two examples are isopropyl myristate and vitamin E TPGS [45,46]. Both emulsifiers produced a burst release period. The formulation using vitamin E TPGS decreased the release time compared to particles made with the surfactant poly(vinyl alcohol) (PVA) [47].
Controlled release systems may be particularly useful for agents produced by biotechnology, such as the proteins and nucleic acids that are needed for immunotherapy or gene therapy. The rare instances of complete recovery from malignant gliomas typically have been attributed to an immune response. This observation triggered the search for compounds that could cause immune cell-mediated cytotoxicity. Type 1 interferons show antitiumor activity but have a short half-life and high dose toxicity [48]. The short half-life necessitates a continuous delivery for the molecule to be active, but the continuous delivery will pose the risk of side effects. Encapsulating these agents protects them from elimination and provides a means to control the amount released. Interleukin-18, which can be used in this manner, can be encapsulated in PLGA microspheres and is active upon release [49]. PLGA can also encapsulate and release viable adenovirus [50]. The controlled release approach improved in vivo efficacy.
Polymeric-controlled release provides a biocompatible, tunable platform in which drug loading, release rate, longevity, and form can be altered via polymer combination and processing. The method of delivery, as witnessed by Gliadel®, adds minimal complexity, requires no additional surgical procedures, and minimizes solubility limitations. Furthermore, the controlled release of the drug slows elimination and increases duration of exposure. This increases the amount of drug administered without exposing the tissue to high concentrations that could cause tissue damage. Most importantly, polymeric-controlled release has a clinical history of beneficial use.
While controlled release systems can deliver drugs for long periods of time locally in the brain, local penetration of the drug is frequently limited by diffusion [21]. When low diffusion coefficients are coupled with a high rate of elimination, drug distance from the delivery locus can be limited to millimeters [51]. The extracellular matrix (ECM) of the brain also imposes its own limits on diffusive transport. The ECM of the brain is a hydrated environment with a low volume fraction, a high degree of tortuosity, and small pore size [52-55]. The local environment of a tumor limits diffusion even further [56]. These factors conspire to confine a high drug concentration to within 3 mm of the delivery site [24]. This distance is exceeded by invasive glioma. Controlled release also is limited by size. The polymer delivery system must be large enough or be used in enough amount to deliver clinically relevant drug dosage.
Convection-enhanced delivery
Convection-enhanced delivery (CED) was developed to deliver compounds throughout the brain to overcome the diffusion barrier seen with polymeric-controlled release [57]. CED utilizes an applied external pressure gradient to induce fluid convection in the brain. Fluid is typically administered via a small catheter using a pump [58]. Because moderately high pressures (up to 70 mmHg) must be used to drive convective flows, such procedures are plagued by backflow along the catheter and often subject the tissue to high pressures. The increase in pressure is due to occlusions that may block the catheter from delivering an adequate flow rate. The use of microfabricated silicon probes reduces those concerns and improves on the general delivery method [59]. The outlet of the silicon probes is along an axis perpendicular to the insertion direction; this geometry prevents tissue from plugging the orifice. A controlled pressure source in stream with a fluid reservoir ensures operation at constant pressure. This prevents the pressure spikes that accompany constant volume delivery. An added advantage to constant pressure delivery is the ease with which fluid properties can be determined using simple mathematical models.
CED and drug delivery
The main benefit to administering drug via CED is the greater distribution volume. Studies that have compared a bolus injection to CED have noted a larger distribution and a greater efficacy [60,61]. The benefits are derived from the greater distribution and a continued exposure due to the long infusion time. The technique is inherently flexible and can be used in chemotherapy, gene therapy, and immune therapy. Methods of drug delivery using free drug have been studied in animals and clinically. The drugs administered run the gamut from nontraditional compounds to typical chemotheraputic drugs. Typical clinical use of CED is for salvage therapy after a recurrence of glioma and follows one of two protocols. Patients are either given local infusions after surgical resection or given infusions directly into the tumor. Drugs used clinically include both targeted toxins and traditional chemotherapeutics (Table 1). Studies using the drug paclitaxel have observed the highest response rates, which are around 75 percent. Typical response rates for targeted toxin treatment were lower [62]. Response rate and the occurrence of adverse events have been observed to be dose dependent [63]. Ongoing clinical trials include the CED of IL13-PE38QQ, and the CED of topotecan.
The drugs listed in Table 1 reflect only a small sample of the therapeutic agents that have been evaluated in animal models — which include the traditional chemotherapy drugs BCNU and topotecan [64,65]. However, the drugs available for delivery via CED are still limited by solubility. A way to avoid solubility issues and protect antineoplastic agents from elimination is to encapsulate them. Recent research studied nanoliposomal encapsulated CPT-11 in a rat glioma model [66]. The study showed that CPT-11 benefited from the increased distribution via CED and a longer residence time.
Large particles, with severely restricted diffusion, stand to benefit most from CED. Accordingly, several research groups are determining and improving the penetration of particles within the brain delivered via CED [Figure 4]. Studies looking at the size of particle infusate concluded that smaller particles, around 20 nm, distributed further than larger particles [67]. The same behavior was observed using liposomes [68]. Interestingly, particles 40nm and larger had similarly restricted distributions. The surface properties of the small particles play a significant role in their volume distribution. Surfaces that were neutral, negatively charged, or coated with poly(ethylene glycol) (PEG) or bovine serum albumin (BSA) maximized distribution volume [69]. Coatings that were positively charged severely restricted particle distribution. Adjuvants have been used to improve distribution regardless of particle properties. A hyperosmolar infusion of mannitol significantly increased volume distribution [68]. When the infusate experiences specific cell binding, an infusion of excess unencapsulated ligands can increase the distribution [70].
Figure 4.
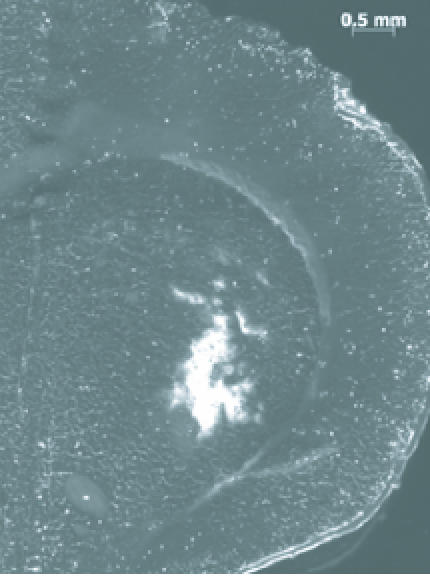
20 nm Polystyrene beads delivered via CED into the caudate of a rat. Image shows local nanoparticle penetration after a 5 µL infusion (Sawyer AJ, Neeves KB, Foley CP, Olbricht WL, and Saltzman WM, unpublished data).
One objective of the previous particle infusion studies was to model the delivery of either viral vectors that could be used for gene therapy or drug loaded particles. CED has the potential to improve gene therapy by increasing the available concentration of vectors. Two studies looked at the distribution of adeno-associated vectors (AAVs) administered via CED [71,72]. Each realized a greater and more homogeneous distribution of transduction by combining AAVs with CED, which was further improved with adjuvant heparin.
CED and imaging
CED has the ability to improve both the imaging and treatment of brain tumors. This allows for a combination approach that can monitor the distribution of drugs as they are being delivered [73]. To observe the distribution via CED, liposomes were labeled with the MRI contrast agent gadolinium. The gadolinium liposomes can then be infused with particles of similar size and monitored using MRI. This approach was validated in a primate model [74]. The distributions observed via MRI completely coincided with distributions determined using fluorescence. Administering contrast agent along with chemotherapeutic drugs allows for the initial volume distribution to be observed and for tissue necrosis to be monitored within that distribution area [75].
CED is limited by its invasiveness and by the anatomical influences on drug distribution. CED requires the insertion of a catheter several centimeters deep into the brain, which can cause tissue damage and may induce air bubbles [76]. The anatomy of the brain affects the distribution of drugs. White matter tracks provide areas of comparatively high fluid conductivity. If the catheter is inserted through white matter, for example the corpus callosum, the high conductivity combined with reflux up the catheter siphons drug away from parenchyma. The perivascular spaces have also been observed to collect infusate during CED [77]. The unpredictable flow can lead to collection of drug either in the perivascular spaces, wound track, or under the scalp. This has caused incidences of edema and wound dehiscence. The localized high dose area caused the same symptoms during the delivery of viral vectors [78].
Clinical trials
A Phase III randomized clinical trial (PRECISE TRIAL) comparing the outcome between Gliadel and CED of IL13-PE38QQR has been completed (unpublished data). This study randomized patients with GBM for treatment after they had failed conventional therapy (surgery, radiotherapy, +/- chemotherapy). This study revealed no significant difference in outcome. Median survival following tumor recurrence was 35.3 weeks for the Gliadel and 36.4 weeks for IL13-PE38QQR. It is anticipated that inaccurate catheter placement and unresolved problems with adequate drug distribution may have contributed to the absence of benefit for the CED arm. These data support the need for continued research in improving drug distribution and better delivery methods.
Conclusions
Local delivery to brain tumors can be accomplished using the two strategies of polymeric-controlled release and convection-enhanced delivery. Each technique strives to address the need for controllable intracranial drug delivery. The two technologies offer unique benefits and suffer distinctive limitations, which are listed in Table 2. The principle disadvantage to controlled release is the restricted drug distribution. The limitations imposed by diffusion within the brain limit drug penetration to a region smaller than the typical invasive area of malignant gliomas.
CED’s strength lies in the potential for large distribution volume of infused drugs. The long infusion times and unpredictable distribution have also caused an abundance of side effects in recent clinical trials. Trials using paclitaxel have noticed a high rate of drug related adverse events that include wound dehiscence, inflammation, and edema [79,80]. These side effects are attributed to drug backflow along the catheter, and drug localization in the perivascular and subarachnoid spaces. Infusing an encapsulated drug may decrease the incidence of these adverse events.
Outlook
CED was developed as a method to address the shortcomings of polymeric-controlled release. Coincidentally, many of the flaws in CED are the strengths of a polymeric-controlled release approach. Encapsulation of drug would limit the reflux and promote better wound healing after delivery. Delivering encapsulated drug could shorten the infusion time. A shorter infusion time would deliver a lower maximum drug concentration which could address the instances of edema and inflammation. Moreover, a combined treatment strategy could truly localize the delivery by restricting it to a prescribed area. Nanoparticle technology has advanced such that high levels of targeting ligand can be expressed on the surface of the degradable particles [81]. These particles could then be targeted to a subpopulation of cells.
A combined approach is not without its potential pitfalls. The polymer particles would have to be large enough to deliver a clinically relevant dose. The increase in size could restrict the distribution of particles in the parenchyma. While the adjuvants could improve the distribution, a multi-focal delivery strategy may be necessary to circumvent the transport limitations.
Local delivery to brain tumors has already provided a modest increase in survival when used in addition to surgery and radiotherapy. Despite the advances, GBM remains fatal regardless of the mode of treatment. There is untapped potential in both of the local delivery strategies discussed here. Polymeric-controlled release and CED can be used as techniques to administer the standard agents, combinations of drugs, as well as new methods of anti-tumor therapy.
Table 1. Therapeutic agents evaluated in clinical trials for glioma treatment.
| TF-CRM107 |
| cpIL-4-PE |
| IL13-PE38QQR |
| TGFα-PE38 (TP-38) |
| LIPO-HSV-1-tk |
| Paclitaxel |
| Cotara® |
Adapted from [62].
Table 2. Advantages and disadvantages of polymeric-controlled release and convection-enhanced drug delivery.
| Advantages | Disadvantages | |
| Polymeric-controlled release | Sustained drug release | Poor drug penetration |
| Define release kinetics | Drug dosage limited by implant size | |
| Tunable release properties | ||
| Low invasiveness | ||
| Low peak drug release limits tissue damage | ||
| Biocompatible | ||
| Localized delivery | ||
| Convection-enhanced delivery | Large drug distribution volume | Invasive |
| Flexible therapy protocol | Long infusion times | |
| Consistent drug concentration | Potential high intracranial pressures | |
| Unpredictable drug distribution |
Abbreviations
- GBM
Glioblastoma multiforme
- CNS
Central nervous system
- EVAc
Polyethylene-co-vinyl acetate
- BBB
Blood-brain barrier
- p(CPP-SA)
Polybis(p-carboxyphenoxy)propane-sebacic acid
- BCNU
Carmustine
- 4-HC
4-hydroperoxycyclophosph amide
- PLA
Polylactic acid
- PGA
Polyglycolic acid
- PLGA
Polylactic-co-glycolic acid
- TM
Temozolomide
- PVA
Polyvinyl alcohol
- CED
Convection-enhanced delivery
- PEG
Polyethylene glycol
- AAVs
Adeno-associated vectors
References
- Deorah S, Lynch CF, Sibenaller ZA, Ryken TC. Trends in brain cancer incidence and survival in the United States: Surveillance, Epidemiology, and End Results Program, 1973 to 2001. Neurosurg Focus. 2006;20(4):E1. doi: 10.3171/foc.2006.20.4.E1. [DOI] [PubMed] [Google Scholar]
- Enam SA, Eisenberg AD, Norman D, Rosenblum ML. Patterns of Spread and Recurrence of Glioma: Studies by Neuroimaging. In: Mikkelsen T, et al., editors. Brain Tumor Invasion: Biological, Clinical, and Therapeutic Considerations. New York: Wiley-Liss; 1998. pp. 133–160. [Google Scholar]
- Demuth T, Berens ME. Molecular mechanisms of glioma cell migration and invasion. J Neurooncol. 2004;70(2):217–228. doi: 10.1007/s11060-004-2751-6. [DOI] [PubMed] [Google Scholar]
- Groothuis DR. The blood-brain and blood-tumor barriers: a review of strategies for increasing drug delivery. Neuro-oncol. 2000;2(1):45–59. doi: 10.1093/neuonc/2.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason WP, Cairncross JG. Drug Insight: temozolomide as a treatment for malignant glioma — impact of a recent trial. Nat Clin Pract Neurol. 2005;1(2):88–95. doi: 10.1038/ncpneuro0045. [DOI] [PubMed] [Google Scholar]
- Medical Research Council Brain Tumor Working Party. Randomized trial of procarbazine, lomustine, and vincristine in the adjuvant treatment of high-grade astrocytoma: a Medical Research Council trial. J Clin Oncol. 2001;19(2):509–518. doi: 10.1200/JCO.2001.19.2.509. [DOI] [PubMed] [Google Scholar]
- Chang CH, Horton J, Schoenfeld D, et al. Comparison of postoperative radiotherapy and combined postoperative radiotherapy and chemotherapy in the multidisciplinary management of malignant gliomas. A joint Radiation Therapy Oncology Group and Eastern Cooperative Oncology Group study. Cancer. 1983;52(6):997–1007. doi: 10.1002/1097-0142(19830915)52:6<997::aid-cncr2820520612>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Zunkeler B, Carson RE, Olson J, et al. Quantification and pharmacokinetics of blood-brain barrier disruption in humans. J Neurosurg. 1996;85(6):1056–1065. doi: 10.3171/jns.1996.85.6.1056. [DOI] [PubMed] [Google Scholar]
- Fortin D, Desjardins A, Benko A, Niyonsega T, Boudrias M. Enhanced chemotherapy delivery by intraarterial infusion and blood-brain barrier disruption in malignant brain tumors: the Sherbrooke experience. Cancer. 2005;103(12):2606–2615. doi: 10.1002/cncr.21112. [DOI] [PubMed] [Google Scholar]
- Misra A, Ganesh S, Shahiwala A, Shah SP. Drug delivery to the central nervous system: a review. J Pharm Pharm Sci. 2003;6(2):252–273. [PubMed] [Google Scholar]
- Sheardown H, Saltzman WM. Novel drug delivery systems for posterior segment ocular disease. In: Barnstable C, Tombran-Tink J, editors. Ocular Angiogenesis: diseases, mechanisms, and therapeutics. New Jersey: Humana Press; 2006. pp. 393–408. [Google Scholar]
- Saltzman WM. In: Drug Delivery: Engineering Principles for Drug Therapy. Gubbins KE, editor. New York: Oxford University Press; 2001. [Google Scholar]
- Luo D, Woodrow-Mumford K, Belcheva N, Saltzman WM. Controlled DNA delivery systems. Pharm Res. 1999;16(8):1300–1308. doi: 10.1023/a:1014870102295. [DOI] [PubMed] [Google Scholar]
- Saltzman WM. Antibodies for treating and preventing disease: the potential role of polymeric-controlled release. Crit Rev Ther Drug Carrier Syst. 1993;10(2):111–142. [PubMed] [Google Scholar]
- Wahlberg LU, Almqvist PM, Glantz MJ, Boethius J. Polymeric controlled-release amsacrine chemotherapy in an experimental glioma model. Acta Neurochir (Wien) 1996;138(11):1323–1329. discussion 9–30. doi: 10.1007/BF01411063. [DOI] [PubMed] [Google Scholar]
- Saini M, Roser F, Hussein S, Samii M, Bellinzona M. Intralesional mitoxantrone biopolymer-mediated chemotherapy prolongs survival in rats with experimental brain tumors. J Neurooncol. 2004;68(3):225–232. doi: 10.1023/b:neon.0000033381.96370.6b. [DOI] [PubMed] [Google Scholar]
- Mak M, Fung L, Strasser JF, Saltzman WM. Distribution of drugs following controlled delivery to the brain interstitium. J Neurooncol. 1995;26(2):91–102. doi: 10.1007/BF01060215. [DOI] [PubMed] [Google Scholar]
- DiMeco F, Li KW, Tyler BM, Wolf AS, Brem H, Olivi A. Local delivery of mitoxantrone for the treatment of malignant brain tumors in rats. J Neurosurg. 2002;97(5):1173–1178. doi: 10.3171/jns.2002.97.5.1173. [DOI] [PubMed] [Google Scholar]
- Olivi A, Ewend MG, Utsuki T, et al. Interstitial delivery of carboplatin via biodegradable polymers is effective against experimental glioma in the rat. Cancer Chemother Pharmacol. 1996;39(1-2):90–96. doi: 10.1007/s002800050542. [DOI] [PubMed] [Google Scholar]
- Hsu W, Lesniak MS, Tyler B, Brem H. Local delivery of interleukin-2 and adriamycin is synergistic in the treatment of experimental malignant glioma. J Neurooncol. 2005;74(2):135–140. doi: 10.1007/s11060-004-6597-8. [DOI] [PubMed] [Google Scholar]
- Fung LK, Ewend MG, Sills A, et al. Pharmacokinetics of interstitial delivery of carmustine, 4-hydroperoxycyclophosphamide, and paclitaxel from a biodegradable polymer implant in the monkey brain. Cancer Res. 1998;58(4):672–684. [PubMed] [Google Scholar]
- Fung LK, Shin M, Tyler B, Brem H, Saltzman WM. Chemotherapeutic drugs released from polymers: distribution of 1,3-bis(2-chloroethyl)-1-nitrosourea in the rat brain. Pharm Res. 1996;13(5):671–682. doi: 10.1023/a:1016083113123. [DOI] [PubMed] [Google Scholar]
- Buahin KG, Brem H. Interstitial chemotherapy of experimental brain tumors: comparison of intratumoral injection versus polymeric controlled release. J Neurooncol. 1995;26(2):103–110. doi: 10.1007/BF01060216. [DOI] [PubMed] [Google Scholar]
- Fleming AB, Saltzman WM. Pharmacokinetics of the carmustine implant. Clin Pharmacokinet. 2002;41(6):403–419. doi: 10.2165/00003088-200241060-00002. [DOI] [PubMed] [Google Scholar]
- Westphal M, Hilt DC, Bortey E, et al. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro-oncol. 2003;5(2):79–88. doi: 10.1215/S1522-8517-02-00023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinberg LR, Weingart J, Burger P, et al. Clinical course and pathologic findings after Gliadel and radiotherapy for newly diagnosed malignant glioma: implications for patient management. Cancer Invest. 2004;22(1):1–9. doi: 10.1081/cnv-120027575. [DOI] [PubMed] [Google Scholar]
- Engelhard HH. The role of interstitial BCNU chemotherapy in the treatment of malignant glioma. Surg Neurol. 2000;53(5):458–464. doi: 10.1016/s0090-3019(00)00211-1. [DOI] [PubMed] [Google Scholar]
- Brem H, Piantadosi S, Burger PC, et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. The Polymer-brain Tumor Treatment Group. Lancet. 1995;345(8956):1008–1012. doi: 10.1016/s0140-6736(95)90755-6. [DOI] [PubMed] [Google Scholar]
- Brem H, Mahaley MS Jr., Vick NA, et al. Interstitial chemotherapy with drug polymer implants for the treatment of recurrent gliomas. J Neurosurg. 1991;74(3):441–446. doi: 10.3171/jns.1991.74.3.0441. [DOI] [PubMed] [Google Scholar]
- Brem H, Gabikian P. Biodegradable polymer implants to treat brain tumors. J Control Release. 2001;74(1-3):63–67. doi: 10.1016/s0168-3659(01)00311-x. [DOI] [PubMed] [Google Scholar]
- Buahin KG, Judy KD, Hartke C, et al. Controlled Release of 4-Hydroxyperoxycyclophosphamide from the Fatty Acid Dimer-Sebacic Acid Coploymer. Polymers for Advanced Technologies. 1992;3:311–316. [Google Scholar]
- Judy KD, Olivi A, Buahin KG, et al. Effectiveness of controlled release of a cyclophosphamide derivative with polymers against rat gliomas. J Neurosurg. 1995;82(3):481–486. doi: 10.3171/jns.1995.82.3.0481. [DOI] [PubMed] [Google Scholar]
- Bala I, Hariharan S, Kumar MN. PLGA nanoparticles in drug delivery: the state of the art. Crit Rev Ther Drug Carrier Syst. 2004;21(5):387–422. doi: 10.1615/critrevtherdrugcarriersyst.v21.i5.20. [DOI] [PubMed] [Google Scholar]
- Hans ML, Lowman AM. Biodegradable nanoparticles for drug delivery and targeting. Current Opinion in solid State & Materials Science. 2002;6:319–327. [Google Scholar]
- Mu L, Seow PH, Ang SN, Feng SS. Study on surfactant coating of polymeric nanopartcles for controlled delivery of anticancer drug. Colloid Polym Sci. 2004;283:58–65. [Google Scholar]
- Xie J, Wang CH. Electrospun Micro- and Nanofibers for Sustained Delivery of Paclitaxel to Treat C6 Glioma in Vitro. Pharm Res. 2006;8:1817–1826. doi: 10.1007/s11095-006-9036-z. [DOI] [PubMed] [Google Scholar]
- Mairs RJ, Wideman CL, Angerson WJ, et al. Comparison of different methods of intracerebral administration of radioiododeoxyuridine for glioma therapy using a rat model. Br J Cancer. 2000;82(1):74–80. doi: 10.1054/bjoc.1999.0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, An TK, Chae GS, et al. Evaluation of in vitro and in vivo antitumor activity of BCNU-loaded PLGA wafer against 9L gliosarcoma. Eur J Pharm Biopharm. 2005;59(1):169–175. doi: 10.1016/j.ejpb.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Seong H, An TK, Khang G, Choi SU, Lee CO, Lee HB. BCNU-loaded poly(D, L-lactide-co-glycolide) wafer and antitumor activity against XF-498 human CNS tumor cells in vitro. Int J Pharm. 2003;251(1-2):1–12. doi: 10.1016/s0378-5173(02)00543-4. [DOI] [PubMed] [Google Scholar]
- Zambaux MF, Bonneaux F, Gref R, et al. Influence of experimental parameters on the characteristics of poly(lactic acid) nanoparticles prepared by a double emulsion method. J Control Release. 1998;50(1-3):31–40. doi: 10.1016/s0168-3659(97)00106-5. [DOI] [PubMed] [Google Scholar]
- Mu L, Feng SS. PLGA/TPGS nanoparticles for controlled release of paclitaxel: effects of the emulsifier and drug loading ratio. Pharm Res. 2003;20(11):1864–1872. doi: 10.1023/b:pham.0000003387.15428.42. [DOI] [PubMed] [Google Scholar]
- Lin R, Shi Ng L, Wang CH. In vitro study of anticancer drug doxorubicin in PLGA-based microparticles. Biomaterials. 2005;26(21):4476–4485. doi: 10.1016/j.biomaterials.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Zhang H, Gao S. Temozolomide/PLGA microparticles and antitumor activity against Glioma C6 cancer cells in vitro. Int J Pharm. 2006;329:122–128. doi: 10.1016/j.ijpharm.2006.08.027. [DOI] [PubMed] [Google Scholar]
- Kumar Naraharisetti P, Ong BYS, Xie J, Lee TKY, Wang CH, Sahinidis NV. In vivo performance of implantable biodegradable preparations delivering Paclitaxel and Etanidazole for the treatment of glioma. Biomaterials. 2007;28:886–894. doi: 10.1016/j.biomaterials.2006.09.044. [DOI] [PubMed] [Google Scholar]
- Mu L, Feng SS. A novel controlled release formulation for the anticancer drug paclitaxel (Taxol): PLGA nanoparticles containing vitamin E TPGS. J Control Release. 2003;86(1):33–48. doi: 10.1016/s0168-3659(02)00320-6. [DOI] [PubMed] [Google Scholar]
- Mo Y, Lim LY. Preparation and in vitro anticancer activity of wheat germ agglutinin (WGA)-conjugated PLGA nanoparticles loaded with paclitaxel and isopropyl myristate. J Control Release. 2005;107(1):30–42. doi: 10.1016/j.jconrel.2004.06.024. [DOI] [PubMed] [Google Scholar]
- Win KY, Feng SS. In vitro and in vivo studies on vitamin E TPGS-emulsified poly(D,L-lactic-co-glycolic acid) nanoparticles for paclitaxel formulation. Biomaterials. 2006;27(10):2285–2291. doi: 10.1016/j.biomaterials.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Streck CJ, Dickson PV, Ng CY, et al. Antitumor efficacy of AAV-mediated systemic delivery of interferon-beta. Cancer Gene Ther. 2006;13(1):99–106. doi: 10.1038/sj.cgt.7700878. [DOI] [PubMed] [Google Scholar]
- Lagarce F, Garcion E, Faisant N, et al. Development and characterization of interleukin-18-loaded biodegradable microspheres. Int J Pharm. 2006;314(2):179–188. doi: 10.1016/j.ijpharm.2005.07.029. [DOI] [PubMed] [Google Scholar]
- Davidson BL, Hilfinger JM, Beer SJ. Extended release of adenovirus from polymer microspheres: potential use in gene therapy for brain tumors. Adv Drug Deliv Rev. 1997;27(1):59–66. [PubMed] [Google Scholar]
- Haller MF, Saltzman WM. Localized delivery of proteins in the brain: can transport be customized? Pharm Res. 1998;15(3):377–385. doi: 10.1023/a:1011911912174. [DOI] [PubMed] [Google Scholar]
- Thorne RG, Nicholson C. In vivo diffusion analysis with quantum dots and dextrans predicts the width of brain extracellular space. Proc Natl Acad Sci USA. 2006;103(14):5567–5572. doi: 10.1073/pnas.0509425103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroh M, Zipfel WR, Williams RM, Webb WW, Saltzman WM. Diffusion of nerve growth factor in rat striatum as determined by multiphoton microscopy. Biophys J. 2003;85(1):581–588. doi: 10.1016/S0006-3495(03)74502-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson C, Sykova E. Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 1998;21(5):207–215. doi: 10.1016/s0166-2236(98)01261-2. [DOI] [PubMed] [Google Scholar]
- Nicholson C. Diffusion and Related Transport Mechanisms in Brain Tissue. Reports on Progress in Physics. 2001;64:815–884. [Google Scholar]
- Zamecnik J. The extracellular space and matrix of gliomas. Acta Neuropathol (Berl) 2005;110(5):435–442. doi: 10.1007/s00401-005-1078-5. [DOI] [PubMed] [Google Scholar]
- Bobo RH, Laske DW, Akbasak A, Morrison PF, Dedrick RL, Oldfield EH. Convection-enhanced delivery of macromolecules in the brain. Proc Natl Acad Sci USA. 1994;91(6):2076–2080. doi: 10.1073/pnas.91.6.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison PF, Chen MY, Chadwick RS, Lonser RR, Oldfield EH. Focal delivery during direct infusion to brain: role of flow rate, catheter diameter, and tissue mechanics. Am J Physiol. 1999;277(4 Pt 2):R1218–R1229. doi: 10.1152/ajpregu.1999.277.4.R1218. [DOI] [PubMed] [Google Scholar]
- Neeves KB, Lo CT, Foley CP, Saltzman WM, Olbricht WL. Fabrication and characterization of microfluidic probes for convection enhanced drug delivery. J Control Release. 2006;111(3):252–262. doi: 10.1016/j.jconrel.2005.11.018. [DOI] [PubMed] [Google Scholar]
- Kawakami K, Kawakami M, Kioi M, Husain SR, Puri RK. Distribution kinetics of targeted cytotoxin in glioma by bolus or convection-enhanced delivery in a murine model. J Neurosurg. 2004;101(6):1004–1011. doi: 10.3171/jns.2004.101.6.1004. [DOI] [PubMed] [Google Scholar]
- Groothuis DR, Benalcazar H, Allen CV, et al. Comparison of cytosine arabinoside delivery to rat brain by intravenous, intrathecal, intraventricular and intraparenchymal routes of administration. Brain Res. 2000;856(1-2):281–290. doi: 10.1016/s0006-8993(99)02089-2. [DOI] [PubMed] [Google Scholar]
- Lopez KA, Waziri AE, Cannoll PD, Bruce JN. Convection-enhanced delivery in the treatment of malignant glioma. Neurol Res. 2006;28(5):542–548. doi: 10.1179/016164106X116836. [DOI] [PubMed] [Google Scholar]
- Hall WA, Rustamzadeh E, Asher AL. Convection-enhanced delivery in clinical trials. Neurosurg Focus. 2003;14(2):e2. doi: 10.3171/foc.2003.14.2.3. [DOI] [PubMed] [Google Scholar]
- Kaiser MG, Parsa AT, Fine RL, Hall JS, Chakrabarti I, Bruce JN. Tissue distribution and antitumor activity of topotecan delivered by intracerebral clysis in a rat glioma model. Neurosurgery. 2000;47(6):1391–1398. discussion 8-9. [PubMed] [Google Scholar]
- Bruce JN, Falavigna A, Johnson JP, et al. Intracerebral clysis in a rat glioma model. Neurosurgery. 2000;46(3):683–691. doi: 10.1097/00006123-200003000-00031. [DOI] [PubMed] [Google Scholar]
- Noble CO, Krauze MT, Drummond DC, et al. Novel nanoliposomal CPT-11 infused by convection-enhanced delivery in intracranial tumors: pharmacology and efficacy. Cancer Res. 2006;66(5):2801–2806. doi: 10.1158/0008-5472.CAN-05-3535. [DOI] [PubMed] [Google Scholar]
- Chen MY, Hoffer A, Morrison PA, et al. Surface properties, more than size, limiting convective distribution of virus-sized particles and viruses in the central nervous system. J Neurosurg. 2005;103(2):311–319. doi: 10.3171/jns.2005.103.2.0311. [DOI] [PubMed] [Google Scholar]
- Mamot C, Nguyen JB, Pourdehnad M, et al. Extensive distribution of liposomes in rodent brains and brain tumors following convection-enhanced delivery. J Neurooncol. 2004;68(1):1–9. doi: 10.1023/b:neon.0000024743.56415.4b. [DOI] [PubMed] [Google Scholar]
- MacKay JA, Deen DF, Szoka FC Jr. Distribution in brain of liposomes after convection enhanced delivery; modulation by particle charge, particle diameter, and presence of steric coating. Brain Res. 2005;1035(2):139–153. doi: 10.1016/j.brainres.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Hamilton JF, Morrison PF, Chen MY, et al. Heparin coinfusion during convection-enhanced delivery (CED) increases the distribution of the glial-derived neurotrophic factor (GDNF) ligand family in rat striatum and enhances the pharmacological activity of neurturin. Exp Neurol. 2001;168(1):155–161. doi: 10.1006/exnr.2000.7571. [DOI] [PubMed] [Google Scholar]
- Nguyen JB, Sanchez-Pernaute R, Cunningham J, Bankiewicz KS. Convection-enhanced delivery of AAV-2 combined with heparin increases TK gene transfer in the rat brain. Neuroreport. 2001;12(9):1961–1964. doi: 10.1097/00001756-200107030-00037. [DOI] [PubMed] [Google Scholar]
- Mastakov MY, Baer K, Kotin RM, During MJ. Recombinant adeno-associated virus serotypes 2- and 5-mediated gene transfer in the mammalian brain: quantitative analysis of heparin co-infusion. Mol Ther. 2002;5(4):371–380. doi: 10.1006/mthe.2002.0564. [DOI] [PubMed] [Google Scholar]
- Saito R, Bringas JR, McKnight TR, et al. Distribution of liposomes into brain and rat brain tumor models by convection-enhanced delivery monitored with magnetic resonance imaging. Cancer Res. 2004;64(7):2572–2579. doi: 10.1158/0008-5472.can-03-3631. [DOI] [PubMed] [Google Scholar]
- Saito R, Krauze MT, Bringas JR, et al. Gadolinium-loaded liposomes allow for real-time magnetic resonance imaging of convection-enhanced delivery in the primate brain. Exp Neurol. 2005;196(2):381–389. doi: 10.1016/j.expneurol.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Mardor Y, Rahav O, Zauberman Y, et al. Convection-enhanced drug delivery: increased efficacy and magnetic resonance image monitoring. Cancer Res. 2005;65(15):6858–6863. doi: 10.1158/0008-5472.CAN-05-0161. [DOI] [PubMed] [Google Scholar]
- Raghavan R, Brady ML, Rodriguez-Ponce MI, Hartlep A, Pedain C, Sampson JH. Convection-enhanced delivery of therapeutics for brain disease, and its optimization. Neurosurg Focus. 2006;20(4):E12. doi: 10.3171/foc.2006.20.4.7. [DOI] [PubMed] [Google Scholar]
- Krauze MT, Saito R, Noble C, et al. Effects of the perivascular space on convection-enhanced delivery of liposomes in primate putamen. Exp Neurol. 2005;196(1):104–111. doi: 10.1016/j.expneurol.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Hall W, Sherr GT. Convection-enhanced delivery: targeted toxin treatment of malignant glioma. Neurosurg Focus. 2006;20(4):E10. [PubMed] [Google Scholar]
- Popperl G, Goldbrunner R, Gildehaus FJ, et al. O-(2-[18F]fluoroethyl)-L-tyrosine PET for monitoring the effects of convection-enhanced delivery of paclitaxel in patients with recurrent glioblastoma. Eur J Nucl Med Mol Imaging. 2005;32(9):1018–1025. doi: 10.1007/s00259-005-1819-7. [DOI] [PubMed] [Google Scholar]
- Mardor Y, Roth Y, Lidar Z, et al. Monitoring response to convection-enhanced taxol delivery in brain tumor patients using diffusion-weighted magnetic resonance imaging. Cancer Res. 2001;61(13):4971–4973. [PubMed] [Google Scholar]
- Fahmy TM, Samstein RM, Harness CC, Saltzman WM. Surface modification of biodegradable polyesters with fatty acid conjugates for improved drug targeting. Biomaterials. 2005;26(28):5727–5736. doi: 10.1016/j.biomaterials.2005.02.025. [DOI] [PubMed] [Google Scholar]