

Abstract
Introduction: Early identification of chemoresistance in patients with ovarian cancer is of utmost importance in order to provide them with the most appropriate therapy. Recently, we described the expression of MyD88 in ovarian cancer cells that were resistant to the cytotoxic agent paclitaxel. In addition to chemoresistance, in MyD88 positive ovarian cancer cells, paclitaxel stimulates growth and production of proinflammatory cytokines. The objective of this study was to determine the correlation of MyD88 expression in primary and recurrent epithelial ovarian cancers with the response to carboplatin and paclitaxel combination chemotherapy. Methods: Tumors are heterogeneous structures that contain different cell populations, thus rendering the identification of specific tumor markers difficult. Using laser capture microdissection, pure cancer cells were isolated from ovarian malignant tumors that were obtained from 20 patients at the time of surgery. The microdissected cells were evaluated for the expression of MyD88, FasL, and XIAP by western blot analysis. Results: Protein expression was observed in samples containing as low as 500 cells. The results were correlated with the clinical course of those patients. It was evident that MyD88 expression in ovarian cancer cells accurately predicts a poor response to paclitaxel chemotherapy as shown by a short progression-free interval and overall survival. Conclusion: We describe for the first time a molecular approach to identify paclitaxel chemoresistance. Toxicity from agents without therapeutic benefit can be avoided by identifying those patients who will not respond to a specific agent. Molecular markers will enable us to design individualized treatments and improve overall survival.
Introduction
Ovarian cancer is the fourth leading cause of cancer-related deaths in women in the United States and the leading cause of gynecologic cancer deaths. The standard first-line chemotherapy for patients with ovarian cancer is a platinum-taxane combination regimen. In excess of 80 percent of patients will respond initially to this treatment. However, less than 10 percent will remain in remission [1].
Recently, we described a sub-group of epithelial ovarian cancer (EOC) cells that express the protein Myeloid Differentiation Protein 88 (MyD88). MyD88 is an adaptor protein that is required for Toll-like receptor (TLR) signaling, a signaling pathway involved in inflammatory response to bacterial and viral products [2]. EOC cells that express MyD88 constitutively secrete pro-inflammatory cytokines and are resistant to the taxane paclitaxel, which is a known TLR-4 ligand. Upon TLR-4 ligation with either LPS or paclitaxel, these cells secrete higher levels of pro-inflammatory cytokines and, more importantly, proliferate in culture [2]. We hypothesized that MyD88 is a specific marker for paclitaxel resistance and is required for paclitaxel-induced cell growth in EOC cells. The objective of this study was to develop an optimized method that can detect MyD88 expression in ovarian cancer tumors and thus function as a biomarker for selection of therapy.
Proteomics has emerged as an important tool to study biological processes in both physiological and pathological circumstances. Identification of specific proteins as biomarkers for a disease or condition may be used for diagnosis and/or therapy. Currently, however, no optimized method exists that can sensitively detect ovarian cancer biomarkers from tumor tissue prior to initiation of therapy. A limiting factor in the discovery and analysis of potential marker from tumor samples are the “contaminating” signals originating from normal cells, including immune cells, that are infiltrating the tumor. In order to overcome this difficulty, several investigators have developed mechanical and/or chemical approaches to separate the cellular components of the tumor. However, the purity of these preparations has never reached the desired levels required for diagnostic.
The use of laser capture microdissection (LCM) has been proposed as an alternative method to obtain pure cell populations. This approach has been used successfully to identify RNA expression and DNA in stored samples [3,4]. However, determination of RNA in cells obtained by LCM is time consuming and requires special measures to prevent RNA degradation. In the present study, we describe a novel approach for the detection of protein expression in as low as 1,000 pure epithelial ovarian cancer cells dissected by LCM. Furthermore, we demonstrate that MyD88 expression in LCM-dissected ovarian cancer cells can be used as a marker for paclitaxel resistance.
Materials and Methods
Patients and samples preparation
Ovarian cancer tissue samples were collected from advanced stage ovarian cancer patients at the time of surgery. All patients signed consent forms and the use of patient samples was approved under Yale University's Human Investigations Committee (HIC #10425).
Tissues were prepared in small aliquots and snap-frozen in liquid N₂. All samples were stored in cryovials at -80°C until future use.
Antibodies and reagents for immunocytochemistry (IHC)
Albumin from bovine serum/BSA was purchased from Sigma Aldrich, St. Louis, Missouri.
The primary antibody was Dako OV-TL mouse monoclonal cytokeratin-7 from Dakocytomation, Carpinteria, California. Primary antibody dilution was 1:150 in 1 percent BSA mixed in wash buffer. The secondary antibody used was biotinylated anti-mouse IgG (H+L) made in horse from Vector Laboratories, Burlingame, California. Dilution was 1:200 in 1 percent BSA/wash buffer. As detection reagent, we used Streptavidin-HRP Conjugate from Zymed, San Francisco, California, in a 1:300 dilution.
Antibodies and reagents for Western blot analysis
Primary antibodies used for Western blot analysis were rabbit polyclonal anti-human and mouse antibody to MyD88 (dilution 1:1000 in PBS-T/1 percent FFPM) from eBioscience (San Diego, California), mouse monoclonal anti-human antibodies to FasL/CD95L (dilution 1:10,000 in PBS-T/1 percent FFPM) from BD Biosciences (San Jose, California), mouse monoclonal anti-human antibodies to hIAL/XIAP (dilution 1:10,000 in PBS-T/1 percent FFPM) from BD Biosciences PharMingen (San Diego, California). Secondary peroxidase-conjugated antibodies, anti-rabbit IgG (H+L) made in goat and anti-mouse IgG (H+L) made in horse, were purchased from Vector Laboratories (Burlingame, California).
Slides preparation
In preparation for Laser capture microdissection, a 5x5 mm tissue sample was embedded in OCT at -20°C. Eight-micron sections were cut with the microtome and collected on Leica glass-foiled PEN-membrane slides. The slides were fixed in 95 percent ethanol for five minutes and stained with H&E for 30 seconds at RT, followed by washing in ddH₂O for 30 seconds. Dehydration is required for successful Laser microdissection, and this was accomplished by immersing the slides in 95 percent ethanol and then in Histosolve, a xylene substitute (Shandon, Inc., Pittsburgh, Pennsylvania), 15 minutes each. The slides were air dried at RT for 15 minutes. Following dehydration, to prevent protein degradation, the samples were either used immediately for microdissection or they were stored in air-tight plastic wrapping at -20°C.
Immunohistochemistry for Laser microdissection
Eight-micron sections were cut with the microtome, collected on Leica glass-foiled PEN-membrane slides and fixed in 95 percent ethanol for five minutes as described above. The slides were then placed in a staining dish with 0.1 percent H₂O₂ in 0.1M PB for 10 minutes to quench for endogenous peroxidase activity and then washed in wash buffer three times. Wash buffer was prepared by adding 0.01 percent Triton X-100 (Sigma Aldrich, St. Louis, Missouri) to 0.1M PB. The area around the tissue sections was scored with a Pap pen to limit the amount of antibodies and reagents used. All steps occurred at room temperature with the slides placed in a moisture chamber to keep the tissue from drying out during the procedure. To block for non-specific background, 100-200 µl of 3 percent BSA made in wash buffer were added to the circumscribed areas and incubated for 20 minutes in the moisture chamber. Next, the primary antibody was incubated for two hours followed by washing with wash buffer (each wash is two to three minutes times three), the specimens were incubated with Biotin labeled secondary antibody for 30 minutes. After this incubation, the slides were washed once with wash buffer then twice with 0.1M Tris (pH 7.5). The tissue was incubated with Streptavidin-HRP conjugate for 30 minutes and then washed with 0.1 M Tris three times. The color was developed with DAB (2.5 mg DAB in 5 ml 0.1 M Tris). Twenty five µl of 0.03 percent H₂O₂ were added to the chromogen just before use. At the end of the procedure, the slides were washed with double distilled water and counterstained with Mayer’s hematoxylin (Sigma Aldrich, St. Louis, Missouri). The specimens were dehydrated and then stored as described above.
Laser Microdissection for Western Blot Analysis
The slides were placed inverted into the holder of a Leica AS LMD Laser Microdissection System with the side containing the tissue facing down (non-contact method). Tumor cells were selected according to their morphology for H&E stained samples or according to their immunoreactivity for CK-7 stained samples. Dissected cells were collected in the cup of 0.5ml Eppendorf tubes containing 35 µl sample buffer (2.5 percent SDS, 10 percent glycerol, 5 percent β-mercapto-ethanol, 0.15M Tris (pH = 6.8) and 0.01 percent bromophenol blue). After a short centrifugation, lysis of the cells was achieved by submitting the samples to five cycles of freezing (liquid N₂) and thawing (95°C) followed by boiling for five minutes, centrifuged again for two minutes and then stored at -80°C until further use.
All procedures were carried out in a timely manner to minimize the activity of proteases. For H&E stained tissues, the interval from tissue frozen section cutting to cell dissection and lysis was under one hour while immunostained (CK-7 or CD45) stained specimens required approximately four hours.
Western blot analysis
Proteins were resolved under reducing conditions on 12 percent SDS-PAGE gels and then transferred onto polyvinylidene difluoride paper (NEN Life Sciences, Boston, Massachusetts) [5]. Membranes were blocked at room temperature for one hour with 5 percent FFPM in PBS/0.05 percent Tween 20 (PBS-T). After three washes for 10 minutes, each with PBS-T, membranes were incubated overnight at 4°C with the primary antibody in PBS-T/1 percent FFPM. After incubation with the primary antibody, membranes were washed three times with PBS-T and then incubated at room temperature with the appropriate secondary antibody conjugated to peroxidase (Vector Laboratories) in PBS-T/1 percent FFPM. After three washes for 10 minutes each with PBS-T and three washes for 10 minutes each with distilled water, the peroxidase-conjugated antibody was detected by enhanced chemoluminiscence (PerkinElmer Life Sciences, Boston, Massachusetts). The signal intensity was analyzed using a Kodak digital imaging analysis system (Kodak Image Station 4000MM) and Kodak Molecular Imaging Software (Scientific Imaging Kodak Company).
Results
Protein detection in isolated cells
Our first objective was to determine whether we could detect protein expression by Western blot analysis in microdissected cells obtained from ovarian tumor samples that were snap-frozen in liquid N2. Thus, we microdissected increasing number of cells with a maximum of 8,000 cells and a minimum of 100 cells from 8 µm ovarian cancer sections and then analyzed them for the expression of MyD88 and other cancer–related proteins such as XIAP [6-8] and FasL [9,10] [Figure 1]. As shown in Figure 2, a strong signal for MyD88 expression was observed in samples containing 8,000 and 5,000 microdissected cells, and no signal was detected for samples containing less than 1,000 cells.
Figure 1.
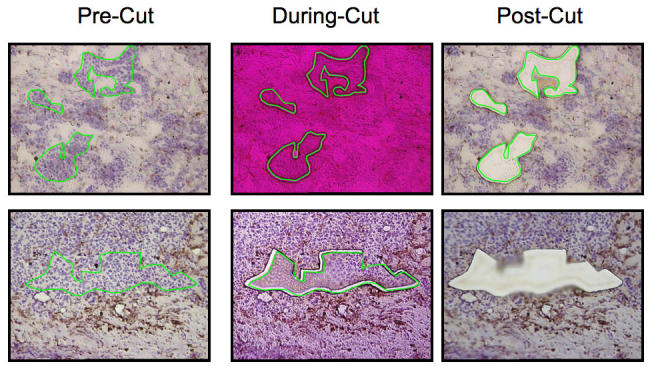
Frozen sections of OC were prepared and stained with H&E. The lines indicate the areas selected for microdissection, then this group of cancer cells is microdissected using a low power pulse laser. The figure shows two different samples before (pre-cut), during (during-cut), and after (post-cut) microdissection.
Figure 2.
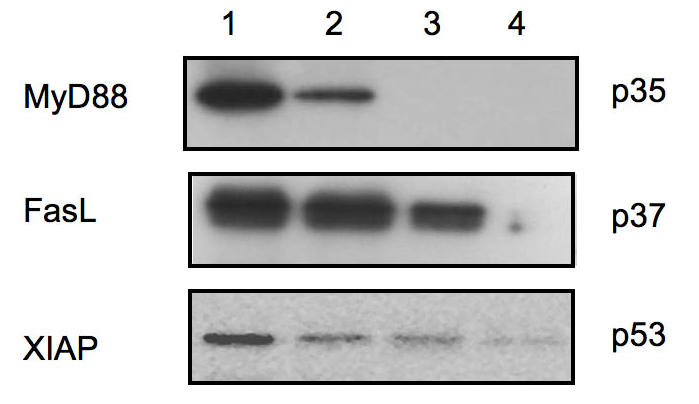
Western blot analysis for microdissected ovarian cancer cells. Eight microns thick frozen sections of epithelial ovarian tumors were prepared and stained with H&E. Different numbers of pure cancer cells were microdissected and evaluated for the expression of FasL, XIAP, and MyD88. 1 = 8,000 cells; 2 = 5,000 cells; 3 = 1000 cells; 4 = 100 cells. As shown, a strong signal was observed for MyD88 expression in samples containing 8,000 and 5, 000 microdissected cells, and no signal was detected for samples containing less than 1000 cells. For FasL and XIAP, a signal can be observed with as little as 1000 cells.
In order to determine whether the sensitivity of the assay remains similar for different proteins, we evaluated the expression of XIAP and FasL using the same number of cells. Contrary to MyD88, XIAP and FasL expression is detected in samples containing as little as 1000 and 100 cells, respectively [Figure 2].
We then evaluated whether the sample thickness affects the signal detected by Western blot. Although increasing the section thickness did increase the amount of protein, it affected cutting time and rendered dissection extremely difficult. Therefore, we standardized the method to eight µm sections. This allows a quick dissection and provides enough protein to be detected by Western blot (data not shown).
MyD88 expression is cell-type specific
Tumors are heterogeneous tissues containing normal supporting cells, immune cells and cancer cells. Immune cells infiltrate the stroma and may “contaminate” the protein profile of the tumor sample [Figure 3]. Indeed, when we analyzed MyD88 expression in H&E stained samples without using appropriate markers for cell type identification, we found that MyD88 was expressed in all samples. The explanation for these results is that the immune cells present in the tumor tissue and surrounding the cancer cells express MyD88 [Figure 3A]. In an H&E stained sample, the selection of specific cells based on their morphology alone is not sufficient to identify intra-tumoral infiltrated cells. An advantage of the LCM is that it affords the possibility of selecting a specific group of cells based on the expression of cellular markers. Therefore, our next objective was to determine whether immunohistochemistry staining affects the detection of proteins in microdissected samples. Thus, we compared the sensitivity of protein detection of cells obtained from H&E stained samples with cells obtained from immuno-stained samples. Tumor tissues were immuno-stained with either antibodies for cytokeratin 7 (epithelial marker) or CD45 (pan-leucocyte marker). Specific staining for cytokeratin 7 and CD45 was observed in these frozen sections [Figure 3A and data not shown]. When we compared MyD88 and XIAP expression in cells obtained from these two preparations, no difference was observed in the signal intensity for these proteins between the two groups, suggesting that the process of IHC does not affect the samples’ integrity [Figure 3B]. Once we established that IHC does not affect protein detection, we evaluated MyD88 expression in ovarian cancer samples after staining for CD45 (panleukocyte antigen). As shown in Figure 4, when we dissected only the cancer cells (CD45 negative cells) and avoided the immune cells (CD45 positive cells), MyD88 was differentially expressed in tumors from different patients with identical pathological diagnoses.
Figure 3.
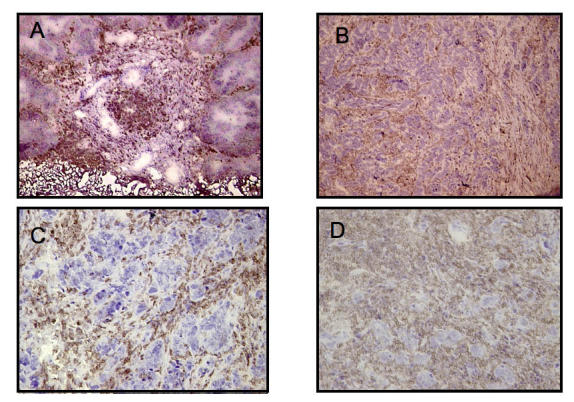
The presence of strong immune infiltrate is recognized in ovarian malignant tumors. The frozen sections were stained with anti-CD45, a specific marker for leukocytes. A. Papillary serous ovarian adenocarcinoma section with a necrotic center. B-D. Immune infiltrate cells (darker stain) can easily be recognized from the cancer cells (nuclei).
Figure 4.
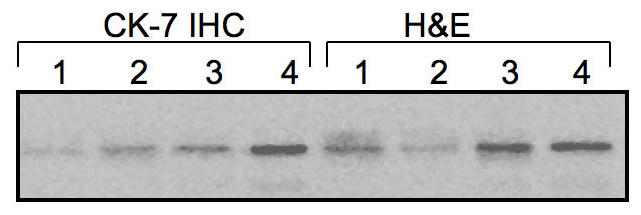
Effect of IHC on protein stability. Frozen sections of ovarian tumors were prepared and stained with H&E alone or with an anti-cytokeratine 7 (CK-7) antibody for epithelial cells. Different number of cells was microdissected and evaluated for MyD88 expression. 1 = 8,000 cells; 2 = 5,000 cells; 3 = 1000 cells; 4 = 100 cells. The original tissue morphology is preserved and contamination from surrounding unwanted cells is avoided. The process does not influence the quantity or quality of proteins contained in the specimen.
Clinical correlation between MyD88 expression and Survival/Progression Free Interval
We evaluated 20 tumor samples from patients with epithelial ovarian cancer. The patient characteristics are summarized in Figure 4. Eleven out of 20 tumors were MyD88 positive and nine tumors were MyD88 negative. Figures 5 and 6 show two representative samples after CD45 staining in preparation for the laser capture microdissection and MyD88 expression in pure cancer cells.
Figure 5.
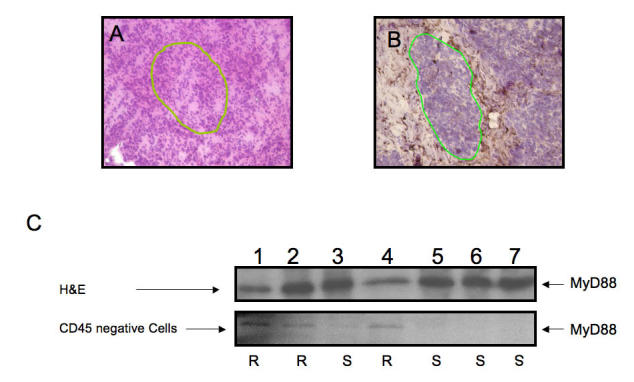
Contamination of samples by CD45+ cells. (A) H&E staining and differentiation of cells based on nuclei morphology alone is not specific enough in frozen samples for obtaining pure cancer cells. The mononuclear immune infiltrate, as evident after CD45 staining, is substantial. The ability to isolate pure cancer cells is greatly increased with IHC Contamination of the sample with immune cells creates a false positive signal. (B) Identification of cancer cells is facilitated after CD45 staining. (C) Microdissection of CD45 negative cells creates a sample pure enough to show differential results between chemosensitve (S) and chemoresistant (R) ovarian cancer patients. Line: area selected for microdissection.
Figure 6.
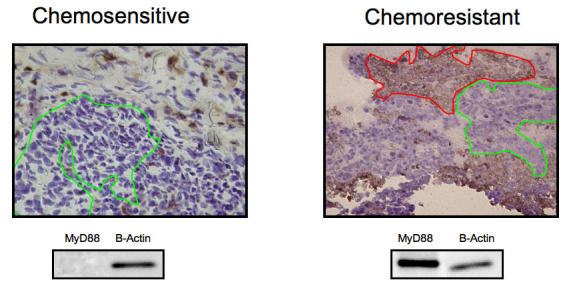
Representative figures for MyD88 positive and negative tumors. Although the histological diagnosis and the clinical stage of the two tumors were identical, the clinical course of the disease for each patient was different and correlated with MyD88 expression. Both received the same treatment, consisting of optimal surgical cytoreduction to no residual disease and combination chemotherapy with carboplatin and paclitaxel. The first tumor was MyD88 negative and the clinical course of the patient was characterized by complete response to Taxol and carboplatin combination therapy. The second tumor proved to be MyD88 negative and the patient had progression of disease during chemotherapy with Taxol and carboplatin. She died of disease 11 months later, despite optimal cytoreductive surgery and chemotherapy with other agents. Line: selected ovarian cancer cells.
From the MyD88 positive tumors, 7/11 were obtained at the time of initial surgery from patients who received neoadjuvant chemotherapy with carboplatin and paclitaxel. These patients had persistent or progression of disease during neoadjuvant chemotherapy. Even after neoadjuvant chemotherapy and optimal tumor debulking, these patients failed postoperative chemotherapy. Their progression-free interval was zero, and four patients died of disease in less than one year. In this group, mean survival was 16 months.
From the MyD88 negative tumor samples, 5/7 were obtained at the time of initial surgery from patients with poorly differentiated serous papillary adenocarcinomas. Three patients were stage IIIC, one was stage IIIB, and one was stage IIC. In this group, all patients were treated with carboplatin and paclitaxel and the mean progression-free interval was 35 months. The mean overall survival in this group was 45 months, and three patients are alive at the time of this report.
Four of eleven MyD88 positive tumors were obtained from patients with recurrent disease who underwent optimal secondary cytoreductive surgery. All four patients (two with recurrent stage IC, one with recurrent stage IIA, and one with recurrent stage IIC) received carboplatin and paclitaxel. Their mean progression-free interval after completion of chemotherapy was three months.
Four MyD88 negative tumor samples were obtained from patients with recurrent disease at the time of the secondary cytoreduction. Postoperatively, these patients were treated with paclitaxel and carboplatin combination chemotherapy. One patient with stage IIIC recurred 33 months after primary treatment and one patient with stage IVA recurred 20 months after primary treatment. Mean progression-free interval in this group was 14 months.
A statistical significant correlation between assay prediction of response and progression-free interval (PFI) was observed in all these cases [Figure 7]. Furthermore, as shown by the Kaplan-Meier curve in Figure 8, the patients that had MyD88 negative tumors responded better to treatment and had a significantly longer survival when compared with the patients that had MyD88 positive tumors [Figure 8]
Figure 7.

Kaplan-Meier curves illustrate the duration of progression-free interval and overall survival for patients with MyD88+ and MyD88- primary tumors. The pathological diagnosis was papillary serous adenocarcinoma of the ovary for all patients. However, the clinical course and the response to combination chemotherapy with carboplatin and paclitaxel were markedly different and correlated with MyD88 expression in the analyzed tumors. A. Mean progression-free interval was zero for patients with MyD88+ tumors (n = 7) and 35 months for patients with MyD88- tumors (n = 5) treated with carboplatin and paclitaxel. This difference was statistically significant (log rank statistical test p = 0.009). B. Overall survival according to the MyD88 status of the analyzed tumors shows divergent Kaplan-Meier curves and was statistically significant (-2 log likelihood ratio test p = 0.022).
Figure 8.
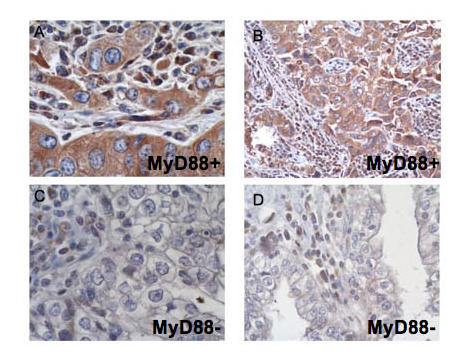
Immunohistochemistry for MyD88 on paraffin sections. While the immune infiltrate is positive for MyD88 in all specimens, the cytoplasm of the cancer cells shows intense staining for MyD88 in A and B (Type I, MyD88 positive EOC) and none in C and D (Type II, MyD88 negative EOC).
Discussion
This study challenges the current approach of prescribing chemotherapy by relying solely on the pathological diagnosis. Instead, we propose that determination of molecular markers be undertaken and incorporated into the diagnostic process prior to treating patients with neoplastic diseases of the ovary. We show that the status of MyD88 expression may have a significant impact in chemo-responsiveness and therefore may be an indispensable piece of information to obtain prior to commencing treatment.
Although the link between inflammation, NF-κB, and cancer progression has been well characterized, the specific signaling pathway upstream of NF-κB is currently not well elucidated. As mentioned above, MyD88 is one of the proteins required in TLR4-mediated early phase NF-κB activation [11]. Our previous studies [2] showed that Type I epithelial ovarian cancer (EOC) cells constitutively secrete IL-6, IL-8, GROα, and MCP-1. In response to LPS stimulation of TLR4, these cells proliferated and showed marked increase in the secretion of these cytokines via the activation of NF-κB. In contrast, type II EOC cells do not constitutively secrete cytokines, and no changes in cell proliferation or cytokine production were observed following treatment with LPS. The characteristic pro-inflammatory environment generated by Type I EOC cells was lost upon the knockdown of MyD88, suggesting that an active MyD88-dependent TLR4 signaling is responsible for the LPS-induced, NF-κB-mediated EOC cell proliferation and cytokine secretion.
As mentioned above, paclitaxel, a first-line chemotherapeutic agent used in the treatment of ovarian cancer [12-14], is a known TLR4 ligand [2,15,16]. Thus, we also determined if paclitaxel treatment would induce the same response in type I EOC cells as was seen with LPS. Our results showed that similar to LPS, paclitaxel also induced NF-κB activation, increased secretion of pro-inflammatory cytokines, and proliferation of type I EOC cells. In contrast, no cytokine secretion was observed in type II EOC cells, and more importantly, they underwent apoptosis in response to paclitaxel. These results suggest that an activated MyD88-dependent TLR4 signaling pathway to NF-κB confers EOC cells the ability to promote a pro-inflammatory environment and the development of paclitaxel resistance. It is noticeable of the specificity of the signaling pathway to paclitaxel, since treatment of type I EOC cells with carboplatin, a chemotherapeutic drug which is not a TLR4 ligand, did induce neither proliferation nor cytokine production [17]. The mechanism that regulates MyD88 expression in these two types of cells, and the correlation of MyD88 expression and clinical response to paclitaxel in patients with EOC, however, still remains to be determined.
Moreover, endogenous ligands of TLR4 such as HMGB1, heparan sulfate and polysaccharide fragments of hyaluronan, which are released from the damaged tissue and necrotic cells, may also contribute to cancer progression and paclitaxel resistance [18-20].
Tumors contain different cell populations that are incorporated in a heterogeneous structure. These different cell types can be studied after meticulous isolation. LCM represents an alternative approach to obtaining pure cancer cells from fresh-frozen cancer tissue, thus providing an accurate snapshot of the tumor and its microenvironment in vivo. After lysis, these cells can be analyzed and their protein expression characterized by Western blot, ELISA, and Luminex assays.
Early identification of chemoresistance in patients with ovarian cancer has important treatment implications. It allows tailoring of therapy to individual patients and prevents the use of cytotoxic drugs with no therapeutic benefit while avoiding toxicities. Unfortunately, no available method exists to predict the chemoresponse of cancer cells prior to initiation of therapy.
The standard treatment for ovarian cancer is the combination of carboplatin and paclitaxel. Unfortunately, approximately 15 percent of newly diagnosed patients will not respond to this combination, and their disease will progress during or shortly after completion of chemotherapy. This percentage of non-responders increases to 65 to 75 percent for recurrent cancers.
Based on these studies, we hypothesized that MyD88 expression can be used as a marker for paclitaxel-resistance. However, we found that a limiting factor in the detection of MyD88 in whole tumors was the presence of intra-tumoral immune infiltrate. Immune cells, mainly macrophages, express high levels of MyD88 and may give a false positive result.
Utilizing LCM and immunostaining protocols that allow clear identification of the desired cell types can overcome this obstacle. In our experiments, CD45 staining proved to be highly useful for the identification and selection of pure cancer cells from tumor tissue and classification of tumors as either MyD88 positive or negative.
We found that proteins expressed in high-levels, such as FasL or XIAP, can be detected with as low as 1,000 cells. However, for proteins with lower expression levels, like MyD88, a minimum of 5,000 cells was required for detection.
The advent of the LCM, used in conjunction with protein or gene detection techniques, has enabled us to gain a better perception of the complex interactions that occur within and among cells in living tissues. In this study, we have found that fresh-frozen tissue is a suitable material for this procedure. We have demonstrated in this report how proteins involved in the apoptotic cascade and chemoresistance of epithelial ovarian cancer cells can be accurately detected using a three-step approach. First, tumor specimens are snap-frozen in liquid nitrogen and eight µm thick specimens are stained to facilitate the selection of pure tumor cells. Second, target cells are identified based on their immunostaining and collected with the LCM system. The third step involves lysis of the collected cells followed by Western blot or Luminex multiplex assay.
The protocol presented here provides a fast and easy method for analyzing protein expression in tissues after LCM. We have used this method to evaluate the correlation between MyD88 expression and clinical outcome, and we have found that all patients who had MyD88 positive tumors presented with poor progression-free interval and overall survival after chemotherapy with carboplatin and paclitaxel, while the patients with MyD88 negative tumors had an excellent response to chemotherapy. A prospective clinical study to validate the specificity and sensitivity of this test aimed at identifying paclitaxel-resistant patients is under way. We have recently developed an immunohistochemistry protocol for MyD88 that is faster, less labor intensive, and can be applied to paraffin sections [Figure 8].
In summary, we describe a sensitive method to determine MyD88 as a potential marker for chemoresponse to paclitaxel. We have identified a specific subclass of cancer patients who, based on their MyD88 expression, are paclitaxel resistant. The use of molecular markers like MyD88 will enable us to design an individualized of treatment, thereby eliminating toxicity from agents without therapeutic benefit or even potential harm, as evidenced by the stimulating effect of paclitaxel on MyD88 positive EOC.
Acknowledgments
This study was supported in part by grants from the Sands Family Foundation and Nicholas Brady.
Abbreviations
- EOC
Epithelial ovarian cancer
- MyD88
Myeloid Differentiation Protein 88
- TLR
Toll-like receptor
- LCM
Laser capture microdissection
- IHC
immunocytochemistry
References
- Schwartz PE. Current diagnosis and treatment modalities for ovarian cancer. Cancer Treat Res. 2002;107:99–118. doi: 10.1007/978-1-4757-3587-1_4. [DOI] [PubMed] [Google Scholar]
- Kelly MG, Alvero AB, Chen R, Silasi DA, Abrahams VM, Chan S, et al. TLR-4 signaling promotes tumor growth and paclitaxel chemoresistance in ovarian cancer. Cancer Res. 2006;66(7):3859–3868. doi: 10.1158/0008-5472.CAN-05-3948. [DOI] [PubMed] [Google Scholar]
- Ding Y, Xu L, Chen S, Jovanovic BD, Helenowski IB, Kelly DL, et al. Characterization of a method for profiling gene expression in cells recovered from intact human prostate tissue using RNA linear amplification. Prostate Cancer Prostatic Dis. 2006;9:379–391. doi: 10.1038/sj.pcan.4500888. [DOI] [PubMed] [Google Scholar]
- Matsuzaki S, Canis M, Pouly JL, Botchorishvili R, Dechelotte PJ, Mage G. Differential expression of genes in eutopic and ectopic endometrium from patients with ovarian endometriosis. Fertil Steril. 2006;86(3):548–553. doi: 10.1016/j.fertnstert.2006.02.093. [DOI] [PubMed] [Google Scholar]
- Alvero AB, O'Malley D, Brown D, Kelly G, Garg M, Chen W, et al. Molecular mechanism of phenoxodiol-induced apoptosis in ovarian carcinoma cells. Cancer. 2006;106(3):599–608. doi: 10.1002/cncr.21633. [DOI] [PubMed] [Google Scholar]
- Asselin E, Mills GB, Tsang BK. XIAP regulates Akt activity and caspase-3-dependent cleavage during cisplatin-induced apoptosis in human ovarian epithelial cancer cells. Cancer Res. 2001;61(5):1862–1868. [PubMed] [Google Scholar]
- Holcik M, Gibson H, Korneluk RG. XIAP: apoptotic brake and promising therapeutic target. Apoptosis. 2001;6(4):253–261. doi: 10.1023/a:1011379307472. [DOI] [PubMed] [Google Scholar]
- Sapi E, Chen W, O'Malley D, Hao X, Dwipoyono B, Garg M, et al. Resistance of ovarian cancer cells to docetaxel is XIAP dependent and reversible by phenoxodiol. Anti-Cancer Drugs. 2004;14:567–578. doi: 10.3727/0965040042707943. [DOI] [PubMed] [Google Scholar]
- Gutierrez L, Eliza M, Niven-Fairchild T, Naftolin F, Mor G. The Fas/Fas-Ligand system: A mechanism for immune evasion in human breast carcinomas. Breast Cancer Research and Treatment. 1999;54(3):245–253. doi: 10.1023/a:1006102601215. [DOI] [PubMed] [Google Scholar]
- Abrahams V, Straszewski S, Kamsteeg M, Hanczaruk B, Schwartz P, Rutherford T, et al. Epithelial ovarian cancer cells secrete functional Fas Ligand. Cancer Res. 2003;63:5573–5581. [PubMed] [Google Scholar]
- Takeda K, Akira S. TLR signaling pathways. Semin Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Ozols RF, Bundy BN, Greer BE, et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2003;21:3194–3200. doi: 10.1200/JCO.2003.02.153. [DOI] [PubMed] [Google Scholar]
- Neijt JP, Engelholm SA, Tuxen MK, Sørensen PG, Hansen M, Sessa C, et al. Exploratory phase III study of paclitaxel and cisplatin versus paclitaxel and carboplatin in advanced ovarian cancer. J Clin Oncol. 2000;18:3084–3092. doi: 10.1200/JCO.2000.18.17.3084. [DOI] [PubMed] [Google Scholar]
- Bookman MA, Greer BE, Ozols RF. Optimal therapy of advanced ovarian cancer: carboplatin and paclitaxel vs. cisplatin and paclitaxel (GOG 158) and an update on GOG0 182-ICON5. Int J Gynecol Cancer. 2003;13(6):735–740. doi: 10.1111/j.1525-1438.2003.13602.x. [DOI] [PubMed] [Google Scholar]
- Byrd-Leifer CA, Block EF, Takeda K, Akira S, Ding A. The role of MyD88 and TLR4 in the LPS-mimetic activity of taxol. Eur J Immunol. 2001;31:2448–2457. doi: 10.1002/1521-4141(200108)31:8<2448::aid-immu2448>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Ding AH, Porteu F, Sanchez E, Nathan CF. Shared actions of endotoxin and taxol on TNF receptors and TNF release. Science. 1990;248:370–372. doi: 10.1126/science.1970196. [DOI] [PubMed] [Google Scholar]
- Chen R, Alvero AB, Silasi DA, Mor G. Inflammation, cancer and chemoresistance: taking advantage of the Toll-like receptor signaling pathway. Am J Reprod Immunol. 2007;57:93–107. doi: 10.1111/j.1600-0897.2006.00441.x. [DOI] [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, et al. Species-specific recognition of single stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Schlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- Tsan MF. Toll-like receptors, inflammation and cancer. Semin Cancer Biol. 2006;16:32–37. doi: 10.1016/j.semcancer.2005.07.004. [DOI] [PubMed] [Google Scholar]