

Abstract
The G-protein coupled receptors (GPCRs) are a class of membrane proteins that trigger cellular responses to external stimuli, and are believed to be targets for nearly half of all pharmaceutical drugs on the market. However, little is known regarding their folding and cellular interactions, as well as what factors are crucial for their activity. Further structural characterization of GPCRs has largely been complicated by problems with expression, purification, and preservation of activity in vitro. Previously, we have demonstrated high-level expression (~4 mg/L of culture) of functional human adenosine A2a receptor fused to a green fluorescent protein (A2aR-GFP) from Saccharomyces cerevisiae. In this work we re-engineered A2aR with a purification tag, developed an adequate purification scheme, and performed biophysical characterization on purified receptors. Milligram amounts per liter of culture of A2aR and A2aR-GFP were functionally expressed in S. cerevisiae, with a C-terminal deca-histidine tag. Lysis procedures were developed for optimal membrane protein solubilization and recovery through monitoring fluorescence of A2aR-GFP-His10. One-step purification of the protein was achieved through immobilized metal affinity chromatography. After initial solubilization in n-dodecyl-β-D-maltoside (DDM), a combination of added cholesterol hemisuccinate (CHS) in 3-(3-cholamidopropyl)-dimethylammoniopropane sulfonate (CHAPS) was required to stabilize the functional state of the protein. Isolated A2aR under these conditions was found to be largely alpha-helical, and properly incorporated into a mixed-micelle environment. The A2a-His10 receptor was purified in quantities of 6 +/− 2 mg/L of culture, with ligand-binding yields of 1 mg/L, although all protein bound to xanthine affinity resin. This represents the highest purified total and functional yields for A2aR yet achieved from any heterologous expression system.
INTRODUCTION
The G-Protein Coupled Receptors (GPCRs) are alpha helical seven transmembrane proteins that are embedded within the cellular plasma membrane. GPCRs are capable of binding to a diverse set of extracellular ligands, which upon ligand binding, undergo a conformational change in the interaction with trimeric G-proteins contributing to intracellular signal transduction. This event allows for further downstream signaling to be brought about through several available cellular pathways (Hill, 2006).
Since cellular communication largely relies on GPCR-mediated signaling, these proteins have emerged as desirable targets for medicinal therapies. GPCRs have been found to play vital roles in maintaining normal function in yeast, insect, and mammalian cells, and the Human Genome Project has identified upwards of 1000 GPCRs in humans (Howard et al., 2001; Venter et al., 2001). In addition to their direct roles in the mammalian visual and olfactory system, these proteins also have been linked to various acute and chronic diseases including cancer, HIV infection, diabetes, and heart disease (Flower, 1999; Jacobson et al., 2002; Pausch, 1997). More detailed structural characterization of individual GPCRs would arguably advance structure-based drug design, as greater than 50% of pharmaceuticals on the market are estimated to affect these proteins in some way (Loll, 2003).
Unfortunately, membrane proteins have proven notoriously difficult to study due to difficulties with their overexpression, purification, and maintenance of proper structure in vitro. These roadblocks have hindered high-resolution structure determination for most known membrane proteins, causing information about them to lag considerably behind their soluble counterparts (Wiener, 2004). Furthermore, problems with low natural expression of mammalian membrane proteins hinder important basic biophysical characterization through circular dichroism or fluorescence spectroscopy. One of the most notable exceptions is rhodopsin, a light-activated GPCR, which exists in extremely high purity from native tissues. Due to its natural abundance and stability in a wide range of different surfactants, rhodopsin is currently the only GPCR for which a high-resolution structure has been determined (Palczewski et al., 2000). Although studies on rhodopsin have yielded valuable information regarding GPCR characteristics in general, they have not provided the vital structural information needed for individual GPCRs (Flower, 1999).
In order to facilitate the study of other mammalian GPCRs, many attempts have been made to circumvent the problem of low natural abundance by using heterologous, non-native expression systems to produce these proteins in sufficient quantities for characterization (Loll, 2003; Sarramegna et al., 2003; Tate and Grisshammer, 1996). Through these techniques, GPCRs have been functionally expressed in various types of mammalian, insect, fungal, and bacterial hosts, yet no one system has emerged as superior for the expression of all GPCRs (Sarramegna et al., 2006; Sarramegna et al., 2003; Tate and Grisshammer, 1996). A recent review by Sarramegna (Sarramegna et al., 2006) cites typical GPCR expression levels in these hosts ~ μg/L of culture, yet a few unusual successes for heterologous expression have been achieved. These include the rat neurotensin receptor in E. coli (Grisshammer and Tucker, 1997), the human adenosine A2a receptor in S. cerevisiae (Niebauer and Robinson, 2006), the human cannabinoid CB1 receptor in insect cells (Akermoun et al., 2005), and hamster β2-adrenergic receptor in mammalian cells (Chelikani et al., 2006). However, high-level expression does not always lead to successful functional purification, as techniques for membrane protein purification are not yet well-developed (Booth, 2003; Keller et al., 2005). Furthermore, several receptors have been expressed functionally on the cell surface, but do not retain their activity post-purification (Feng et al., 2002; Kim et al., 2005).
Yeast, specifically S. cerevisiae, offer an advantage to mammalian GPCR expression since this system combines rapid, inexpensive growth with cellular machinery similar to that of higher eukaryotes. Like mammalian cells, yeast also use GPCRs and G-proteins naturally to communicate with their environment (Pausch, 1997). Also, they have the ability to perform most post-translational protein modifications, which may be essential for proper protein function (Eckart and Bussineau, 1996). Specifically, for the expression of GPCRs in yeast, advances have made use of sequence information gathered from the endogenous STE2 yeast receptor, in order to properly target the protein to the membrane (Reilander and Weiss, 1998; Sarramegna et al., 2003). While yeast have somewhat been overlooked for membrane protein over-expression, their use has undergone a recent resurgence. In fact, of the relatively few published membrane protein crystal structures obtained from recombinant sources, most have resulted from yeast expression systems (White, 2006). Combined with their inexpensive operation and ease of use, yeast expression systems present an attractive alternative to more complicated mammalian or insect cell cultures for the expression of authentic mammalian GPCRs.
Previously, we have developed a yeast expression system in S. cerevisiae which produces milligram amounts per liter of culture of membrane-localized functional human adenosine A2a receptor (A2aR) (Niebauer and Robinson, 2006). A2aR is a 45 kDa protein, and one of the four known subtypes of the adenosine receptor family (A1, A2a, A2b, and A3). It has known roles in cardioprotection, inflammation, and has been linked to heart disease (Jacobson et al., 2002; Mubagwa and Flameng, 2001; Poulsen and Quinn, 1998). The full-length human adenosine A2a receptor has been widely expressed in different host systems (Fraser, 2006; Kamiya et al., 2003; Niebauer and Robinson, 2006; Weiss and Grisshammer, 2002). However, at this point in time, no high-resolution structure has been determined for A2aR, and relatively little information exists pertaining to its folding, stability, and detailed structure (Thevenin et al., 2005).
In our previous studies, we sought to develop a yeast expression system for A2aR, and quantify the amount of active and total protein which could be produced. A high-expressing A2aR-GFP clonal isolate was identified through the use of flow cytometry, and numerous methods were employed to quantify its production levels (Niebauer and Robinson, 2006). Cellular chaperone manipulation was also explored in order to increase proper folding and heightened expression of A2aR produced in this system (Butz et al., 2003). Furthermore, culture conditions which may affect total and functional expression levels were also extensively analyzed (Wedekind et al., 2006). Overall, our previous work has shown that expression of A2aR in S. cerevisiae undergoes a per-cell maxima over time (~25 hours of expression), which has been attributed to a translational or post-translational expression bottleneck (Niebauer et al., 2004). Due to our adequate understanding of the expression system, we have been able to identify conditions which correlate well with the highest total and functional expression levels of A2aR.
Methods we have developed to achieve high-level expression of the A2a receptor have well prepared us for purification of this GPCR in order to study its structure, interactions, and stability in vitro. Here, we demonstrate our ability to produce, purify, and characterize functional human adenosine A2a receptor with an added purification tag as expressed from S. cerevisiae. Using these techniques, we are able to routinely produce milligram amounts of total and functional A2aR from batch culture for structural studies. Applying spectroscopic methods to purified protein has shown that it is entrapped within a micellar environment, and has native-like secondary structure. Finally, the role of the endogenous lipid for the functionality and proper folding of A2aR are also considered.
MATERIALS AND METHODS
Sub-cloning of A2aR –His10 and A2aR -GFP-His10
The multi-integrating pITy4-wt plasmid (Parekh et al., 1996), previously used for overexpression of A2aR in S. cerevisiae, was also employed in these studies. pITy contains a Gal1-10 promotor for inducible expression upon addition of galactose in the growth medium, a synthetic pre-pro leader sequence which directs the protein for secretion (Clements et al., 1991; Parekh et al., 1995), and the yeast alpha terminator.
pITy4-β (Smith and Robinson, 2002) was digested with AflII and EagI restriction enzymes in order to remove the β-glucosidase gene, and the empty plasmid was recovered through gel extraction using a PCR-cleanup kit (Promega, Madison, WI). Complementary oligonucleotides containing the sequence 5′-CGGCCGGACGTCCCGCGGCACGTGTCATCACATCATCATCATCATCATCATCATCATCATTAATAACCTTAAG-3′ were obtained from Operon (Huntsville, AL) and annealed together for 1 minute at 90°C and then allowed to cool at room temperature for 1 hour. When translated into an amino acid sequence, this cassette within the pITy vector encodes peptides E-A-R-P-D-V upstream of the A2aR gene, and P-R-H-V-S-S between the end of the gene and the decahistidine tag. This double-stranded DNA was flanked by EagI (5′) and AflII (3′) restriction sites and introduced the unique multiple cloning sites (EagI – AatII – SacII – PmlI), a small peptide linker, a 10-histidine tag, and two stop codons to halt translation after the histidine tag. Once annealed together, these nucleotides were subsequently digested with AflII and EagI, and ligated into the gel-extracted pITy plasmid. Ligated DNA was transformed in E. coli DH5a, and cells containing the pITy vector were selected based on kanamycin resistance. Propagated DNA was extracted and collected from DH5a using mini-preps (Promega, Madison, WI). Candidate DNA, containing the inserted multiple cloning site, was identified through a decrease in mobility on a 0.8% agarose gel compared to parental pITy plasmids. Candidates containing the insert were renamed pITy-MC-His10 and were further verified through DNA sequencing.
Separately, polymerase chain reaction (PCR) was used to amplify the A2aR gene from a previously created pt23b-A2aR-His6 plasmid. Primer sequences which flanked the gene were the forward primer 5′-ATGGCCGGCCGATGCCCATCATGGGCTCC-3′ which introduced an AatII site and the reverse primer 5′-GCCATCCGCGGGGA CACTCCTGCTCCATC-3′which introduced a SacII site. The amplified PCR product was run on a 0.8% agarose gel to verify correct amplification. The A2aR PCR product and the pITy-MC-His10 plasmid were then digested with AatII and SacII and ligated together. Ligation products were transformed into E. coli DH5a, selected through kanamycin resistance, and their DNA was extracted. Candidates containing the insert were identified by decreased mobility on a 0.8% agarose gel. Proper ligation and creation of the pITy- A2aR –His10 plasmid was verified through DNA sequencing.
Similar techniques were used to create the pITy-A2aR-GFP-His10 plasmid. The previously-created pET23b-A2aR-GFP-His6 template was used as the template for A2aR and enhanced GFP amplification. Forward and reverse primer sequences were 5′-ATG GCCGGCCGATGCCCATCATGGGCTCC-3′and 5′- GCCATCCGCGGCTTGTACAGCTCGTCCAT-3′, respectively and introduced the same restriction sites as for the previously-constructed plasmid. Peptide linkers in the final translated protein are E-A-R-P-upstream of the A2aR gene, G-S-E-F between the A2aR and eGFP genes, and P-R-H-V-S-S between the end of the eGFP gene and the decahistidine tag. Restriction digests, ligation, and candidate screening were carried out as described above. After sequencing verification, the desired construct was renamed pITy-A2aR-GFP-His10.
Strains and Culture Conditions
The yeast S. cerevisiae strain BJ5464 (MATa ura3-52 trp1 leu2? 1 his? 200 pep4::HIS3 prb1? 1.6R canI GAL), obtained from the American Type Culture Collection, was used as host for all A2aR expression. pITy-A2aR-His10 and pITy-A2aR-GFP-His10 plasmids were linearized by digestion with BsaBI, then ethanol precipitated to yield approximately 1μg/μL. 5μL of this linearized DNA was electroporated into BJ5464 using a Bio-Rad Gene-Pulser. Transformed cells were immediately combined with 1M sorbitol/YPD mix and were allowed to recover for 4 hours to develop resistance to G418. These cells were then plated on high-concentration (2.0 mg/mL) G418/YPD plates containing 1 M sorbitol to bias only the survival of high-expressing transformed cells.
Cultures were grown and expression was induced essentially as described previously (Niebauer and Robinson, 2006). Transformed cells were allowed to proliferate in either 5 mL culture tubes or flasks containing 25 mL YPD media (2% Bacto peptone, 2% glucose, 1% yeast extract) at 30°C in a shaking water bath at 275 rpm. After these cultures reached late exponential phase or saturation (OD600 > 13), cells were centrifuged at 2,000g and the supernatant was removed. Cells were resuspended in YPG media (2% Bacto peptone, 2% galactose, 1% yeast extract) to an OD600 of 0.5 in order to induce expression of A2aR. Routinely, 25 mL, 100 mL, or 500 mL batch cultures were prepared in this fashion. Expressing cells were harvested by centrifugation at 2,000g 25 hours post-induction, as this time point was shown to exhibit a per cell maxima for A2aR expression in S. cerevisiae (Niebauer et al., 2004).
Western Blotting and Staining
Western blotting was performed essentially according to the method described previously (Butz et al., 2003). Briefly, equal quantities of 1 OD-mL whole cells were collected, resuspended in STE10 (10 mM sucrose, 10 mM Tris-HCl, 10 mM EDTA, pH 7.6) supplemented with protease inhibitors (Roche), and lysed on a mini BeadBeater (Biospec). After unlysed cells were removed, the lysate was incubated with 3x SDS-loading buffer (175 mM Tris-HCl, pH 6.8, 0.25 mg/mL Bromophenol Blue, 25 mg/mL SDS, 40% glycerol). Alternatively, samples that were obtained from purification were incubated with an appropriate amount of SDS-loading buffer and run directly on SDS-PAGE and analyzed by Western blot.
Samples were separated on 12% SDS-PAGE with sample volumes of approximately 20 μL per lane. The resulting gel was either stained with Coomassie Blue or blotted onto a nitrocellulose membrane using a Trans-Blot Cell (Bio-Rad, Hercules, CA) for Western blotting. Primary antibodies to detect A2aR and A2aR-GFP were Rabbit anti-A2aR (Molecular Probes, Eugene, OR) and Chicken anti-GFP (Chemicon, Temecula, CA), which were used at dilutions of 1:200 and 1:5000, respectively. Secondary antibodies were anti-rabbit and anti-chicken HRP conjugated antibodies used at a dilution of 1:10000, and 1:10000, respectively. The ECL Plus Western Blotting Detection System (Amersham, Piscataway, NJ) was used to detect proteins of interest. Chemiluminescence was detected on a Typhoon 9400 Variable Mode Imager (Amersham) set to excite at 457 nm using a 520BP40 filter.
Selection of High-Expressing Clones
After transformation, cells were screened for their ability to express either A2aR-His10 or A2aR-GFP-His10, since the pITy vector is capable of integrating 1–30 times within different locations of the yeast chromosome (Parekh et al., 1996). Transformed BJ5464 containing integrated A2aR-His10 DNA were selected from individual colonies, grown to saturation, and expression was induced. After 25 hours, approximately 1 OD-mL of BJ5464 containing A2aR-His10 DNA were lysed and analyzed by Western blot using an anti-A2aR primary antibody. Western blot band intensity for A2aR was compared between different transformed cells, and also to BJ A2aRGFP high-expressing cells used in previous studies (Niebauer and Robinson, 2006). Transformants that exhibited more pronounced A2aR bands, especially compared to BJ A2aRGFP, were selected and analyzed in more detail. The transformed cells that were found to yield the highest expression levels as detected by Western blot were named BJ A2aRHis10 and used in all subsequent A2aR-His10 expression.
Screening for high-expressing A2aR-GFP-His10 cells was facilitated by the presence of the attached green fluorescent protein. In previous studies, GFP fluorescence was found to correlate well with expression levels of A2aR in S. cerevisiae (Niebauer and Robinson, 2006). BJ5464 cells containing integrated A2aR-GFP-His10 DNA were selected from individual colonies, grown, and expressed. At 25 hours of expression, 1 OD-mL quantities of different transformed cells were spun down at 2,000g and the supernatant was decanted. Cells were then resuspended in approximately 2 mL of Phosphate Buffered Saline (PBS) solution (1.44 g/L Na2HPO4, 0.24 g/L KH2PO4, 0.2 g/L KCl, 8.0 g/L NaCl, pH 7.4), and placed into a quartz cuvette. GFP fluorescence of these whole cells was measured using a Hitachi Fluorimeter with an excitation wavelength of 489 nm, and emission was detected at 511 nm.
Lysis and Solubilization of A2aR-expressing Whole Cells
Lysis of yeast cells was accomplished using two different methods. The first method solubilized A2aR while mechanically lysing the cells (Figure 1). Cells were harvested from shake flasks at 25 hours post-induction, spun down at 2,000g, and the supernatant was decanted. The cells were then washed in PBS to remove residual media, spun down again, and resuspended in Purification buffer (50 mM NaH2PO4, 300 mM NaCl, 10% glycerol, pH 8.0) containing 2% n-dodecyl-β-D-maltoside (DDM) (Sigma, St. Louis, MO), 1% 3-(3-cholamidopropyl)-dimethylammoniopropane sulfonate (CHAPS) (Anatrace, Maumee, OH), and 0.2% cholesterol hemisuccinate (CHS) (Anatrace) and supplemented with Complete EDTA-free protease inhibitors (Roche Applied Science, Indianapolis, IN) and 1 mM PMSF. This cell/surfactant mixture was combined with 0.5 mm zirconia/silica beads (BioSpec, Bartlesville, OK) and lysed on a mini-vortexer for 6 pulses of 120 seconds on the vortex and 120 seconds on ice in between pulses. The beads were separated from the mixture using a Kontes separation column. Crude lysate was spun at 3,220g for 1 hour at 4°C in order to spin out unlysed cells and heavy cellular debris. The supernatant containing solubilized A2aR was separated from the mixture and stored at 4°C until further use.
Figure 1. Western blot screening reveals the relative total expression levels of A2aR-His10 in transformed BJ5464 cells.

Lanes labeled 1–11 represent different isolated clones that have been transformed with pITy-A2aR-His10 DNA and induced for 25 hours of expression. BJAG represents BJ A2aRGFP high-expressing cells (Niebauer and Robinson, 2006) also induced for 25 hours of expression. BJ represents the parental yeast strain, BJ5464 cells which underwent electroporation in the absence of pITy-A2aR-His10 DNA, treated under the same expression conditions. Rabbit anti-A2aR primary antibody was used to detect A2aR for all samples. Arrows indicate the migrations of A2aR-His10 monomer and dimer, as well as A2aR-GFP monomer and dimer bands.
Alternatively, A2aR was recovered and solubilized from isolated yeast membranes. For this method, cells were also harvested after 25 hours of expression and washed in PBS. Pelleted cells were then resuspended in Purification buffer supplemented with Complete EDTA-free protease inhibitors and 1 mM PMSF. The cell mixture was lysed using an Avistin Emulsiflex, which lyses the cells through high-pressure homogenization. S. cerevisiae cells were lysed using 3–4 passes through the instrument, with homogenizing pressures above 22,500 psi as per the manufacturer’s recommendation. Once the cells were sufficiently lysed, the mixture was centrifuged for 5 minutes at 3,220g to remove unlysed cells, and the supernatant was recovered and further centrifuged for 30 minutes at 10,000g to remove cellular debris generated during lysis. This supernatant was further spun at 100,000g for 30 minutes in order to pellet yeast membranes. The supernatant was decanted, and the membrane pellet was incubated with 2% DDM/1% CHAPS/0.2% CHS in Purification buffer. Solubilization of A2aR from the membranes was allowed to proceed with gentle agitation for 2–3 hours on a rotating mixer. After solubilization, the solution was again centrifuged at 100,000g for 30 minutes to pellet the membranes, and the supernatant containing solubilized A2aR was saved at 4°C until further use.
Purification of A2aR Through Immobilized Metal Affinity Chromatography
Immobilized metal affinity chromatography (IMAC) was the primary method used in these studies to purify A2aR from S. cerevisiae. Approximately 250 μL of settled Ni-NTA Superflow resin (Quiagen, Valencia, CA) was used per 100 mL yeast culture, pre-equilibrated with at least 5 column volumes of Purification buffer containing either 2% DDM alone or a combination of 2% DDM/1% CHAPS/0.2% CHS, depending on the conditions of the A2aR sample being analyzed. Lysate containing solubilized, polyhistidine-tagged A2aR obtained either from bead-vortexing or high-pressure homogenization, was combined with pre-equilibrated resin and allowed to bind in batch for at least 6 hours with gentle mixing on an end-over-end mixer at 4°C. Imidazole was also added to a concentration of approximately 15 mM to prevent non-specific binding of other proteins to the resin.
Non-specifically bound material was removed from the resin with several washes of low-concentration imidazole. These washes consisted of at least five column-volumes of decreased surfactant and/or additive concentrations; either 0.1% DDM in purification buffer or 0.1% DDM/0.1% CHAPS/0.02% CHS, depending on the conditions of the sample being analyzed. Imidazole wash concentrations were 20 mM, 30 mM, and 50 mM. A small amount (typically 500 μL – 1 mL) of 500 mM imidazole solution in the appropriate buffer was used to both elute and concentrate the solubilized A2aR. Aliquots were collected throughout the purification process, and analyzed on SDS-PAGE to assess purification. Before any biophysical characterization, purified protein samples were extensively dialyzed into 10 mM NaH2PO4, pH 7.0, which contained either 0.1% DDM or 0.1% DDM/0.1% CHAPS/0.02% CHS, depending on the conditions of the sample being analyzed.
Purification of Active A2aR
Both active A1 and A2a adenosine receptors have been purified by affinity methods via an immobilized xanthine ligand (Nakata, 1989; Weiss and Grisshammer, 2002). In this study, ligand affinity media was prepared by the method of Weiss and Grisshammer (2002). Briefly, xanthine amine congener (XAC, Sigma Aldrich, St. Louis, MO) was dissolved in 96 mL dimethylsulfoxide at a concentration of 0.5 mg mL−1. Eight mL packed Affigel-10 (Bio-Rad Laboratories, Hercules, CA) was washed thoroughly with ice cold isopropanol and then dimethylsulfoxide before exposing it to the XAC solution. Ligand incorporation was achieved over 20 hrs under conditions of constant stirring and room temperature. The amount of covalently incorporated XAC was estimated to be approximately 11 μmol per mL packed resin by monitoring the absorbance at 310 nm in 10 mM HCl of the XAC solution before and after the incorporation reaction. The reaction was quenched by extensively washing the media with dimethylsulfoxide and then 50 mM Tris-HCl, pH 7.4, before being washed and stored in 20% ethanol.
Synthesized XAC resin was packed into a Tricorn 5/50 column (GE Healthcare) that ultimately contained approximately 3.7 mL of packed resin for automated purification on an ÄKTA Purifier (GE Healthcare). Prior to affinity purification, approximately 1–2 mL of concentrated IMAC-purified protein was passed through a PD-10 column (GE Healthcare) to remove imidazole. The sample was diluted to a volume of approximately 10 – 12 mL in Purification/Lysis Buffer containing 0.1% DDM/0.1% CHAPS/0.02 % CHS, which was loaded onto the affinity column at a flow rate of 0.1 mL/min. Unbound protein was washed from the column at a flow rate of 0.2 mL/min for 2 column volumes, then at a flow rate of 0.4 mL/min for 6 column volumes. Ligand-bound A2aR was eluted from the column through the addition of 20 mM theophylline at a flow rate of 0.2 mL/min for 25 column volumes. 4 mL fractions were collected, and samples were run undiluted on 12% SDS-PAGE and subsequently silver-stained to track purification.
Ligand Binding
Saturation ligand binding on whole yeast cells was performed essentially as described in previous studies (Wedekind et al., 2006). Whole cells were tested 25 hours post-induction at an in-well concentration of approximately 1 OD-mL, in 96-well Millipore (Bedford, MA) glass fiber filter B plates. For all whole cell experiments, a ligand binding buffer consisting of 50 mM MES/Tris and 10 mM MgCl2 at pH 5.8 was used.
Ligand binding experiments performed on purified A2aR were carried out with the protein immobilized on nickel resin. Previous experiments have shown that this technique works well for analyzing activity of GPCRs, and mimics proper solution conditions for ligand binding (Berger et al., 2005). After the second 50 mM imidazole wash during batch purification, approximately 50 μL of settled resin was separated from the mixture, centrifuged at 3,220g, and the supernatant was decanted. This separated resin was thoroughly resuspended in ligand binding buffer (50 mM Tris-HCl, 10 mM MgCl2, 1 mM EDTA, pH 7.4) containing either 0.1% DDM or 0.1% DDM/0.1% CHAPS/0.02%CHS. The resin was further diluted in ligand binding buffer containing the appropriate amount of surfactant and additives to obtain approximately 2 μL settled resin per well in a 96-well plate. Samples were allowed to incubate with ligand solutions approximately 1.5 – 2 hours with gentle agitation. As with whole cells, Millipore glass fiber filter B plates were used for all purified protein experiments, and binding was measured through radioactive counts (cpms) read by a MicroBeta Jet (Perkin-Elmer, Wellesly, MA). Non-specific binding was determined through measuring binding to the same amount of resin in the absence of bound protein.
All ligand binding data was fit to a one-site binding model. Samples were typically run in triplicate, and plotted as an average, with error bars representing standard error. KaleidaGraph 3.5 was used to fit the data, and determine relevant parameters such as Bmax and Kd. R2 values for the fit were typically greater than 0.98.
Circular Dichroism
Secondary structure of A2aR reconstituted in micelles was characterized by circular dichorism (CD) spectroscopy. The protein was solubilized in 0.1% n-dodecyl-β-D-maltoside (DDM) micelles, in the presence or absence of 0.1% CHAPS and 0.02 % cholesterol hemisuccinate (CHS).
Far-UV CD spectra of the A2aR reconstituted in micelles were recorded on a Jasco J-810 spectropolarimeter, equipped with a Peltier thermally controlled cuvette holder. All measurements were performed at 25°C, using a 0.1 cm path length quartz cuvette, from 260 to 190 nm, at a 1 nm step resolution and with an integration time of 3 seconds. To minimize effects of scattering, several precautions were taken as previously described (Lazarova et al., 2004). In all measurements, the appropriate buffer absorbance values were recorded and the reference CD spectra have been subtracted from representative CD spectra presented.
Fluorescence Spectroscopy
Fluorescence measurements were performed on an ISS PC-1 spectrofluorimeter, operating in photon-counting mode, using 10 mm × 10 mm or 2 mm × 10 mm quartz cuvettes at 25°C, unless otherwise specified. To minimize light scattering effects, the scans were performed with the emission polarizer oriented at 0°and the excitation polarizer at 90°. In the experiments, appropriate reference spectrum was subtracted from the sample spectrum. In order to prevent inner filter effects the concentration of A2aR reconstituted in micelles was chosen to give an absorbance at 280 nm (A280) of no more than 0.1.
Calculation of Protein Concentration and Yields
Purified protein concentrations were determined using a variety of different methods. UV absorbance at 280nm (A280) measurements were carried out for purified solubilized protein, with the appropriate buffer blanks subtracted from the absorbance of the sample. Any scatter in the near-UV region was subtracted from the reading. Extinction coefficients used in the determination of protein concentration were 53910 M−1cm−1 for A2aR-His10, and 73680 M−1cm−1 for A2aR-GFP-His10, as determined from the primary sequence of both proteins (Stoscheck, 1990). Additionally, a Bio-RAD DC Protein Assay was also used to determine total purified protein concentrations, with BSA (Sigma) as a standard. For A2aR-GFP-His10, fluorescence spectroscopy supplemented the above methods, utilizing a GFP calibration curve to convert fluorescence values to protein concentration as described in our prior studies (Niebauer and Robinson, 2006). Active protein yields for whole cells and purified protein were determined as described previously (Wedekind et al., 2006). 1 OD600 was estimated to correspond to 2.5 × 107 cells/mL.
RESULTS AND DISCUSSION
Selection of High-Expressing Yeast Cells
Since the pITy integrating vector is capable of integrating between 1–50 copies within the yeast chromosome, and at varying location (Parekh et al., 1996), screening of transformed cells was necessary to achieve adequate GPCR expression levels. Previously, flow cytometry was successfully implemented to isolate a high-expressing A2aR-GFP yeast cell (~4 mg/L of culture active protein) from a population of transformed cells (Niebauer et al., 2004). In those studies the GFP fusion allowed for rapid sorting of expressing cells. However, the presence of a large additional tag on the protein might interfere with the interpretation of spectroscopic data used to structurally characterize the GPCR.
In order to allow purification and structural characterization of the wild type human adenosine A2aR, a small 10-histidine tag (His10) was placed on the C-terminus of the protein, as it is unlikely that this extremely small addition would appreciably impact protein structure (Carson et al., 2007). Similar purification tactics have been found to work well for the isolation of membrane proteins (David et al., 1997; Feng et al., 2002; Grisshammer and Tucker, 1997; Mohanty and Wiener, 2004; Theis et al., 2001). However, flow cytometry does not lend itself well to the rapid sorting and isolation of high-expressing cells that do not have a quantifiable tag on the expressed protein. Thus, an alternative method was needed to screen transformants and identify high-expressing A2aR-His10 clones.
Western blotting provided for the direct comparison of total expression levels between transformed cells, and primarily was used to screen A2aR-His10 transformants. Over 45 transformed cells were compared to each other, as well as to the previously identified high-expressing A2aR-GFP cells (BJ A2aRGFP) (Niebauer and Robinson, 2006). As seen in Figure 1, many of these cells exhibit similar-intensity A2aR bands to each other, and also on the same order of magnitude to BJ A2aRGFP. Both monomer and dimer bands for A2aR are visible, and the monomer mobility correlates to the expected migration of ~ 45 kDa for A2aR-His10. Furthermore, the appearance of the more pronounced dimer band for highly expressing cells may be a result of concentration-dependent dimerization.
Transformant #6 was selected as having one of the highest Western blot band intensities, and was analyzed in more detail through ligand binding studies. This clone was determined to produce approximately 1.5 mg/L of culture active A2aR-His10 on the cell membrane through whole cell saturation ligand binding. Kd for the expressed A2aR-His10 receptor was found to be 49 +/− 5 nM (data not shown) upon binding to 3H-CGS-21,680.
In contrast, screening for high expression levels of A2aR-GFP-His10 from transformed cells was more easily carried out through whole cell fluorescence. Although the aim of this work was not to produce the fluorescently-tagged protein for extensive biophysical characterization, this protein provided helpful insights for the purification of histidine-tagged A2aR. Monitoring the fluorescently-tagged protein throughout the purification facilitated the development and refinement of the overall process. Also, these clones provided an easy way to compare relative expression levels between transformants, and thus evaluate the ability to isolate a relatively high-expressing cell without using flow cytometry. About 45 transformants were screened, and found to have an average fluorescence of 260 +/− 65 which corresponds to total expression levels of approximately 10 mg/L of culture as determined from a GFP standard curve. The lowest expressing cell had a fluorescence of 130 (4.5 mg/L), and the highest achieved 380 (15 mg/L). Ultimately, this highest-expresser was used in all subsequent expression of A2aR-GFP-His10 experiments. From saturation ligand binding studies, it was confirmed that BJ A2aR-GFP-His10 produces approximately 2.0 mg/L of culture functional receptor on the cell surface. Kd for the expressed A2aR-GFP-His10 receptor was verified to be 39 +/− 4 nM (data not shown).
Relatively little variation in total expression levels of A2aR was observed among transformed cells using both screening methods. In fact, it was a rare occurrence to find clones that were low expressers (Figure 1), as observed by comparison of Western blot band intensities. In further characterizing these clones, we find that the ratio of functional membrane-localized A2aR to protein retained within the cell is similar to transformed cells with high Western blot band intensities. The ability to isolate a large amount of high-expressing clones is attributed to selection on 2.0 mg/mL G418 plates, a condition which is stringent enough to select primarily for cells which have integrated multiple copies of the DNA in suitable locations within the yeast genome. High concentration G418 selection has been previously correlated to increased pITy integration and expression of bovine pancreatic trypsin inhibitor in S. cerevisiae (Parekh et al., 1996).
While the previously reported ~4 mg/L active A2aR expression level in the plasma membrane (Niebauer and Robinson, 2006) was not achieved for histidine-tagged protein, these methods still allow for the identification of cells which have functional yields on the same order of magnitude as those isolated by flow cytometry. Thus, when sorting of transformed cells with the aid of a fluorescent fusion partner is neither desirable nor possible, screening through more basic and low-throughput methods like Western blotting will identify clones that are still sufficient for over-expression as long as selective pressure via a high concentrations of antibiotic is used to enrich for these clones. In this case relatively few cells needed to be screened (~50) compared to approximately ~104 cells which were sorted in previous efforts through flow cytometry. Importantly, functional expression levels for histidine-tagged A2aR of 1.5–2 mg/L will readily facilitate further purification and characterization of this GPCR.
Purification of A2aR from Batch Culture
Suitable lysis procedures were developed on a small-scale (~5 mL batch cultures) and scaled-up to handle larger A2aR expressing cultures (~100–500 mL batch cultures). Previously for this expression system, we have found that in addition to its presence within the membrane, a large amount of A2aR is retained within the cell (Niebauer et al., 2004) Therefore, in order to maximize recovery of A2aR, cell lysis and protein solubilization were done concurrently. The effectiveness of variables within a given process were analyzed through a design of experiments approach, and evaluated by measuring recovery of fluorescence of the GFP fusion tag from lysed BJ A2aR-GFP or BJ A2aR-GFP-His10 cells (manuscript in preparation). Fluorescence of recovered lysate at the end of a given process was measured and compared to initial whole cell fluorescence before lysis. Any loss in fluorescence was attributed to protein degradation or ineffective processing. Ultimately, a sufficient process was identified as detailed in Materials and Methods. As a comparison, protein was also solubilized from fractionated membranes rather than whole cells. Lysate containing detergent-solubilized A2aR was purified using immobilized metal affinity chromatography in both cases.
Since several laboratories have cited difficulties in maintaining receptor functionality outside the plasma membrane, various solution conditions were tested during the purification process. Initially, solubilization on a small scale was attempted using digitonin. Though digitonin has been successfully used for the functional purification of other adenosine receptors (Berger et al., 2005; Nakata, 1989), its heterogeneity compromises spectroscopic characterization, and it tends to recrystallize over time. The surfactant n-dodecyl-β-D-maltoside (DDM) was substituted for digitonin, since it has been extensively cited as a useful surfactant for maintaining proper structure of membrane proteins, and many solved membrane protein structures have been solubilized in DDM (Protein Data Bank). Contrary to digitonin, DDM proved to be a much more user-friendly surfactant, as it did not present any known hindrance to the purification process or downstream characterization. Furthermore, in some cases throughout solubilization and purification, a small amount of cholesterol hemisuccinate (CHS) dissolved in CHAPS was added. This addition has proven useful for promoting the activity of GPCRs in vitro, specifically for the A2a receptor heterologously expressed in E. coli (Weiss and Grisshammer, 2002).
Protein was purified through either bead-vortexing solubilization or membrane solubilization, and the effectiveness of these methods was assessed by SDS-PAGE. As seen in Figure 2, relatively pure protein results from one-step purification for both processes. For bead-vortexing solubilization, both monomer and dimer bands are visible for purified A2aR-His10, while only a monomer band is seen using membrane fractionation and solubilization. Since less protein was obtained through the membrane solubilization method, this observation could be due to concentration-dependent dimerization. However, it remains unclear whether this band results from A2aR dimers in solution or if this oligomeric state is an artifact of SDS-PAGE, as has been observed previously in both yeast and mammalian species (Sander et al., 1994; Shenoy et al., 2001). Current work aims to characterize the oligomeric state of A2aR in solution, as this protein is known to dimerize in native tissues (Kamiya et al., 2003). In addition, N-terminal sequencing of the purified protein (data not shown), verifies that the N-terminal pre-pro signal sequence has been properly processed and removed from the protein.
Figure 2. Immobilized metal affinity chromatography enables purification of A2aR from batch culture using two lysis methods.
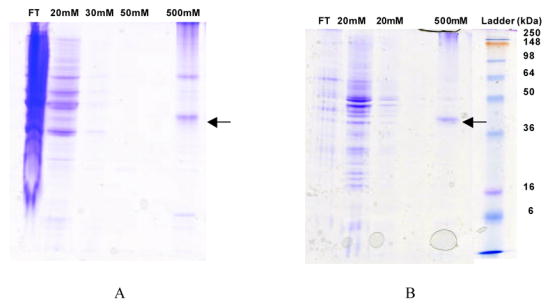
Coomassie-stained 12% SDS-PAGE gels illustrate the purification process. (A) A2aR-His10 was eluted using 500 mM imidazole as recovered using the bead-vortexing lysis method. (B) A2aR-His10 was also purified using the membrane solubilization method. Similar results were obtained for the purification using both methods. All samples were solubilized in purification buffer containing 2% DDM, 1% CHAPS, and 0.2% CHS and supplemented with protease inhibitors and 1 mM PMSF. Wash steps were carried out in purification buffer containing 0.1% DDM, 0.1% CHAPS, and 0.02% CHS supplemented with protease inhibitors and 1 mM PMSF. This cell/surfactant flow-through (FT) denotes proteins that were present in the crude lysate, and did not bind to the resin. 20 mM–50 mM lanes show proteins which were removed from the resin during washes with low-concentration imidazole. The ladder is See-BluePlus2 protein molecular weight standard, with molecular weights indicated. A2aR-His10 monomer is denoted with the arrows.
The Effect of Cholesterol on A2aR Functionality in vitro
Ligand binding experiments were performed on purified A2aR-His10 to assess the ability of the GPCR to bind its ligand in a reconstituted micellar environment. Initially, no appreciable activity was observed for protein suspended in DDM micelles (Figure 3). With this finding, several other conditions including pH, buffer compositions, and protein concentration were altered throughout the purification process to preserve protein activity. However, ligand-binding activity was neither preserved nor recovered using these methods (data not shown).
Figure 3. DDM/CHAPS/CHS solubilized A2aR constitutes active, functional protein whereas DDM solubilized A2aR is inactive.
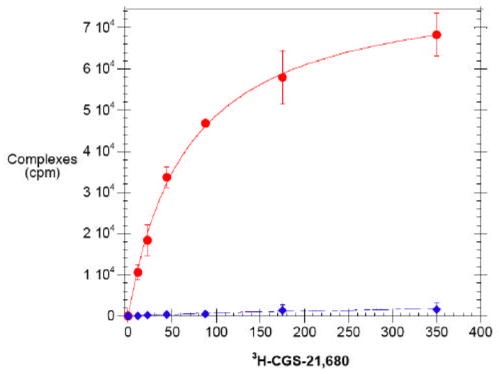
Saturation ligand binding on purified A2aR solubilized in DDM/CHAPS/CHS (circles, solid) and DDM (diamonds, dashed) using 3H-CGS-21,680 ligand. Points indicate experimentally determined data, while lines indicate the best fit to a single-site model, where Kd for the active receptor was determined to be 66 +/− 4 nM. Bmax was 92 +/− 21 pmol/mg from the best fit of this data to a single-site binding model.
Noting the differences between the reconstituted micellar environment of the purified protein and its native environment within the plasma membrane, new strategies were formulated to preserve A2aR activity by better mimicking the mammalian plasma membrane. For this reason, mammalian lipids were explored to determine their impact on A2aR functionality. A soluble form of the mammalian lipid cholesterol, cholesterol hemisuccinate (CHS), was the first candidate. It was found that CHS dissolved in a stock solution of CHAPS was adequate for maintaining solubility of the lipid in solution, and easily facilitated its addition to DDM surfactant. Upon purification and subsequent radioligand binding in the presence of DDM/CHAPS/CHS, purified A2aR was found to retain a large degree of its activity in vitro (Figure 3).
It is important to note that the main sterol within yeast membranes is ergosterol, and the main sterol in mammalian cells is cholesterol (Opekarova and Tanner, 2003; Pucadyil and Chattopadhyay, 2006). Thus, the environment of the receptor in a more primitive system, even within the plasma membrane, is quite different from the native system. During solubilization, the co-extraction of yeast lipids like ergosterol with the membrane protein may not be sufficient to stabilize the active state of the receptor in a non-native micelle. Due to the different lipids present in the mammalian plasma membrane, the addition of endogenous mammalian lipids like cholesterol during and throughout purification may be necessary to maintain proper folding of mammalian membrane proteins expressed in microbial systems. The importance of particular lipids for proper conformation also has been demonstrated for other membrane proteins (Opekarova and Tanner, 2003).
Purification of Active A2aR
In order to further identify the active population of A2a receptors, protein purified via immobilized metal affinity chromatography was applied to a xanthine-agarose ligand affinity column. This technique was first developed to allow for the separation of functional adenosine A1 receptors (Nakata, 1989) and has also been successfully used to separate active adenosine A2a receptors expressed in E. coli (Weiss and Grisshammer, 2002). Active A2aR binds to an = 3.2 × 10−8 M) while inactive protein and other agarose-immobilized xanthine ligand (Ki contaminants are removed through column washes. Active protein is subsequently eluted from the column through competition with theophylline, a low-affinity adenosine receptor antagonist (Ki=2.7 × 10−6 M, Weiss and Grisshammer, 2002).
When fractions collected from the affinity purification of A2a-GFPHis10 were separated via SDS-PAGE and silver-stained, the entire population of full-length protein purified through IMAC was eluted from the column only upon theophylline addition (Figure 4). Most of the protein eluted from the column within 8 column volumes (~30 mL) of elution buffer (20 mM theophylline, 50 mM Tris-HCl, 10 mM MgCl2, 1 mM EDTA, pH 7.4). Staining of the wash steps illustrates the removal of a low molelcular weight material (~ 30–35 kDa) from the column, later confirmed to be a C-terminal protease fragment containing GFP by N-terminal sequencing. This C-terminal protease cleavage is consistent with that previously observed for the A2a receptor (Weiss and Grisshammer, 2002). No inactive full-length receptor was visualized in these fractions, in contrast to that observed by Weiss and Grisshammer for the E. coli-produced receptor (2002). Further analysis of these wash fractions through Western blotting failed to show any trace of full-length A2aR (data not shown), indicating that all of the purified pool of full-length A2aR is in fact in its active, ligand-binding conformation, or is a proteolytically-cleaved fragment. Since protein was purified both from the plasma membrane and from the intracellular localized protein population, this finding further illustrates that even internalized A2aR is in a ligand-binding conformation within the protein-detergent complex. Similar results were also found for the ligand affinity purification of A2aHis10.
Figure 4. Xanthine affinity chromatography shows full-length A2aR is active in ligand-binding.
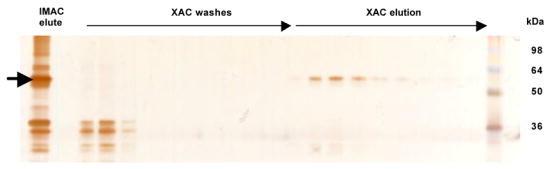
Silver-stained 12% SDS-PAGE gel tracks purification of an IMAC purified population of A2aGFPHis10 through xanthine ligand affinity chromatography. The IMAC elute lane denotes protein loaded onto the XAC column which has been desalted and diluted in purification buffer containing 0.1% DDM/0.1% CHAPS/0.02% CHS as described in Materials and Methods. XAC wash lanes represent undiluted samples from 4 mL fractions collected as unbound material from the column. XAC elute lanes represent undiluted samples collected from 4 mL fractions and show active A2aGFPHis10 that was eluted from the column with 20mM theophylline.
Total and Functional A 2aR Yields
Large amounts of both total and functional, purified A2aR-His10 were achieved through purification in repeated studies. Through A280 measurements of protein obtained from 100 mL of yeast culture, it was determined that 6 mg of A2aR-His10 per liter of culture was purified, whereas for the A2aR-GFP-His10 protein, approximately 10 mg/L of culture was purified. Additionally, fluorescence spectroscopy indicated that approximately 2 – 4 mg/L of A2aR-GFP-His10 was purified. Subsequent preparations of 500 mL and 1 L yeast cultures have yielded similar results to those predicted from smaller scale preparations.
In terms of functional yields for purified protein as determined by saturation ligand binding to 3H-CGS-21,680 (KD = 50 nM), we find that 0.7–1.12 mg/L of culture A2aR-His10 protein are routinely obtained. However, from the xanthine ligand affinity chromatography step it appears that all full-length purified A2aR is active as determined by ligand binding. The discrepancy between these findings most likely arises from the limitations associated with our radioligand binding assay, since the affinities of the ligands are similar. Conducting radioligand binding assays with solubilized protein on a 96-well filter plate vacuum manifold requires immobilization of A2aR on nickel resin so as to not lose protein through the 2-μm filter. Thus, results from our radioligand binding experiments are likely to be an underestimate of active A2aR yields.
Overall, these data imply that approximately 30 – 40% of the total expressed protein can be readily purified, and nearly all of this protein is in an active, ligand-binding conformation. Importantly, total protein yields are among the highest achieved for the A2a receptor. Furthermore, the active yields reported are the highest yet achieved for the human A2a receptor from any expression system. Current efforts are underway to further increase our protein recoveries on a larger scale to maximize the amount of A2aR that can be purified from our expression system.
A2aR is Largely Helical and Properly Reconstituted in a Micellar Environment
Purification of milligram amounts of A2aR allowed spectroscopic characterization of the receptor to be performed through techniques such as circular dichroism (CD), and intrinsic fluorescence. The solubilized A2aR-His10 and A2aR-GFP-His10 fusion protein samples in DDM micelles and CHAPS/CHS exhibit UV-CD spectra characteristic of predominantly alpha helical structure (data not shown for A2aR-GFP-His10), as seen by the presence of two distinct absorbance bands at 222nm and 202nm (Figure 5). Interestingly, the absence of CHAPS/CHS during purification enabled the formation of some visible aggregates, characterized by precipitated protein after elution from nickel resin. The CD spectrum of A2aR under these conditions displays a loss of helical qualities, and only one maximum at about 208nm, possibly representative of beta sheet structure as seen in Figure 5. These finding are in agreement with radiolabeled ligand binding experiments, which demonstrate proper activity of samples containing the added lipid and a loss of ligand-binding activity in samples lacking it. Similar spectroscopic results were obtained for purified A2aR-His10 from both lysis/solubilization techniques.
Figure 5. Purified A2aR exhibits alpha-helical structure in the presence of CHS/CHAPS.

CD spectrum of purified A2aR-His10 in DDM/CHAPS/CHS micelles (solid) and purified A2aR-His10 in DDM micelles without CHAPS/CHS (dotted). Both spectra are representative of three separate samples.
Fluorescence spectroscopy was used to probe whether the protein was properly incorporated into micelles. Since it was observed that A 2aR activity fluctuated as a function of lipid additives, we wanted to confirm that the protein was properly incorporated into micelles under both conditions to rule out improper folding due to solvent exposure. The sensitivity of tryptophan (Trp) fluorescence emission to the polarity of the environment allows us to use the Trp residues within A2aR as reporter groups. It is well known that the emission spectrum of the Trp amino acid is strongly dependent on the environment, as a blue maximum at about 320 – 330 nm is reflects a mainly hydrophobic environment, whereas a maximum at 350 nm is reflects a hydrophilic environment. The fluorescence spectra of A2aR reconstituted in DDM micelles, in the presence as well as in the absence of CHS, show similar emission florescence maxima at about 325 nm and 323 nm, respectively (Figure 6). This indicates the presence of a hydrophobic environment around the protein’s shielded tryptophans. Blue-shifted fluorescence emission indicates that in the absence of CHS, A2aR is also incorporated in micelles; however the protein fails to fold correctly.
Figure 6. Fluorescence spectroscopy verifies that solubilized A2aR-His 10 in DDM both with CHS/CHAPS and without is properly incorporated into micelles.

Intrinsic fluorescence spectra for A2aR-His10 in DDM micelles without CHAPS/CHS (dashed) and with CHAPS/CHS (solid). Fluorescence maxima are listed on the spectra.
CONCLUSIONS
In these studies, we aimed to capitalize on our ability to express large amounts of the human adenosine A2a receptor, isolate high-expressing clones using a purification tag, and purify the receptor for structural studies. A deca-histidine tag was added to the A2aR in order to allow purification through immobilized metal affinity chromatography. Through the development of suitable lysis and solubilization techniques, histidine-tagged A2aR was readily purified from S. cerevisiae in large enough quantities to allow for spectroscopic characterization.
At present time, two other reports have cited over-expression and purification of A2aR from other heterologous systems. E. coli have been used to produce functional A2aR fused to a maltose binding protein in quantities ~150 μg/L of culture (Weiss and Grisshammer, 2002), and an automated purification scheme has been developed involving large-scale fermentation and elaborate purification and concentration steps. Expression of a glycosylation-deficient version of A2aR in Pichia pastoris has also accomplished expression levels ~200 μg/L of culture (Fraser, 2006) in a series of batch processes. However, without the ability to readily purify mg/L amounts of active protein, these methods prove expensive and require extensive equipment for large-scale production.
The functional, purified yields for the A2aR presented in this work are the highest achieved for this receptor from any expression system. Furthermore, our functional yields are comparable to the highest yet achieved for other GPCRs. Currently, only a handful of other human GPCRs have been able to be readily purified in functional form at or above milligram amounts per liter of culture (Sarramegna et al., 2006; Sarramegna et al., 2003). Certainly this work will help alleviate the lack of structural knowledge concerning the human A2aR. Currently, methods are being developed to further automate the purification process, and larger-scale fermentation will be carried out. With the ability to produce and purify milligram amounts of A2aR, we have begun extensive studies on the receptor in order to gain a better sense of its stability, folding, and interactions. These studies will not only yield valuable information concerning the behavior of A2aR in vitro, but will also contribute to its crystallization and high-resolution structure determination.
Table 1.
Sample purification of A2aGFPHis10 protein through immobilized metal affinity chromatography as monitored via fluorescence spectroscopy. Results for mg/L based on estimation extrapolated from quantification of protein purified from a 25 mL culture.
| Total Protein | Solubilized Material* | IMAC Flow Through * | IMAC Wash Flow Though* | IMAC elute | Radio-ligand binding | |
|---|---|---|---|---|---|---|
| mg/L of culture | 7.4 +/− 0.5 | 4.6 +/− 0.5 | 1.0 +/− 0.9 | 0.6 +/− 0.5 | 2.3 +/− 0.2 | 0.43 +/−0.02 |
| pmol/mg† | 480 +/− 32 | 300 +/− 33 | 64 +/− 60 | 39 +/− 30 | 150 +/− 14 | 26 +/− 1.0 |
Based on values obtained from similar experiments where fluorescence of these fractions was monitored.
Based on the estimation of molecules/cell to pmol/mg described previously (Sarramegna et al., 2003)
Acknowledgments
This research was supported by NIH RR15588, NSF IGERT (MAO), and NASA-JPFP (MAO). The authors thank Dr. K. Dane Wittrup for the generous donation of the pITy plasmid, Dr. Marlene Jacobson for providing the human A2a receptor gene, Yu Cue Huang for N-terminal sequencing of the purified A2a receptor, and Rachael Lewus for useful discussions and technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akermoun M, Koglin M, Zvalova-Iooss D, Folschweiller N, Dowell SJ, Gearing KL. Characterization of 16 human G protein-coupled receptors expressed in baculovirus-infected insect cells. Protein Expression and Purification. 2005;44:65–74. doi: 10.1016/j.pep.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Berger BW, Garcia RY, Lenhoff AM, Kaler EW, Robinson CR. Relating surfactant properties to activity and solubilization of the human adenosine A(3) receptor. Biophysical Journal. 2005;89:452–464. doi: 10.1529/biophysj.104.051417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth PJ. The trials and tribulations of membrane protein folding in vitro. Biochimica Et Biophysica Acta-Biomembranes. 2003;1610:51–56. doi: 10.1016/s0005-2736(02)00714-9. [DOI] [PubMed] [Google Scholar]
- Butz JA, Niebauer RT, Robinson AS. Co-expression of molecular chaperones does not improve the heterologous expression of mammalian G-protein coupled receptor expression in yeast. Biotechnology and Bioengineering. 2003;84:292–304. doi: 10.1002/bit.10771. [DOI] [PubMed] [Google Scholar]
- Carson M, Johnson DH, McDonald H, Brouillette C, Delucas LJ. His-tag impact on structure. Acta Crystallogr D Biol Crystallogr. 2007;63:295–301. doi: 10.1107/S0907444906052024. [DOI] [PubMed] [Google Scholar]
- Chelikani P, Reeves PJ, Rajbhandary UL, Khorana HG. The synthesis and high-level expression of a beta(2)-adrenergic receptor gene in a tetracycline-inducible stable mammalian cell line. Protein Science. 2006;15:1433–1440. doi: 10.1110/ps.062080006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JM, Catlin GH, Price MJ, Edwards RM. Secretion of Human Epidermal Growth-Factor from Saccharomyces-Cerevisiae Using Synthetic Leader Sequences. Gene. 1991;106:267–272. doi: 10.1016/0378-1119(91)90209-t. [DOI] [PubMed] [Google Scholar]
- David NE, Gee M, Andersen B, Naider F, Thorner J, Stevens RC. Expression and purification of the Saccharomyces cerevisiae alpha-factor receptor (Ste2p), a 7-transmembrane-segment G protein-coupled receptor. Journal of Biological Chemistry. 1997;272:15553–15561. doi: 10.1074/jbc.272.24.15553. [DOI] [PubMed] [Google Scholar]
- Eckart MR, Bussineau CM. Quality and authenticity of heterologous proteins synthesized in yeast. Current Opinion in Biotechnology. 1996;7:525–530. doi: 10.1016/s0958-1669(96)80056-5. [DOI] [PubMed] [Google Scholar]
- Feng W, Cai H, Pierce WM, Song ZH. Expression of CB2 cannabinoid receptor in Pichia pastoris. Protein Expression and Purification. 2002;26:496–505. doi: 10.1016/s1046-5928(02)00569-7. [DOI] [PubMed] [Google Scholar]
- Flower DR. Modelling G-protein-coupled receptors for drug design. Biochimica Et Biophysica Acta-Reviews on Biomembranes. 1999;1422:207–234. doi: 10.1016/s0304-4157(99)00006-4. [DOI] [PubMed] [Google Scholar]
- Fraser NJ. Expression and functional purification of a glycosylation deficient version of the human adenosine 2a receptor for structural studies. Protein Expression and Purification. 2006 doi: 10.1016/j.pep.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Grisshammer R, Tucker J. Quantitative evaluation of neurotensin receptor purification by immobilized metal affinity chromatography. Protein Expression and Purification. 1997;11:53–60. doi: 10.1006/prep.1997.0766. [DOI] [PubMed] [Google Scholar]
- Hill SJ. G-protein-coupled receptors: past, present and future. British Journal of Pharmacology. 2006;147:S27–S37. doi: 10.1038/sj.bjp.0706455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard AD, McAllister G, Feighner SD, Liu QY, Nargund RP, Van der Ploeg LHT, Patchett AA. Orphan G-protein-coupled receptors and natural ligand discovery. Trends in Pharmacological Sciences. 2001;22:132–140. doi: 10.1016/s0165-6147(00)01636-9. [DOI] [PubMed] [Google Scholar]
- Jacobson KA, Jarvis MF, Williams M. Purine and pyrimidine (P2) receptors as drug targets. Journal of Medicinal Chemistry. 2002;45:4057–4093. doi: 10.1021/jm020046y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya T, Saitoh O, Yoshioka K, Nakata H. Oligomerization of adenosine A(2A) and dopamine D-2 receptors in living cells. Biochemical and Biophysical Research Communications. 2003;306:544–549. doi: 10.1016/s0006-291x(03)00991-4. [DOI] [PubMed] [Google Scholar]
- Keller T, Elfeber M, Gorboulev V, Reilander H, Koepsell H. Purification and functional reconstitution of the rat organic cation transporter OCT1. Biochemistry. 2005;44:12253–12263. doi: 10.1021/bi050676c. [DOI] [PubMed] [Google Scholar]
- Kim TK, Zhang RD, Feng W, Cai H, Pierce W, Song ZH. Expression and characterization of human CB1 cannabinoid receptor in methylotrophic yeast Pichia pastoris. Protein Expression and Purification. 2005;40:60–70. doi: 10.1016/j.pep.2004.10.026. [DOI] [PubMed] [Google Scholar]
- Lazarova T, Brewin KA, Stoeber K, Robinson CR. Characterization of peptides corresponding to the seven transmembrane domains of human adenosine A(2)a receptor. Biochemistry. 2004;43:12945–12954. doi: 10.1021/bi0492051. [DOI] [PubMed] [Google Scholar]
- Loll PJ. Membrane protein structural biology: the high throughput challenge. Journal of Structural Biology. 2003;142:144–153. doi: 10.1016/s1047-8477(03)00045-5. [DOI] [PubMed] [Google Scholar]
- Mohanty AK, Wiener MC. Membrane protein expression and production: effects of polyhistidine tag length and position. Protein Expression and Purification. 2004;33:311–325. doi: 10.1016/j.pep.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Mubagwa K, Flameng W. Adenosine, adenosine receptors and myocardial protection: An updated overview. Cardiovascular Research. 2001;52:25–39. doi: 10.1016/s0008-6363(01)00358-3. [DOI] [PubMed] [Google Scholar]
- Nakata H. Purification of A1 Adenosine Receptor from Rat-Brain Membranes. Journal of Biological Chemistry. 1989;264:16545–16551. [PubMed] [Google Scholar]
- Niebauer RT, Robinson AS. Exceptional total and functional yields of the human adenosine (A2a) receptor expressed in the yeast Saccharomyces cerevisiae. Protein Expression and Purification. 2006;46:204–211. doi: 10.1016/j.pep.2005.09.020. [DOI] [PubMed] [Google Scholar]
- Niebauer RT, Wedekind A, Robinson AS. Decreases in yeast expression yields of the human adenosine A2a receptor are a result of translational or post-translational events. Protein Expression and Purification. 2004;37:134–143. doi: 10.1016/j.pep.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Opekarova M, Tanner W. Specific lipid requirements of membrane proteins - a putative bottleneck in heterologous expression. Biochimica Et Biophysica Acta-Biomembranes. 2003;1610:11–22. doi: 10.1016/s0005-2736(02)00708-3. [DOI] [PubMed] [Google Scholar]
- Palczewski K, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Parekh R, Forrester K, Wittrup D. Multicopy Overexpression of Bovine Pancreatic Trypsin-Inhibitor Saturates the Protein-Folding and Secretory Capacity of Saccharomyces-Cerevisiae. Protein Expression and Purification. 1995;6:537–545. doi: 10.1006/prep.1995.1071. [DOI] [PubMed] [Google Scholar]
- Parekh RN, Shaw MR, Wittrup KD. An integrating vector for tunable, high copy, stable integration into the dispersed Ty delta sites of Saccharomyces cerevisiae. Biotechnology Progress. 1996;12:16–21. doi: 10.1021/bp9500627. [DOI] [PubMed] [Google Scholar]
- Pausch MH. G-protein-coupled receptors in Saccharomyces cerevisiae: high-throughput screening assays for drug discovery. Trends in Biotechnology. 1997;15:487–494. doi: 10.1016/S0167-7799(97)01119-0. [DOI] [PubMed] [Google Scholar]
- Poulsen SA, Quinn RJ. Adenosine receptors: New opportunities for future drugs. Bioorganic & Medicinal Chemistry. 1998;6:619–641. doi: 10.1016/s0968-0896(98)00038-8. [DOI] [PubMed] [Google Scholar]
- Pucadyil TJ, Chattopadhyay A. Role of cholesterol in the function and organization of G-protein coupled receptors. Progress in Lipid Research. 2006;45:295–333. doi: 10.1016/j.plipres.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Reilander H, Weiss HM. Production of G-protein-coupled receptors in yeast. Current Opinion in Biotechnology. 1998;9:510–517. doi: 10.1016/s0958-1669(98)80038-4. [DOI] [PubMed] [Google Scholar]
- Sander P, Grunewald S, Maul G, Reilander H, Michel H. Constitutive expression of the human D2S-dopamine receptor in the unicellular yeast Saccharomyces cerevisiae. Biochim Biophys Acta. 1994;1193:255–62. doi: 10.1016/0005-2736(94)90161-9. [DOI] [PubMed] [Google Scholar]
- Sarramegna V, Muller I, Milon A, Talmont F. Recombinant G protein-coupled receptors from expression to renaturation: a challenge towards structure. Cellular and Molecular Life Sciences. 2006;63:1149–1164. doi: 10.1007/s00018-005-5557-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarramegna V, Talmont R, Demange P, Milon A. Heterologous expression of G-protein-coupled receptors: comparison of expression systems from the standpoint of large-scale production and purification. Cellular and Molecular Life Sciences. 2003;60:1529–1546. doi: 10.1007/s00018-003-3168-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science. 2001;294:1307–13. doi: 10.1126/science.1063866. [DOI] [PubMed] [Google Scholar]
- Smith JD, Robinson AS. Overexpression of an archaeal protein in yeast: Secretion bottleneck at the ER. Biotechnology and Bioengineering. 2002;79:713–723. doi: 10.1002/bit.10367. [DOI] [PubMed] [Google Scholar]
- Stoscheck CM. Quantitation of Protein. Methods in Enzymology. 1990;182:50–68. doi: 10.1016/0076-6879(90)82008-p. [DOI] [PubMed] [Google Scholar]
- Tate CG, Grisshammer R. Heterologous expression of G-protein-coupled receptors. Trends in Biotechnology. 1996;14:426–430. doi: 10.1016/0167-7799(96)10059-7. [DOI] [PubMed] [Google Scholar]
- Theis S, Doring F, Daniel H. Expression of the myc/His-tagged human peptide transporter hPEPT1 in yeast for protein purification and functional analysis. Protein Expression and Purification. 2001;22:436–442. doi: 10.1006/prep.2001.1455. [DOI] [PubMed] [Google Scholar]
- Thevenin D, Roberts MF, Lazarova T, Robinson CR. Identifying interactions between transmembrane helices from the adenosine A(2A) receptor. Biochemistry. 2005;44:16239–16245. doi: 10.1021/bi051422u. [DOI] [PubMed] [Google Scholar]
- Venter JC, et al. The sequence of the human genome. Science. 2001;291:1304. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Wedekind A, O’Malley MA, Niebauer RT, Robinson AS. Optimization of the human adenosine A2a receptor yields in Saccharomyces cerevisiae. Biotechnology Progress. 2006 doi: 10.1021/bp050431r. [DOI] [PubMed] [Google Scholar]
- Weiss HM, Grisshammer R. Purification and characterization of the human adenosine A(2a) receptor functionally expressed in Escherichia coli. European Journal of Biochemistry. 2002;269:82–92. doi: 10.1046/j.0014-2956.2002.02618.x. [DOI] [PubMed] [Google Scholar]
- White S. Membrane Protein Resources 2006 [Google Scholar]
- Wiener MC. A pedestrian guide to membrane protein crystallization. Methods. 2004;34:364–372. doi: 10.1016/j.ymeth.2004.03.025. [DOI] [PubMed] [Google Scholar]