

Abstract
A novel method for the fabrication of polymeric particles on the order of tens of nanometers to several microns is described. This imprint lithographic technique called PRINT™ (Particle Replication In Non-wetting Templates), takes advantage of the unique properties of elastomeric molds comprised of a low surface energy perfluoropolyether network, allowing the production of monodisperse, shape-specific nanoparticles from an extensive array of organic precursors. This engineered nature of particle production has a number of advantages over the construction of traditional nanoparticles such as liposomes, dendrimers, and colloidal precipitates. The gentle “top down” approach of PRINT enables the simultaneous and independent control over particle size and shape, composition, and surface functionality, and permits the loading of delicate cargos such as small organic therapeutics and biological macromolecules. Thus, this single tool serves as a comprehensive platform for the rational design and investigation of new nanocarriers in medicine, having applications ranging from therapeutics to advanced diagnostics. Preliminary in vitro and in vivo studies were conducted, demonstrating the future utility of PRINT particles as delivery vectors in nanomedicine. Monodisperse 200 nm poly(ethylene glycol)-based (PEG) particles were fabricated using PRINT methodology and characterized via scanning electron microscopy and dynamic light scattering. Incubation with HeLa cells showed very little cytotoxicity, even at high concentrations. The biodistribution and pharmacokinetics of [125I]-labeled particles were studied in healthy mice following bolus tail vein administration. The particles were distributed mainly to the liver and the spleen with an apparent distribution t1/2 of approximately 17 min followed by slow redistribution with a t1/2 of 3.3 h. The volume of distribution for the central and peripheral compartments was found to be approximately 3 mL and 5 mL, respectively.
Keywords: PRINT, biodistribution, nanoparticles, pharmacokinetics, nanomedicine
1. Introduction
Despite continued progress in the identification, characterization, and synthesis of advanced therapeutics, the full potential of such innovations can only be achieved with the concomitant realization of in vivo profiles ideal for pharmacological intervention. In the realm of drug discovery, the hindrance to obtaining such a profile may be as simple as poor solubility in biological media. For instance, it has been reported that ten percent of marketed drugs suffer from solubility problems, over a third of pipeline drugs are poorly soluble, and almost two-thirds of drugs coming from early pre-clinical development have low solubility [1]. As such, almost forty percent of all possible drug targets fail early due to poor solubility characteristics. More complex problems with drug candidates may include unfavorable pharmacokinetics, and high systemic toxicity. Particular attention must also be given when considering the delivery of biochemically labile substances such as siRNA and other oligonucleotides for gene therapy, as these sensitive cargos need to be protected during circulation. In addition, they need to be delivered to the appropriate tissue or organ, and released intracellularly into the cytosol or nucleus to be effectively used as therapeutics. Finally, the efficient delivery of detection and imaging agents is an extremely important step in the early diagnosis and treatment of disease.
The development and utilization of nanocarriers in response to many of the aforementioned problems encountered in vivo has led to dramatic improvements in the biological profile of important therapies. Intense research in drug delivery over the past few decades has seen the design and construction of valuable nanocarriers such as liposomes, micelles, dendrimers, polymer particles, and colloidal precipitates [2-9]. However, only a handful of drugs and imaging agents delivered using these classical approaches have made it into the clinic [1,8]. One underlying reason for the delayed development is that none of these approaches offers the ability to comprehensively, simultaneously, and independently address several different design criteria. The ability to meet such demanding parameters is quintessential to the design of effective delivery vectors and has been the focus of intense research in our laboratory. Herein, we report the utilization of recent breakthroughs in the nanofabrication of polymeric particles to develop an effective platform delivery system for use in nanomedicine.
Our technique, called PRINT (Particle Replication In Non-wetting Templates), is based on the exploitation of the low surface energy of novel fluoropolymeric molds (Fig. 1) [10-12]. The molds are derived from liquid perfluoropolyether (PFPE) precursors, which can be photochemically crosslinked at room temperature. The resulting elastomeric solids enable high-resolution imprint lithography, an emerging technique from the microelectronics industry, to fabricate a variety of organic particles. PRINT is therefore an adaptation of the highly precise mass production technologies used today for the fabrication of nanoscale, silicon-based devices found in the microelectronics field. By developing lithographic technologies that are able to fabricate transistors that are smaller and smaller, companies have been able to increase the number of transistors on a microprocessor to over one billion: this represents an increase of over six orders of magnitude since the early 1970’s [13,14]. The minimum feature size currently found on each individual transistor is under 100 nm (approximately the size of an individual virus particle) [15,16]. The state-of-the-art in the microelectronics industry has finally reached the size scale appropriate for the ideal drug delivery vector. It is now timely to envision and propose the use of lithographic techniques found in the microelectronics industry to fabricate carriers of precise size for use in nanomedicine. Indeed, transistors in today’s microelectronic devices are even smaller than traditional drug delivery carriers that have been studied over the last 25 years: micelles, liposomes, and polymer particles.
Fig. 1. Illustration of PRINT.
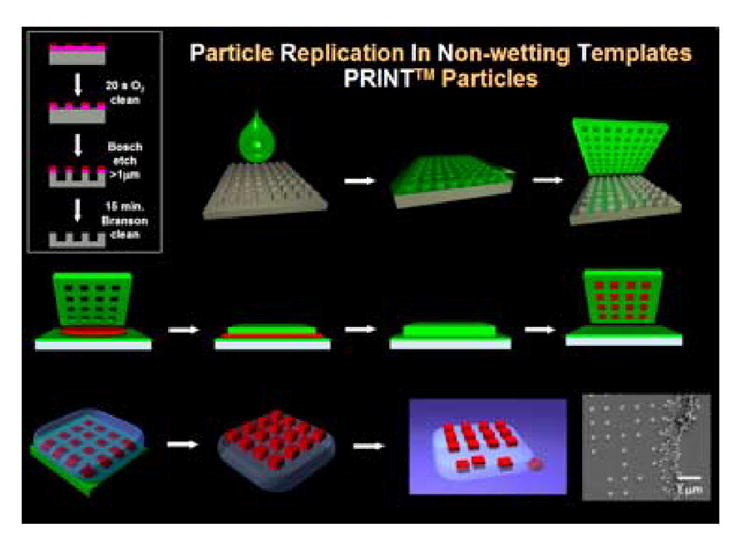
Fabrication of the silicon master template (box, upper left); Wetting of the silicon master with (green) liquid fluoropolymer, followed by curing (top row); PFPE elastomeric mold produced with nanoscale features from the master (upper right); Confining (red) organic liquid to cavities by applying pressure between mold and a PFPE surface (middle row); Removal of organic particles from mold with adhesive layer (bottom left); Dissolution of adhesive layer producing free particles (bottom right)
The nature of PRINT technology takes drug delivery for the first time into the uncharted realm of engineered drug therapies given its á la carte approach and versatility. This innovative tool allows for the simultaneous control over all of the parameters that are essential in the rational design of conventional delivery vectors in nanomedicine. PRINT allows for the precise control over particle size (20 nm to >100 μm) through use of an appropriately designed master template. The advanced imprint lithography of PRINT ensures replication of the identical master features to afford particles that are truly monodisperse. Particle shape is also controlled through the judicious choice of a master, and geometries such as spheres, cylinders, discs, and toroids with defined aspect ratios can be accommodated. The composition of particles made using PRINT is also readily tunable and amendable to the inclusion of a variety of organic matrices including albumin, hydrogels, PLLA, PLGA, etc. (Fig. 2). Moreover, the porosity, texture, and modulus of the particles can be altered in a logical fashion through careful alteration of the matrix formulation. In view of the fact that PRINT is compatible with a wide range of chemistries and the gentle nature in which particles are fabricated, this technology enjoys the straightforward incorporation of a variety of cargos. The inclusion of hydrophilic or hydrophobic therapeutic molecules, biologicals, peptides, proteins, oligonucleotides, siRNA, contrast agents, radiotracers, and fluorophores can be accommodated through inclusion in the particle matrix. The concentration of such cargos in the particles can be exactly chosen to meet specific needs and standards since PRINT does not rely on the kinetic trapping of external molecules during particle fabrication as is the case with liposomes and micelles. Finally, particle surface properties are readily modified though either the matrix composition or post-functionalization with surface moieties. Thus, particle surfaces are amenable to decoration with targeting peptides, antibodies, aptamers, avidin/biotin complexes, cationic/anionic charges, and “stealth” poly(ethylene glycol) (PEG) chains for steric stabilization. We believe that PRINT is the only technology that can independently design in these attributes to create truly engineered nanovectors for drug therapies. For the first time, key therapeutic parameters such as bioavailability, biodistribution, and target-specific cell penetration can be simultaneously designed into a therapy. In this report we document the first in vivo study of PRINT particles administered intravenously into healthy mice. The promising biodistribution profile and blood pharmacokinetics of 200 nm non-targeted radiolabeled PEG-based nanogels fabricated using PRINT methodology are discussed.
Fig. 2. Results of the PRINT Process.

Top row, left to right: A) SEM of an etched silicon wafer master template of 3 μm posts having a height of 1.7 μm; B) Cured PFPE mold of the master template shown in A; C) PFPE mold containing PEG particles prior to harvesting; D) Harvested and dispersed PEG PRINT particles. Bottom row, left to right: E) SEM of an etched silicon wafer patterned with approximately 400 billion posts that are 100 nm in diameter and 400 nm tall; F) A cured PFPE mold of the silicon master template shown in E; G) 100 nm PEG particles made using PRINT and transferred to a medical adhesive layer for surface functionalization and subsequent harvesting
2. Materials and Methods
2.1 Materials
Fluorocur™, the perfluoropolyether used as the molding material in the PRINT process, was purchased from Liquidia Technologies (Product # 2M-140). Trimethylolpropane ethoxylate triacrylate (Mn = 428 g/mol) (Aldrich), was passed through a short plug of alumina prior to use to remove inhibitor. Poly(ethylene glycol) monomethyl ether monomethacrylate (Mn = 1,000 g/mol) (Polysciences), para-hydroxystyrene (Alfa Aesar, 10% (w/w) in propylene glycol) and 2,2-diethoxyacetophenone (Aldrich) were used as received without further purification. Iodogen® pre-coated tubes were purchased from Pierce Biotechnology, Inc., and radioactive iodine (Na125I) was purchased from Perkin Elmer Life and Analytical Sciences, Inc. as 100 mCi/mL in 10-5 M NaOH. HeLa cells and all cell culture media (MEM, OptiMEM) were purchased from the tissue culture facility at The University of North Carolina at Chapel Hill. CellTiter 96* AQueous One Solution Cell Proliferation Assay (MTS) was purchased from Promega Corporation. The lysis agent used for negative controls in in vitro viability studies with HeLa cells was 9% w/v solution of Triton® X-100 in water. Silicon templates used as masters were obtained from Benchmark Technologies. C57BL/6J mice were purchased from The Jackson Laboratory. Ketamine HCl (100 mg/mL) was purchased from Abbott Laboratories. Cholesterol and 1,2-Distearoyl-sn-Glycero-3-Phosphocholine (DSPC) were purchased from Avanti Polar Lipids, Incorporated. All animal experiments were conducted in accordance with guidelines set forth by The University of North Carolina at Chapel Hill, and approval was obtained for the completion of these experiments.
2.2 Preparation of PRINT nanoparticles
The fabrication of patterned Fluorocur™ molds has been described elsewhere [10]. Briefly, 20 mL of Fluorocur™ resin containing 0.1% (w/w) of 2,2-diethoxyacetophenone was pooled in the center of an 8 inch patterned master (with feature sizes of 200 nm) which was set up inside an enclosed UV chamber. Ten minutes was allowed to pass so that the Fluorocur™ resin was spread out over the entire 8 inch wafer. The entire system was then purged with nitrogen for 3 minutes. Following this, the coated wafer was exposed to UV irradiation (λ = 365 nm, power > 20 mW/cm2) for 2 minutes to cure the Fluorocur™ resin. The elastomeric mold was then removed from the master template by gently peeling it away from the silicon surface.
In these experiments, the PRINT particles were derived from a mixture composed of 78% (w/w) PEG triacrylate, 20% (w/w) PEG monomethyl ether monomethacrylate, 1% (w/w) 2,2-diethoxyacetophenone, and 1% (w/w) para-hydroxystyrene. A 10% (w/v) solution of this mixture in 2-propanol (filtered through a 0.22 μm PTFE filter) was prepared. This solution (1 mL) was then sprayed onto a Fluorocur™ patterned mold using an air brush and residual 2-propanol was allowed to evaporate over 10 minutes. A poly(ethylene) sheet (American Plastics Co.) was then placed over the 8 inch (diameter) mold ensuring that the entire active area was covered. This poly(ethylene) sheet was then peeled back at a rate of approximately 2.5 cm/min. Following this, the mold was placed in a UV curing chamber. The chamber was purged with nitrogen for 3 minutes and UV irradiation was applied (λ = 365 nm, power > 20 mW/cm2) for 2 minutes.
To facilitate removal of the particles from the mold, a physical means for harvesting the particles was utilized. Specifically, a 2 mL aliquot of acetone (filtered through a 0.22 μm PTFE filter) was placed on the particle-filled mold and this drop of acetone was gently moved along the surface of the mold using a glass slide. The movement of the glass slide facilitated release of the particles from the mold. The suspended particles were collected in a 50 mL Falcon tube and diluted to the 50 mL mark with filtered acetone after particle collection was complete. The suspension was vortexed for 10 minutes and was centrifuged at 3200 rpm for 30 minutes using a IEC CENTRA CL2 Centrifuge (Thermo Electron Corporation). The supernatant was removed via aspiration and the particle pellet was redispersed in 50 mL of fresh acetone by vortexing for 10 minutes followed by centrifugation for an additional 30 minutes. This process was repeated once more and after aspiration the particles were redispersed in 5 mL of distilled water by sonicating the dispersion for 15 minutes. The particle dispersion was filtered through a 20 μm filter into a fresh 50 mL Falcon tube, and diluted to the 50 mL mark with acetone. This particle suspension was then centrifuged for one hour. The supernatant was removed via aspiration and the particle pellet was redispersed in 50 mL of fresh acetone by vortexing for 10 minutes followed by centrifugation for an additional 30 minutes. This washing process was repeated once more (with acetone) and after aspiration the particles were redispersed in a minimal amount of acetone, transferred to a tarred Eppendorf tube, and centrifuged in a microfuge (Fisher Scientific) for 20 minutes. The supernatant was removed and the pellet was dried in a vacuum oven overnight, massed, and dispersed in the appropriate amount of sterile water to make a 10 mg/mL dispersion of particles.
2.3 Preparation of liposomes
Lipids (DSPC:Cholesterol, 55:45 mol) were dissolved in chloroform and evaporated to dryness in a rotary evaporator under reduced pressure at 50 °C. After leaving the lipid film overnight under reduced pressure, the film was hydrated with PBS at pH 7.0. Unilamellar liposomes were formed by extrusion with 20 passes through a double-stacked polycarbonate membrane (Whatman Nucleopore) with a pore size of 200 nm, resulting in a liposome diameter of 177 nm with a polydispersity of 0.026 as determined by dynamic light scattering [17,18].
2.4 Particle size analysis of PRINT nanoparticles using scanning electron microscopy (in the dry state)
The size of PRINT nanoparticles was analyzed via scanning electron microscopy (Hitachi model S-4700). Particle dispersions were prepared at concentrations of 0.5 mg/mL, and a drop of this solution was placed on a glass slide. The drop was then allowed to dry, and the glass slide was coated with 1.5 nm of Pd/Au alloy using a Cressington 108 auto sputter coater (Cressington Scientific Instruments Ltd.). The Pd/Au coated glass slide was then adhered to the sample holder using double-sided adhesive tape, and placed inside the vacuum chamber of the SEM and observed under low vacuum (10-6 Torr).
2.5 Particle size analysis of PRINT nanoparticles, and liposomes, using dynamic light scattering (in suspension)
The size and polydispersity of PRINT nanoparticles was analyzed via dynamic light scattering (DLS) using a 90Plus Particle Size Analyzer (Brookhaven Instruments Corporation). The particles were dispersed in PBS at a concentration of 0.5 mg/mL and measured without filtration at 25 °C and 37 °C. The DSPC:CHOL liposomes were diluted with PBS to a concentration of 0.5 mg/mL, and were measured under the same conditions as PRINT particles.
2.6 Zeta potential measurements
The zeta potential of PRINT nanoparticles was measured using a ZetaPlus Zeta Potential Analyzer (Brookhaven Instruments Corporation). The nanoparticles were dispersed in water at a concentration of 0.3 mg/mL and the zeta potential was measured.
2.7 Radiolabeling of PRINT nanoparticles with 125I
PRINT nanoparticles were radiolabeled using Iodogen™ solid phase oxidant in the presense of Na125I. Briefly, 10 mg of PRINT particles in 1 mL of H2O, 53 μL of phosphate buffered saline, and 1 mCi of Na125I in 10 μL of 10-5 M NaOH were added to an Iodogen™ pre-coated tube (50 μg of Iodogen™ reagent) and the tube was swirled every other minute for 15 minutes. The radiolabeled particle solution was then transferred to a pre-weighed 1.5 mL Eppendorf tube. The original reaction tube was rinsed with one 20 μL portion of 1 mM KI and with two 100 μL portions of water and the rinsing solutions were added to the tube containing radiolabeled particles followed by the addition of NaHSO3 (1 μmol/10 μL). The particle dispersion was then centrifuged for 10 minutes at 15,000 × g using an Eppendorf centrifuge 5415 D (Eppendorf). The supernatant was removed and the particles were washed with four 500 μL portions of water (until the radiation in the supernatant was no greater than the background, ensuring complete removal of non-specifically bound 125I) and evaporated to dryness in a SpeedVac SC100 (Savant Instruments). The total mass recovered was 9.45 mg. The specific activity was measured with a Beckman Gamma 5500B gamma counter (Laboratory Technologies) and found to be 5.5 μCi/mg PRINT particles.
2.8 In vitro cytotoxicity
HeLa cells were seeded in 100 μL of media [Minimum Essential Medium (MEM) containing Earle’s salts and supplemented with 1 mM sodium pyruvate and non-essential amino acids] at a density of 5 × 103 cells per cm2 into a 96-well microtitre plate. Cells were allowed to adhere for 24 h before MEM was replaced with Opti-MEM (90 μL per well) and the particle preparation (10 μL per well in PBS). Positive controls contained PBS alone. HeLa cells were incubated with the PRINT particles for 4 h at 37 °C in a humidified 5% CO2 atmosphere. After the 4 h incubation period, negative controls were prepared by the addition of 2 μL of lysis solution to a few wells containing cells only. After 2 minutes, the MTS assay solution was added (20 μL per well) into each well. The cells were then incubated for an additional 1 h at 37 °C in a humidified 5% CO2 atmosphere. The optical density at 450 nm was measured using a BioRad Model 3550 microplate reader (BioRad Laboratories). The viability of the cells exposed to neutral PRINT particles was expressed as a percentage of the viability of cells grown in the absence of particles.
2.9 Biodistribution of [125I]-labeled PRINT particles
C57Bl/6J mice were housed under specific pathogen-free conditions for one week and were used at 8 weeks of age (~18 g). Animals were injected intravenously via bolus tail vein administration with 0.32 mg of [125I]-labeled PRINT particles with a specific activity of 4.3 μCi/mg in 100 μL of PBS (phosphate buffered saline, Sigma-Aldrich). At 10 min, 30 min, 1, 3, and 8 h after dosing, groups of four mice were anesthetized by intraperitoneal injection of 100 μL of ketamine HCl solution in PBS (50 mg/mL). Blood was collected via cardiac puncture. Samples of blood and organs harvested (liver, kidneys, spleen, lungs, and heart) were weighed and counted to determine the total radioactivity in a Beckman Gamma 5500B gamma counter (Laboratory Technologies). An additional four animals were kept in a metabolic cage after injection of the [125I]-labeled PRINT nanoparticles. At 24 h post-injection, the accumulated urine and feces were collected for radioactivity measurements. After these animals were sacrificed (as described above) the blood as well as the organs were removed, weighed, and assayed for radioactivity.
3. Results and Discussion
3.1 The Print Process
In the PRINT process (Fig. 1), the permanent silicon master template is fabricated using advanced lithographic techniques. The liquid PFPE fluoropolymer is then added to the surface of the master template. A positive spreading coefficient allows the perfluoropolyether to wet the nanoscale features of the master template with extremely high fidelity. After the fluoropolymer has wet the master template it is photochemically cross-linked and peeled away to generate a precise mold having nanoscale cavities. The low surface energy and high gas permeability of the PRINT mold enables the organic liquid precursor to the drug carrier particles to fill the cavities through capillary action, but it does not form an inter-connecting “flash” layer of liquid wetting the land area between the cavities. Such specific wetting and filling enables the fabrication of freestanding and harvestable particles that have the same precise shape of the silicon master template from which they were derived. Once the liquid in the mold cavities is converted to a solid using a wide range of gentle chemistries, the array of organic particles can be removed from the mold either by physical methods or by bringing the mold in contact with an adhesive layer (e.g. surgical adhesive/water soluble excipient layer). Physical methods were used herein to harvest the PRINT particles, which were then purified, characterized, and radiolabeled for biodistribution studies.
3.2 Characterization of PRINT Nanoparticles
The particle composition was engineered to produce biologically relevant delivery vectors. Several monomers were added to introduce specific functionality into the hydrogel nanoparticles: poly(ethylene glycol) triacrylate, poly(ethylene glycol) monomethyl ether monomethacrylate, and p-hydroxystyrene (PHS) (Fig. 3). Poly(ethylene glycol) derivatives have long been known to impart biocompatibility, solubility, stability, and increased circulation times to proteins, liposomes, and particles [19-23]. The phenol-containing monomer, PHS, was selected as a chemical handle so that gentle radioiodination of the particles was possible. Since mPEG1000 monomethacrylate is a solid, 2-propanol was used to obtain a homogeneous solution of the monomer precursors, so that a thin film of monomer mixture could be sprayed onto the PFPE mold. To this point, any solvent (or combinations of solvents) can be used in the PRINTing process, as long as the contact angle of the resultant solution onto the PFPE mold is less than 90°. Alternatives to this method include a melt PRINT process, where the monomers are heated to above their melting point, and the PRINT process is carried out, allowing a solvent-free process.
Fig. 3. Chemical structures of monomers and a partial structure of the PRINT nanogel.
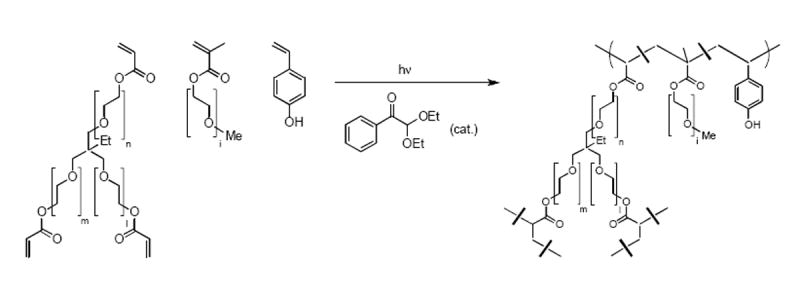
p-Hydroxystyrene was introduced for radioiodination at 1% (w/w). Throughout the present study, it is assumed that iodinated PRINT nanogels behave the same as unmodified particles.
The uniformity of size and shape of the hydrogels was confirmed using scanning electron microscopy (SEM, Fig. 4). The micrographs show the isolation of thousands of virtually identical cylindrical particles, thus verifying PRINT’s ability to copy the nanoscale features of a patterned wafer with high precision. The diameter of the particles in the dry state was determined from the scanning electron micrographs to be 201 ± 10 nm and the height was determined to be 155 ± 10 nm. A dry sample is required for SEM and thus the micrographs obtained do not represent the dispersion the particles have once in solution.
Fig. 4.

Scanning electron micrographs of 200 nm PRINT particles used in the present study
The nanocarriers used most frequently today for the delivery of therapeutics in the clinic can best be classified under the general heading of self-assembled structures. Nanovectors fitting this description include micelles, liposomes, and protein aggregates. As a result of their inherent dynamic nature, these nanoparticles derived from the self-assembly of small molecules would be expected to undergo structural changes once in vivo. As such, it is difficult to make a direct connection between the particle formulation prior to administration and the therapeutic outcome. As an example of this behavior, the size and polydispersity of liposomes and PRINT particles in water were investigated via dynamic light scattering at room temperature (25 °C) and then at physiological temperature (37 °C) over the course of several hours (Fig. 5). At 25 °C both liposomes and PRINT particles have a low polydispersity immediately upon formation or dispersion. Upon heating to 37 °C, the mean diameter and polydispersity of liposomes changes dramatically as a function of time. Over the time course of 6 h at physiological temperature, the PRINT particles remain stable as observed by a steady size and polydispersity (234 ± 12 nm, PDI = 0.005). This could be an important attribute in the design of nanocarriers, since it is expected that the size and polydispersity of dynamic structures may change even more rapidly once in the serum. The zeta potential of these PRINT nanoparticles was measured to be-30.4 ± 1.5 mV.
Fig. 5.
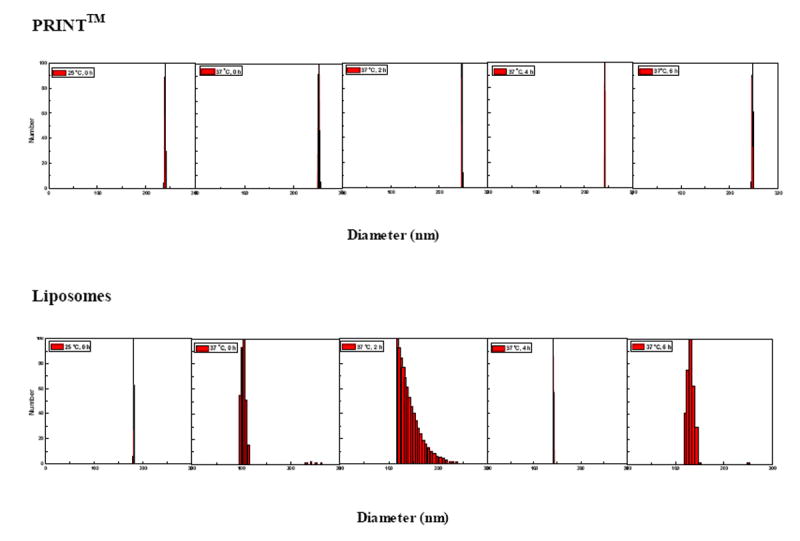
A time-dependent study of the mean diameter, polydispersity and stability of PRINT particles (top), and liposomes (bottom) using dynamic light scattering
3.3 Cytotoxicity studies in vitro
The cytotoxicity of the PRINT nanoparticles was assessed in a cursory manner using a MTS cell viability assay [24]. This particular assay is a colorimetric evaluation that determines the number of living cells by quantifying the amount of formazan product present, which is directly proportional to the number of viable cells. Figure 6 shows high viability relative to the negative control, supporting the non-toxic nature of the nanoparticles, even at high concentrations (100 μg/mL is equivalent to 800,000 particles/cell). Based on the toxicity data obtained, the PRINT nanoparticles appear biocompatible and suitable for future in vivo studies.
Fig 6.
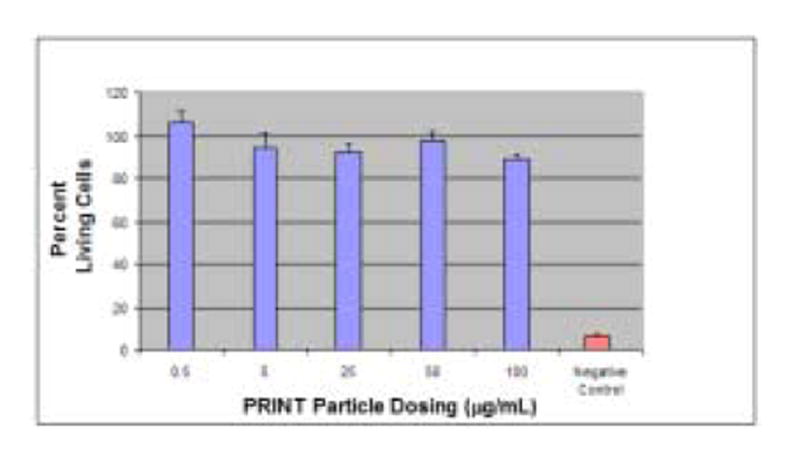
MTS assay depicting the non-toxic nature of 200 nm PRINT particles incubated with HeLa cells. Approximately 103 cells were plated per 1 cm2. Cells were exposed to varying concentrations of PRINT particles in 0.1 mL media for 4 h at 37 °C before the MTS assay was performed. Control wells, where the cells were exposed to only PBS serve as 100% in normalization. Vertical bars stand for one SD with n = 5.
3.4 In vivo biodistribution studies of PRINT particles in healthy mice
In this study, 200 nm [125I]-labelled PRINT particles were administered into healthy C57BL/6J mice via tail vein bolus injection at a dose of 20 mg/kg. The particles were radiolabeled using Iodogen® following the procedure recommended by the supplier (Pierce). The tissue distribution was monitored using a gamma counter, and the percent recovery of injected dose was calculated from the measured radioactivity. Figure 7 shows the tissue distribution of 200 nm PRINT nanoparticles at 10 min, 30 min, 1 h, 3 h, 8 h, and 24 h post-injection. Throughout the time-course of the study, the 200 nm PRINT particles were distributed mainly in the liver and spleen. The total recovery from these two organs amount to as much as 30% over the 24 h study period. This observation is consistent with the fact that the sinusoidal walls of these organs are lined with discontinuous endothelium that allows for passive entrapment of foreign particulates [25,26]. Although the present study does not provide any direct evidence for or against it, it is likely that these particles are eventually taken up by the resident macrophages possibly subsequent to opsonization with serum proteins [27].
Fig. 7.
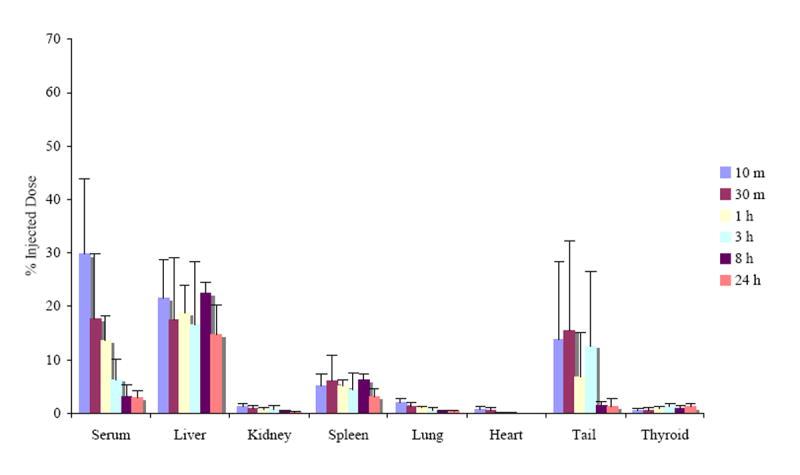
Biodistribution of 200 nm [125I]-labeled PRINT particles in healthy mice subsequent to bolus tail vein injection at a dose of 20 mg/kg. The organ accumulation is expressed as a percent of injected dose after animals were sacrificed at 10 min, 30 min, 1, 3, 8, and 24 h post-intravenous injection. The organ data is presented as the mean ± SD with n = 4. The recovery found in the blood assumes a blood volume of 2.18 mL/25 g mouse [34].
The particle accumulation was not significant in other organs harvested, often ~ 1% of the injected dose was found in the kidneys, heart, and lungs. The extent of particle accumulation reported in these organs may slightly over-represent the actual accumulation since the organs were not thoroughly rinsed to remove residual blood. Tails were collected in an effort to monitor the amount of nanoparticles that were retained at the injection site. A significant amount of injected particles were found in the tail, especially during the initial 3 h period. This may be due to the rupture of blood vessels upon rapid injection of a large number of fine particles in a short period of time (< 5 seconds), creating endothelial gaps for particle retention. Finally, a trace but significant amount of radioactivity was observed in thyroid gland, possibly indicating the biodegradation of particles, yielding radioactive I2. Production of iodine from biochemical degradation of iodinated proteins or peptides is not uncommon and is also consistent with the observation that total recovery decreased steadily over the 24 h period studied (vide infra) [28].
Total recovery of the radioiodinated particles was found to decrease with time, beginning with an 81 ± 6 % recovery at 10 minutes post-injection and ending with a 24 ± 7 % recovery after 24 h (data not shown). Here, the total recovery was calculated from radioactivity measured only from the blood, liver, kidneys, spleen, lungs, heart, tail, bone marrow, and thyroid. In an effort to achieve a full mass balance, four mice were kept in metabolic cages for 24 h so that urine and feces could be collected and analyzed. For these mice, additional tissues and body parts such as fat, muscle, head, legs, intestines, and the remainder of the body were also analyzed for additional radioactivity. It was found that with these additional measurements, the total recovery at 24 h post-injection improved from 24 ± 7 %, with the main organs only, to 58 ± 4 %. The appearance of radioactivity in the thyroid gland throughout the study period (approximately 1% of injected dose) and in urine 24 h after dosing (as much as 8.6%) may suggest particle degradation [29]. This is also consistent with a hypothesis that loss of volatile radioactive iodine via the lungs could have contributed to the time-dependent decrease in total recovery of radioactivity observed.
As shown in Figure 8, the disappearance of PRINT particles from circulation was bi-exponential. The data set was fitted to a two-compartmental pharmacokinetic model with reversible distribution between central and peripheral compartments and with elimination from the central compartment. The pharmacokinetic parameters obtained using WinNonLin 5.0.1 (Pharsight Corporation) show the initial phase of rapid distribution with an apparent t1/2 of 17 min. The rapid distribution is not surprising considering that the steric coat on the PRINT particles is only a low molecular weight PEG chain (9 mol %, 20% w/w of 1-kDa PEG monomethyl ether). It has been suggested that the optimal coating for the creation of long-circulating liposomes is 3-7 mol % of 2-5 kDa PEG [30]. The shorter PEG chains used in the current particle formulation may not offer a radius of protection that is sufficient to effectively block the adsorption of opsonic proteins. The subtle nuances that both the degree of PEG incorporation and the molecular weight of PEG play in prolonging the circulatory t1/2 are well documented for other types of nanocarriers [30-32].
Fig. 8. Blood pharmacokinetic profile of PRINT particles in healthy C57BL/6J mice.
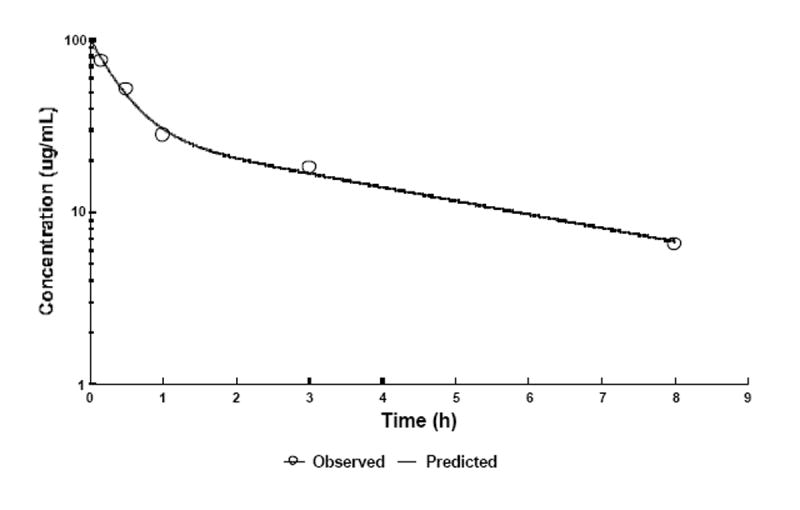
At given time intervals, four animals were sacrificed and blood was collected via cardiac puncture. Radioactivity observed was converted to particle concentration using the specific radioactivity measured and assuming a total blood volume of 2.18 mL/25 g mouse [34]. The data was subject to two-compartmental analysis (WinNonlin) resulting in pharmacokinetic parameters discussed in the text. For simplicity, data obtained 24 h post-injection are not shown in the figure or used for PK parameter determination.
Volumes of distribution of central and peripheral compartments were found to be 3 and 5 mL, respectively. Considering the blood volume of approximately 1.7 mL/20 g mouse, liver of 0.8 g, and spleen < 0.1 g, these values appear to be somewhat exaggerated, however, they certainly rule out any significant extravasation. Since the stability of the radiolabel is not well established in the present study and since it is the radioactivity that is monitored, it is difficult to unambiguously interpret the slow phase of radioactivity decay in the later time-points with an apparent t1/2 of 3.3 h. The appearance of radioactivity in urine strongly suggests that these PRINT particles and/or their degradation products must be cleared rapidly from the blood. Thus, the slow elimination phase may well represent slow re-distribution of particles or particle remnants between the blood and organs/tissues.
The AUC, a measure of total availability of particles in the circulation for organ distribution extrapolated to infinite time, was determined to be 191 μg·h/mL. Unmodified, conventional liposomes show dose-dependent pharmacokinetic parameters, including AUC upon i.v. administration. A liposome dose equivalent to the dose in the present study, 20 mg/kg, shows AUC values of approximately 70 to 700 μg·h/mL. Thus, the present data are certainly in agreement with that reported for liposomes [33].
Conclusions
This paper is concerned with the characterization of nanofabricated particles that are monodisperse in size and shape. It includes the first pharmacokinetic evaluation of PRINT-derived cylinders of 200 nm. PRINT allows for powerful realizations regarding pharmacokinetics of nanoparticles since intersubject variation can be ascribed fully to the subject and not to variations of the nanocarrier. This is a significant step in pharmacokinetic analysis since it eliminates the effect that polydisperse samples can have on biodistribution. This in turn allows us to be able to precisely demonstrate the effect of size, composition, the addition of cargo, modulus, and functionalization on biodistribution, which has never been possible until now. Current efforts are focused on the creation of long-circulating PRINT particles for the eventual use in engineered drug therapies. Results will be reported in due course.
PRINT is the first general, singular method capable of forming organic nanoparticles in which critical design parameters can be precisely and independently tailored bringing a greater understanding of cause-and-effect to the field of nanomedicine. With the unprecedented ability of PRINT technology to control particle size, shape, composition, modulus, cargo, and surface properties, questions such as “what interrelated role does shape, size and mechano-chemico functionality play on the biodistribution of carriers in vivo?” and, “how can this understanding translate into more efficacious detection, diagnosis, therapeutic and prevention strategies?” can finally begin to be answered. As such, PRINT is a significant scientific and technological breakthrough, which will allow the fabrication of heretofore inaccessible populations of nanobiomaterials which are poised to revolutionize and accelerate our translational understanding, detection, and treatment of disease.
Acknowledgments
The authors would like to thank Michael J. Barrett for his gracious help with light scattering, Kevin P. Herlihy for his artistic rendition of the PRINT process, CHANL (especially Carrie Donley), Ginger Denison-Rothrock (Liquidia Technologies) and Mary Napier for useful discussions, and John An for his help with cell work. This work was supported in part by the STC Program of the NSF (CHE-9876674), NIH P01-GM59299, NIH U54-CA119343 (the Carolina Center of Cancer Nanotechnology Excellence), the William R. Kenan Professorship of the University of North Carolina at Chapel Hill, and Liquidia Technologies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Riley S. Innovation in Drug Delivery: The future of nanotechnology and non-invasive protein delivery. Business Insights Ltd; 2006. pp. 25–26. [Google Scholar]
- 2.Moghimi SM, Hunter AC, Murray JC. Nanomedicine: current status and future prospects. FASEB J. 2005;19(3):311–330. doi: 10.1096/fj.04-2747rev. [DOI] [PubMed] [Google Scholar]
- 3.Barenholz Y. Liposome application: problems and prospects. Curr Opin Colloid Interface Sci. 2001;6(1):66–77. [Google Scholar]
- 4.Kwon GS, Kataoka K. Block copolymer micelles as long-circulating drug vehicles. Adv Drug Delivery Rev. 1995;16:295–309. [Google Scholar]
- 5.Lee CC, MacKay JA, Frechet JMJ, Szoka FC. Designing dendrimers for biological applications. Nat Biotechnol. 2005;23(12):1517–1526. doi: 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]
- 6.Torchilin VP. Targeted polymeric micelles for delivery of poorly soluble drugs. Cell Mol Life Sci. 2004;61:2549–2559. doi: 10.1007/s00018-004-4153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vicent MJ, Duncan R. Polymer conjugates: nanosized medicines for treating cancer. Trends Biotechnol. 2006;24(1):39–47. doi: 10.1016/j.tibtech.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 8.Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discovery. 2003;2(5):347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 9.McAllister K, Sazani P, Adam M, Cho MJ, Rubinstein M, Samulski RJ, DeSimone JM. Polymeric Nanogels Produced via Inverse Microemulsion Polymerization as Potential Gene and Antisense Delivery Agents. J Am Chem Soc. 2002;124(51):15198–15207. doi: 10.1021/ja027759q. [DOI] [PubMed] [Google Scholar]
- 10.Rolland JP, Maynor BW, Euliss LE, Exner AE, Denison GM, DeSimone JM. Direct fabrication and harvesting of monodisperse, shape-specific nanobiomaterials. J Am Chem Soc. 2005;127(28):10096–10100. doi: 10.1021/ja051977c. [DOI] [PubMed] [Google Scholar]
- 11.Rolland JP, Hagberg EC, Denison GM, Carter KR, DeSimone JM. High-resolution soft lithography: enabling materials for nanotechnologies. Angew Chem Int Ed Engl. 2004;43(43):5796–5799. doi: 10.1002/anie.200461122. [DOI] [PubMed] [Google Scholar]
- 12.Euliss LE, DuPont JA, Gratton S, DeSimone J. Imparting size, shape, and composition control of materials for nanomedicine. Chem Soc Rev. 2006;35(11):1095–1104. doi: 10.1039/b600913c. [DOI] [PubMed] [Google Scholar]
- 13.Patt YN, Patel SJ, Evers M, Friendly DH, Stark J. One Billion Transistors, One Uniprocessor, One Chip, Computer. 1997;30(9):51–57. [Google Scholar]
- 14.Hiremane R. From Moore’s Law to Intel Innovation-Prediction to Reality. Technology@Intel Magazine. 2005:2–9. [Google Scholar]
- 15.Baker TS, Olson NH, Fuller SD. Adding the third dimension to virus life cycles: three-dimensional reconstruction of icosahedral viruses from cryo-electron micrographs. Microbiol Mol Biol Rev. 1999;63(4):862–922. doi: 10.1128/mmbr.63.4.862-922.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. [1/22/07]; www.intel.com.
- 17.Huang Z, Guo X, Li W, MacKay JA, Szoka FC., Jr Acid-triggered transformation of diortho ester phosphocholine liposome. J Am Chem Soc. 2006;128(1):60–61. doi: 10.1021/ja057024w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grit M, Crommelin DJA. The effect of aging on the physical stability of liposome dispersions. Chemistry and Physics of Lipids. 1992;62(2):113–22. doi: 10.1016/0009-3084(92)90089-8. [DOI] [PubMed] [Google Scholar]
- 19.Woodle MC. Controlling liposome blood clearance by surface-grafted polymers. Adv Drug Delivery Rev. 1998;32:139–152. doi: 10.1016/s0169-409x(97)00136-1. [DOI] [PubMed] [Google Scholar]
- 20.Papisov MI. Theoretical considerations of RES-avoiding liposomes: Molecular mechanics and chemistry of liposome interactions. Adv Drug Delivery Rev. 1998;32:119–138. doi: 10.1016/s0169-409x(97)00135-x. [DOI] [PubMed] [Google Scholar]
- 21.Allen TM, Hansen CB, de Menezes DEL. Pharmacokinetics of long-circulating liposomes. Adv Drug Delivery Rev. 1995;16:267–284. [Google Scholar]
- 22.Gregoriadis G. Engineering liposomes for drug delivery: progress and problems. Trends Biotechnol. 1995;13(12):527–37. doi: 10.1016/S0167-7799(00)89017-4. [DOI] [PubMed] [Google Scholar]
- 23.Torchilin VP. Polymer-coated long-circulating microparticulate pharmaceuticals. J Microencapsulation. 1998;15(1):1–19. doi: 10.3109/02652049809006831. [DOI] [PubMed] [Google Scholar]
- 24.Barltrop JA, Owen TC, Cory AH, Cory JG. 5-(3-Carboxymethoxyphenyl)-2-(4,5-dimethylthiazolyl)-3-(4-sulfophenyl)tetrazolium, inner salt (MTS) and related analogs of 3-(4,5-dimethylthiazolyl)-2,5-diphenyltetrazolium bromide (MTT) reducing to purple water-soluble formazans as cell-viability indicators. Bioorg Med Chem Lett. 1991;1(11):611–614. [Google Scholar]
- 25.Wisse E, De Leeus AM. In: Structural Elements Determining Transport and Exchange Processes in the Liver. Davis SS, Illum L, McVie JG, Tomlinson E, editors. Elsevier Scientific; Amsterdam: 1984. pp. 1–23. [Google Scholar]
- 26.Senior JH. Fate and behavior of liposomes in vivo: a review of controlling factors. Crit Rev Ther Drug Carrier Syst. 1987;3(2):123–193. [PubMed] [Google Scholar]
- 27.Chonn A, Semple SC, Cullis PR. β2-glycoprotein I is a major protein associated with very rapidly cleared liposomes in vivo, suggesting a significant role in the immune clearance of “non-self” particles. J Biol Chem. 1995;270(43):25845–25849. doi: 10.1074/jbc.270.43.25845. [DOI] [PubMed] [Google Scholar]
- 28.Wolf H, Marschall F, Scheffold N, Clausen M, Schramm M, Henze E. Iodine-123 labelling of atrial natriuretic peptide and its analogs: initial results. Eur J Nucl Med Mol Imag. 1993;20(4):297–301. doi: 10.1007/BF00169804. [DOI] [PubMed] [Google Scholar]
- 29.Takakura Y, Hashida M. Macromolecular carrier systems for targeted drug delivery: pharmacokinetic considerations on biodistribution. Pharm Res. 1996;13(6):820–831. doi: 10.1023/a:1016084508097. [DOI] [PubMed] [Google Scholar]
- 30.Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles Theory to practice. Pharmacol Rev. 2001;53(2):283–318. [PubMed] [Google Scholar]
- 31.Owens DE, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307(1):93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 32.Drummond DC, Meyer O, Hong K, Kirpotin DB, Papahadjopoulos D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 1999;51(4):691–743. [PubMed] [Google Scholar]
- 33.Allen TM, Hansen C. Pharmacokinetics of stealth versus conventional liposomes: effect of dose. Biochim Biophys Acta. 1991;1068(2):133–141. doi: 10.1016/0005-2736(91)90201-i. [DOI] [PubMed] [Google Scholar]
- 34.Kataoka K, Matsumoto T, Yokoyama M, Okano T, Sakurai Y, Fukushima S, Okamoto K, Kwon GS. Doxorubicin-loaded poly(ethylene glycol)-poly(b-benzyl-L-aspartate) copolymer micelles: their pharmaceutical characteristics and biological significance. J Controlled Release. 2000;64:143–153. doi: 10.1016/s0168-3659(99)00133-9. [DOI] [PubMed] [Google Scholar]