

Abstract
Studies in the yeast S. cerevisiae have provided a framework for understanding how eukaryotic cells replicate their chromosomal DNA to ensure faithful transmission of genetic information to their daughter cells. In particular, S. cerevisiae is the first eukaryote to have its origins of replication mapped on a genomic scale, by three independent groups using three different microarray-based approaches. Here we describe a new technique of origin mapping via detection of single-stranded DNA in yeast, this method not only identified the majority of previously discovered origins, but also detected new ones. We have also shown that this technique can identify origins in S. pombe, illustrating the utility of this method for origin mapping in other eukaryotes.
1. Introduction
Microarrays have proven to be a powerful tool for mapping the genomic locations of replication origins. In S. cerevisiae they have been used in combination with a variety of molecular techniques including density transfer, Chromatin Immunoprecipitation (ChIP), and DNA copy number changes [1–3]. There are challenges to overcome in applying these techniques to other organisms of interest, particularly to organisms with large genomes. Density transfer and ChIP techniques are technically demanding. While the detection of copy number changes appears straight forward, it requires reliably detecting differences in hybridization signal of less than two-fold. We have developed a straightforward technique for origin mapping that measures ssDNA abundance across the genome in response to replication stress induced by hydroxyurea (HU), a drug inhibitor of ribonucleotide reductase. ssDNA accumulation is largely limited to origins of replication in genetic backgrounds in which an essential protein kinase for the DNA replication checkpoint is rendered checkpoint-defective by mutations (rad53 in S. cerevisiae and cds1 in S. pombe) [4].
2. Overview of experimental design: strain requirements and growth conditions
The idea for identifying replication origins via ssDNA detection is based on the following observations. First, in vivo, ssDNA is a natural intermediate during DNA replication due to the discontinuous nature of replication on the lagging strand. Second, when cellular pools of dNTPs are reduced by the presence of HU, replication forks accumulate larger regions of ssDNA than in the absence of HU [5]. Third, HU treatment of a checkpoint-deficient mutant such as rad53 results in aberrant, increased stretches of ssDNA that do not appear to expand with time [5]. Therefore, we reasoned that if we could map the locations of these single-stranded genomic regions we would be able to follow origin activation and the possible progression of fork movement.
To map the locations of genomic ssDNA we took advantage of the fact that, in vitro, double-stranded DNA (dsDNA) is an inefficient template for primer extension by DNA polymerases unless it is first denatured. Under non-denaturing conditions any single-stranded portion of genomic DNA would preferentially serve as a template for random primed synthesis compared to the double stranded portion of the same DNA. Hybridization of the in vitro synthesized fragments to microarrays would identify which regions of the genome were single-stranded in vivo. In WT cells synchronously entering S phase in the presence of HU, we predicted that ssDNA would appear first at origins at the beginning of S phase and then migrate to the neighboring regions as forks move. In contrast, based on the model for Rad53’s role in maintaining the integrity of replication forks during HU-challenge [5–7], we expected that ssDNA would remain localized to the origins of replication over time in a rad53 mutant.
Based on the rationale just described, we devised a strategy (Fig. 1) to identify ssDNA locations on a genome-wide scale as cells (both wild type and rad53 mutant) progress through S phase in the presence of HU. Genomic DNA isolated from an S phase sample and a control G1 phase sample were differentially labeled with cyanine (Cy)-conjugated dUTPs through random priming and synthesis without denaturing the template DNA, and co-hybridized to a DNA microarray. We quantified the amount of ssDNA at a given genomic location by calculating the fluorescent signal from the channel representing the S phase sample relative to that from the channel representing the control sample. In parallel with microarray hybridization, we also determined the total percentage of ssDNA in both the S phase and G1 control samples by slot blotting undenatured chromosomal DNA and hybridizing with a genomic DNA probe. The total percentage of ssDNA was used to normalize the microarray data.
Figure 1.
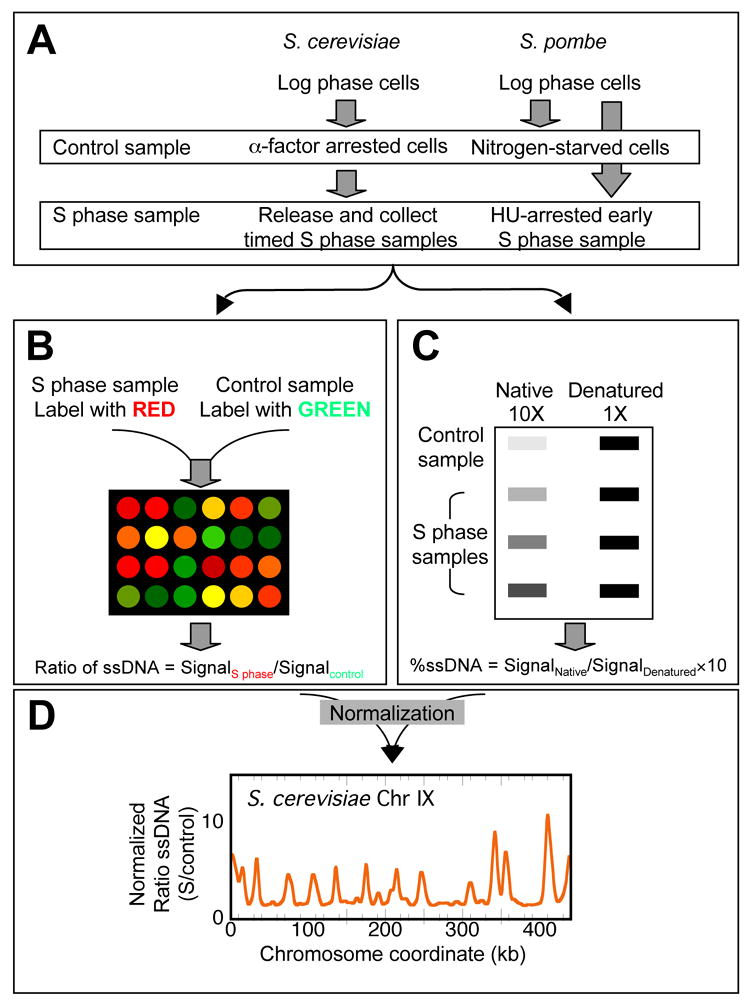
Schematic representations of experimental procedures for microarray analysis with both S. cerevisiae and S. pombe samples.
Encouraged by the success of our method in identifying origins in S. cerevisiae, we tested our methodology for its ability to map origins in a distantly related organism, S. pombe, exploiting the conserved mechanism of checkpoint activation by HU through the Rad53 homolog Cds1. In this modified procedure (Fig. 1A, right panel), we treated asynchronous fission yeast cells (WT and Δcds1) with either nitrogen starvation or treatment with HU to arrest cells in G1 and early S phase, respectively. We isolated and labeled genomic DNA and performed microarray analysis in a similar fashion as described above for S. cerevisiae (Fig. 1B–D).
3. Methods
3.1 Preparation of template DNA
3.1.1 Growth conditions
S. cerevisiae cell cultures are grown in synthetic complete medium at 30°C unless temperature sensitive alleles are present. Exponential phase cells (at OD660 of 0.2) are synchronized in G1 by the addition of alpha factor at 200 nM, followed by incubation for approximately one and a half generations, or until the unbudded cell population reaches at least 90%. This concentration of alpha factor is applied to strains that contain the bar1 mutation. For strains that are BAR1, 200 μM of alpha factor should be used. HU is added at 200 mM, and cells are released from the G1 arrest by the addition of pronase at 0.2 mg/ml.
S. pombe cell cultures are grown in Edinburgh minimal medium at 30°C. Nitrogen starvation of cells is achieved by growing exponential phase cells in Edinburgh minimal medium lacking a nitrogen source for approximately 20 hours at 25°C (recommended temperature for nitrogen starvation rather than 30°C, Paul Nurse lab manual). The early S phase sample is obtained by growing exponential phase cells in Edinburgh minimal medium containing 12 mM HU for 3 hours at 30°C.
3.1.2 Cell collection
Three hundred ml of both the G1 control (alpha factor arrested) culture and the S phase culture at discrete time points are collected by quick mixing with 3 ml of 10% NaN3 and a 60 ml frozen pellet of 0.2 M EDTA, pH 8.0. After centrifugation at 5,000 rpm in a Sorvall SLA-3000 rotor for 5 minutes, cell pellets are washed with ice-cold water. Washed cell pellets are then stored at −20°C until DNA preparation. The same procedures are used for collecting S. pombe cultures.
3.1.3 Chromosomal DNA preparation
The isolation of genomic DNA from yeast cells is adapted from a procedure originally described by Huberman et al. [8], later modified by Bonny Brewer. The detailed procedure has been published online at http://fangman-brewer.genetics.washington.edu/DNA_prep.html. All steps are the same as described except that the Hoechst dye is omitted during the preparation of the CsCl gradient. Also, after the ultracentrifugation, instead of “pulling the band” of DNA through the side of the tube using a needle, the gradient is drip-fractionated from the bottom of the tube into approximately 40 fractions of 12 drops each. Two microliters of each fraction are denatured and slot-blotted, and the blot is hybridized with a genomic DNA probe to identify the fractions that contain genomic DNA [9]. Once the genomic DNA fractions are identified, they are pooled for purification of DNA. Usually six to seven fractions are pooled, starting from roughly the tenth fraction from the bottom of the tube. We avoid pooling too many fractions from the tails of the DNA peak, particularly from the upper tail (towards the top of the tube) since these fractions contain considerable amounts of mitochondrial DNA. The pooled fractions are then mixed with 3 volumes of cold 70% ethanol to precipitate the DNA. The DNA pellet is resuspended in 200 μl of 10 mM Tris-HCl, pH 8.0, 0.05 mM EDTA, 100 mM NaCl and stored at 4°C for two to three days to allow DNA to dissolve completely. Once dissolved, the DNA is used for labeling as soon as possible to minimize in vitro generation of single-stranded template. From here on, the DNA is pipetted only through wide-bore tips to minimize shearing of the DNA.
3.2 Labeling of DNA samples for microarray hybridization
Prior to labeling the DNA for hybridization, it is important to decide how much template DNA to use for labeling in order to achieve appropriate levels of fluorescent signal on a microarray. Based on pilot experiments, we estimated that up to 10% by mass of an HU-treated S phase DNA sample from a checkpoint-deficient rad53 strain could be ssDNA. In our experiments we therefore used 5 μg of genomic DNA assuming that 500 ng would be single-stranded and therefore available to act as a template for labeling without denaturation. When used at this quantity for both the G1 control and S phase samples, this methodology has given satisfactory hybridization results and allows normalization of microarray data (see below).
3.2.1 Digesting the chromosomal DNA for labeling
We routinely obtain 10 to 30 μg of DNA in a standard DNA preparation from 300 ml of culture. Because the DNA is of very high molecular weight, we first digest the chromosomal DNA with the restriction enzyme EcoRI to reduce the average size of the genomic fragments. In general, each digestion reaction contains 8 μg of DNA--5 μg for microarray analysis and 3 μg for slot blot hybridization to determine the total percentage of ssDNA (see below). For G1 control samples that will be used as controls for multiple S phase samples in microarray hybridizations, we simply scale up the DNA for each reaction.
To digest 8 μg of DNA in a volume of 200 μl, add the appropriate volume of DNA to a reaction tube containing:
20 μl of 10X reaction buffer for EcoRI (New England BioLabs, NEB)
8 μl of EcoRI (NEB, 20 U/μl)
distilled H2O to bring the volume to 200 μl
The digestion reaction is allowed to take place at 37°C for 3 hours. The DNA is then precipitated by the addition of 20 μl (1/10 volume) of 3M NaAc followed by 400 μl of cold 100% ethanol. Precipitation takes place at −20°C overnight. The precipitated DNA is washed with 70% ethanol followed by drying under vacuum.
3.2.2 Assessment of reaction specificity: control labeling reactions with and without random primers
The use of primers in the labeling scheme with undenatured genomic DNA as template is crucial for the outcome of the experiment. The very reason that ssDNA persists at normal replication forks in vivo is that the replication machinery is waiting for a primer to start the next Okazaki fragment: there is no available 3’-hydroxyl to act as a primer for synthesis from the single-stranded template (Fig. 2A). However, there are other potential sources of ssDNA in the genomic DNA preparation that would not require a primer for in vitro synthesis: single-stranded gaps, such as those at ends of molecules, could serve as templates for in vitro synthesis by extending an existing 3’-hydroxyl (Fig. 2A). Therefore, prior to labeling DNA for microarray analysis, we asked to what extent the labeling reaction was dependent on the addition of random primers. For template DNA, we used an S phase rad53 DNA sample isolated at 1 hr post release from the G1 arrest into medium containing HU and labeled it in the presence or absence of the random hexameric primers. The reaction composition is shown below:
Figure 2.
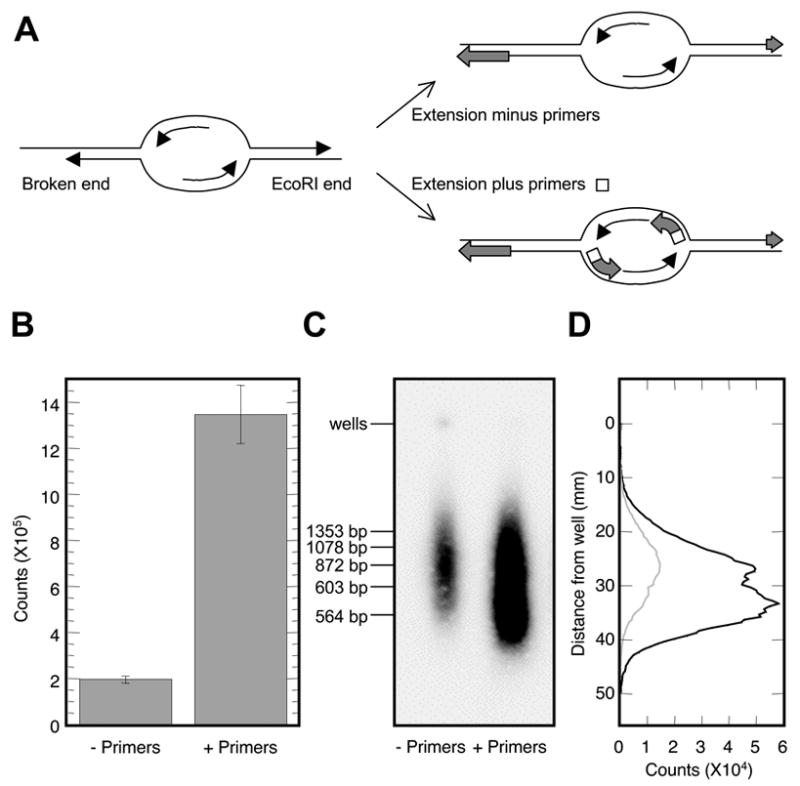
Specificity of the random-primed labeling reactions. (A) Schematic representation of different types of genomic ssDNA and their potential to act as templates during primed and non-primed in vitro synthesis. (B) A rad53 S phase DNA sample isolated at 2 hr post release from alpha factor arrested into medium containing HU was used as template and labeled with 5’-[α-32P]-dATP in the presence (+ Primers) or the absence (− Primers) of random hexameric primers. (C) Agarose gel electrophoresis of heat-denatured, labeled ssDNA in (B) with and without primers. The gel image was produced on an autoradiographic Instant Imager (Packard). Positions of dsDNA markers are as indicated. (D) Quantification of the relative abundance of DNA fragments in the agarose gel image for reaction without primers (gray profile) and with primers (black profile). The profiles were generated by ImageQuant (Packard).
2.5 μg of EcoRI-digested template DNA in a volume of 23.5 μl
20 μl of 2.5X reaction buffer (125 mM Tris-HCl, pH 6.8; 12.5 mM MgCl2; 25 mM β-mercaptoethanol; with or without 750 μg/ml random hexamers)
5 μl of 10X dNTP mix (1.2 mM of each dATP, dCTP, and dGTP; 0.6 mM dTTP; 10 mM Tris-HCl, pH 8.0)
1 μl of Klenow (exonuclease deficient) (Roche, 2 U/μl)
0.5 μl of 5’-[α-32P]-dATP (Perkin Elmer, 6000 Ci/mmole, 10 mCi/ml)
The labeling reactions were allowed to incubate at 37°C for 2.5 hours prior to TCA (trichloroacetic acid) precipitation. TCA precipitation of the completed labeling reaction mixture was performed in triplicate. Three μl of the reaction mixture was added to 3 ml of ice-cold 10% TCA. The mixture was then filtered through a 25 mm-diameter glass fiber filter (Schleicher & Schuell, 0.2 μm). The filter was washed three times with 1 ml of 5% TCA and three times with 1 ml of 95% ethanol, followed by drying at room temperature. Quantification of the incorporation of 5’-[α-32P]-dATP in the DNA was performed by an autoradiographic Instant Imager (Packard). The results (Fig. 2B) demonstrated that the presence of random primers enhanced the labeling reaction 6.7-fold for S phase DNA. The effect of primer addition on labeling of the G1-arrested DNA was limited to 1.6-fold (data not shown). Moreover, when equal masses of DNA were used from both the G1 and S phase samples as template, the S phase sample yielded higher levels of labeling than the G1 phase sample (data not shown). These results suggest that much of the labeling that occurs in the G1 phase sample is due to the filling-in single-stranded gaps, such as staggered ends generated by EcoRI cleavage and/or random fragmentation during DNA isolation (Fig. 2A). The analysis of the size of the DNA products on an agarose gel confirmed the identity of these two classes of labeled products (Fig. 2C&D). The predominant size of labeled ssDNA fragments synthesized in the presence of primers was skewed to smaller sizes, migrating in the gel between 500 and 600 bps (Fig. 2C&D).
3.2.3 Labeling reaction for microarray analysis
The EcoRI-digested DNA described in 3.2.1 is resuspended in 33.6 μl of 10 mM Tris-HCl, pH 8.5. Out of the resuspended DNA, 12.6 μl is set aside for slot blotting (see 3.3) and the remaining 21 μl is used for the labeling reaction (see below).
To complete a labeling reaction, follow the steps below:
To 5 μg of DNA in a volume of 21 μl, add 20 μl of the 2.5X reaction buffer (125 mM Tris-HCl, pH 6.8; 12.5 mM MgCl2; 25 mM β-mercaptoethanol; 750 μg/ml random hexamers)
Add 5 μl of 10X dNTP mix (1.2 mM of each dATP, dCTP, and dGTP; 0.6 mM dTTP; 10 mM Tris-HCl, pH 8.0)
Add 3 μl of Cy-conjugated dUTP (Amersham)
Add 1 μl of Klenow (exonuclease deficient) (50 U/μl, NEB)
Incubate at 37°C in the dark for approximately 2.5 hours
Add 5 μl of 0.2 M EDTA, pH 8.0 to stop the reaction
Add 60 μl of 1 mg/ml tRNA
Purify the mixture through a Sephadex G50 (Sigma) column
Add 20 μl of 1 mg/ml poly dA::dT and precipitate the DNA with one tenth volume of 3 M NaAc and two volumes of cold 100% ethanol
Wash the DNA pellet with 70% ethanol and vacuum dry the DNA (the DNA will be dissolved in solutions suitable for hybridization according to the microarray manufacturer’s recommendation)
3.3 Determination of total amount of ssDNA in DNA samples via slot-blot hybridization
It is crucial to determine the total percentage of ssDNA present in the samples prior to microarray hybridization in order to normalize the data from the microarray hybridization. The protocol is based on the concept that when undenatured DNA is blotted onto a membrane, only the single-stranded portion of the DNA is available for hybridization. Thus, by slot blotting the S phase and G1 control DNA samples onto membranes without prior denaturation (along with a control in which 10% of the same DNA sample is fully denatured) followed by hybridization with a genomic DNA probe, one can calculate the total percentage of single-stranded DNA template present in a given sample. The slot blotting procedures are the same as described previously [7].
4. Hybridization of Cy-conjugated DNA to microarrays
4.1 Pairing sample DNAs for co-hybridization (designing internal controls)
There are several control samples one could choose to combine with an experimental sample for microarray co-hybridization (Fig. 3A). For example, one could combine an undenatured G1 DNA sample with a denatured portion of the same DNA for differential labeling and co-hybridization, thereby identifying regions of the genome that are prone to “breathe” and more readily act as templates for priming and labeling reactions. We have conducted such an experiment with the rad53 G1 phase sample (Fig. 3A, Method 1). As anticipated, the ssDNA profile plotting the relative ratio of ssDNA (native/denatured) revealed that there were some regions of the genome that accumulated more ssDNA than other regions, such as the ssDNA peaks at 78 and 542 kb on chromosome V (Fig. 3B). One could then repeat the same experiment with the S phase samples and ascertain the single-stranded regions in the S phase sample. By comparing the two profiles for G1 and S phase samples, one could then normalize the S phase sample profile and determine the regions of the genome that become single-stranded specifically in S phase, thus eliminating the aforementioned ssDNA peaks on Chr V. However, this method requires twice as many microarray slides and involves three normalization steps. Therefore, we chose to perform a direct comparison between G1 and S phase samples by labeling an equal mass (5 μg) of undenatured DNA from both samples for co-hybridization. Any regions that are prone to “breathe” independent of DNA replication in the S phase sample should be equally prone to “breathe” in the G1 phase sample, and therefore would not appear as regions that have elevated ssDNA level in the S phase sample. We have used this design in all of our experiments (Fig. 3A, Method 2). The result for such experiments are shown in Fig. 1D and Fig. 3C. Note that the spurious peaks of ssDNA at 78 and 542 kb on Chr V were no longer identified as ssDNA peaks in the S phase sample (Fig. 3C).
Figure 3.
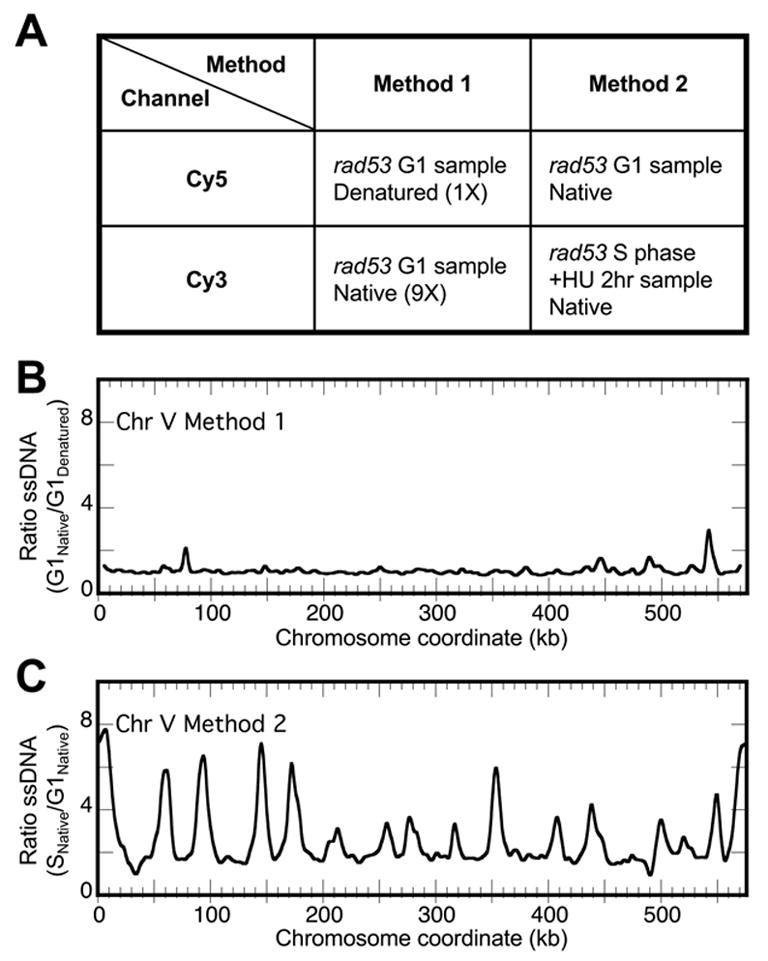
Comparison of two methods for selecting control vs. experimental samples for microarray analysis. (A) Strategies for labeling DNA samples with Cy5 or Cy3-conjugated dUTPs, conditions of template DNA during labeling (native/undenatured vs. denatured), and choices of co-hybridizing DNA samples. In Method 1, 10% of the G1 sample DNA was used for labeling with denaturation and 90% of the same DNA was used for labeling without denaturation. In Method 2, 5 μg of both G1 and S phase DNA were used for labeling without denaturation. (B)&(C) ssDNA profiles for the indicated method of co-hybridization. Normalized ratios of ssDNA (Cy3 channel/Cy5 channel) were plotted against chromosome coordinate for Method 1 (B) and Method 2 (C).
4.2 Microarray hybridization and scanning
We have tested our labeling method with a collection of DNA microarrays, including the yeast oligo ORF array (Agilent) as well as locally printed PCR products of yeast ORF and Intergenic arrays (Microarray Facility at Fred Hutchinson Cancer Research Center (FHCRC)). The results obtained from these microarrays have been consistent with each other.
Microarray hybridization and scanning are performed at the Center for Expression Array and the Microarray Facility at FHCRC for the Agilent and locally printed microarrays, respectively, according to the manufacturer’s recommendations.
5. Data analysis
5.1 Data extraction and normalization of microarray data
Microarray data extraction is performed with GenePix Pro 4.0 software (Axon Instruments). We obtain background-extracted, median fluorescence signals from the Cy-5 (G1 control sample) and the Cy-3 (S phase sample) channels for each spot on the microarray. We first remove those spots that contain repetitive sequences from further calculations. We then average signals for duplicated spots. For a given ORF spot on the microarray, the relative amount of ssDNA can be expressed by the apparent ratio (Rapparent) of fluorescence signal from the Cy-3 channel (S phase sample) to that from the Cy-5 channel (G1 control sample). We then calculate the real ratio of ssDNA (Rreal) after normalization, which takes into account hybridization differences as well as the intrinsic amount of ssDNA in each sample, using the following equation:
where “total % ssDNA” for the S phase and G1 control samples are the values measured by slot-blotting and hybridization of undenatured DNA samples and “∑ Cy-3 signal” and “∑ Cy-5 signal” are the sums of fluorescent signals from the Cy-3 (S phase sample) and Cy-5 (G1 control sample) channels, respectively. Once we obtain the Rreal value, or the relative amount of ssDNA, for every spot on the microarray, we then plot it against chromosomal coordinates to generate ssDNA profiles for each chromosome.
5.2 Smoothing of ssDNA profiles and identification of origins and termini
The raw ssDNA profiles are smoothed to eliminate small peaks that arise from noise in the signals. We employ Fourier Transformation to smooth the ssDNA profile across each chromosome at a specified window size (4 kb) at a step size of 1 kb. The detailed procedures of this method have been described previously [1]. We smoothed the S. pombe data at a 12 kb window size due to lower probe density on the S. pombe microarray. For an organism for which no prior origin information is known, the investigator could make the choice of greater stringency versus greater sensitivity as seems appropriate. Once a smoothed profile is obtained for each chromosome, we proceed to identify the origin locations (local maxima) by using an algorithm named ExtremaFinder developed in our lab [4]. Other algorithms that allow the identification of local maxima and minima can also be used.
To determine whether a given local maximum likely represents a bona fide replication origin, we evaluate the height differential from the local maximum to its two flanking local minima, i.e., we examine the magnitude of a given peak relative to its neighboring regions. The criteria set for the height differential may be arbitrary due to different types of data sources. For the S. cerevisiae rad53 checkpoint mutant exposed to HU, we have required the height differential to be at least three times the standard deviation calculated from all of the data points below the median value.
6. Concluding remarks
Prior to this study, genomic mapping of replication origins in yeast had been done by at least three independent approaches. While two of these studies measured the quantities of DNA during replication, the other measured protein-DNA binding during initiation of DNA replication. We have described a method that measures a necessary intermediate in DNA replication, namely ssDNA. In particular, we took advantage of the fact that HU treatment slows down replication and allows more expansive formation of ssDNA at replication forks. We have not yet fully explored the potential of detecting ssDNA at replication forks during a normal S phase without HU treatment. It will be interesting to determine whether our method is sensitive enough for such detection. Obviously, the feasibility of the method also depends on the probe density on the microarray.
Since we have applied this technology to map origins in two distantly-related organisms, we are optimistic about the potential of our method to be applied in other eukaryotes such as humans to determine the location of their replication origins. Single-stranded DNA is also an important intermediate in other chromosomal processes such as DNA repair and recombination. The assessment of genomic locations of ssDNA during these processes is at once a challenge and a necessity. We believe our method has the potential to shed light on these different molecular processes.
Acknowledgments
We thank G. Alvino and L. Danielson for their support and technical assistance. We are grateful to G. Findlay for helpful comments in critiquing the manuscript. We also thank the staff at the microarray facilities at FHCRC and CEA for microarray slide hybridization and scanning. This work was supported by NIGMS grant 18926 to W. L. Fangman, B. J. Brewer and M. K. Raghuraman. W. Feng was supported by a Ruth L. Kirschstein Postdoctoral Fellowship from NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Raghuraman MK, Winzeler EA, Collingwood D, Hunt S, Wodicka L, Conway A, Lockhart DJ, Davis RW, Brewer BJ, Fangman WL. Science. 2001;294:115–121. doi: 10.1126/science.294.5540.115. [DOI] [PubMed] [Google Scholar]
- 2.Wyrick JJ, Aparicio JG, Chen T, Barnett JD, Jennings EG, Young RA, Bell SP, Aparicio OM. Science. 2001;294:2357–2360. doi: 10.1126/science.1066101. [DOI] [PubMed] [Google Scholar]
- 3.Yabuki N, Terashima H, Kitada K. Genes Cells. 2002;7:781–789. doi: 10.1046/j.1365-2443.2002.00559.x. [DOI] [PubMed] [Google Scholar]
- 4.Feng W, Collingwood D, Boeck ME, Fox LA, Alvino GM, Fangman WL, Raghuraman MK, Brewer BJ. Nat Cell Biol. 2006;8:148–155. doi: 10.1038/ncb1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sogo JM, Lopes M, Foiani M. Science. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- 6.Tercero JA, Diffley JF. Nature. 2001;412:553–557. doi: 10.1038/35087607. [DOI] [PubMed] [Google Scholar]
- 7.Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M. Nature. 2001;412:557–561. doi: 10.1038/35087613. [DOI] [PubMed] [Google Scholar]
- 8.Huberman JA, Spotila LD, Nawotka KA, el-Assouli SM, Davis LR. Cell. 1987;51:473–481. doi: 10.1016/0092-8674(87)90643-x. [DOI] [PubMed] [Google Scholar]
- 9.Garvik B, Carson M, Hartwell L. Mol Cell Biol. 1995;15:6128–6138. doi: 10.1128/mcb.15.11.6128. [DOI] [PMC free article] [PubMed] [Google Scholar]