

Abstract
Lactoperoxidase (LPO) is believed to serve as a mediator of host defense against invading pathogens. The protein is more abundant in body fluids such as milk, saliva and tears. Lactoperoxidase is known to mediate the oxidation of halides and (pseudo)halides in the presence of hydrogen peroxide to reactive intermediates presumably involved in pathogen killing. More recently, LPO has been shown to oxidize a wide diversity of thiol compounds to thiyl free radicals, which ultimately lead to the formation of a protein-radical characterized by DMPO-immunospin trapping. In the same study by our group the authors claimed that a consequence of this protein radical formation was the inactivation of LPO (Guo et al., J. Biol. Chem., 279, 13272–13283). Here we demonstrate that although thiyl radical formation does lead to LPO-radical production, the formation of such radical is unrelated to the enzyme’s activity. As the source of this misleading interpretation we suggest the binding of GSH to ELISA plates, which interfere with ABTS and guaiacol oxidation. In addition, DMPO-GSH nitrone adducts bind to ELISA plates, leading to ambiguities of interpretation since we have demonstrated that DMPO-GSH nitrone does not bind to LPO, and only LPO-protein-DMPO nitrone adducts can be detected by Western Blot.
INTRODUCTION
Lactoperoxidase (LPO) is a member of the mammalian peroxidase superfamily II, in which the heme group is covalently attached to the enzyme. Lactoperoxidase is abundant in body fluids such as milk, tears and saliva and is believed to play an important role in host defense against invading microorganisms [1–4]. Several substrates such as (pseudo)halide anions can be oxidized by LPO in the presence of H2O2, which leads to the production of reactive intermediates [5–7]. In addition, LPO has been shown to oxidize thiol compounds to thiyl free radicals both in the presence and in the absence of hydrogen peroxide [2, 8–11]. The oxidation of thiols, specifically GSH, by LPO triggers a series of events in which O2 ●− and its unavoidable product H2O2 are produced as demonstrated by oxygen uptake and EPR experiments [11], a feature shared by other peroxidases as well [12–15]. Nevertheless, particular to LPO, the oxidation of GSH to GS● directly or indirectly induces the formation of a LPO-protein radical, which can be trapped by DMPO [11]. This radical seems to be different from the one produced when LPO is exposed to excess H2O2 alone [16], based on the different trapping abilities of DMPO to trap the radicals produced from LPO interactions with H2O2 or GSH (discussed in 11). In the case of H2O2, it has been demonstrated that although radical formation occurs on LPO, the enzyme is not significantly deprived of its peroxidase activity regardless of the structural changes that occur as a consequence of radical formation [16]. In the case of GSH, in a previous study from our group Guo and co-workers claimed that the radical formation on LPO induced by GSH exposure efficiently leads to enzyme inactivation [11]. Here we demonstrate through several lines of evidence that although GSH induces the formation of DMPO-trappable enzyme radicals on LPO, this does not correlate at all with loss of peroxidase activity.
EXPERIMENTAL PROCEDURES
Chemicals
Lactoperoxidase from bovine milk, GSH and the spin trap 5,5-dimethyl-1-pyrroline N-oxide (DMPO) were purchased from Sigma (St. Louis, MO). DMPO was purified by vacuum distillation at ambient temperature and stored at −80 °C until use. Sodium phosphate was purchased from Mallin Krodt Aker Inc. (Paris, KY). Chelex 100 resin was purchased from Bio-Rad Laboratories (Hercules, CA). Glutathione peroxidase was purchased from Roche (Indianapolis, IN). Catalase (from beef liver, 65,000 U/mg) (EC 1.11.1.6) and superoxide dismutase (from bovine erythrocytes, 5,000 U/mg) (EC 1.15.1.1) were purchased from Roche Molecular Biochemicals (Indianapolis, IN). Anti-glutathione was purchased from Calbiochem (La Jolla, CA) All the reactions were carried out in 100 mM sodium phosphate buffer, pH 7.4. The buffer was treated with Chelex 100 resin to remove traces of transition metal ions to minimize the possibility of trace-metal interference.
Electron Paramagnetic Resonance Experiments
EPR spectra were recorded on a Bruker EMX EPR spectrometer (Billerica, MA) operating at 9.81 GHz with a modulation frequency of 100 kHz and equipped with an ER 4122 SHQ cavity. All experiments were performed at room temperature with a quartz flat cell.
H2O2 measurements
H2O2 uptake experiments were performed using a H2O2 electrode attached to an Apollo 4000 Free radical analyzer unit [World Precision Instruments (WPI), Sarasota, FL]. Amperometric detection of H2O2 was carried out using poise voltage of +400mV. The oxidation of H2O2 at the surface of the sensor produced an electric current in the nA range. The reaction was constantly stirred throughout the course of the experiment.
Visible Spectrometry studies
All optical measurements were carried out with a Varian Cary 100 Bio spectrophotometer.
ELISA
The DMPO-GSH-nitrone adducts’ ability to bind to ELISA plates was determined using a standard ELISA in 96 well plates (Greiner Labortechnik, Frickenhausen, Germany). LPO (5 μM) was incubated with GSH (5 mM) in the presence of DMPO (100 mM) for 2h at 37 °C. Then incubations were passed through Millipore ultrafree mass cut filters, and both high and low molecular weight fractions were analyzed. In addition, pre-synthesized, HPLC-purified and mass spectrometry-characterized DMPO-GSH-nitrone derivative was incubated on the ELISA plates. The DMPO-GSH-nitrone adduct was synthesized by the thermal decomposition of the nitrosothiol GSNO in the presence of DMPO [17] and was used in its isolated purified form. After incubation with the samples, the plates were washed once with 1X TBS washing buffer (0.05% Tween-20, 0.05% casein, and 0.05% BSA, pH 7.4) and blocked with coating buffer (0.1 M bicarbonate buffer, pH 9.6, containing 2.5% casein and 2.5% BSA) for 90 min. Thereafter, the rabbit anti-DMPO serum (1:5,000) in washing buffer was added and incubated for 60 min. After 4 washes, the secondary antibody, anti-rabbit IgG-alkaline phosphatase (Pierce Chemical Co., Rockford, IL), 1:5,000 in washing buffer, was added and incubated for 60 min. After 4 more washes, the antigen-antibody complexes were analyzed by using a chemiluminescence system (CDP-Star, Roche Molecular Biochemicals, IN), and the light emitted was recorded in arbitrary units using Xfluor Software (Tecan US, Research Triangle Park, NC).
RESULTS AND DISCUSSION
Lactoperoxidase and other animal or plant peroxidases share in common the ability to rapidly react with H2O2, generating a hypervalent state referred to as compound I. This intermediate is highly reactive and currently accepted as consisting of a ferryl type Fe(IV)=O component with a valence of + 4 and a delocalized porphyrin radical cation. The enzyme decays back to its resting state by oxidizing two molecules of substrates, producing substrate-derived radical according to the reactions (1–3) below:
| (reaction 1) |
| (reaction 2) |
| (reaction 3) |
Thiols dissolved in aerated buffers oxidize at varying rates, producing the trace amounts of H2O2 required to initiate reaction 1 (see above). In order to gauge the effect of GSH on LPO radical formation, lactoperoxidase (5 μM) was incubated with DMPO (100 mM) in the presence and in the absence of GSH (5 mM) at 37 °C for 1 h. In addition to GSH, we evaluated the effects of scavengers of H2O2 (glutathione peroxidase and catalase) and O2●− (Cu, Zn-SOD) upon LPO/GSH/DMPO incubations. As shown in Fig. 1, GSH addition significantly enhanced the production of LPO-DMPO-nitrone adducts as demonstrated by Western Blot in accordance with previously reported observations [11]. Superoxide dismutase alone or in conjunction with the H2O2 scavenging enzymes (catalase and glutathione peroxidase), greatly enhanced the formation of LPO-nitrone adducts, suggesting a role for O2●− in the inhibition of LPO activity [18–20]. Indeed, compound III formation was detected in incubations of LPO with GSH when trace amounts of phenol were added (data not shown). Superoxide dismutase’s remarkable stimulation of LPO-nitrone adduct formation suggests the production of appreciable O2●− even in the absence of phenol. This result is not surprising since O2●− is a known inhibitor of peroxidases and is effectively disproportionated by superoxide dismutases. It clearly demonstrates that LPO radical formation depends on the enzyme’s peroxidase activity. As expected, catalase and glutathione peroxidase, two H2O2 scavengers, greatly inhibited LPO-nitrone adduct formation, implying a role for H2O2 in the formation of protein radicals as previously observed [16].
Fig. 1.
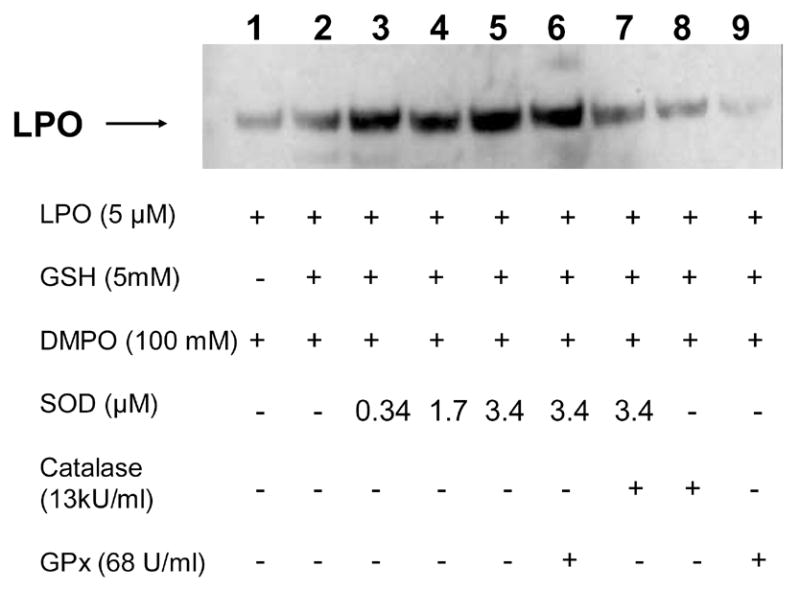
Detection of lactoperoxidase-DMPO protein-nitrone adducts induced by GSH. Lactoperoxidase (5 μM) was incubated with DMPO (100 mM) in the presence or in the absence of GSH (5 mM) for 1 h at 37 °C. All reactions were performed in phosphate buffer (0.1M, pH 7.4). Lane 1, in the absence of GSH; Lane 2, in the presence of GSH (5 mM); Lane 3, in the presence of GSH and SOD (340 nM); Lane 4, in the presence of GSH and SOD (1.7 μM); Lane 5, in the presence of GSH and SOD (3.4 μM); Lane 6, in the presence of GSH and SOD (3.4 μM) and GPx (68 U/ml); Lane 7, in the presence of GSH and SOD (3.4 μM) and catalase (13,000 U/ml); Lane 8, in the presence of GSH and catalase; Lane 9; in the presence of GSH and GPx. Reactions were stopped by the addition of NEM (75 mM).
EPR studies confirm O2●− during LPO-mediated GSH oxidation
Next we studied low molecular weight free radical production by LPO/GSH incubations using DMPO as the spin trap. As shown in Fig. 2A, LPO (2.5 μM) incubated with GSH (2.5 mM) in the presence of the spin trap DMPO (100 mM) produced an EPR spectrum consistent with a mixture of DMPO/●SG (aN= 15.13 G and aHβ = 16.14 G)[21] and DMPO/●OH radical adducts (aN = 14.9 G and aHβ = 14.9) [22]. In the absence of GSH no signal could be observed, as previously reported (Fig. 2B) [11]. Interestingly, Cu, Zn-SOD (5 μM) addition led to the detection of a pure and considerably more intense EPR spectrum attributable to DMPO/●SG radical adduct (Fig. 2C). This result not only demonstrated O2●− production in this system but also confirmed this species as the actual inhibitor of LPO. As expected, the scavenging of O2●− by Cu, Zn-SOD greatly increased the oxidation of GSH to GS● by LPO. Thiyl radical production catalyzed by SOD-thiol oxidase activity [23] was much inferior to that mediated by LPO at the concentrations used (Fig. 2F). Catalase at the concentrations tested in this experiment had marginal effects upon thiyl radical production (Fig 2D). Fig. 2E shows the effect of adding both Cu, Zn-SOD and catalase to the incubations. To determine H2O2 stimulatory effects on the rate of GSH oxidation by LPO, experiments using glutathione peroxidase (an efficient, non-heme H2O2 scavenger) were performed. As shown in Fig. 3, addition of glutathione peroxidase to LPO incubations with GSH greatly inhibited thiyl radical production, confirming the stimulatory role for H2O2 in mediating LPO compounds I and II formation which react with GSH producing thiyl radicals. The inhibition was concentration-dependent, and thiyl radical production was barely detectable at glutathione peroxidase concentrations over 100 U/ml. This result strongly supported the H2O2-dependent oxidation of GSH by LPO. Also, the EPR results further support the production of O2●− in the system and its inhibitory effects upon LPO activity, which suggests that LPO-nitrone adduct formation is a consequence of the enzyme’s activity.
Fig. 2.
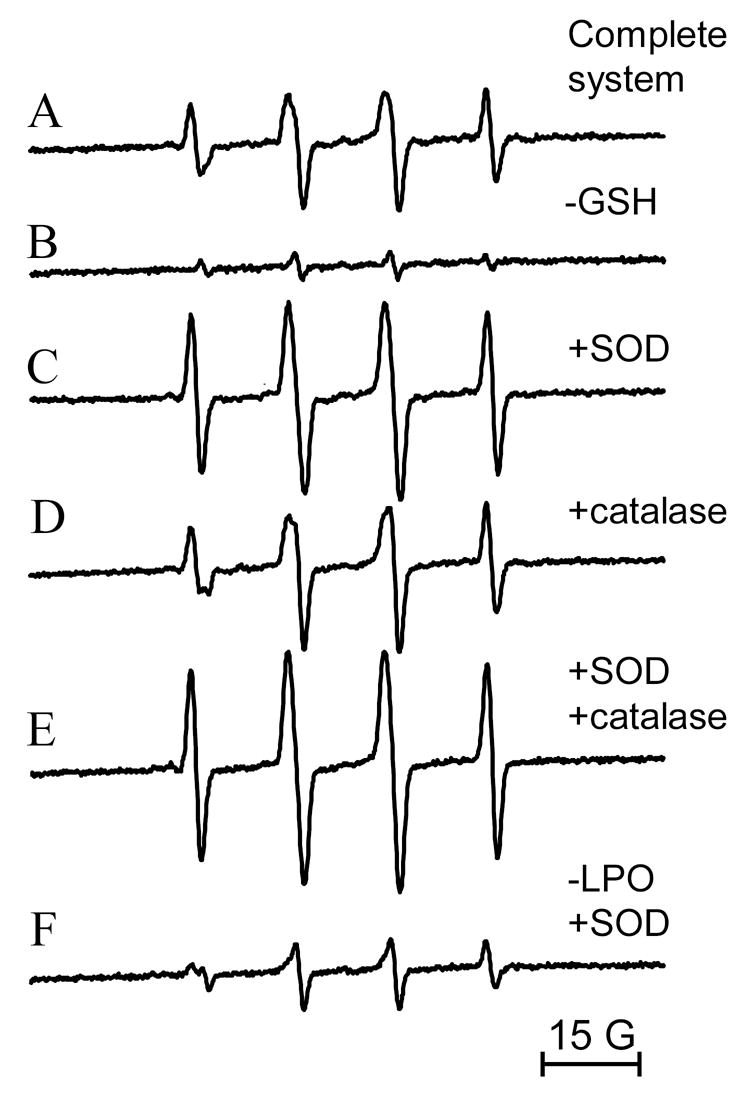
EPR spectra of lactoperoxidase (2.5 μM) incubations with GSH (2.5 mM) in the presence of DMPO (100 mM). (A) No further additions; (B) in the absence of GSH; (C) in the presence of GSH and Cu, Zn-SOD (10 μM); (D) in the presence of GSH and catalase (650 U/ml); (E) in the presence of GSH, Cu, Zn-SOD and catalase; (F) in the absence of LPO and in the presence of Cu, Zn-SOD (10 μM). Instrumental conditions were modulation frequency, 100 kHz; modulation amplitude, 1 G; microwave power, 20 mW; receiver gain, 6 × 104; time constant, 164 ms; conversion time, 164 ms; total scan time 168 s.
Fig. 3.
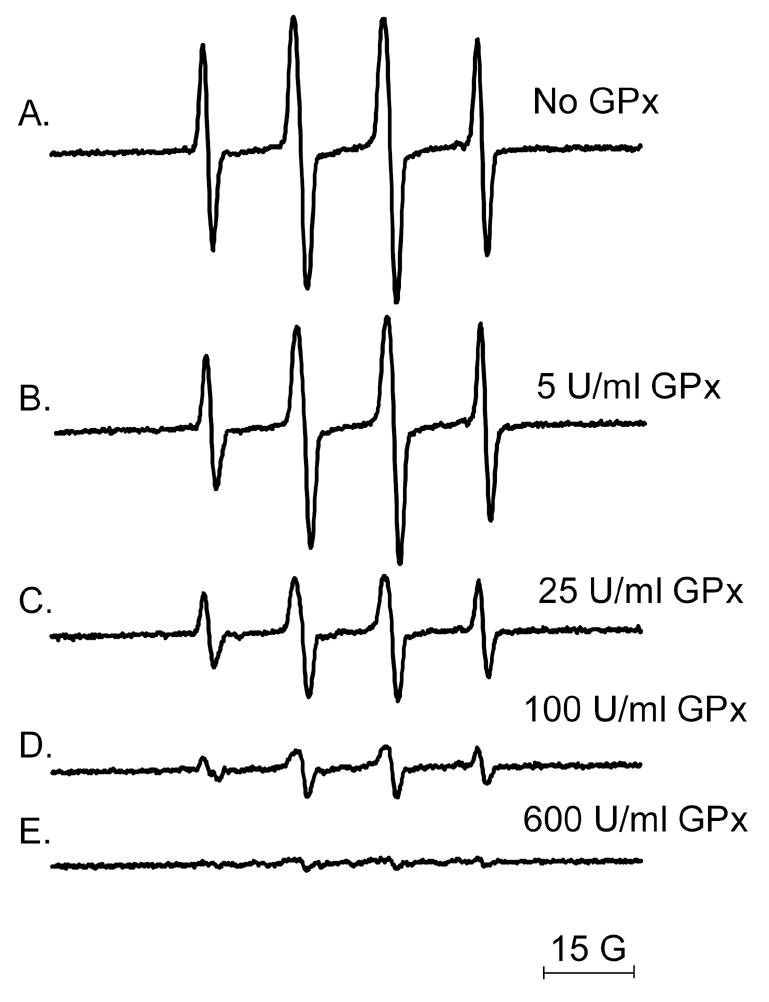
Effect of glutathione peroxidase on the production of DMPO/●SG radical adducts. LPO (5 μM) was incubated with GSH (5 mM) in the presence of DMPO (100 mM) at room temperature in phosphate buffer (0.1 M, pH 7.4) in the absence (A) or in the presence of different glutathione peroxidase (GPx) concentrations: (B) 5 U/ml; (C) 25 U/ml; (D) 100 U/ml; (E) 600 U/ml. Instrumental conditions were modulation frequency, 100 kHz; modulation amplitude, 1 G; microwave power, 20 mW; receiver gain, 6 × 104; time constant, 164 ms; conversion time, 164 ms; total scan time 168 s.
Non-covalent interaction of DMPO/thiyl adducts with LPO
To test the hypothesis that carboxy-bearing DMPO-GSH adducts would interact non-covalently with the positively charged LPO surface and generate misleading immunochemical signals on Western Blots, we performed experiments in which LPO was either incubated in the presence of DMPO and GSH to produce the DMPO/● SG adducts or added to purified DMPO-GSH-nitrone solutions (see materials and methods for details). According to Fig. 4, LPO incubated in the presence of DMPO (100 mM) and GSH (0–2 mM) led to a significant concentration-dependent production of LPO-DMPO nitrone adducts as revealed by Western Blot analysis using anti-DMPO antibody. The replacement of GSH and DMPO by authentic, pre-synthesized DMPO-GSH-nitrone adduct did not lead to the detection of protein-bound DMPO-nitrone derivatives, thus convincingly demonstrating that DMPO traps LPO-protein radicals generated when the protein is exposed to GSH in solution. The observed Western Blot signals were derived not from the interaction of the negatively charged DMPO-GSH-nitrone adduct molecule with the positively charged LPO surface, but rather from the trapping of actual protein-bound radicals produced during the incubation of LPO with GSH.
Fig. 4.
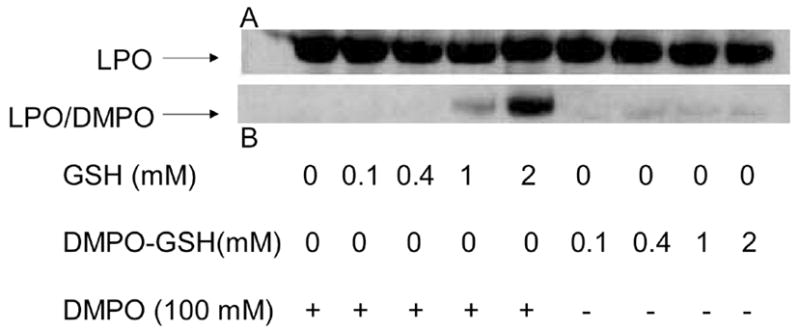
Detection of DMPO-nitrone moieties bound to LPO. LPO (15 μM) was reacted with GSH and DMPO or DMPO-GSH nitrone adduct (0.1–2 mM) for 2 h at 37 °C. When present, DMPO was 100 mM. Panel A represents the protein loading control which was accessed by Western Blot analysis using anti-LPO antibody. Panel B, is Western Blot analysis of DMPO nitrone-derivatives bound to the protein. For this experiment LPO was incubated with GSH in the presence of DMPO at the indicated concentrations, or alternatively, LPO was exposed to the pre-synthesized DMPO-GSH nitrone adduct at the indicated concentrations to demonstrate that low molecular weight nitrone-derivatives do not stick tightly to the protein.
Peroxidase activity assayed through H2O2 electrode
The activity of LPO was assayed by its ability to catalyze the H2O2-mediated SCN- oxidation after the incubation of the enzyme with several GSH concentrations. Indeed, data have been accumulating in the literature highlighting SCN− oxidation as a main physiological role for LPO [4, 24]. According to the data shown in Fig. 5, the addition of H2O2 (100 μM) to phosphate buffer as a control generated a rapid increase in the electrode current measuring H2O2 concentration (points of H2O2 addition are marked on the figure with arrows), while addition of SCN− (2 mM) alone did not cause any change in H2O2 concentration, as expected. The subsequent addition of LPO that had been previously incubated in the absence or in the presence of GSH (0.5–2 mM) for 2 h at 37 °C caused a rapid H2O2 uptake (Fig. 5). Addition of more H2O2 (100 μM) yielded increases in the electrode current which did not reach the maximum obtained in the absence of LPO. Indeed, H2O2 was rapidly consumed by the incubation mixtures within seconds after its addition to the reaction chamber, proving that LPO retained full activity after incubation with GSH. To further ascertain that LPO was responsible for the H2O2 uptake, azide (1 mM), a known peroxidase inhibitor [25, 26], was added to the reaction chamber prior to H2O2. In this case, a plateau approximately on the level of the first H2O2 addition could be observed, as expected, from complete inhibition of LPO activity by azide.
Fig. 5.
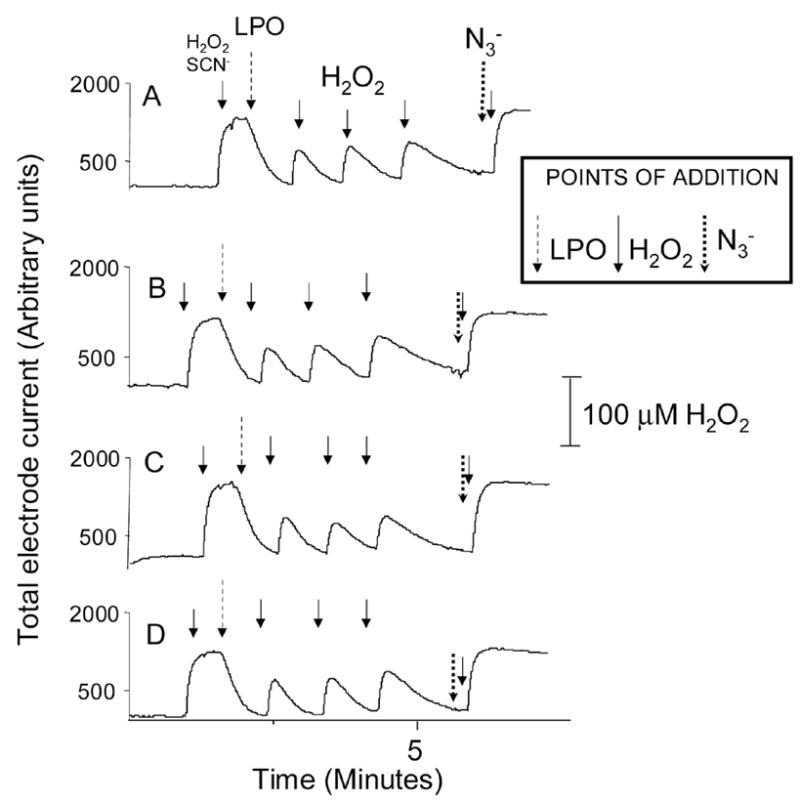
Representative H2O2 uptake traces by LPO (5 μM) incubated with different GSH concentrations (0–2 mM) for 2 hs at 37 °C in phosphate buffer (pH 7.4, 0.1M). Two-hour incubations of LPO with GSH were stopped by rapidly freezing the samples at −80 °C. For the experiments, samples were hand-thawed and diluted into the reaction chamber. Experiments were performed in triplicate. Panel A represents the traces obtained with control LPO incubated in phosphate buffer in the absence of GSH. Panels B–D are the traces obtained when LPO was incubated with 0.15, 0.5 and 2.0 mM GSH, respectively. For those experiments, pulses of 100 μM H2O2 were used. NaSCN (2 mM) was added at the point marked. NaN3 (1 mM) was added to the reaction chamber to inhibit LPO. Its point of addition is indicated as well.
Interference of trace amounts of GSH in the measurement of LPO activity with guaiacol or ABTS
Lactoperoxidase incubations in the presence and the absence of GSH were also evaluated in regard to their ability to oxidize guaiacol or ABTS. As shown in Figs. 6 and 7, LPO additions to incubations containing H2O2 and guaiacol (Fig. 6) or ABTS (Fig. 7) produced a marked production of guaiacol or ABTS-derived chromophores monitored at 470 or 415 nm, respectively. Prior incubation of LPO with GSH for 2 h at 37 °C failed to inhibit the enzyme’s peroxidase activity in accordance with H2O2 measurements (Fig. 5). Nevertheless, although there was a small initial increase in the rate of tetraguaicol formation at the lowest concentration, the addition of trace amounts of GSH to incubations of LPO/H2O2/guaiacol generally inhibited the oxidation of guaiacol to its chromophore. The initial stimulation of the tetraguaiacol formation is possibly due to the propagation of guaiacol oxidation by the lactoperoxidase-mediated production of thiyl radicals and H2O2. Nevertheless, the thiol-mediated reduction of the guaiacol phenoxyl radical (an obligatory precursor of tetraguaiacol formation) inhibited the formation of the chromophore in a concentration-dependent manner (Fig. 6 traces D–F).
Fig. 6.
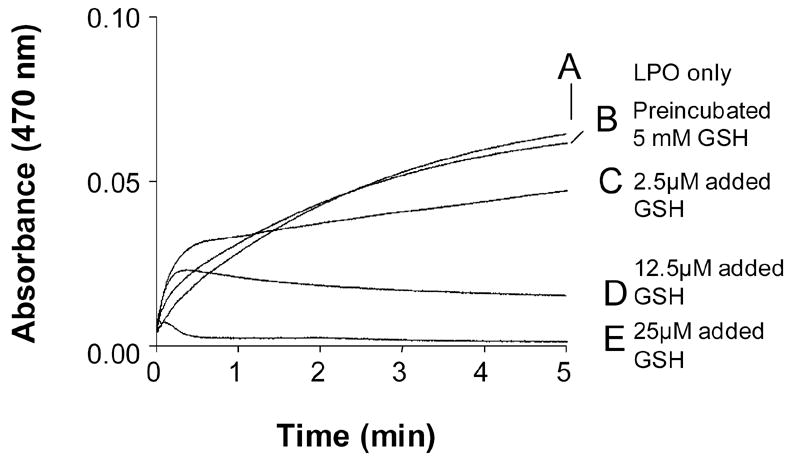
Representative kinetic traces of guaiacol (9 mM) oxidation by H2O2 (0.9 mM) mediated by LPO (5 μM) previously incubated in the absence (Trace A) or in the presence of 5 mM GSH (Trace B) for 2 h at 37 °C. After that, the incubations were diluted 10,000 times in the spectrophotometer cuvette containing phosphate buffer, guaiacol and finally H2O2 were then added. Experiments were performed in triplicate. For traces C–E fresh LPO was used. For these experiments GSH at the concentrations indicated was added prior to H2O2. Trace C, GSH 2.5 μM was added; Trace D, GSH 12.5 μM was added; Trace E, GSH 50 μM was added.
Fig. 7.
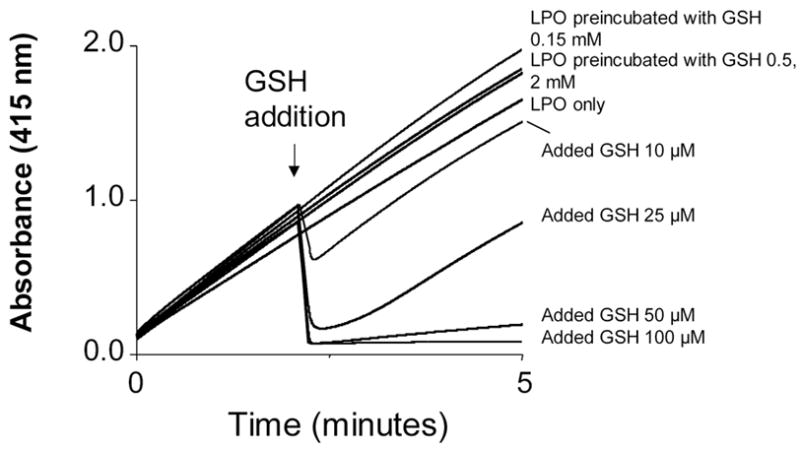
Representative kinetic traces of ABTS (0.5 mM) oxidation by H2O2 (0.1 mM) mediated by LPO previously incubated in the absence (upper trace) or in the presence of different GSH concentrations (remaining traces). LPO was 5 μM before dilution into the spectrophotometer’s cuvette. Reactions of LPO and GSH were stopped by NEM addition to the incubations at a final 20 mM concentration. Experiments were performed in triplicate. LPO was preincubated with the following concentrations of GSH: 0.15 mM; 0.5 mM, and 2 mM. Alternatively, GSH (10 – 100 μM) was added as indicated.
A similar inhibitory GSH effect was observed when the thiol was added to LPO/H2O2/ABTS incubations. At lower doses GSH instantly scavenged ABTS●+, which strongly absorbs at 415 nm, in a concentration-dependent manner, demonstrating the prompt reduction of the molar equivalents of the cromophore at the point of addition by GSH. At higher doses GSH not only reduced ABTS●+, but also diminished the rate of ABTS oxidation, once more reflecting the scavenging of ABTS●+ by remaining GSH (Fig. 7).
Binding of DMPO-GSH and GSH to ELISA plates as an explanation for the misleading interpretations about LPO activity
Although our results convincingly demonstrate that LPO retains its activity after incubation with GSH regardless of protein radical formation, the binding of GSH to ELISA plates was also investigated as the possible source of error that led Guo et al . [11] to associate LPO incubation with GSH with peroxidase activity inhibition. In that paper the authors measured LPO peroxidase activity after incubating the reaction mixtures (containing LPO, DMPO, GSH and presumably DMPO/●SG-derived products) on ELISA plates. The success of this method relies on the assumption that only LPO will bind to the plates and at approximately the same relative concentration in all wells, and all other components will be washed away. However, it has been demonstrated that dithiothreitol adducts of DMPO bind to ELISA plates [27] and can be assayed through the immunospin trapping technique, which strongly suggests that other DMPO-thiol compounds could do the same. To determine whether DMPO-GSH-nitrone adduct also binds to ELISA plates, we incubated LPO (5 μM) with GSH (5 mM) in the presence of DMPO (100 mM) for 2 h at 37 °C and passed the incubations through mass cut filters (Millipore, ultrafree mass cut filter, 30 kDa) to separate protein and low molecular weight products (Fig. 8). The retained protein fractions were washed twice with phosphate buffer, and both fractions were applied onto ELISA microplates. As a standard, HPLC-purified and mass-spectrometrically characterized DMPO-GSH-nitrone was also incubated on the ELISA plate. According to Fig. 8, DMPO-GSH-nitrone adduct, produced under the same conditions as reported by Guo et al., efficiently bound to the ELISA. Of note, a considerably higher response was obtained from the low molecular weight fractions in comparison to the fractions containing LPO. Similarly, GSH-binding to ELISA plates was demonstrated using anti-GSH to detect residual GSH adhering to the microplates after incubation of different concentrations of GSH in the wells for 2 h (Figure 9). In addition to GSH and its nitrone adducts, we also measured LPO binding to ELISA plates in the presence of GSH at various concentrations (0–5 mM) using an anti-LPO antibody. These results indicated that GSH at higher concentrations tends to displace LPO from binding to the microplates, but such displacements never led to more than a 15 % decrease in the amount of bound LPO under our experimental conditions (data not shown). Although LPO displacement by low molecular weight compounds can not be accounted as the sole reason that led Guo et al. to conclude that LPO is inactivated by GSH, we have demonstrated that fairly low concentrations of GSH can drastically reduce both the rate of formation and the total yield of ABTS and guaiacol-derived chromophores. This reaction is the reason why Guo et al. mistakenly interpreted their results as an inhibition of peroxidase activity. Therefore, the results presented here clearly demonstrate that LPO is not inactivated by GSH, and previous conclusions were due to artifact related to GSH binding to ELISA plates.
Fig. 8.
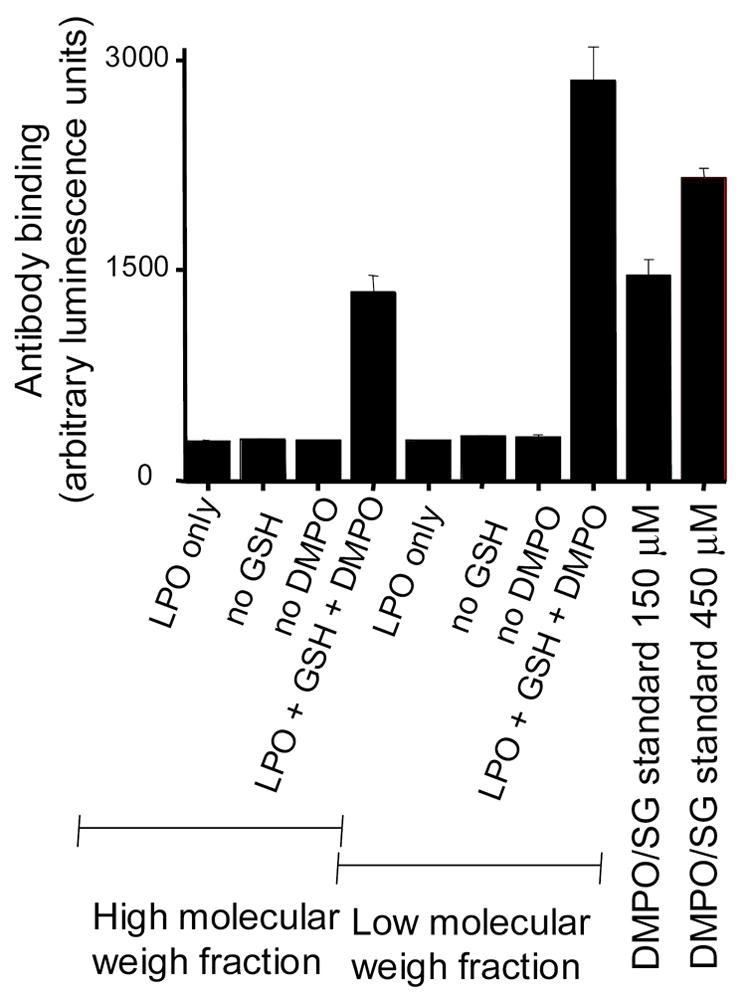
Immunochemical detection of the DMPO/GS-nitrone adduct by ELISA. LPO (5 μM) was incubated with GSH (5 mM) and DMPO (100 mM) for 2h at 37 °C. After samples were filtered through Millipore ultrafree mass cut filters (30 kDa), both low and high molecular weight fractions were analyzed through ELISA. Protein fractions were re-suspended and washed twice with phosphate buffer prior to transferring to the ELISA plate. DMPO-GSH standards were also incubated on the ELISA plates at the concentrations indicated on the figure. Each column is the average of three independent experiments.
Fig. 9.
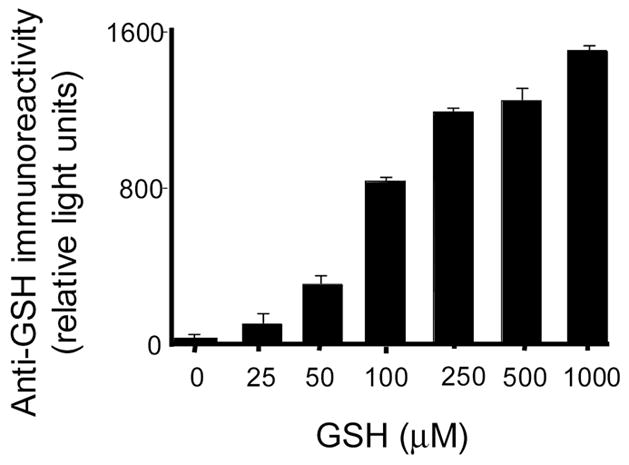
Immunochemical detection of the remaining GSH bound to microplates by ELISA. GSH was incubated for 2 h (37 °C) on ELISA plates at the indicated concentrations in phosphate buffer, pH 7.4. After this time, ELISA plates were washed as described under materials and methods, blocked with BSA/casein buffer and developed using polyclonal anti-GSH antibody. Each column is the average of three independent experiments.
SUMMARY
The results presented herein demonstrate that although GSH oxidation to GS● mediated by LPO induces the formation of protein radicals on the enzyme, this event is unrelated to the inhibition of the peroxidase activity (Figs. 5–7) in contrast to a previous suggestion [11]. Indeed, LPO protein radical formation seems to depend on the enzyme’s activity rather than causing the enzyme’s inactivation as demonstrated by the higher yields of LPO-DMPO-nitrone adducts detected in incubations of LPO/GSH/DMPO in which Cu, Zn-SOD was added. This result is also consistent with O2●−, which is produced in this system (see reactions 4-6), being the actual inhibitor of LPO [18–20]. This result is corroborated by the fact that a robust generation of GS● takes place when Cu, Zn-SOD is present (Figure 2, Traces C and E). Catalase did inhibit LPO-nitrone adduct production, suggesting the intermediacy of H2O2 on LPO-protein radical formation (Fig. 1). Also, at high enough concentrations, catalase significantly inhibited the oxidation of GSH to GS● mediated by LPO, demonstrating that this process is stimulated by H2O2 (data not shown).
| (reaction 4) |
| (reaction 5) |
| (reaction 6) |
This fact has been further confirmed by replacing catalase with another H2O2 scavenger, glutathione peroxidase (Figure 3). Furthermore, the results presented here demonstrate that GSH is a major source of interference during the assessment of peroxidase activity measured through classical guaiacol and ABTS methods (Figs. 6 and 7). This interference is a consequence of the reduction of the chromophores or their precursors by contaminant GSH, which remain bound to ELISA microplates under the conditions employed here and in reference 11. This fact, led Guo et al. [11] to incorrect interpretations regarding the GSH effects on LPO peroxidase activity. Our studies focused on lactoperoxidase. However the observations presented in regard to bound-GSH interference on peroxidase activity measurement through ELISA technology can certainly be extend to other peroxidases such as myeloperoxidase and horseradish peroxidase and pseudo-peroxidases such as myoglobin. Therefore, our results demonstrate the importance of measuring peroxidase activity in the absence of reduced thiols, which interfere with the commonly used guaiacol and ABTS assays. In summary, the production of LPO-protein radicals during the incubation of LPO with GSH does not inactivate the enzyme, and the original conclusions of Guo et al. [11] about the suicidal inactivation of LPO by GSH are based on an artifact.
Acknowledgments
The authors would like to acknowledge Dr. Marilyn Ehrenshaft and Dr. Olivier M. Lardinois for helpful discussions and Dr. Ann Motten and Mrs. Mary J. Mason for their valuable assistance in the preparation of this manuscript. Supported by the intramural research program of the NIH, NIEHS.
ABBREVIATIONS
The abbreviations used are:
- ABTS
2,2′-azino-bis-[3-ethylbenzothiazoline]-6-sulfonic acid
- Cu, Zn-SOD
copper, zinc superoxide dismutase
- DMPO
5,5-dimethyl-1-pyrroline N-oxide
- EPR
electron paramagnetic resonance
- DMPO-GSH
5,5-dimethyl-1-pyrroline-N-oxide-glutathione nitrone adduct
- ELISA
enzyme-linked immuno-sorbent assay
- GPx
glutathione peroxidase
- GSH
reduced glutathione
- GS●
glutathione thiyl radical
- GSSG
oxidized glutathione, or glutathione dithiol
- GSSG●−
glutathione disulfide radical
- HRP
horseradish peroxidase
- LPO
bovine milk lactoperoxidase
- NEM
N-ethylmaleimide
- Guaiacol
2-methoxyphenol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Klebanoff SL. In: Peroxidases in Chemistry and Biochemistry. Everse J, Everse K, Grisham MB, editors. Vol. 1. CRC Press; Boca Raton, FL: 1991. 1991. pp. 1–35. [Google Scholar]
- 2.Svensson BE. Abilities of peroxidases to catalyse peroxidase-oxidase oxidation of thiols. Biochem J. 1988;256:757–762. doi: 10.1042/bj2560757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferrari RP, Ghibaudi EM, Traversa S, Laurenti E, De Gioia L, Salmona M. Spectroscopic and binding studies on the interaction of inorganic anions with lactoperoxidase. J Inorg Biochem. 1997;68:17–26. doi: 10.1016/s0162-0134(97)00003-2. [DOI] [PubMed] [Google Scholar]
- 4.Ferrari RP, Laurenti E, Cecchini PI, Gambino O, Sondergaard I. Spectroscopic investigations on the highly purified lactoperoxidase Fe(III)-heme catalytic site. J Inorg Biochem. 1995;58:109–127. doi: 10.1016/0162-0134(94)00041-8. [DOI] [PubMed] [Google Scholar]
- 5.Kohler H, Taurog A, Dunford HB. Spectral studies with lactoperoxidase and thyroid peroxidase: interconversions between native enzyme, compound II, and compound III. Arch Biochem Biophys. 1988;264:438–449. doi: 10.1016/0003-9861(88)90309-8. [DOI] [PubMed] [Google Scholar]
- 6.Metodiewa D, Reszka K, Dunford HB. Evidence for a peroxidatic oxidation of norepinephrine, a catecholamine, by lactoperoxidase. Biochem Biophys Res Commun. 1989;160:1183–1188. doi: 10.1016/s0006-291x(89)80128-7. [DOI] [PubMed] [Google Scholar]
- 7.Metodiewa D, Reszka K, Dunford HB. Oxidation of the substituted catechols dihydroxyphenylalanine methyl ester and trihydroxyphenylalanine by lactoperoxidase and its compounds. Arch Biochem Biophys. 1989;274:601–608. doi: 10.1016/0003-9861(89)90475-x. [DOI] [PubMed] [Google Scholar]
- 8.Knecht KT, Mottley C, Mason RP. Thiyl free radical metabolites of thiol drugs and glutathione. Basic Life Sci. 1988;49:75–79. doi: 10.1007/978-1-4684-5568-7_11. [DOI] [PubMed] [Google Scholar]
- 9.Mottley C, Toy K, Mason RP. Oxidation of thiol drugs and biochemicals by the lactoperoxidase/hydrogen peroxide system. Mol Pharmacol. 1987;31:417–421. [PubMed] [Google Scholar]
- 10.Bandyopadhyay U, Bhattacharyya DK, Chatterjee R, Banerjee RK. Irreversible inactivation of lactoperoxidase by mercaptomethylimidazole through generation of a thiyl radical: its use as a probe to study the active site. Biochem J. 1995;306(Pt 3):751–757. doi: 10.1042/bj3060751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo Q, Detweiler CD, Mason RP. Protein radical formation during lactoperoxidase-mediated oxidation of the suicide substrate glutathione: immunochemical detection of a lactoperoxidase radical-derived 5,5-dimethyl-1-pyrroline N-oxide nitrone adduct. J Biol Chem. 2004;279:13272–13283. doi: 10.1074/jbc.M310034200. [DOI] [PubMed] [Google Scholar]
- 12.Svensson BE, Graslund A, Strom G, Moldeus P. Thiols as peroxidase substrates. Free Radic Biol Med. 1993;14:167–175. doi: 10.1016/0891-5849(93)90007-h. [DOI] [PubMed] [Google Scholar]
- 13.McGirr LG, Jatoe SD, O'Brien PJ. Myeloperoxidase catalysed cooxidative metabolism of methimazole: oxidation of glutathione and NADH by free radical intermediates. Chem Biol Interact. 1990;73:279–295. doi: 10.1016/0009-2797(90)90009-c. [DOI] [PubMed] [Google Scholar]
- 14.Svensson BE, Lindvall S. Myeloperoxidase-oxidase oxidation of cysteamine. Biochem J. 1988;249:521–530. doi: 10.1042/bj2490521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harman LS, Mottley C, Mason RP. Free radical metabolites of L-cysteine oxidation. J Biol Chem. 1984;259:5606–5611. [PubMed] [Google Scholar]
- 16.Lardinois OM, de Montellano PR. H2O2-mediated cross-linking between lactoperoxidase and myoglobin: elucidation of protein-protein radical transfer reactions. J Biol Chem. 2001;276:23186–23191. doi: 10.1074/jbc.M102084200. [DOI] [PubMed] [Google Scholar]
- 17.Stoyanovsky DA, Goldman R, Jonnalagadda SS, Day BW, Claycamp HG, Kagan VE. Detection and characterization of the electron paramagnetic resonance-silent glutathionyl-5,5-dimethyl-1-pyrroline N-oxide adduct derived from redox cycling of phenoxyl radicals in model systems and HL-60 cells. Arch Biochem Biophys. 1996;330:3–11. doi: 10.1006/abbi.1996.0219. [DOI] [PubMed] [Google Scholar]
- 18.Kono Y, Fridovich I. Superoxide radical inhibits catalase. J Biol Chem. 1982;257:5751–5754. [PubMed] [Google Scholar]
- 19.Winterbourn CC, Garcia RC, Segal AW. Production of the superoxide adduct of myeloperoxidase (compound III) by stimulated human neutrophils and its reactivity with hydrogen peroxide and chloride. Biochem J. 1985;228:583–592. doi: 10.1042/bj2280583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Metodiewa D, Dunford HB. The reactions of horseradish peroxidase, lactoperoxidase, and myeloperoxidase with enzymatically generated superoxide. Arch Biochem Biophys. 1989;272:245–253. doi: 10.1016/0003-9861(89)90216-6. [DOI] [PubMed] [Google Scholar]
- 21.Harman LS, Carver DK, Schreiber J, Mason RP. One- and two-electron oxidation of reduced glutathione by peroxidases. J Biol Chem. 1986;261:1642–164. [PubMed] [Google Scholar]
- 22.Lloyd RV, Hanna PM, Mason RP. The origin of the hydroxyl radical oxygen in the Fenton reaction. Free Radic Biol Med. 1997;22:885–888. doi: 10.1016/s0891-5849(96)00432-7. [DOI] [PubMed] [Google Scholar]
- 23.Winterbourn CC, Peskin AV, Parsons-Mair HN. Thiol oxidase activity of copper, zinc superoxide dismutase. J Biol Chem. 2002;277:1906–1911. doi: 10.1074/jbc.M107256200. [DOI] [PubMed] [Google Scholar]
- 24.Furtmuller PG, Jantschko W, Regelsberger G, Jakopitsch C, Arnhold J, Obinger C. Reaction of lactoperoxidase compound I with halides and thiocyanate. Biochemistry. 2002;41:11895–11900. doi: 10.1021/bi026326x. [DOI] [PubMed] [Google Scholar]
- 25.Ohtaki S, Nakagawa H, Nakamura S, Nakamura M, Yamazaki I. Characterization of hog thyroid peroxidase. J Biol Chem. 1985;260:441–448. [PubMed] [Google Scholar]
- 26.Clark RA, Klebanoff SJ, Einstein AB, Fefer A. Peroxidase-H2O2-halide system: Cytotoxic effect on mammalian tumor cells. Blood. 1975;45:161–170. [PubMed] [Google Scholar]
- 27.Guo Q, Gao G, Qian SY, Mason RP. Novel identification of a sulfur-centered, radical-derived 5,5-dimethyl-1-pyrroline N-oxide nitrone adduct formed from the oxidation of DTT by LC/ELISA, LC/electrospray ionization-MS, and LC/tandem MS. Chem Res Toxicol. 2004;17:1481–1490. doi: 10.1021/tx049837o. [DOI] [PubMed] [Google Scholar]